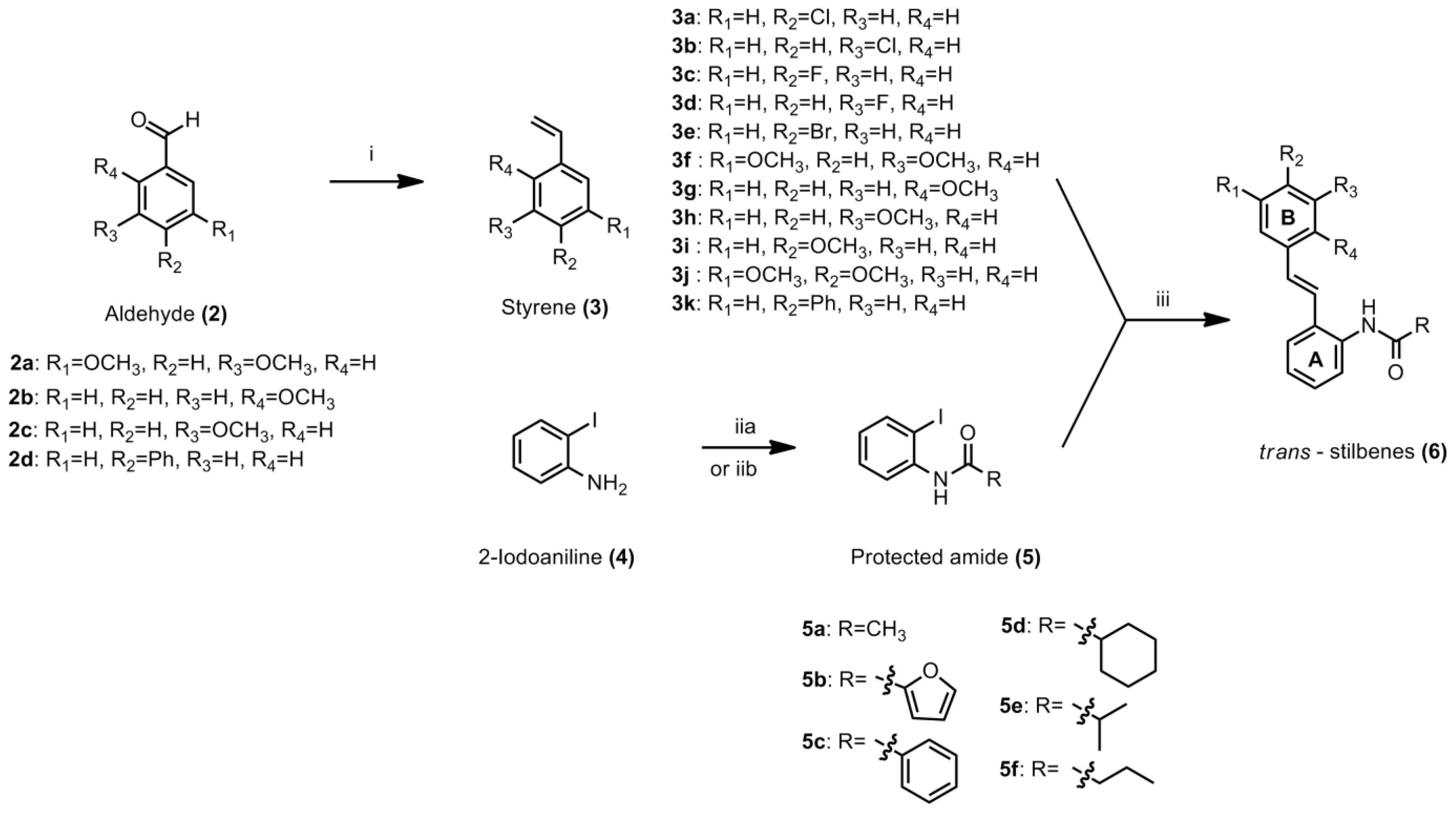
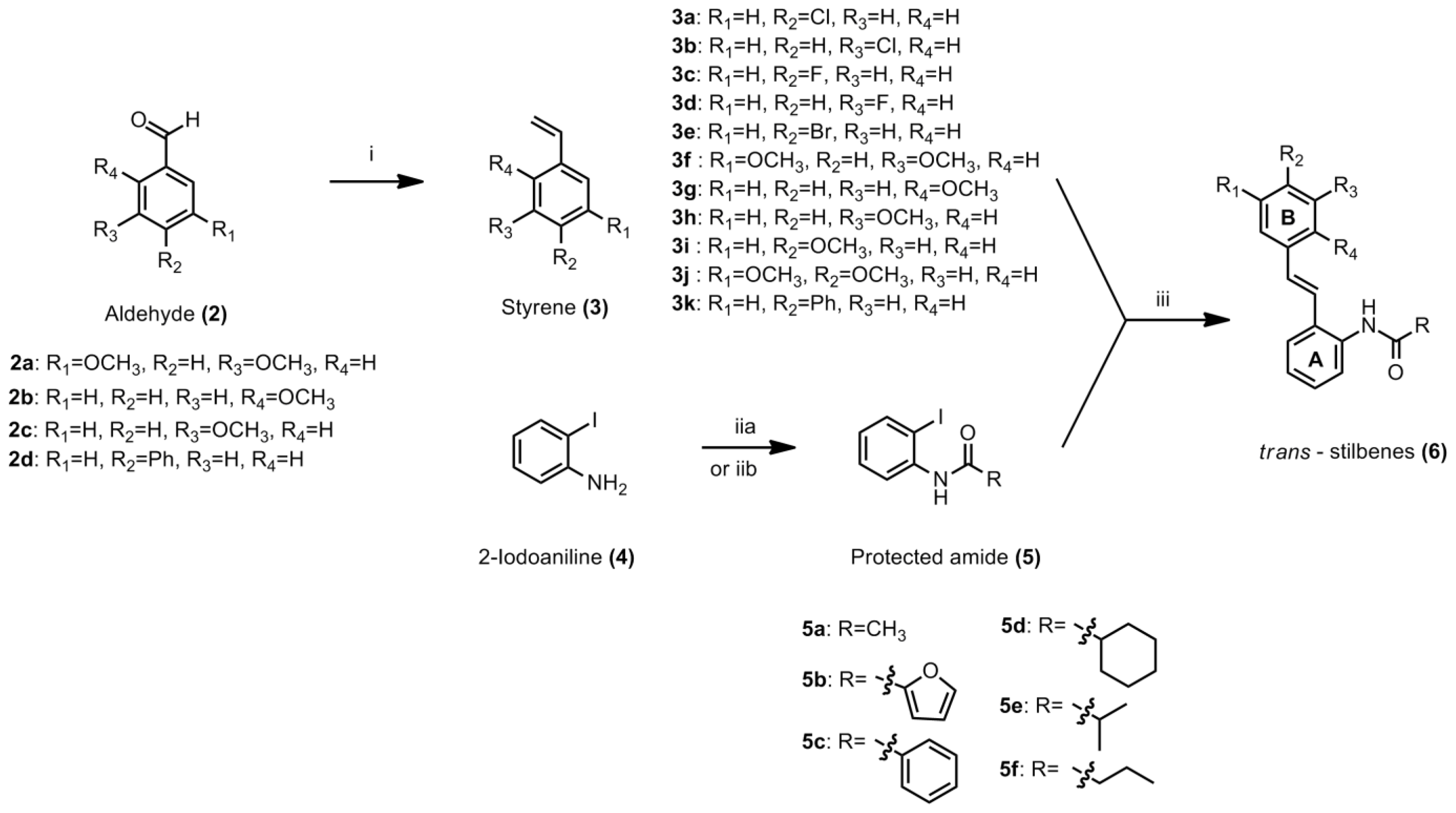
3.4.1. General Procedure for Styrene
To a suspension of methyltriphenylphosphonium iodide (1 equiv) in dry THF (25 mL), potassium tert-butoxide (1 equiv) was added in one portion. The mixture was stirred for 1 h at −70 °C to −80 °C. The appropriate aldehyde (1 equiv) was added to the solution. The ice bath was removed and the mixture was allowed to warm to room temperature. After consumption of starting material to form the product, the reaction was quenched with saturated ammonium chloride solution. The mixture was extracted with ethyl acetate and washed with plenty of distilled water. The resulting organic fractions were combined and solvent was removed under reduced pressure to yield crude product. Purification by column chromatography afforded the desired product. The procedure for compounds
