Lessons from Mouse Models of High-Fat Diet-Induced NAFLD

Abstract
:1. Introduction
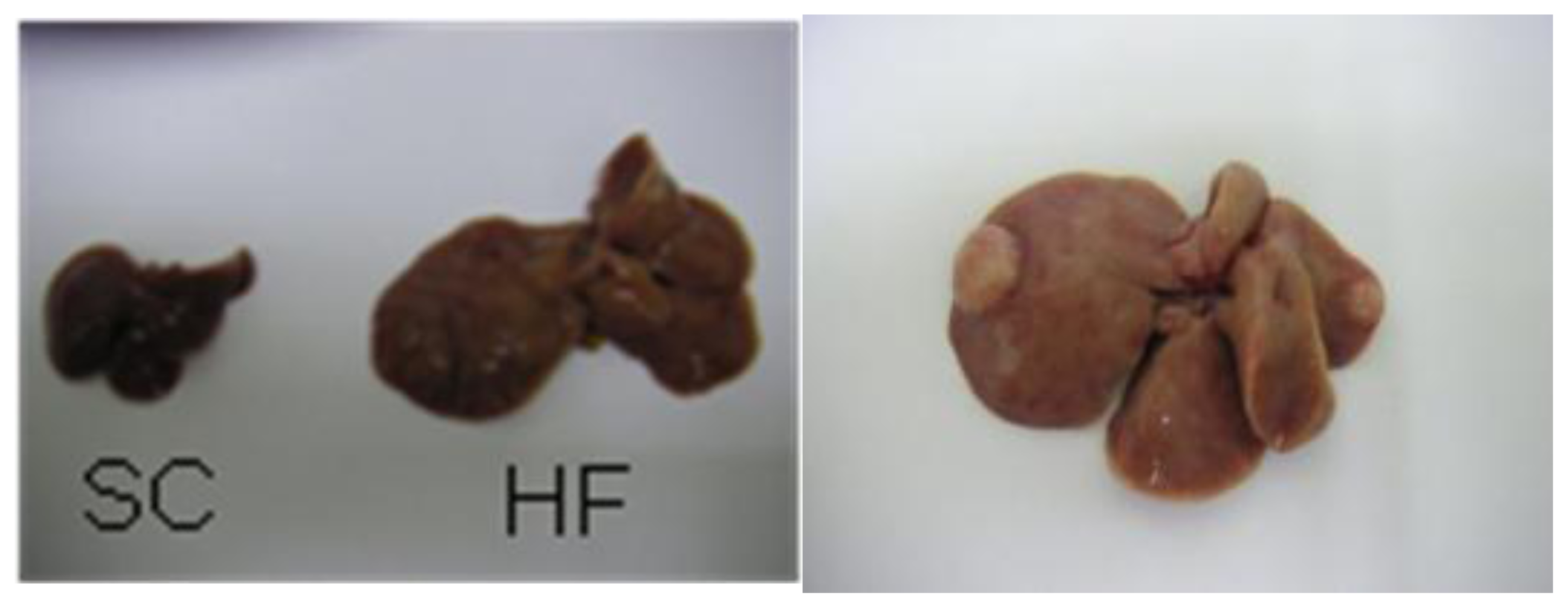
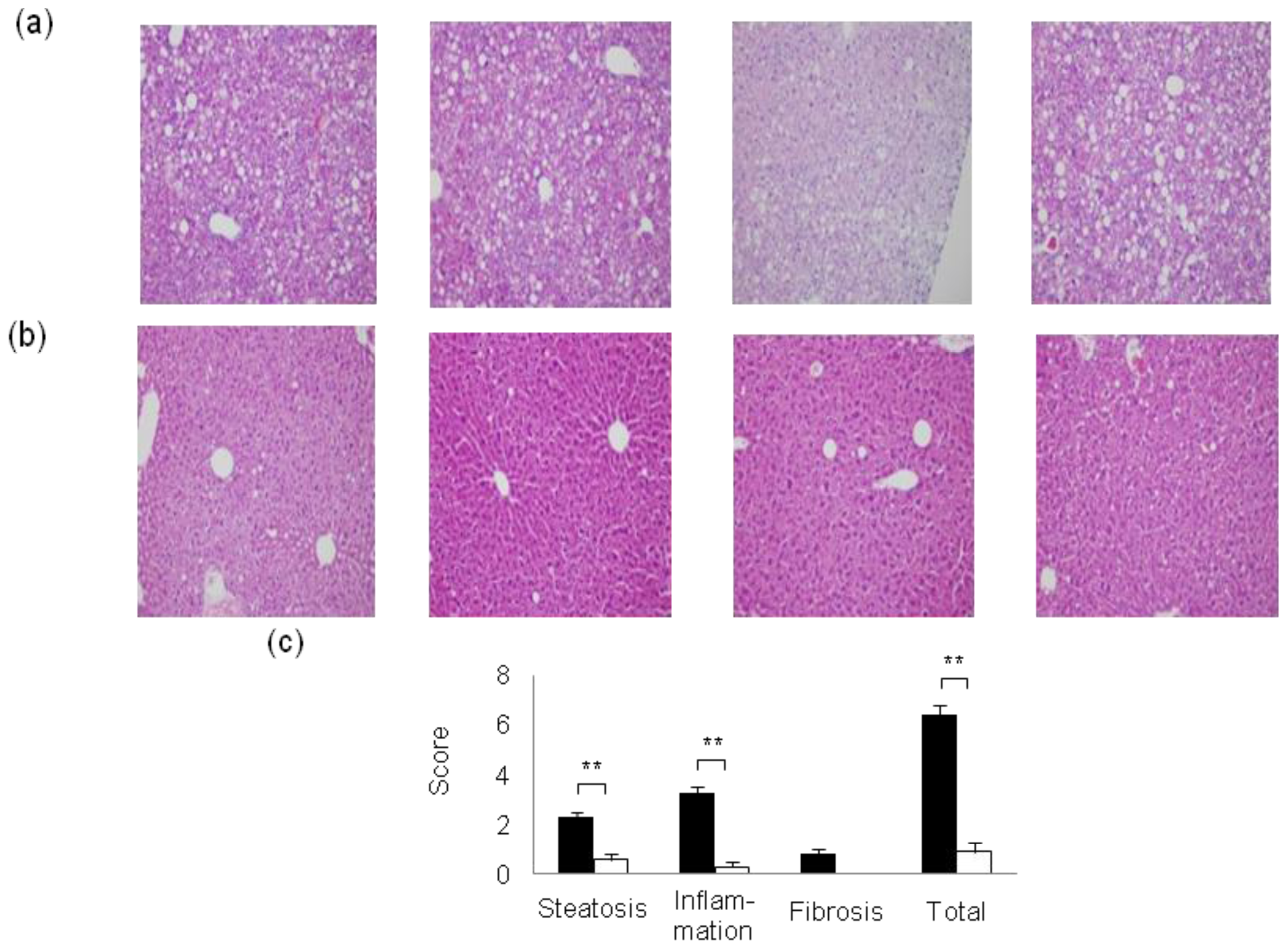
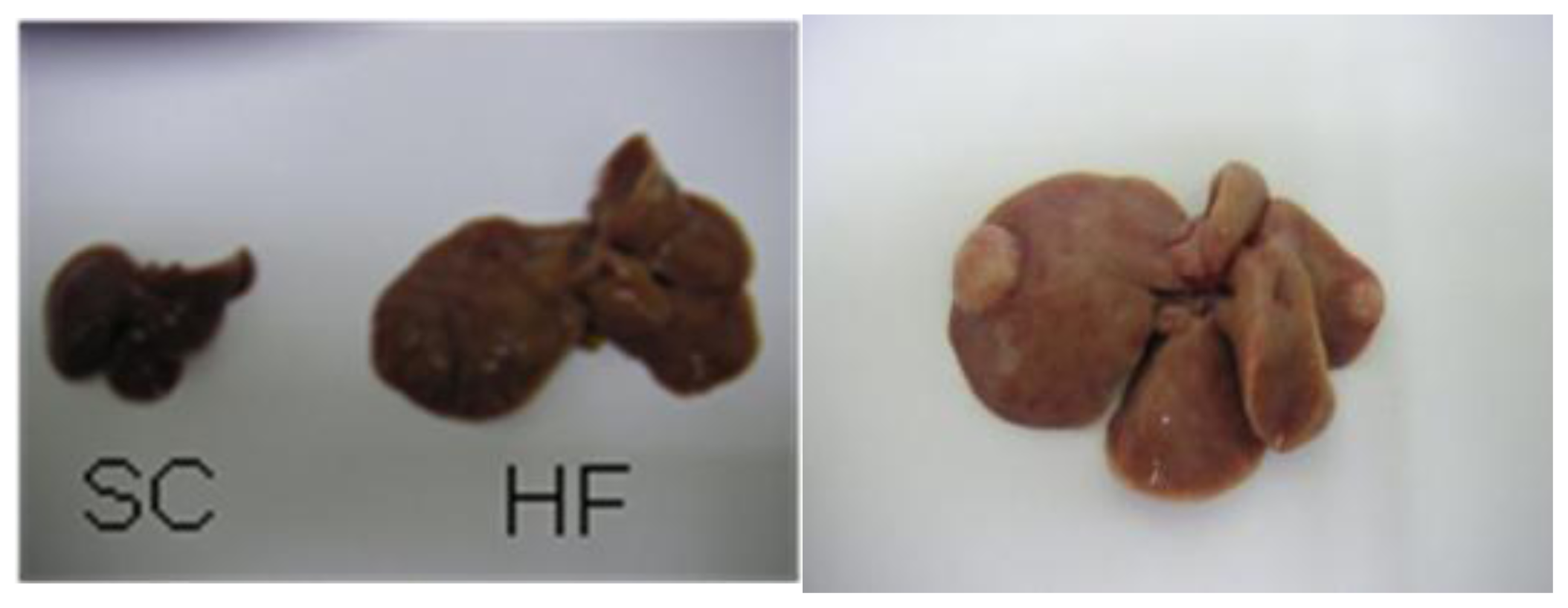
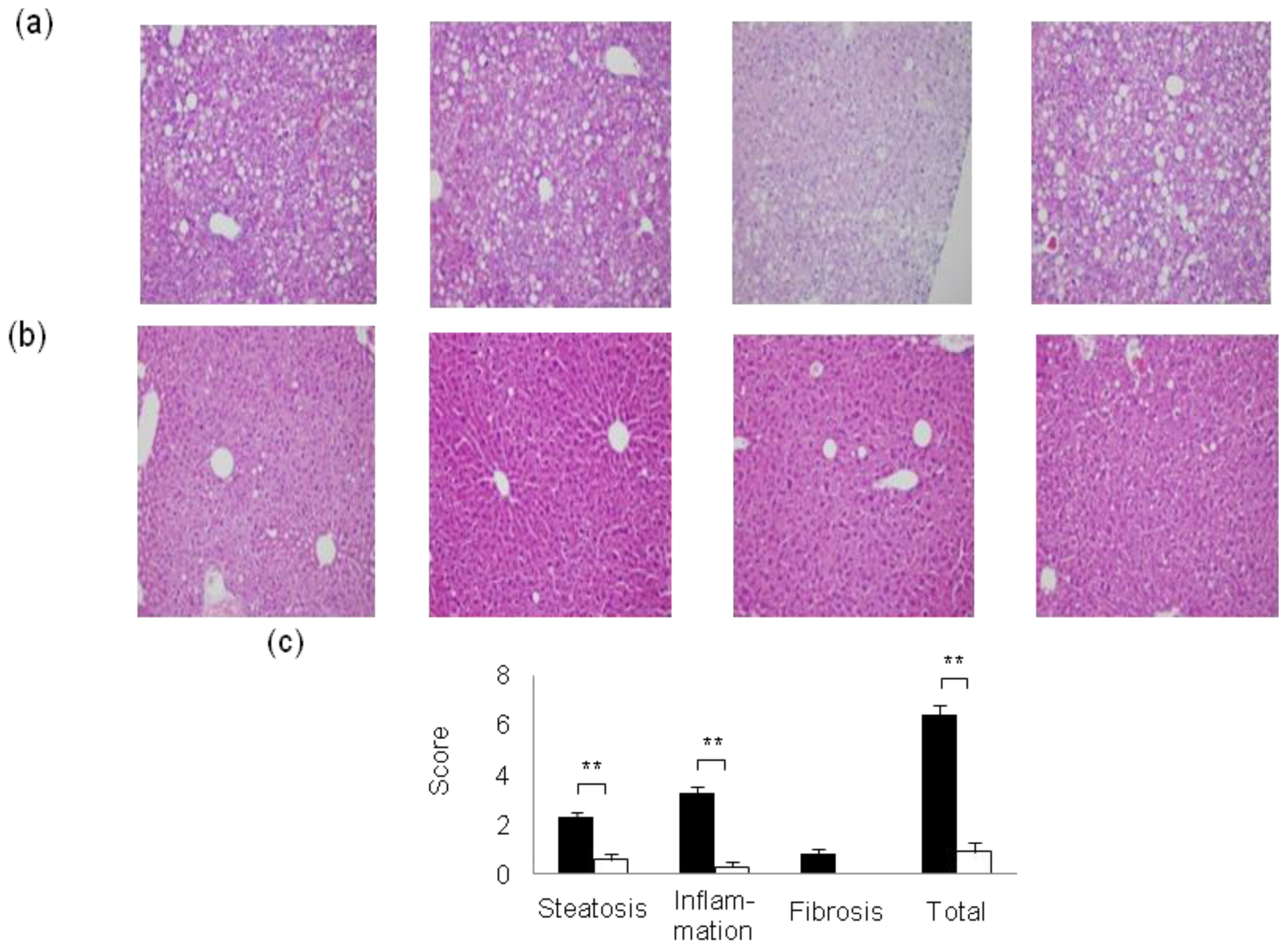
2. Mouse Models of High-Fat Diet-Induced NAFLD/NASH
3. Role of Insulin Signaling in HF Diet-Induced NAFLD/NASH
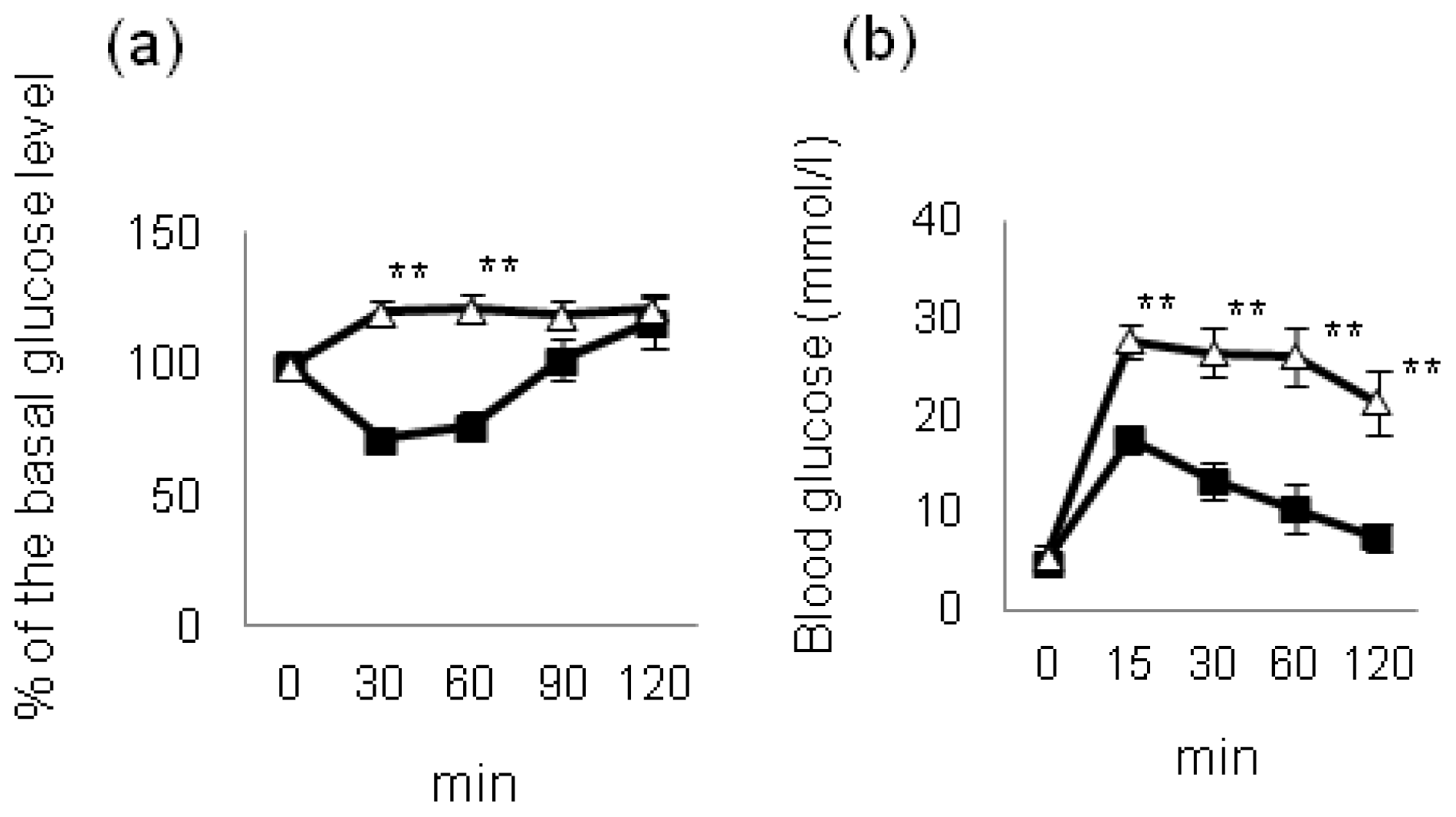
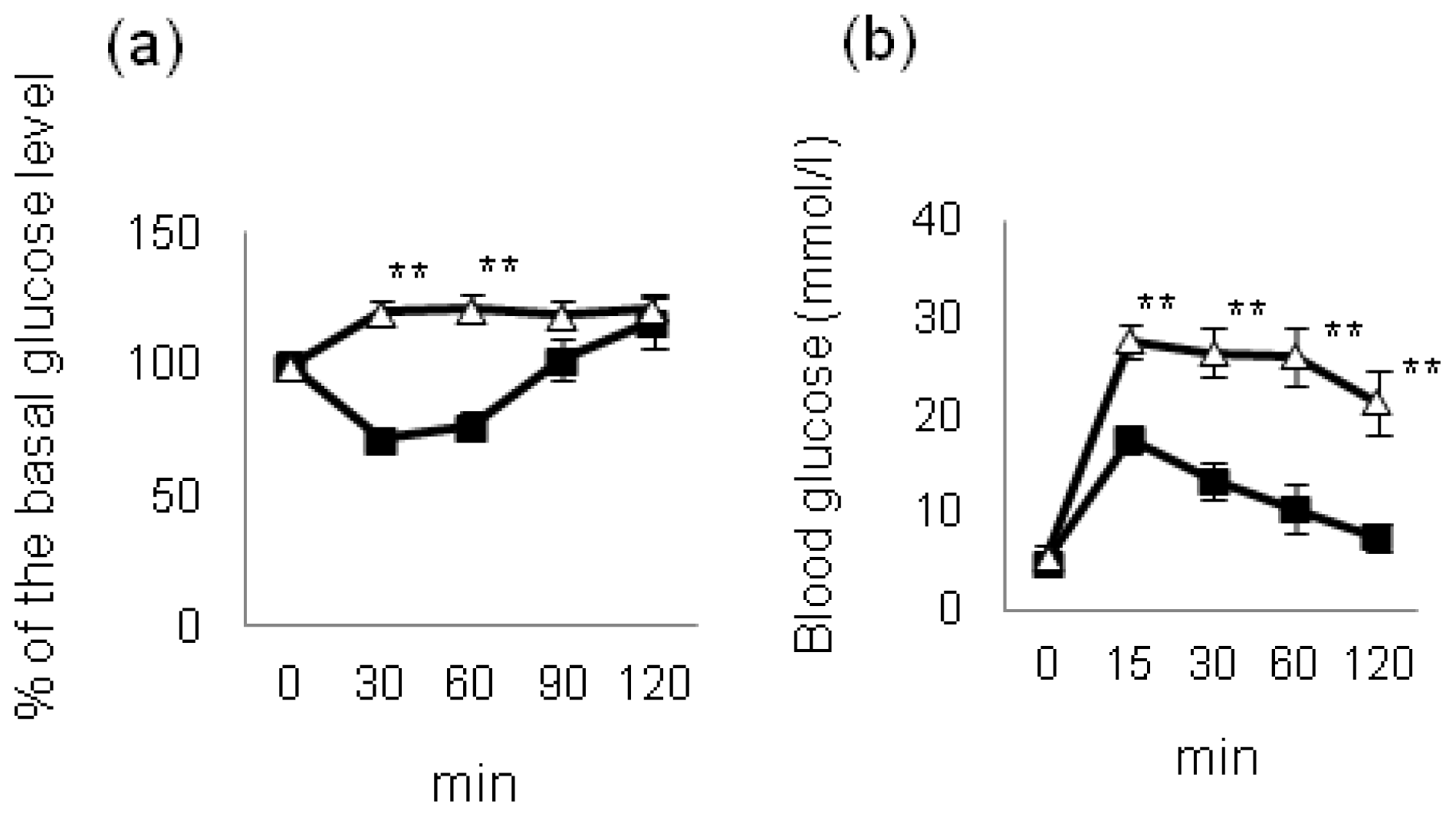
4. Effect of Hyperglycemia on HF Diet-Induced NAFLD/NASH
5. Impact of Current Treatments on HF Diet-Induced NAFLD/NASH
5.1. Lifestyle Modification
5.2. Insulin-Sensitizers: Thiazolidinediones
5.3. Insulin-Sensitizers: Metformin
5.4. Lipid-Lowering Drugs
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
| SC | HF | |
|---|---|---|
| Moisture (%) | 7.9 | 6.2 |
| Crude protein (%) | 23.1 | 25.5 |
| Crude fat (%) | 5.1 | 32.0 |
| Crude fiber (%) | 2.8 | 2.9 |
| Crude ash (%) | 5.8 | 4.0 |
| Nitrogen-free extract (%) | 55.3 | 29.4 |
| Total calories (kcal/100 g) | 359.0 | 507.6 |
Acknowledgments
Conflicts of Interest
References
- Tarantino, G.; Savastano, S.; Colao, A. Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol 2010, 16, 4773–4783. [Google Scholar]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis 2008, 28, 339–350. [Google Scholar]
