Crosstalk between Beta-Catenin and Snail in the Induction of Epithelial to Mesenchymal Transition in Hepatocarcinoma: Role of the ERK1/2 Pathway

Abstract
:1. Introduction
2. Results and Discussion
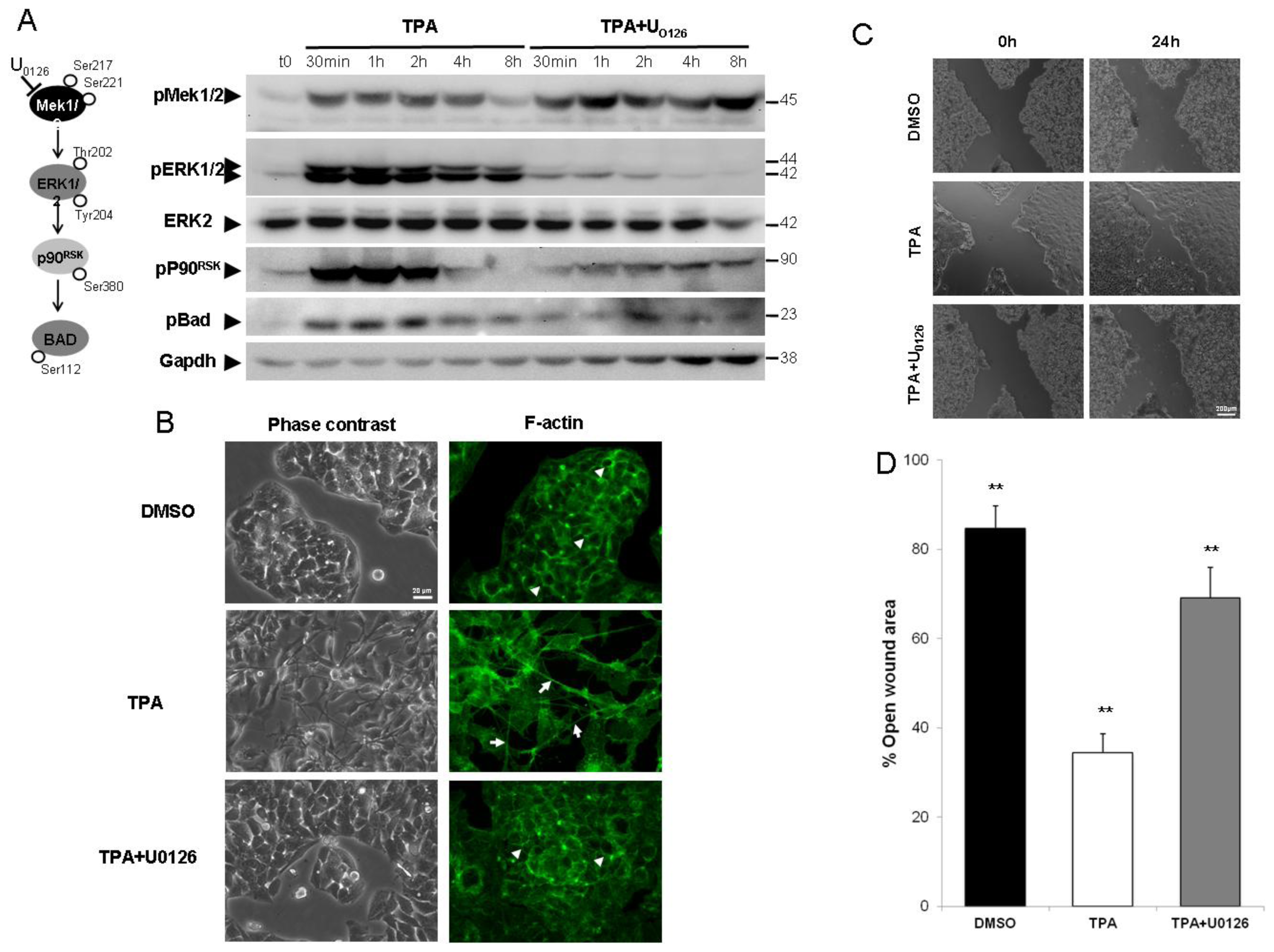
2.1. ERK1/2 Activation Is Critical for TPA-Mediated Hepatoblastoma Cell Migration
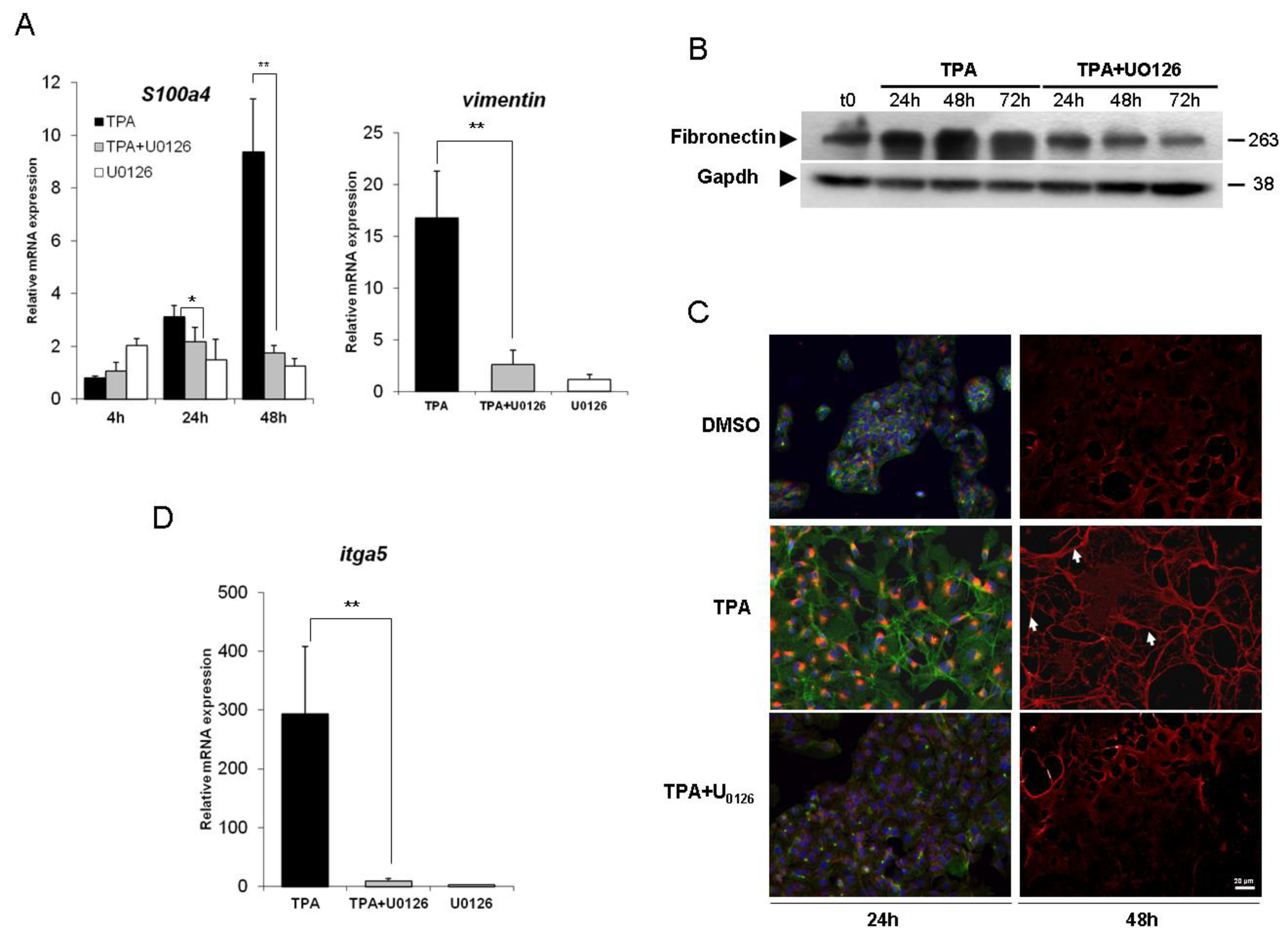