3f,
3g,
3h and
3k is attached in
Appendix.
1,3-methoxy-5-vinylbenzene, 3f. 1H NMR (CDCl3, 400 MHz) δ: 6.66 (dd, J = 17.5, 10.7 Hz, 1H), 6.59 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 5.75 (dd, J = 17.6, 1.0 Hz, 1H), 5.26 (dd, J = 10.9, 1.0 Hz, 1H), 3.80 (s, 6H, 2 × OCH3); 13C NMR (CDCl3, 100 MHz): 160.8, 139.5, 136.8, 114.2, 104.2, 99.9, 55.1 (2 × OCH3).
1-methoxy-2-vinylbenzene, 3g. 1H NMR (CDCl3, 400 MHz) δ: 7.46 (dd, J = 7.8, 1.8 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 7.04 (dd, J = 17.8, 11.4 Hz, 1H), 6.93 (t, J = 7.3 Hz, 1H), 6.87 (d, J = 8.2 Hz, 1H), 5.73 (dd, J = 17.8, 1.4 Hz, 1H), 5.25 (dd, J = 9.1, 1.4 Hz, 1H), 3.84 (s, 3H, OCH3); 13C NMR (CDCl3, 100 MHz): 156.9, 131.9, 129.0, 126.9, 126.7, 120.8, 114.6, 111.0, 55.6.