- Tarantino, G.; Finelli, C. What about non-alcoholic fatty liver disease as a new criterion to define metabolic syndrome? World J. Gastroenterol 2013, 19, 3375–3384. [Google Scholar]
- Bhatia, L.S.; Curzen, N.P.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease: A new and important cardiovascular risk factor? Eur. Heart J 2012, 33, 1190–1200. [Google Scholar]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Farzadfar, F.; Khang, Y.H.; Stevens, G.A.; et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 27 million participants. Lancet 2011, 378, 31–40. [Google Scholar]
- Guariguata, L. By the numbers: New estimates from the IDF Diabetes Atlas Update for 2012. Diabetes Res. Clin. Pract 2012, 98, 524–525. [Google Scholar]
- Smith, B.W.; Adams, L.A. Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nat. Rev. Endocrinol 2010, 7, 456–465. [Google Scholar]
- Targher, G.; Bertolini, L.; Padovani, R.; Poli, F.; Scala, L.; Tessari, R.; Zenari, L.; Falezza, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar]
- Adams, L.A.; Harmsen, S.; St. Sauver, J.L.; Charatcharoenwitthaya, P.; Enders, F.B.; Therneau, T.; Angulo, P. Nonalcoholic fatty liver disease increases risk of death among patients with diabetes: A community-based cohort study. Am. J. Gastroenterol 2010, 105, 1567–1573. [Google Scholar]