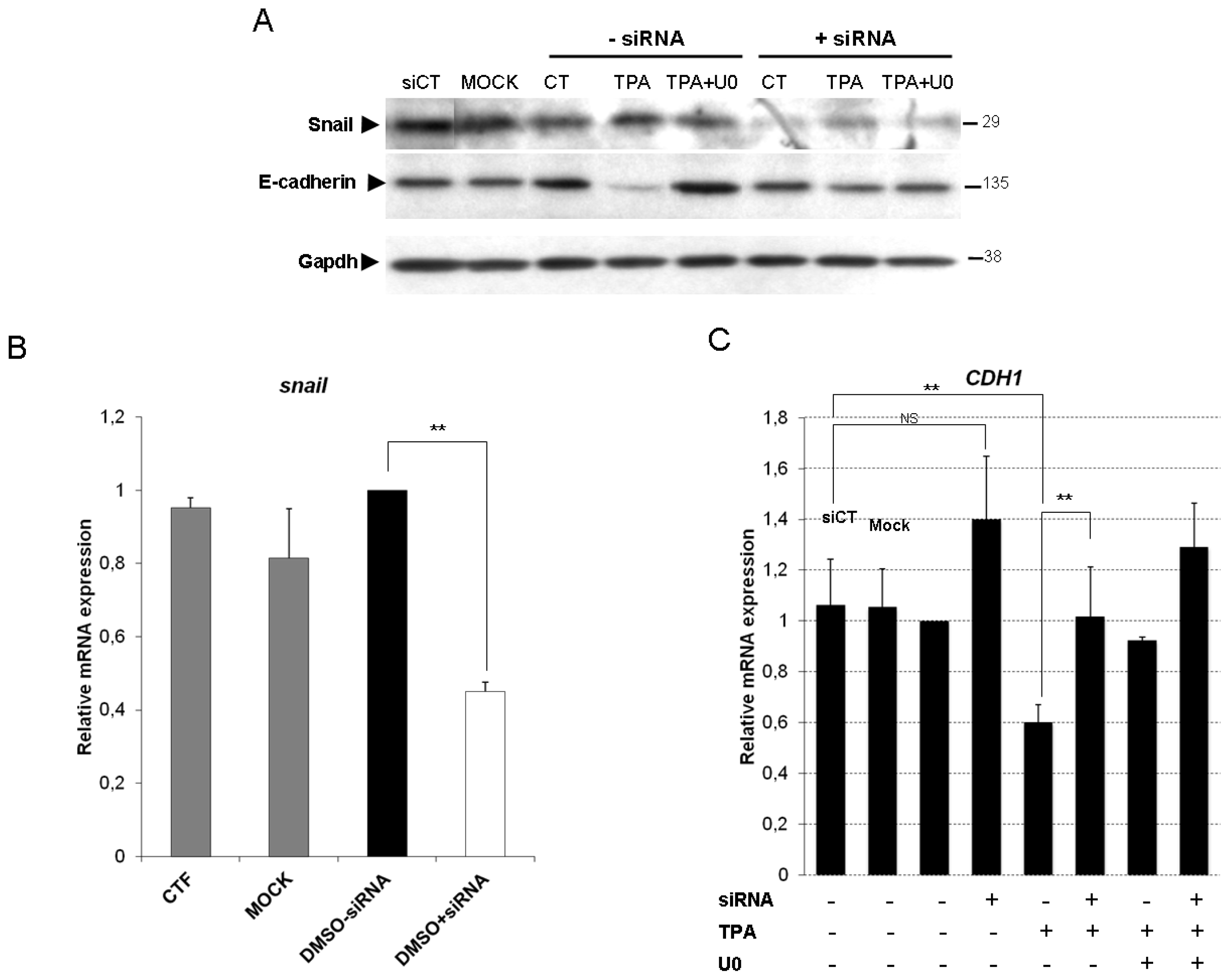
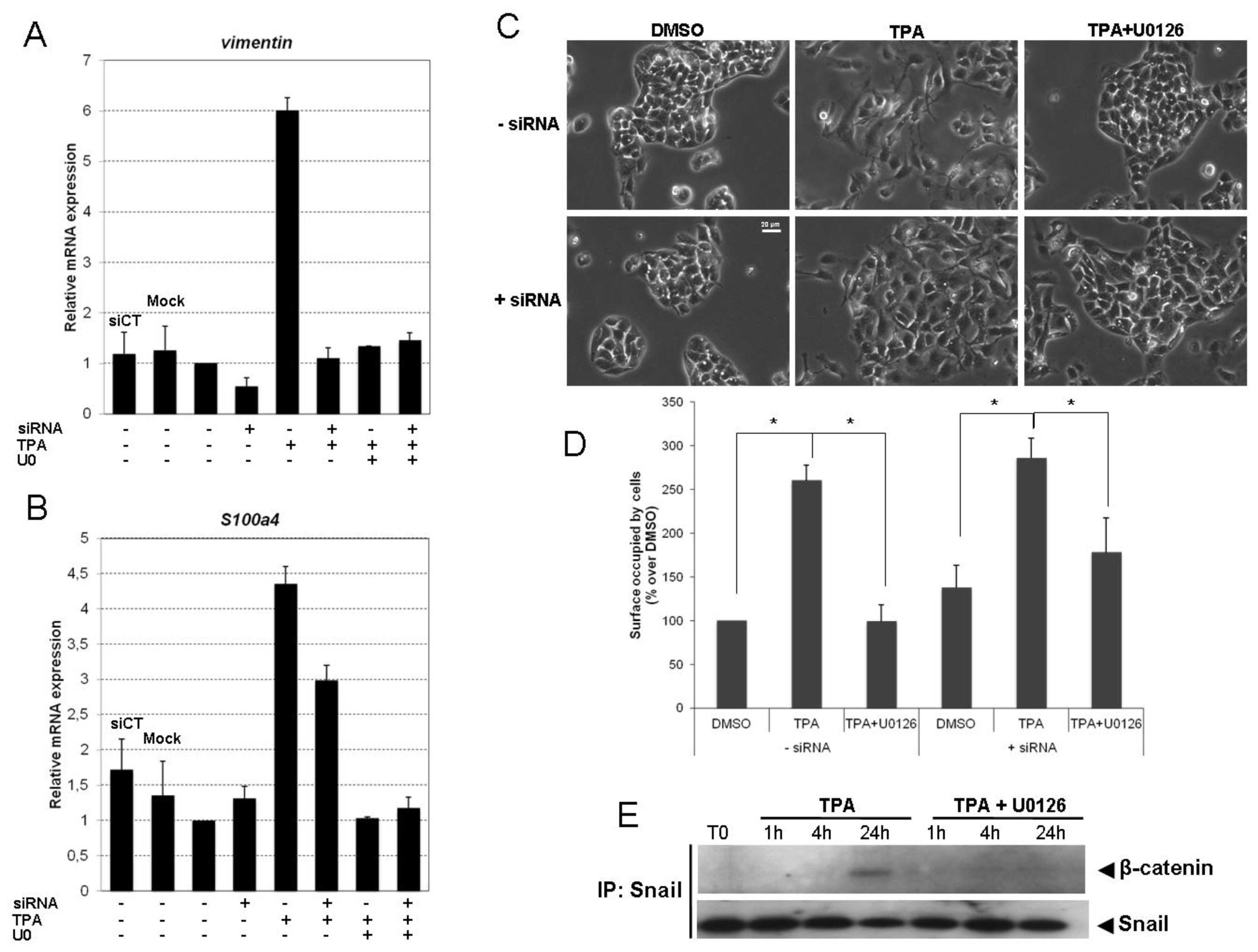
2.2. Inhibition of the ERK1/2 Pathway Abrogates the TPA-Mediated Deregulation of Epithelial and Mesenchymal Markers