1-methoxy-3-vinylbenzene, 3h. 1H NMR (CDCl3, 400 MHz) δ: 7.26 (t, J = 8.0 Hz, 1H), 7.03 (t, J = 7.8 Hz, 1H), 6.98 (s, 1H), 6.83 (dd, J = 8.2 Hz, 2.8Hz, 1H), 6.71 (dd, J = 17.8 Hz, 11.0 Hz, 1H), 5.77 (d, J = 17.8 Hz, 1H), 5.27 (d, J = 11.0 Hz, 1H), 3.83 (s, OCH3, 3H); 13C NMR (CDCl3, 100 MHz): 159.9, 139.1, 136.9, 129.6, 119.1, 114.2, 113.5, 111.6, 55.3 (OCH3).
4-vinyl-1,1′-biphenyl, 3k. 1H NMR (CDCl3, 400 MHz) δ: 7.60 (d, J = 6.8 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.49 (d, J = 8.7 Hz, 2H), 7.44 (t, J = 7.5 Hz, 2H), 7.34 (t, J = 7.3 Hz, 1H), 6.76 (dd, J = 17.8, 11.0 Hz, 1H), 5.80 (d, J = 17.8 Hz, 1H), 5.28 (d, J = 11.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz): 140.9, 140.7, 136.7, 136.5, 128.9, 127.5, 127.4, 127.1, 126.8, 114.0.
3.4.2. General Procedure for the Preparation of N-(2-iodophenyl)acylamide
As described in [
18] a solution of an appropriate acyl chloride (1 equiv) in 5 mL of dry THF was added dropwise to a stirred, cooled (0–5 °C) solution of 2-iodoaniline (1 equiv) and Et
3N (1 equiv) in 20 mL of dry THF. The ice bath was then removed and the mixture stirred vigorously overnight at room temperature. Solid Et
3N·HCl was then filtered off and washed with THF (3 × 5 mL). The resulting organic fractions were combined and THF was removed under reduced pressure to yield crude amides. Recrystallization from hexanes/chloroform and drying in vacuum produced the desired product. The procedure for compounds
5a–
5f is attached in
Appendix.
N-(2-iodophenyl)acetamide, 5a. Mp 103–105 °C. 1H NMR (CDCl3, 400 MHz) δ: 8.17 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.40 (br s, 1H, NH), 7.32 (t, J = 7.3 Hz, 1H), 6.82 (t, J = 7.4 Hz, 1H), 2.22 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz): 168.1, 138.8, 138.2, 129.2, 126.0, 122.1, 90.0, 24.8 (CH3).
N-(2-iodophenyl)furan-2-carboxamide, 5b. Mp 80–81 °C; IR (NaCl): 3364, 1683, 1582, 1526, 1430, 1304, 1162, 1010, 750 cm−1; UV (MeOH)max nm: 257, 231; 1H NMR (CDCl3, 400 MHz) δ: 8.52 (br s, 1H, NH,), 8.39 (dd, J = 8.7, 1.4 Hz, 1H), 7.80 (dd, J = 7.8, 1.4 Hz, 1H), 7.56 (d, J = 1.4 Hz, 1H), 7.36 (td, J = 8.7, 1.4 Hz, 1H), 7.26 (d, J = 3.7 Hz, 1H), 6.85 (td, J = 7.8, 1.4 Hz, 1H), 6.57 (dd, J = 3.6, 1.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz): 156.1, 147.7, 144.8, 139.0, 138.0, 129.4, 126.1, 121.7, 115.8, 112.8, 89.9; HRMS (+ESI) [M + H]+: 313.9671, C11H9INO2 requires 313.9672.
N-(2-iodophenyl)benzamide, 5c. Mp 135–137 °C; 1H NMR (CDCl3, 400 MHz) δ: 8.46 (d, J = 8.3 Hz, 1H), 8.30 (br s, 1H, NH), 7.96–7.98 (m, 2H), 7.82 (dd, J = 7.9, 1.5 Hz, 1H), 7.51–7.61 (m, 3H), 7.41 (td, J = 7.8, 1.5 Hz, 1H), 6.89 (td, J = 7.7, 1.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz): 165.4, 138.9, 138.4, 134.6, 132.3, 129.5, 129.0, 127.3, 126.2, 121.9, 90.3.
N-(2-iodophenyl)cyclohexanecarboxamide, 5d. Mp 134–146 °C; 1H NMR (CDCl3, 400 MHz) δ: 8.24 (d, J = 8.2 Hz, 1H), 7.75 (dd, J = 8.2, 1.4 Hz, 1H), 7.51 (br s, 1H, NH), 7.32 (td, J = 7.4, 1.4 Hz, 1H), 6.82 (td, J = 7.8, 1.8 Hz, 1H), 2.30 (tt, J = 11.7, 3.4 Hz, 1H), 1.23–2.06 (m, 10H); 13C NMR (CDCl3, 100 MHz): 174.4, 138.8, 138.3, 129.3, 125.9, 122.1, 90.2, 46.7, 29.8 (CH2), 25.8 (CH2).
N-(2-iodophenyl)isobutyramide, 5e. Mp 110–111 °C; 1H NMR (CDCl3, 400 MHz) δ: 8.23 (d, J = 8.0 Hz, 1H,), 7.75 (d, J = 8.1 Hz, 1H), 7.51 (br s, 1H, NH), 7.32 (t, J = 7.6 Hz, 1H), 6.82 (t, J = 7.3 Hz, 1H), 2.59 (septet, J = 7.0 Hz, 1H), 1.30 (d, J = 6.8 Hz, 6H, 2 × CH3); 13C NMR (CDCl3, 100 MHz): 175.3, 138.8, 138.2, 129.4, 125.9, 122.0, 90.2, 37.1, 19.7 (CH3).