- Kawamura, Y.; Arase, Y.; Ikeda, K.; Seko, Y.; Imai, N.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Sezaki, H.; Akuta, N.; et al. Large-scale long-term follow-up study of Japanese patients with non-alcoholic Fatty liver disease for the onset of hepatocellular carcinoma. Am. J. Gastroenterol 2012, 107, 253–261. [Google Scholar]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Masuoka, H.C.; Chalasani, N. Nonalcoholic fatty liver disease: An emerging threat to obese and diabetic individuals. Ann. N.Y. Acad. Sci 2013, 1281, 106–122. [Google Scholar]
- Kanuri, G.; Bergheim, I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci 2013, 14, 11963–11980. [Google Scholar]
- Shimomura, I.; Hammer, R.E.; Richardson, J.A.; Ikemoto, S.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes Dev 1998, 12, 3182–3194. [Google Scholar]
- Nakayama, H.; Otabe, S.; Ueno, T.; Hirota, N.; Yuan, X.; Fukutani, T.; Hashinaga, T.; Wada, N.; Yamada, K. Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism 2007, 56, 470–475. [Google Scholar]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest 2004, 113, 1774–1783. [Google Scholar] [Green Version]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res 2008, 49, 1068–1076. [Google Scholar]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol 2004, 40, 47–51. [Google Scholar]