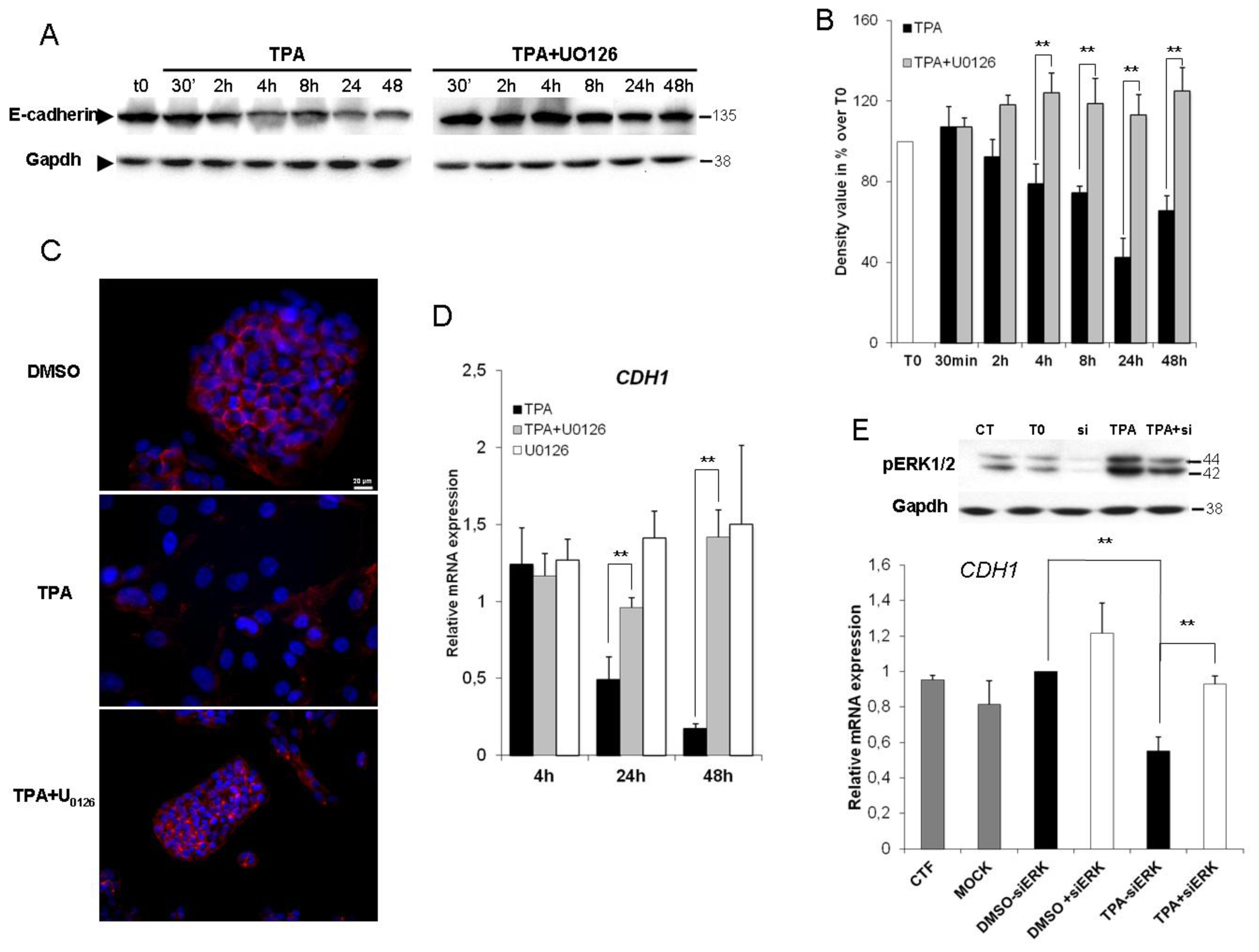
2.3. ERK1/2 Is Critical for TPA-Mediated CDH1 Repression
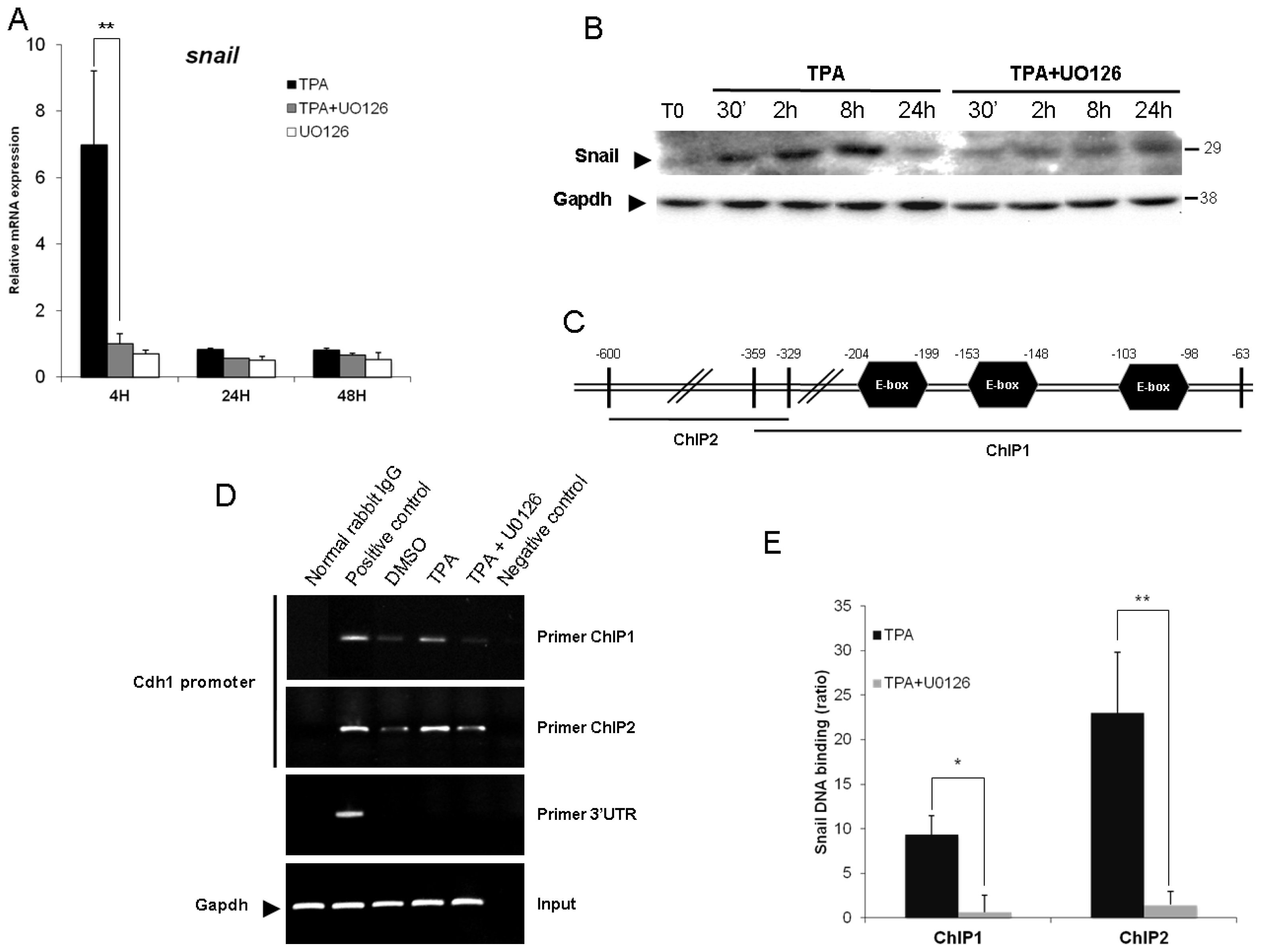
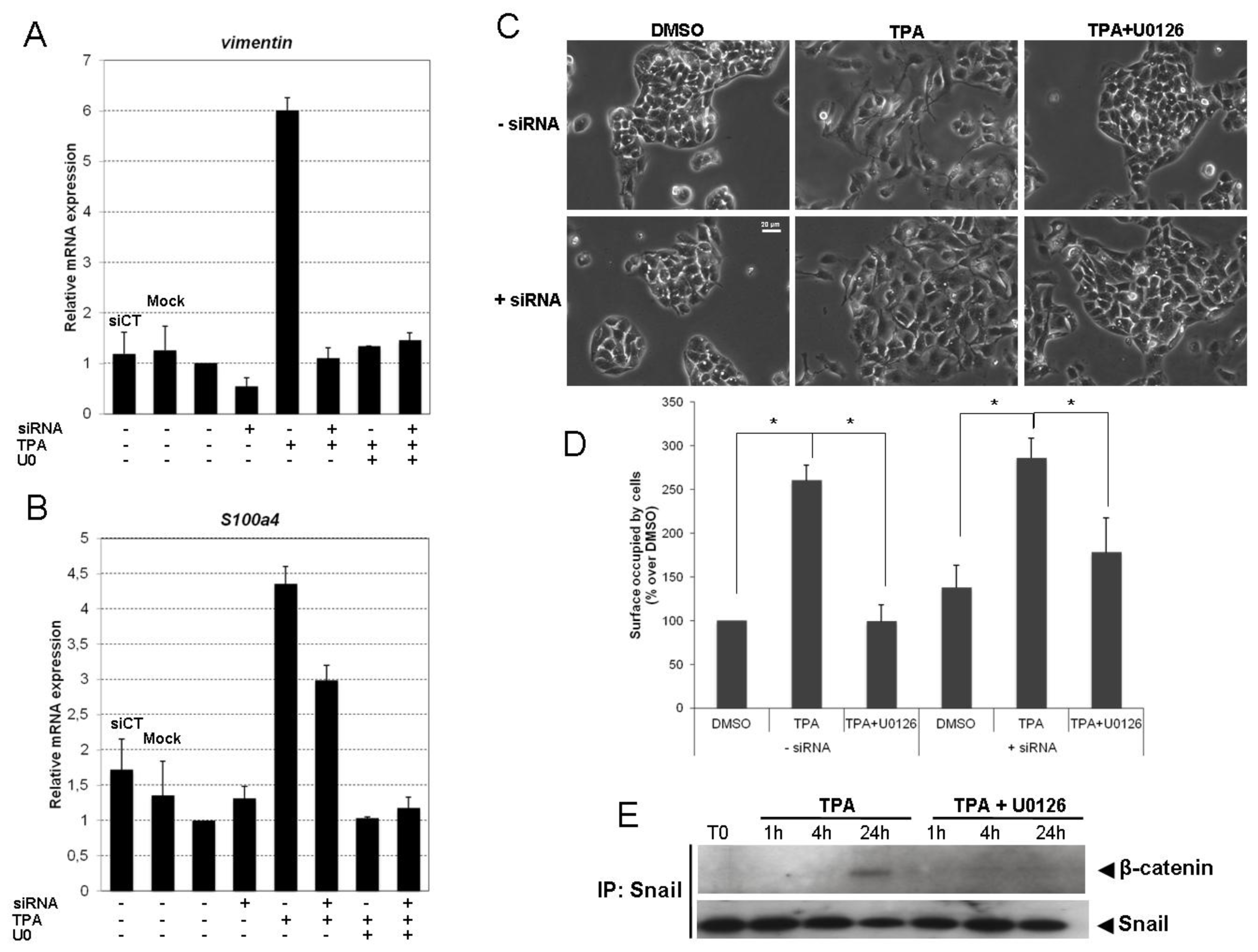