N-(2-iodophenyl)butyramide, 5f. Mp 81–83 °C; 1H NMR (CDCl3, 400 MHz) δ: 8.22 (d, J = 7.8 Hz, 1H), 7.76 (dd, J = 8.0, 1.4 Hz, 1H), 7.43 (br s, 1H, NH,), 7.33 (td, J = 8.0, 1.4 Hz, 1H), 6.82 (t, J = 7.8 Hz, 1H), 2.40 (d, J = 7.6 Hz, 2H), 1.79 (sextet, J = 7.8 Hz, 2H), 1.03 (t, J = 7.3 Hz, 3H, CH3); 13C NMR (CDCl3, 100 MHz): 171.3, 138.8, 138.3, 129.4, 126.0, 122.1, 90.0, 40.0, 19.2, 13.7 (CH3).
3.4.3. General Procedure for Stilbene
In a dry two neck flask, the appropriate
N-(2-iodophenyl)acylamide (1 equiv) was dissolved in dry DMF and stirred under nitrogen. The solution was heated up to 120 °C and refluxed for a few minutes. Palladium (II) acetate (0.01 equiv) was added, followed by triethylamine (3.5 equiv) into the mixture. Lastly, the appropriate styrene (1.2 equiv) was added into the reaction flask. The mixture was heated at 120 °C under nitrogen until consumption of
N-(2-iodophenyl)acylamide (check via TLC). The reaction mixture was quenched with saturated ammonium chloride aqueous solution. It was then extracted with ethyl acetate and washed with plenty of distilled water. The resulting organic fractions were combined, dried over anhydrous sodium sulphate and solvent was removed under reduced pressure to yield crude product. Purification by column chromatography afforded the desired products. The procedure for compounds
6a–
6s is attached in
Appendix.
(
E)
-N-(2-(4-chlorostyryl)phenyl)acetamide,
6a[
22].
Mp 201–202 °C; IR (neat): 3281, 1642, 1531, 1299, 951, 803, 748 cm
−1; 1H NMR (CDCl
3, 400 MHz) δ: 7.42 (d,
J = 8.3 Hz, 2H), 7.33 (d,
J = 8.5 Hz, 2H), 6.95 (d,
J = 16.0 Hz, 1H), 7.11 (d,
J = 16.0 Hz, 1H), 7.75 (d,
J = 8.2 Hz, 1H), 7.18 (t,
J = 7.3 Hz, 1H), 7.52 (d,
J = 7.3 Hz, 1H), 2.21 (s, 3H, CH
3);
13C NMR (CDCl
3, 100 MHz) 135.5, 127.8, 128.9, 133.7, 128.9, 127.8, 130.9, 124.1, 130.4, 134.6, 124.7, 128.5, 125.8, 126.7, 24.2, 168.6 (C=O); HRMS (+ESI) [M + H]
+: 272.0873, C
16H
15ClNO requires 272.0842.
(E)-N-(2-(3-chlorostyryl)phenyl)acetamide, 6b. IR (neat): 3281, 1655, 1530, 1298, 951, 773, 749 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 7.47 (br s, 1H), 7.35–7.24 (m, 4H), 6.92 (d, J = 16.0 Hz, 1H), 7.10 (d, J = 16.0 Hz, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 2.21 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz) 139.0, 126.9, 134.8, 125.0, 128.8, 130.1, 130.8, 125.1, 130.3, 134.8, 124.8, 128.1, 125.9, 126.5, 24.4 (CH3), 168.8 (C=O); HRMS (+ESI) [M + H]+: 272.0800, C16H15ClNO requires 272.0842.
(E)-N-(2-(4-fluorostyryl)phenyl)acetamide, 6c. IR (neat): 3259, 1659, 1527, 1297, 963, 818 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 7.46 (d, J = 6.0 Hz, 2H), 7.01 (d, J = 8.2 Hz, 2H), 6.95 (d, J = 16.5 Hz, 1H), 7.01–7.05 (m, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 6.8 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 2.21 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz) 163.7, 128.1, 115.7, 161.2, 115.5, 128.1, 131.0, 123.2, 130.4, 134.4, 124.5, 128.2, 125.6, 126.6, 24.1 (CH3), 168.6 (C=O); HRMS (+ESI) [M + H]+: 256.1139, C16H15FNO requires 256.1138.
(E)-N-(2-(3-fluorostyryl)phenyl)acetamide, 6d. IR (neat): 3281, 1642, 1531, 1299, 951, 803, 748 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 7.14 (br s, 1H), 6.94 (td, J = 1.4, 8.2 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 7.16–7.05 (m, 3H), 6.81 (d, J = 16.5 Hz, 1H), 7.04–7.10 (m, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 2.10 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz) 139.6, 112.9, 162.0, 113.1, 130.2, 122.8, 130.3, 125.3, 130.4, 134.6, 124.7, 128.5, 125.8, 126.7, 24.2 (CH3), 168.6; HRMS (+ESI) [M + H]+: 256.1810, C16H15FNO requires 256.1138.