- Clapper, J.R.; Hendricks, M.D.; Gu, G.; Wittmer, C.; Dolman, C.S.; Herich, J.; Athanacio, J.; Villescaz, C.; Ghosh, S.S.; Heilig, J.S.; et al. Diet-induced mouse model of fatty liver disease and non-alcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol 2013, 305, G483–G495. [Google Scholar]
- Hill-Baskin, A.E.; Markiewski, M.M.; Buchner, D.A.; Wittmer, C.; Dolman, C.S.; Herich, J.; Athanacio, J.; Villescaz, C.; Ghosh, S.S.; Heilig, J.S.; et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum. Mol. Genet 2009, 18, 2975–2988. [Google Scholar]
- Van Saun, M.N.; Lee, I.K.; Washington, M.K.; Matrisian, L.; Gorden, D.L. High fat diet induced hepatic steatosis establishes a permissive microenvironment for colorectal metastases and promotes primary dysplasia in a murine model. Am. J. Pathol 2009, 175, 355–364. [Google Scholar]
- Nakamura, A.; Tajima, K.; Zolzaya, K.; Sato, K.; Inoue, R.; Yoneda, M.; Fujita, K.; Nozaki, Y.; Kubota, K.C.; Haga, H.; et al. Protection from nonalcoholic steatohepatitis and liver tumorigenesis in high fat-fed insulin receptor substrate-1-knockout mice despite insulin resistance. Diabetologia 2012, 55, 3382–3391. [Google Scholar]