2.4. Snail Is Necessary, but Not Sufficient, for TPA-Induced EMT
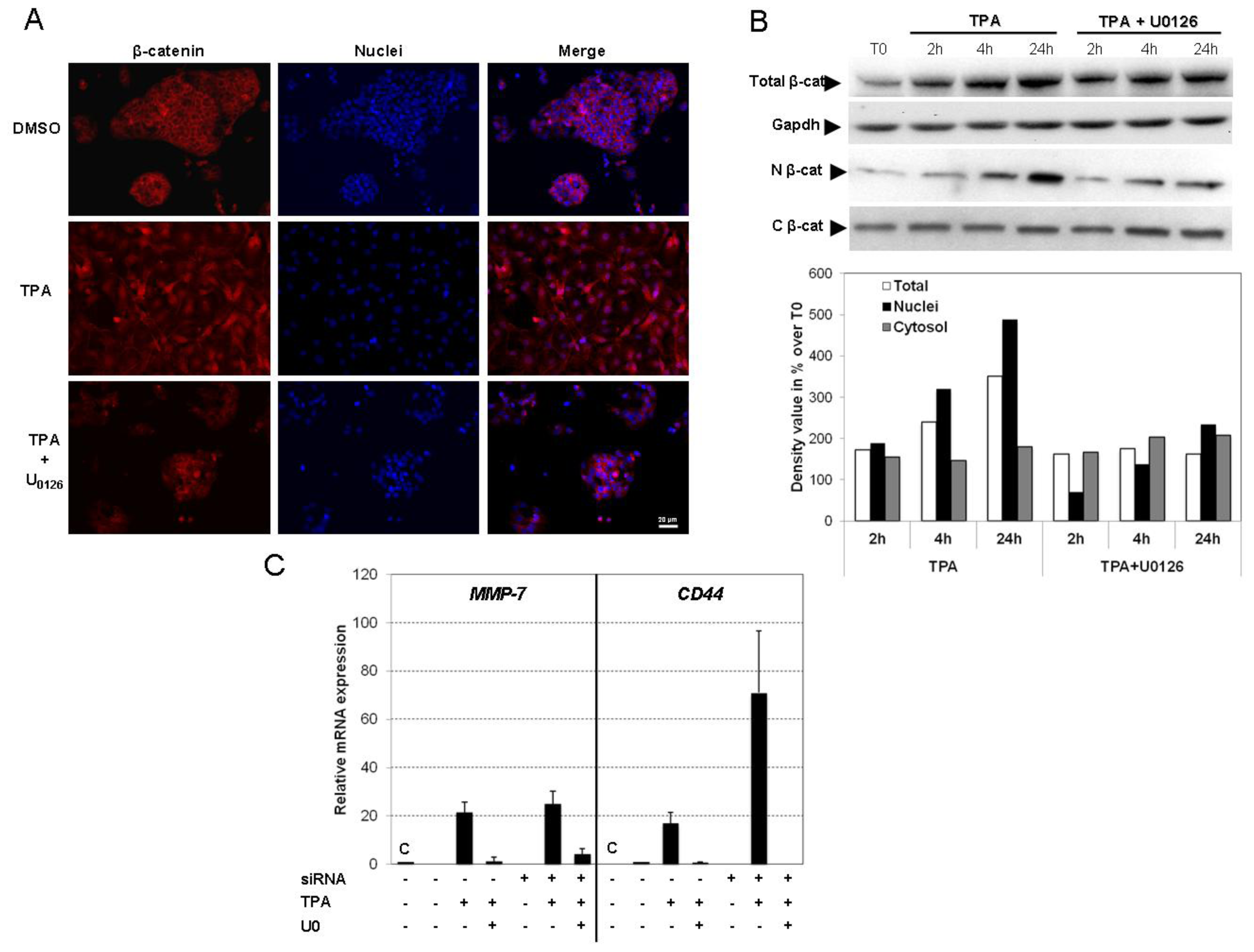
2.5. TPA Activates the Wnt/β-Catenin Signaling Pathway
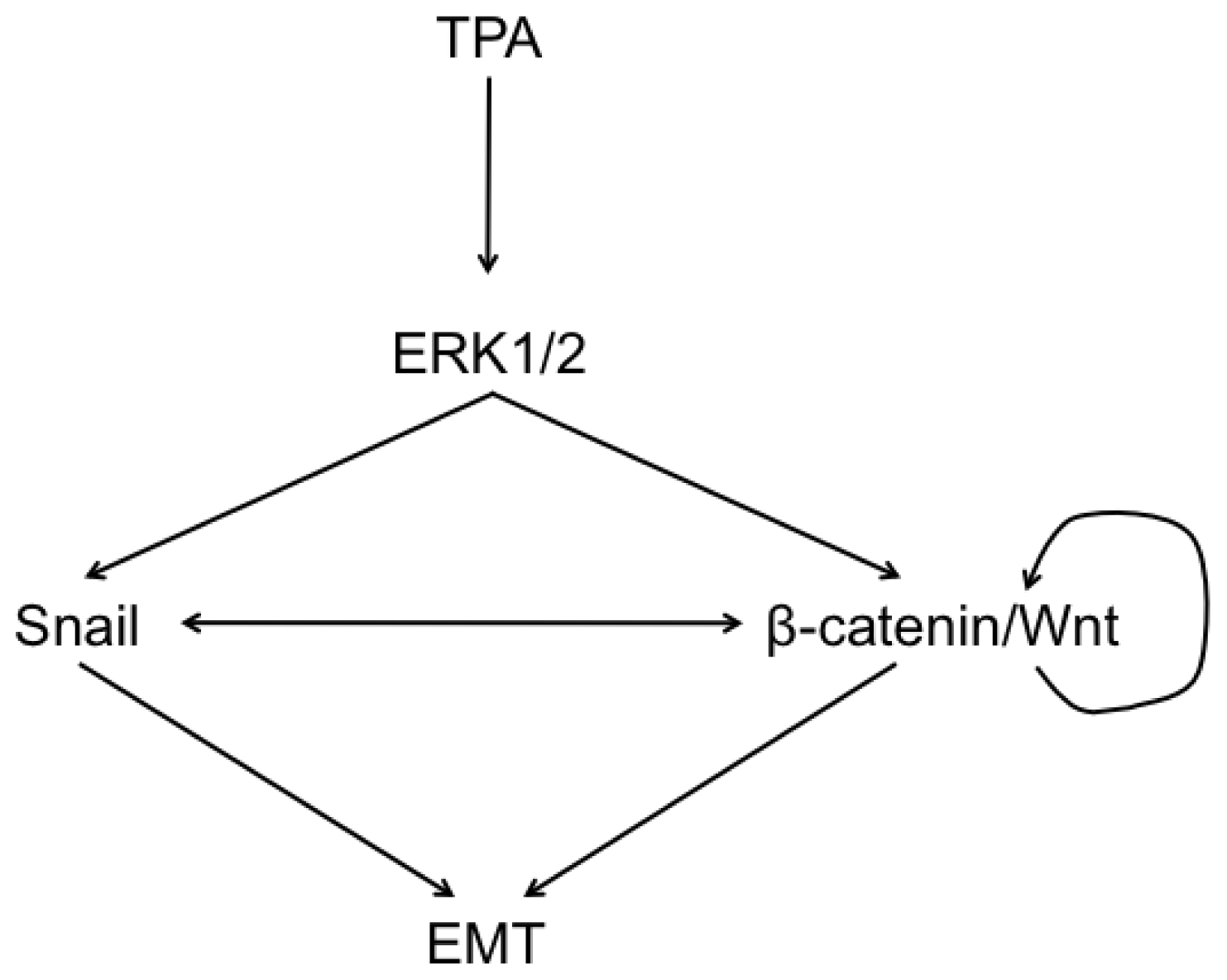