(E)-N-(2-(4-bromostyryl)phenyl)acetamide, 6e. IR (neat): 3315, 1664, 1557, 1259, 1029, 798, 772 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 7.48 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 6.91 (d, J = 16.5 Hz, 1H), 7.12 (d, J = 16.5 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.17 (t, J = 7.8 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 2.19 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz) 136.1, 132.0, 128.2, 121.9, 128.2, 132.0, 130.9, 124.3, 130.5, 134.7, 124.9, 128.6, 126.0, 126.8, 24.3 (CH3), 168.9; HRMS (+ESI) [M + H]+: 316.0334, C16H15BrNO requires 316.0377.
(E)-N-(2-(3,5-dimethoxystyryl)phenyl)acetamide, 6f. IR (NaCl): 3262, 1654, 1153, 773 cm−1. UV (MeOH)max nm: 305, 215; 1H NMR (CDCl3, 400 MHz) δ: 7.77 (d, J = 8.0 Hz, 1H), 7.49 (d J = 8.0 Hz, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.20 (br s, 1H, NH), 7.15 (t, J = 8.0 Hz, 1H), 7.08 (d, J = 16.1 Hz, 1H), 6.88 (d, J = 16.1 Hz, 1H), 6.63 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 3.82 (s, 6H, OCH3), 2.19 (s, 3H, CH3). 13C NMR (CDCl3, 100 MHz): 168.8, 161.1, 139.1, 134.7, 132.4, 130.3, 128.4, 126.9, 125.7, 124.4, 124.2, 105.0, 100.1, 55.4, 24.3; HRMS (+ESI) [M + H]+: 298.1430, C18H20NO3 requires 298.1443.
(
E)-
N-(2-(2-methoxystyryl)phenyl)furan-2-carboxamide,
6g[
15].
Mp 128–129 °C; IR (NaCl): 3285, 1671, 1585, 1304, 1247, 751 cm
−1; UV (MeOH)
max nm: 262, 321;
1H NMR (CDCl
3, 400 MHz) δ: 8.25 (br s, 1H, NH), 8.04 (d,
J = 8.2 Hz, 1H), 7.60 (dd,
J = 7.8, 1.0 Hz, 1H), 7.56 (dd,
J = 7.6, 1.4 Hz, 1H), 7.49 (d,
J = 1.0 Hz, 1H), 7.40 (d,
J = 16.5 Hz, 1H), 7.29 (d,
J = 16.8 Hz, 1H), 7.28–7.33 (m, 3H), 7.20 (t,
J = 7.1 Hz, 1H), 6.98 (t,
J = 7.6 Hz, 1H), 6.91 (d,
J = 8.2 Hz, 1H), 6.55 (dd,
J = 3.7, 1.8 Hz, 1H), 3.85 (s, 3H OCH
3);
13C NMR (CDCl
3, 100 MHz): 157.3, 156.4, 148.0, 144.5, 134.1, 130.7, 129.3, 128.3, 128.2, 127.3, 127.2, 126.2, 125.6, 123.7, 123.5, 120.9, 115.4, 112.7, 111.1, 55.5 (OCH
3); HRMS (+ESI) [M + H]
+: 320.1285, C
20H
18NO
3 requires 320.1287.
(
E)-
N-(2-(3-methoxystyryl)phenyl)furan-2-carboxamide,
6h[
15].
Mp 96–97 °C; IR (NaCl): 3285, 1669, 1585, 1521, 1304, 1163, 755 cm
−1; UV (MeOH)
max nm: 210, 264, 300;
1H NMR (CDCl
3, 400 MHz) δ: 8.17 (br s, 1H, NH), 8.02 (d,
J = 8.2 Hz, 1H), 7.55 (d,
J = 7.8 Hz, 1H), 7.50 (d,
J = 1.0 Hz, 1H), 7.33 (t,
J = 7.2 Hz, 1H), 7.29 (t,
J = 8.0 Hz, 1H), 7.26 (d,
J = 3.2 Hz, 1H), 7.22 (d,
J = 16.0 Hz, 1H), 7.21 (t,
J = 7.6 Hz, 1H), 7.12 (d,
J = 7.8 Hz, 1H), 7.04 (s, 1H), 7.02 (d,
J = 16.5 Hz, 1H), 6.85 (dd,
J = 8.5, 2.3 Hz, 1H), 6.56 (dd,
J = 3.7, 1.8 Hz, 1H), 3.83 (s, 3H, OCH
3);
13C NMR (CDCl
3, 100 MHz): 160.0, 156.4, 147.9, 144.5, 138.5, 134.2, 132.9, 130.1, 129.9, 128.6, 127.3, 125.6, 123.6, 119.4, 115.6, 113.7, 112.7, 112.3, 55.4 (OCH
3); HRMS (+ESI) [M + H]
+: 320.1282, C
20H
18NO
3 requires 320.1287.
(
E)-
N-(2-(4-methoxystyryl)phenyl)furan-2-carboxamide,
6i[
15].