- Farese, R.V., Jr.; Zechner, R.; Newgard, C.B.; Walther, T.C. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 2012, 15, 570–573. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Selective vs. total insulin resistance: A pathogenic paradox. Cell Metab 2008, 7, 95–96. [Google Scholar]
- Semple, R.K.; Sleigh, A.; Murgatroyd, P.R.; Adams, C.A.; Bluck, L.; Jackson, S.; Vottero, A.; Kanabar, D.; Charlton-Menys, V.; Durrington, P.; et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J. Clin. Invest 2009, 119, 315–322. [Google Scholar]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Alemán, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab 2008, 7, 125–134. [Google Scholar]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar]
- Kubota, N.; Kubota, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Mineyama, T.; Ogata, H.; Tokuyama, K.; Ohsugi, M.; et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab 2008, 8, 49–64. [Google Scholar]
- Guo, S.; Copps, K.D.; Dong, X.; Park, S.; Cheng, Z.; Pocai, A.; Rossetti, L.; Sajan, M.; Farese, R.V.; White, M.F. The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol. Cell. Biol 2009, 29, 5070–5083. [Google Scholar]
- Tamemoto, H.; Kadowaki, T.; Tobe, K.; Yagi, T.; Sakura, H.; Hayakawa, T.; Terauchi, Y.; Ueki, K.; Kaburagi, Y.; Satoh, S.; et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 1994, 372, 182–186. [Google Scholar]