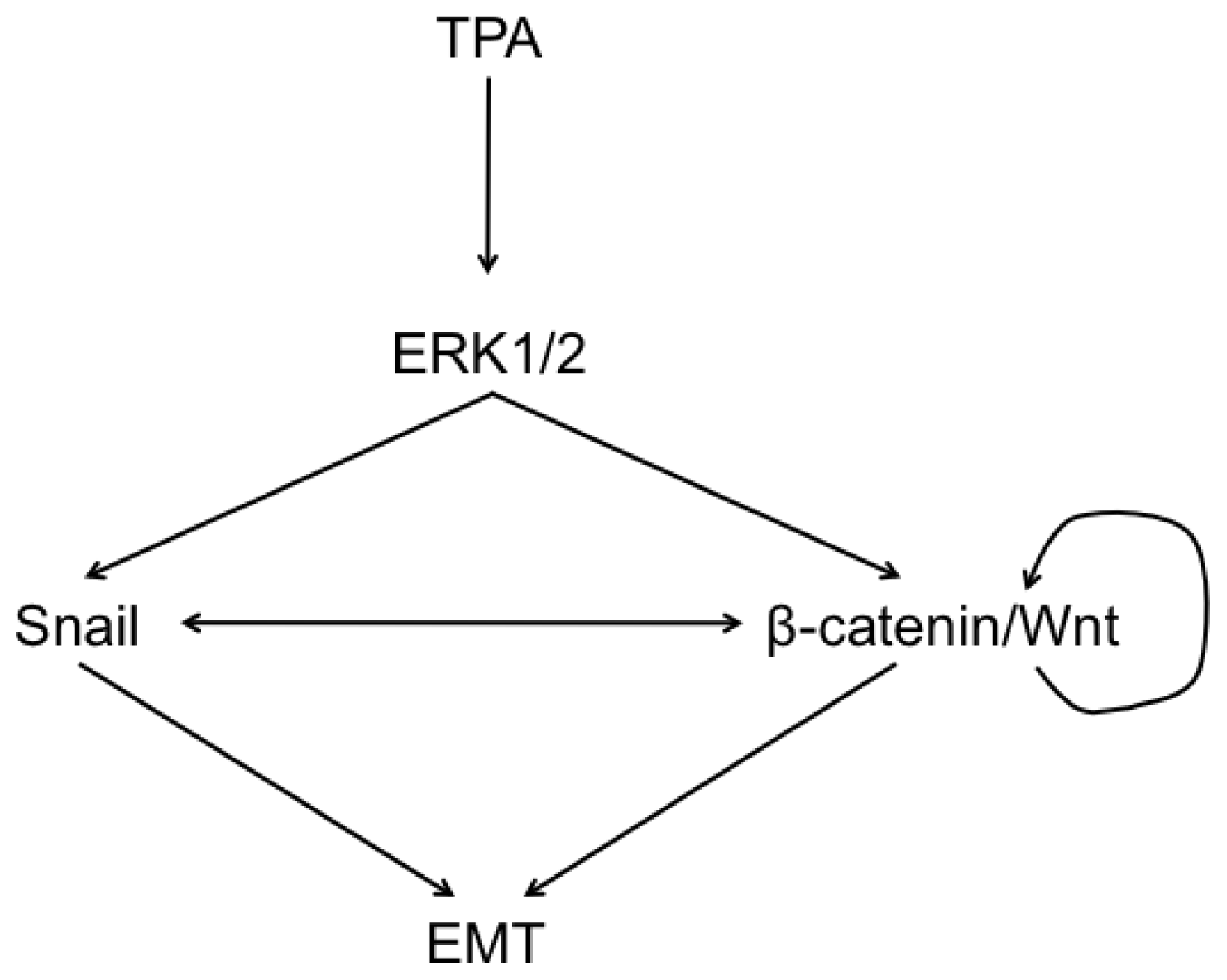
2.6. Discussion
3. Experimental Section
3.1. Materials
3.2. Cell Culture and Drug Treatments
3.3. Western Blot Analysis
3.4. Immunofluorescence Staining