Mp 120–121 °C; IR (NaCl): 3276, 1669, 1584, 1509, 1251, 755 cm
−1; UV (MeOH)
max nm: 271, 323;
1H NMR (CDCl
3, 400 MHz) δ: 8.17 (br s, 1H, NH), 8.04 (d,
J = 8.2 Hz, 1H), 7.54 (d,
J = 7.8 Hz, 1H), 7.50 (d,
J = 1.0 Hz, 1H), 7.47 (t,
J = 7.6 Hz, 2H), 7.31 (td,
J = 7.3, 1.3 Hz, 1H), 7.26 (d,
J = 3.7 Hz, 1H), 7.19 (t,
J = 7.6 Hz, 1H), 7.09 (d,
J = 16.5 Hz, 1H), 7.00 (d,
J = 16.5 Hz, 1H), 6.92 (t,
J = 7.6 Hz, 2H), 6.56 (dd,
J = 3.2, 1.8 Hz, 1H), 3.83 (s, 3H, OCH
3);
13C NMR (CDCl
3, 100 MHz): 159.8, 156.3, 148.0, 144.5, 134.0, 132.7, 130.4, 129.9, 128.2, 128.1, 127.1, 125.6, 123.5, 121.0, 115.5, 114.3, 112.7, 55.5 (OCH
3); HRMS (+ESI) [M + H]
+: 320.1302, C
20H
18NO
3 requires 320.1287.
(
E)-
N-(2-(3,4-dimethoxystyryl)phenyl)furan-2-carboxamide,
6j[
15].
Mp 142–144 °C; IR (NaCl): 3239, 1651, 1575, 1514, 1456, 1265, 1157, 1137, 1027, 751 cm
−1; UV (MeOH)
max nm: 210, 252, 326;
1H NMR (CDCl
3, 400 MHz) δ: 8.19 (br s, 1H, NH), 8.00 (d,
J = 8.2 Hz, 1H), 7.52 (d,
J = 7.8 Hz, 1H), 7.47 (dd,
J = 1.8, 1.0 Hz, 1H), 7.29 (t,
J = 7.8 Hz, 1H), 7.23 (d,
J = 2.8 Hz, 1H), 7.18 (t,
J = 7.8 Hz, 1H), 7.07 (d,
J = 16.5 Hz, 1H), 7.05 (d,
J = 8.9 Hz, 1H), 7.03 (s, 1H), 6.96 (d,
J = 16.5 Hz, 1H), 6.85 (d,
J = 8.2, 2.3 Hz, 1H), 6.54 (dd,
J = 3.4, 1.8 Hz, 1H), 3.89 (s, 3H, OCH
3), 3.88 (s, 3H, OCH
3);
13C NMR (CDCl
3, 100 MHz): 156.4, 149.4, 149.2, 148.0, 144.5, 134.0, 132.7, 130.4, 130.2, 128.2, 127.1, 125.6, 123.7, 121.4, 120.1, 115.4, 112.7, 111.4, 109.3, 56.1 (OCH
3), 56.0 (OCH
3); HRMS (+ESI) [M + H]
+: 320.1411, C
21H
20NO
4 requires 320.1392.
(
E)
-N-(2-(3,5-dimethoxystyryl)phenyl)furan-2-carboxamide,
6k[
15].
Mp 109–110 °C; IR (NaCl): 3282, 1670, 1590, 1520, 1455, 1303, 1204, 1152, 1065, 755 cm
−1; UV (MeOH)
max nm: 239, 269, 308;
1H NMR (CDCl
3, 400 MHz) δ: 8.16 (br s, 1H, NH), 8.04 (d,
J = 7.8 Hz, 1H), 7.54 (dd,
J = 7.8, 1.4 Hz, 1H), 7.50 (d,
J = 1.0 Hz, 1H), 7.33 (td,
J = 7.8, 1.4 Hz, 1H), 7.26 (d,
J = 3.2 Hz, 1H), 7.21 (d,
J = 16.0 Hz, 1H), 7.20 (t,
J = 7.5 Hz, 1H), 6.99 (d,
J = 16.0 Hz, 1H), 6.66 (s, 2H, H-2), 6.56 (dd,
J = 3.6, 1.8 Hz, 1H), 6.43 (t,
J = 1.8 Hz, 1H), 3.81 (s, 6H, 2xOCH
3);
13C NMR (CDCl
3, 100 MHz): 161.1, 156.3, 147.9, 144.5, 139.1, 134.2, 133.0, 129.9, 128.6, 127.3, 125.6, 123.9, 123.6, 115.6, 112.7, 105.0, 100.3, 55.9 (2 × OCH
3); HRMS (+ESI) [M + H]
+: 350.1412, C
21H
20NO
4 requires 350.1392.
(
E)-
N-(2-(2-(biphenyl-4-yl)vinyl)phenyl)furan-2-carboxamide,
6l[
15].