- Araki, E.; Lipes, M.A.; Patti, M.E.; Brüning, J.C.; Haag, B., 3rd; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994, 372, 186–190. [Google Scholar]
- Terauchi, Y.; Iwamoto, K.; Tamemoto, H.; Komeda, K.; Ishii, C.; Kanazawa, Y.; Asanuma, N.; Aizawa, T.; Akanuma, Y.; Yasuda, K.; et al. Development of non-insulin-dependent diabetes mellitus in the double knockout mice with disruption of insulin receptor substrate-1 and beta cell glucokinase genes. Genetic reconstitution of diabetes as a polygenic disease. J. Clin. Invest 1997, 99, 861–866. [Google Scholar]
- Chattopadhyay, M.; Selinger, E.S.; Ballou, L.M.; Lin, R.Z. Ablation of PI3K p110-α prevents high-fat diet-induced liver steatosis. Diabetes 2011, 60, 1483–1492. [Google Scholar]
- Leavens, K.F.; Easton, R.M.; Shulman, G.I.; Previs, S.F.; Birnbaum, M.J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab 2009, 10, 405–418. [Google Scholar]
- Kadowaki, T.; Ueki, K.; Yamauchi, T.; Kubota, N. SnapShot: Insulin signaling pathways. Cell 2012, 148, 624. [Google Scholar]
- Johnson, J.A.; Carstensen, B.; Witte, D. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar]
- Matschinsky, F.M.; Banting, Lecture. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45, 223–241.
- Matschinsky, F.M. Pancreatic beta-cell glucokinase: Closing the gap between theoretical concepts and experimental realities. Diabetes 1998, 47, 307–315. [Google Scholar]
- Terauchi, Y.; Takamoto, I.; Kubota, N.; Matsui, J.; Suzuki, R.; Komeda, K.; Hara, A.; Toyoda, Y.; Miwa, I.; Aizawa, S.; et al. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J. Clin. Invest 2007, 117, 246–257. [Google Scholar]
- Takamoto, I.; Terauchi, Y.; Kubota, N.; Ohsugi, M.; Ueki, K.; Kadowaki, T. Crucial role of insulin receptor substrate-2 in compensatory beta-cell hyperplasia in response to high fat diet-induced insulin resistance. Diabetes Obes. Metab 2008, 10, 147–156. [Google Scholar]
- Nakamura, A.; Terauchi, Y.; Ohyama, S.; Kubota, J.; Shimazaki, H.; Nambu, T.; Takamoto, I.; Kubota, N.; Eiki, J.; Yoshioka, N.; et al. Impact of small-molecule glucokinase activator on glucose metabolism and beta-cell mass. Endocrinology 2009, 150, 1147–1154. [Google Scholar]
- Nakamura, A.; Yoneda, M.; Fujita, K.; Tajima, K.; Kikuchi, K.; Nakajima, A.; Maeda, S.; Terauchi, Y. Impact of glucose tolerance on the severity of non-alcoholic steatohepatitis. J. Diabetes Invest 2011, 2, 483–489. [Google Scholar]
- Johnson, J.A.; Bowker, S.L. Intensive glycaemic control and cancer risk in type 2 diabetes: A meta-analysis of major trials. Diabetologia 2011, 54, 25–31. [Google Scholar]
- Johnson, J.A.; Pollak, M. Insulin, glucose and the increased risk of cancer in patients with type 2 diabetes. Diabetologia 2010, 53, 2086–2088. [Google Scholar]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar]
- Radonjic, M.; Wielinga, P.Y.; Wopereis, S.; Kelder, T.; Goelela, V.S.; Verschuren, L.; Toet, K.; van Duyvenvoorde, W.; van der Werff; van der Vat, B.; Stroeve, J.H.; et al. Differential effects of drug interventions and dietary lifestyle in developing type 2 diabetes and complications: A systems biology analysis in LDLr−/− mice. PLoS One 2013, 8, e56122. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar]
- Chang, C.H.; Lin, J.W.; Wu, L.C.; Lai, M.S.; Chuang, L.M.; Chan, K.A. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes. Hepatology 2012, 55, 1462–1472. [Google Scholar]
- Kus, V.; Flachs, P.; Kuda, O.; Bardova, K.; Janovska, P.; Svobodova, M.; Jilkova, Z.M.; Rossmeisl, M.; Wang-Sattler, R.; Yu, Z.; et al. Unmasking differential effects of rosiglitazone and pioglitazone in the combination treatment with in mice fed a high-fat diet. PLoS One 2011, 6, e27126. [Google Scholar]
- Kong, H.; Lee, S.; Shin, S.; Kwon, J.; Jo, T.H.; Shin, E.; Shim, K.S.; Park, Y.I.; Lee, C.K.; Kim, K. Down-regulation of adipogenesis and hyperglycemia in diet-induced obesity mouse model by Aloe QDM. Biomol. Ther 2010, 18, 336–342. [Google Scholar]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Invest 2006, 116, 581–589. [Google Scholar]
- Hsiao, P.J.; Hsieh, T.J.; Kuo, K.K.; Hung, W.W.; Tsai, K.B.; Yang, C.H.; Yu, M.L.; Shin, S.J. Pioglitazone retrieves hepatic antioxidant DNA repair in a mice model of high fat diet. BMC Mol. Biol 2008, 9, 82. [Google Scholar]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R.; et al. Management of hyperglycemia in type 2 diabetes: A patient-centered approach: Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012, 35, 1364–1379. [Google Scholar]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med 1996, 334, 574–579. [Google Scholar]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.; Gans, R.O.; de Vries, E.G. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar]
- Decensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res 2010, 3, 1451–1461. [Google Scholar]
- Zhang, Z.J.; Zheng, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS One 2012, 7, e33411. [Google Scholar]
- Hassan, M.M.; Curley, S.A.; Li, D.; Kaseb, A.; Davila, M.; Abdalla, E.K.; Javle, M.; Moghazy, D.M.; Lozano, R.D.; Abbruzzese, J.L.; et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 2010, 116, 1938–1946. [Google Scholar]
- Zhang, Z.J.; Zheng, Z.J.; Shi, R.; Su, Q.; Jiang, Q.; Kip, K.E. Metformin for liver cancer prevention in patients with type 2 diabetes: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab 2012, 97, 2347–2353. [Google Scholar]