Mp 142–143 °C; IR (NaCl): 3283, 1671, 1585, 1521, 1487, 1452, 1304, 762 cm
−1; UV (MeOH)
max nm: 204, 267, 323;
1H NMR (CDCl
3, 400 MHz) δ: 8.19 (br s, 1H, NH), 8.04 (d,
J = 7.8 Hz, 1H), 7.58–7.64 (m, 7H), 7.52 (t,
J = 1.0 Hz, 1H), 7.44–7.48 (m, 2H), 7.32–7.38 (m, 2H), 7.21–7.30 (m, 3H), 7.10 (d,
J = 16.5 Hz, 1H);
13C NMR (CDCl
3, 100 MHz): 156.4, 148.0, 144.5, 141.0, 140.6, 136.1, 134.2, 132.5, 130.2, 129.0, 128.6, 127.6, 127.5, 127.3, 127.0, 125.7, 123.7, 123.3, 115.6, 112.7; HRMS (+ESI) [M + Na]
+: 388.1324, C
27H
22NO requires 388.1308. An X-ray of this compound was published in 2008 [
23].
(
E)
-N-(2-(2-([1,1′-biphenyl]-4-yl)vinyl)phenyl)benzamide,
6m[
15].
Mp 167–169 °C; IR (NaCl): 3271, 1648, 1517, 1487, 1302, 764 cm
−1; UV (MeOH)
max nm: 319;
1H NMR (CDCl
3, 400 MHz) δ: 8.05 (br s, 1H, NH), 7.91 (d,
J = 7.3 Hz, 3H), 7.43–7.62 (m, 12H), 7.31–7.36 (m, 2H), 7.22–7.26 (m, 2H), 7.06 (d,
J = 16.5 Hz, 1H);
13C NMR (CDCl
3, 100 MHz): 166.0, 140.9, 140.6, 136.1, 134.8, 134.7, 132.2, 132.1, 131.0, 129.0, 128.5, 127.5, 127.3, 127.2, 127.1, 127.0, 126.0, 124.7, 123.5; HRMS (+ESI) [M + H]
+: 376.1691, C
27H
22NO requires 376.1701.
(
E)-
N-(2-(3,4-dimethoxystyryl)phenyl)benzamide,
6n[
15].
Mp 169–171 °C; IR (NaCl): 3273, 3012, 1646, 1572, 1509, 1265, 749 cm
−1; UV (MeOH)
max nm: 231, 324;
1H NMR (CDCl
3, 400 MHz) δ: 8.03 (br s, 1H, NH), 7.87 (d,
J = 7.8 Hz, 3H), 7.51–7.55 (m, 2H), 7.44 (t,
J = 7.3 Hz, 2H), 7.28 (t,
J = 7.8 Hz, 2H), 7.20 (t,
J = 7.5 Hz, 1H), 7.04 (d,
J = 16.0 Hz, 1H), 6.98 (d,
J = 9.2 Hz, 1H), 6.97 (s, 1H), 6.93 (d,
J = 16.0 Hz, 1H), 6.82 (d,
J = 8.2 Hz, 1H), 3.86 (s, 3H, OCH
3), 3.83 (s, 3H, OCH
3);
13C NMR (CDCl
3, 100 MHz): 166.0, 149.4, 149.2, 134.8, 134.7, 132.5, 132.0, 131.1, 130.2, 128.9, 128.2, 127.3, 127.0, 125.9, 124.5, 121.7, 120.1, 111.3, 109.1, 56.0 (OCH
3), 55.9 (OCH
3); HRMS (+ESI) [M + H]
+: 360.1594, C
23H
22NO
3 requires 360.1600.
(E)-N-(2-(4-methoxystyryl)phenyl)benzamide, 6o. IR (neat): 3283, 1644, 1512, 1482, 1248, 1178, 1027, 749 cm−1; 1H NMR (400MHz, CDCl3) δ: 3.82 (s, 3H. OCH3), 6.87 (dt, J = 3.2, 8.7 Hz, 2H), 6.98 (d, J = 16.0 Hz, 1H), 7.06 (d, J = 16.5 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.41 (t, J = 4.3 Hz, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.54 (t, J = 7.3 Hz 2H), 7.89 (d, J = 7.3 Hz, 2H), 7.97 (d, J = 7.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz): 132.0, 129.0, 127.2, 134.8, 134.6, 124.1, 128.2, 125.8, 127.1, 130.9, 121.1, 132.9, 129.8, 128.1, 114.3, 159.8, 165.5; HRMS (+ESI) [M + H]+: 330.1493, C22H20NO2 requires 330.1494.
(
E)-
N-(2-(3,4-dimethoxystyryl)phenyl)cyclohexanecarboxamide,
6p[
15].