- Tajima, K.; Nakamura, A.; Shirakawa, J.; Togashi, Y.; Orime, K.; Sato, K.; Inoue, H.; Kaji, M.; Sakamoto, E.; Ito, Y.; et al. Metformin prevents liver tumorigenesis induced by high-fat diet in C57Bl/6 mice. Am. J. Physiol. Endocrinol. Metab 2013, in press. [Google Scholar]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W., 2nd; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar]
- Tan, C.Y.; Vidal-Puig, A. Adipose tissue expandability: The metabolic problems of obesity may arise from the inability to become more obese. Biochem. Soc. Trans 2008, 36, 935–940. [Google Scholar]
- Unger, R.H. The physiology of cellular liporegulation. Annu. Rev. Physiol 2003, 65, 333–347. [Google Scholar]
- Wang, M.Y.; Grayburn, P.; Chen, S.; Ravazzola, M.; Orci, L.; Unger, R.H. Adipogenic capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6139–6144. [Google Scholar]
- Duval, C.; Thissen, U.; Keshtkar, S.; Accart, B.; Stienstra, R.; Boekschoten, M.V.; Roskams, T.; Kersten, S.; Müller, M. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57BL/6 mice. Diabetes 2010, 59, 3181–3191. [Google Scholar]
- Nseir, W.; Mahamid, M. Statins in nonalcoholic fatty liver disease and steatohepatitis: Updated review. Curr. Atheroscler. Rep 2013, 15, 305. [Google Scholar]
- Athyros, V.G.; Mikhaildis, D.P.; Didangelos, T.P.; Giouleme, O.I.; Liberopoulos, E.N.; Karagiannis, A.; Kakafika, A.I.; Tziomalos, K.; Burroughs, A.K.; Elisaf, M.S. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: A randomized study. Curr. Med. Res. Opin 2006, 22, 873–883. [Google Scholar]
- Nelson, A.; Torres, D.M.; Morgan, A.E. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J. Clin. Gastroenterol 2009, 43, 990–994. [Google Scholar]
- Altmann, S.W.; Davis, H.R.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar]
- Knopp, R.H.; Dujovne, C.A.; Beaut, A.L.; Lipka, L.J.; Suresh, R.; Veltri, E.P. Ezetimbe Study Group. Evaluation of the efficacy, safety, and tolerability of ezetimibe in primary hypercholesterolaemia: A pooled analysis from two controlled phase III clinical studies. Int. J. Clin. Pract 2003, 57, 363–368. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest 2004, 114, 147–152. [Google Scholar]
- González-Ortiz, M.; Martínez-Abundis, E.; Kam-Ramos, A.M.; Hernández-Salazar, E.; Ramos-Zavala, M.G. Effect of ezetimibe on insulin sensitivity and lipid profile in obese and dyslipidaemic patients. Cardiovasc. Drug Ther 2006, 20, 143–146. [Google Scholar]
- Yoneda, M.; Fujita, K.; Nozaki, Y.; Endo, H.; Takahashi, H.; Hosono, K.; Suzuki, K.; Mawatari, H.; Kirikoshi, H.; Inamori, M.; et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol. Res 2010, 40, 613–621. [Google Scholar]
- Muraoka, T.; Aoki, K.; Iwasaki, T.; Shinoda, K.; Nakamura, A.; Aburatani, H.; Mori, S.; Tokuyama, K.; Kubota, N.; Kadowaki, T.; et al. Ezetimibe decreases SREBP-1c expression in liver and reverses hepatic insulin resistance in mice fed a high-fat diet. Metabolism 2011, 60, 617–628. [Google Scholar]
- Nozaki, Y.; Fujita, K.; Yoneda, M.; Wada, K.; Shinohara, Y.; Takahashi, H.; Kirikoshi, H.; Inamori, M.; Kubota, K.; Saito, S.; et al. Long-term combination therapy of ezetimibe and acarbose for non-alcoholic fatty liver disease. J. Hepatol 2009, 51, 548–556. [Google Scholar]
- Van Rooyen, D.M.; Gan, L.T.; Yeh, M.M.; Haigh, W.G.; Larter, C.Z.; Ioannou, G.; Teoh, N.C.; Farrell, G.C. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J. Hepatol 2013, 59, 144–152. [Google Scholar]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab 2012, 16, 44–54. [Google Scholar]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar]
- Chang, K.Y.; Chang, J.Y.; Yen, Y. Increasing incidence of intrahepatic cholangiocarcinoma and its relationship to chronic viral hepatitis. J. Natl. Compr. Canc. Netw 2009, 7, 423–427. [Google Scholar]
- Palmer, W.C.; Patel, T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J. Hepatol 2012, 57, 69–76. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakamura, A.; Terauchi, Y. Lessons from Mouse Models of High-Fat Diet-Induced NAFLD. Int. J. Mol. Sci. 2013, 14, 21240-21257. https://doi.org/10.3390/ijms141121240
Nakamura A, Terauchi Y. Lessons from Mouse Models of High-Fat Diet-Induced NAFLD. International Journal of Molecular Sciences. 2013; 14(11):21240-21257. https://doi.org/10.3390/ijms141121240
Chicago/Turabian StyleNakamura, Akinobu, and Yasuo Terauchi. 2013. "Lessons from Mouse Models of High-Fat Diet-Induced NAFLD" International Journal of Molecular Sciences 14, no. 11: 21240-21257. https://doi.org/10.3390/ijms141121240