Mp 204–206 °C; IR (NaCl): 2930, 2853, 1682, 1515, 1449, 1269, 1137, 1026, 757 cm
−1; UV (MeOH)
max nm: 217, 300;
1H NMR (CDCl
3, 400 MHz) δ: 7.77 (d,
J = 8.2 Hz, 1H), 7.50 (d,
J = 7.3 Hz, 1H), 7.25 (td,
J = 8.0, 1.4 Hz, 1H), 7.21 (br s, 1H, NH), 7.17 (d,
J = 7.8 Hz, 1H), 7.16 (d,
J = 7.6 Hz, 1H), 7.03 (d,
J = 7.3 Hz, 1H), 7.02 (s, 1H), 6.97 (d,
J = 16.5 Hz, 1H), 6.91 (d,
J = 16.5 Hz, 1H), 3.91 (s, 3H, OCH
3), 3.90 (s, 3H, OCH
3), 2.31 (tt,
J = 11.7, 3.4 Hz, 1H), 1.22–2.01 (m, 10H);
13C NMR (CDCl
3, 100 MHz): 174.5, 149.3, 149.2, 134.6, 132.2, 130.8, 130.2, 128.1, 126.8, 125.6, 124.4, 121.7, 120.1, 111.3, 108.9, 56.1 (OCH
3), 55.9 (OCH
3), 46.3, 29.9, 25.8; HRMS (+ESI) [M + H]
+: 366.2069, C
23H
28NO
3 requires 366.2069.
(
E)-
N-(2-(3,4-dimethoxystyryl)phenyl)isobutyramide,
6q[
15].
Mp 166–168 °C; IR (NaCl): 3274, 1652, 1516, 1269, 1141, 1025, 961, 801, 752 cm
−1; UV (MeOH)
max nm: 210, 236, 325;
1H NMR (CDCl
3, 400 MHz) δ: 7.74 (d,
J = 7.8 Hz, 1H), 7.49 (d,
J = 7.3 Hz, 1H), 7.31 (br s, 1H, NH), 7.24 (t,
J = 7.8 Hz, 1H), 7.15 (t,
J = 7.6 Hz, 1H), 7.01 (d,
J = 7.8 Hz, 1H), 7.00 (s, 1H), 6.97 (d,
J = 16.4 Hz, 1H), 6.90 (d,
J = 16.5 Hz, 1H), 6.85 (d,
J = 8.7 Hz, 1H), 3.89 (s, 6H, 2xOCH
3), 2.54–2.61 (m, 1H), 1.27 (d,
J = 6.9 Hz, 6H);
13C NMR (CDCl
3, 100 MHz): 175.6, 149.3, 149.2, 134.6, 132.1, 130.9, 130.2, 128.1, 126.8, 125.7, 124.5, 121.7, 120.1, 111.3, 108.9, 56.1 (OCH
3), 55.9 (OCH
3), 36.5, 19.9 (2 × CH
3); HRMS (+ESI) [M + H]
+: 326.1759, C
20H
24NO
3 requires 326.1756.
(E)-N-(2-(4-methoxystyryl)phenyl)isobutyramide, 6r. IR (NaCl): 3267, 2966, 1510, 1267, 1033, 750 cm−1; 1H NMR (400MHz, CDCl3) δ: 1.27 (d, J = 6.9 Hz, 6H), 2.53–2.59 (m, 1H), 3.82 (s, 3H, OCH3), 7.79 (d, J = 7.8 Hz, 1H), 7.23 (td, J = 1.0, 8.0 Hz, 1H), 7.14 (d, J = 7.5 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 6.97 (d, J = 16.5 Hz, 1H), 6.92 (m, 1H), 7.41 (d, J = 9.2 Hz, 2H), 6.89 (m, 2H), 7.33 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz): 19.8 (CH3), 36.5, 55.4 (OCH3), 134.6, 124.2, 128.0, 125.5, 126.9, 130.7, 121.3, 132.0, 129.9, 128.0, 114.3, 175.5; HRMS (+ESI) [M + H]+: 296.1649, C19H22NO2 requires 296.1651.
(
E)-
N-(2-(3,4-dimethoxystyryl)phenyl)butyramide,
6s[
15].
Mp 139–140 °C; IR (NaCl): 3280, 2963, 1510, 1267, 1025, 759 cm
−1; UV (MeOH)
max nm: 206, 323;
1H NMR (CDCl
3, 400 MHz) δ: 7.63 (d,
J = 7.8 Hz, 1H), 7.56 (br s, 1H, NH), 7.46 (d,
J = 7.8 Hz, 1H), 7.18 (t,
J = 7.0 Hz, 1H), 7.11 (t,
J = 7.3 Hz, 1H), 6.98 (d,
J = 7.8 Hz, 1H), 6.97 (s, 1H), 6.95 (d,
J = 15.6 Hz, 1H), 6.85 (d,
J = 16.2 Hz, 1H), 6.81 (d,
J = 8.2 Hz, 1H), 3.86 (s, 3H, OCH
3), 3.85 (s, 3H, OCH
3), 2.31 (t,
J = 7.3 Hz, 2H), 1.69–1.75 (m, 2H, CH
2), 0.97 (t,
J = 7.3 Hz, 3H);
13C NMR (CDCl
3, 100 MHz): 172.0, 149.2, 149.1, 134.6, 131.6, 131.1, 130.3, 127.9, 126.5, 125.7, 124.9, 121.8, 120.1, 111.3, 109.1, 56.0 (OCH
3), 55.9 (OCH
3), 39.3, 19.4, 13.9 (CH
3); HRMS (+ESI) [M + H]
+: 326.1755, C
20H
24NO
3 requires 326.1756.




{kind=link}
{kind=link}
{kind=link}
{kind=link}