3.5. Cell Migration Assay
3.6. Reverse Transcription-Quantitative Polymerase Chain Réaction
3.7. Chromatin Immunoprecipitation
3.8. Small Interfering RNA (siRNA)
3.9. Statistical Analysis
4. Conclusions

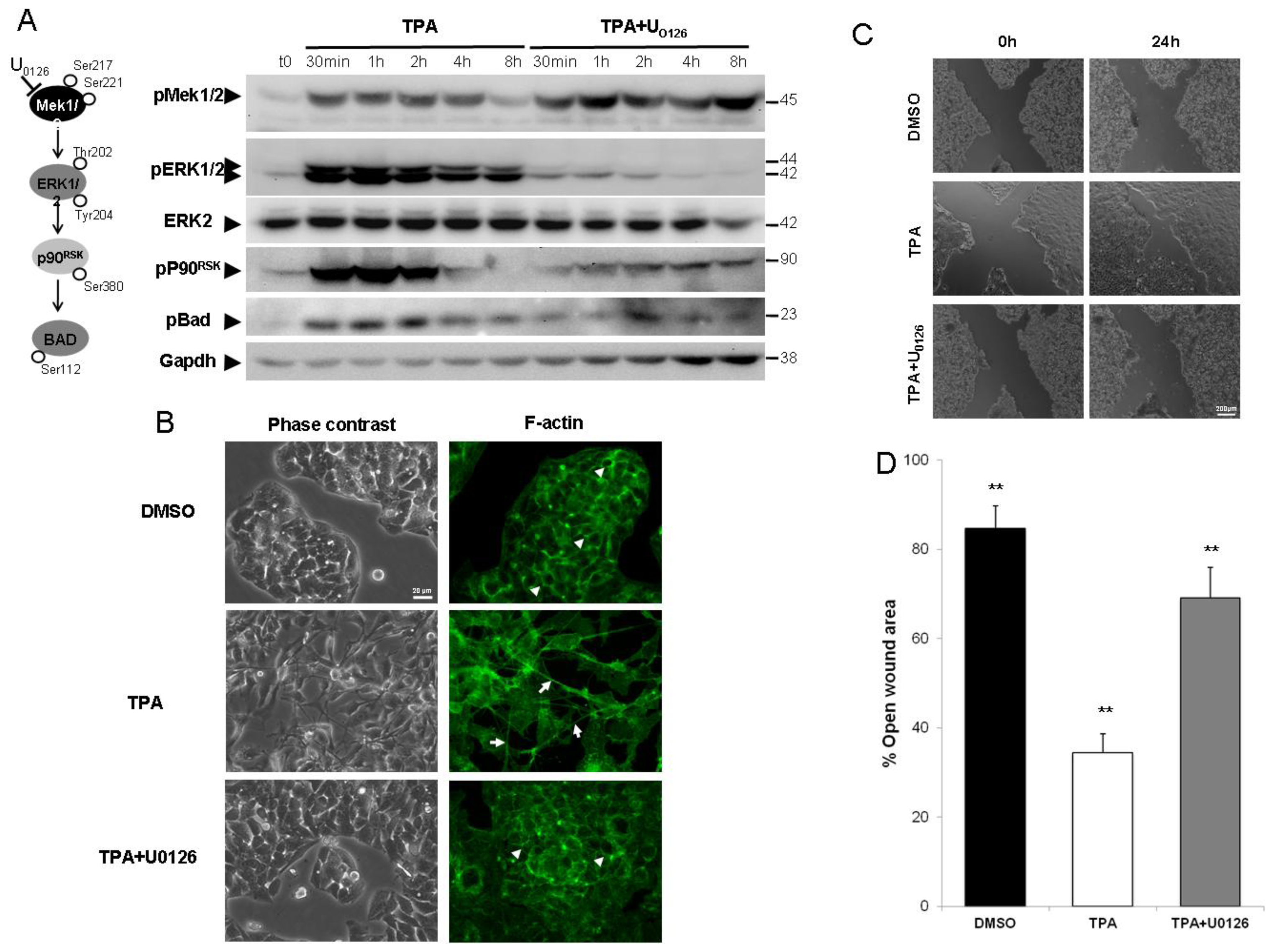{kind=link}
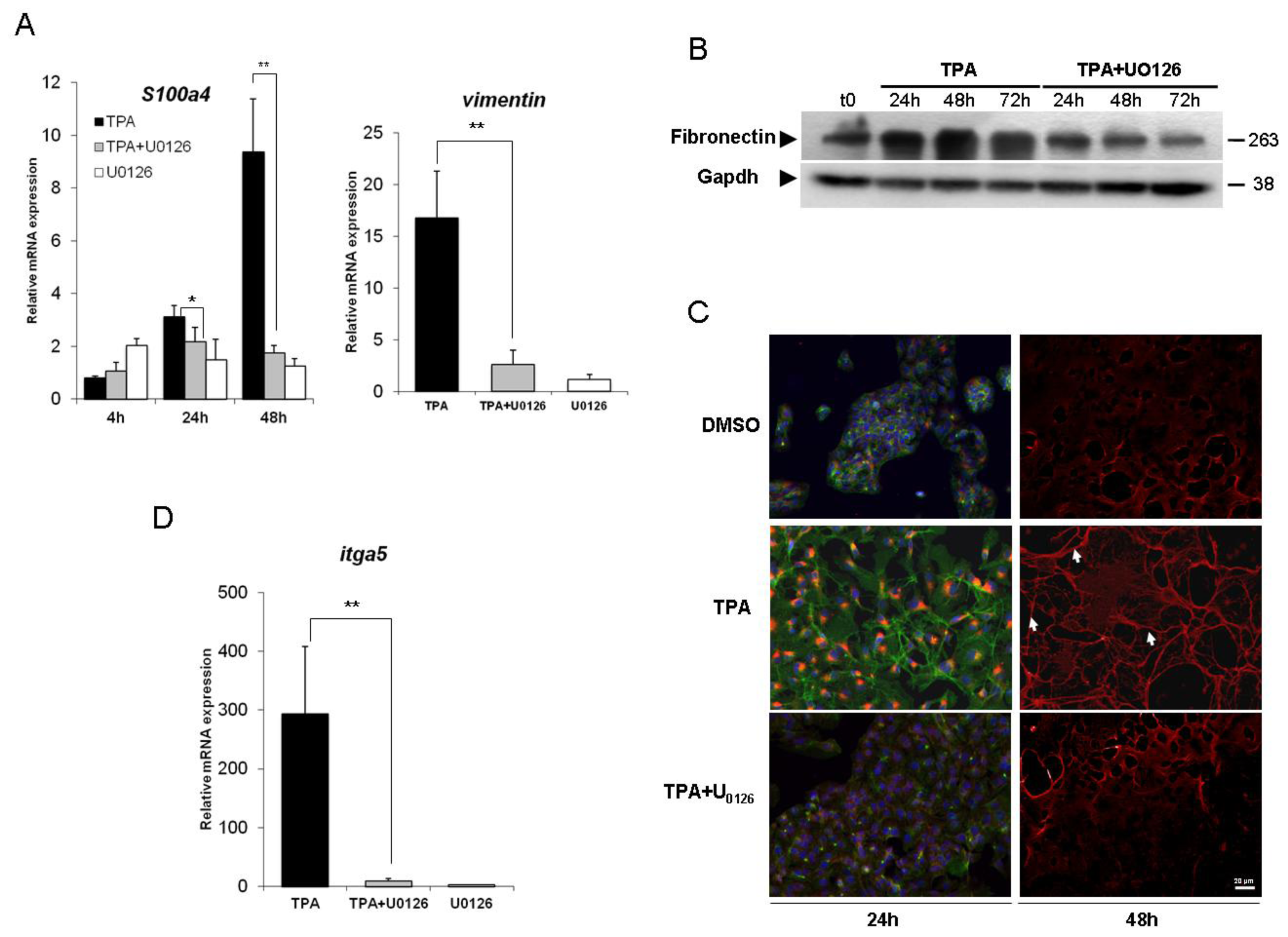{kind=link}
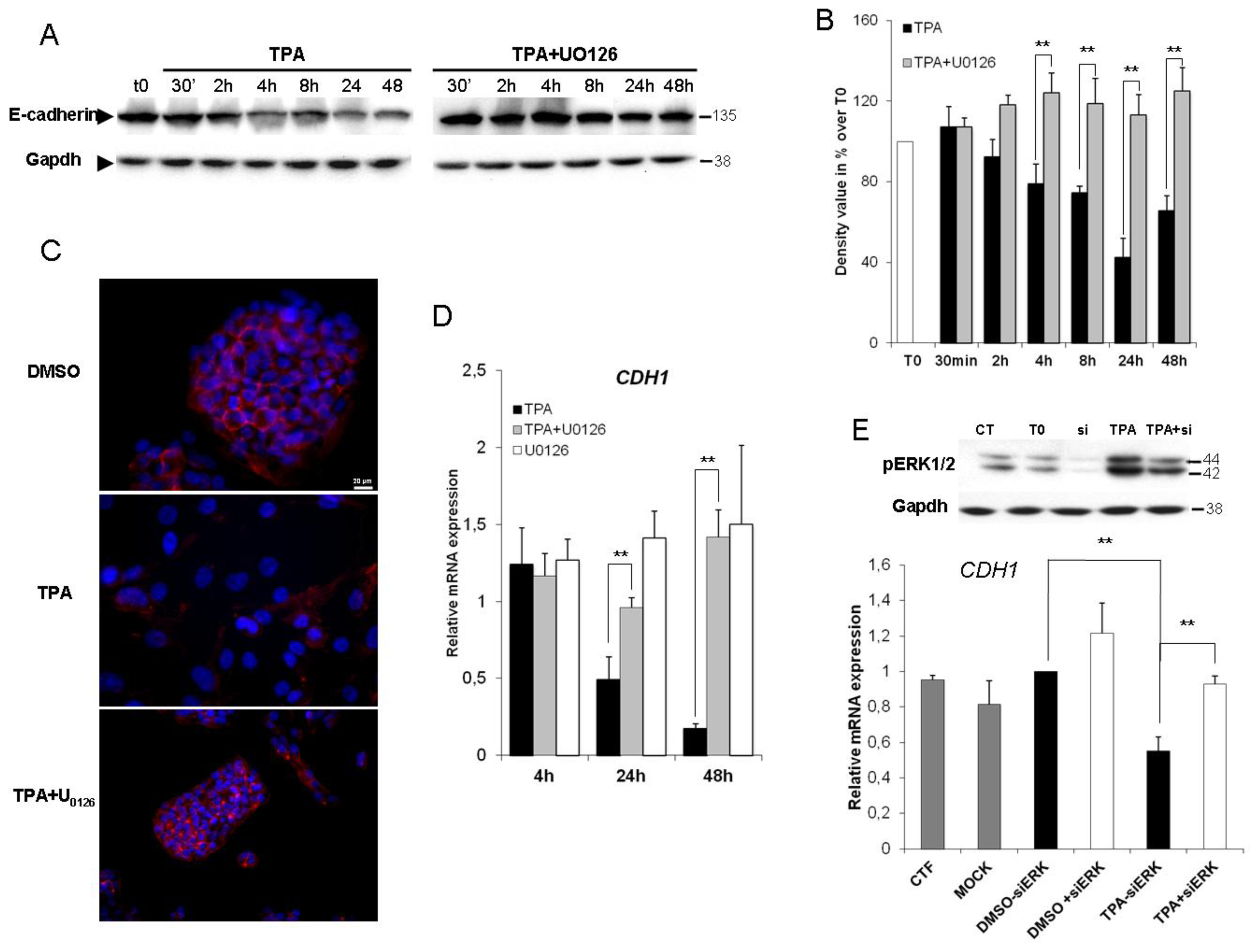{kind=link}
{kind=link}
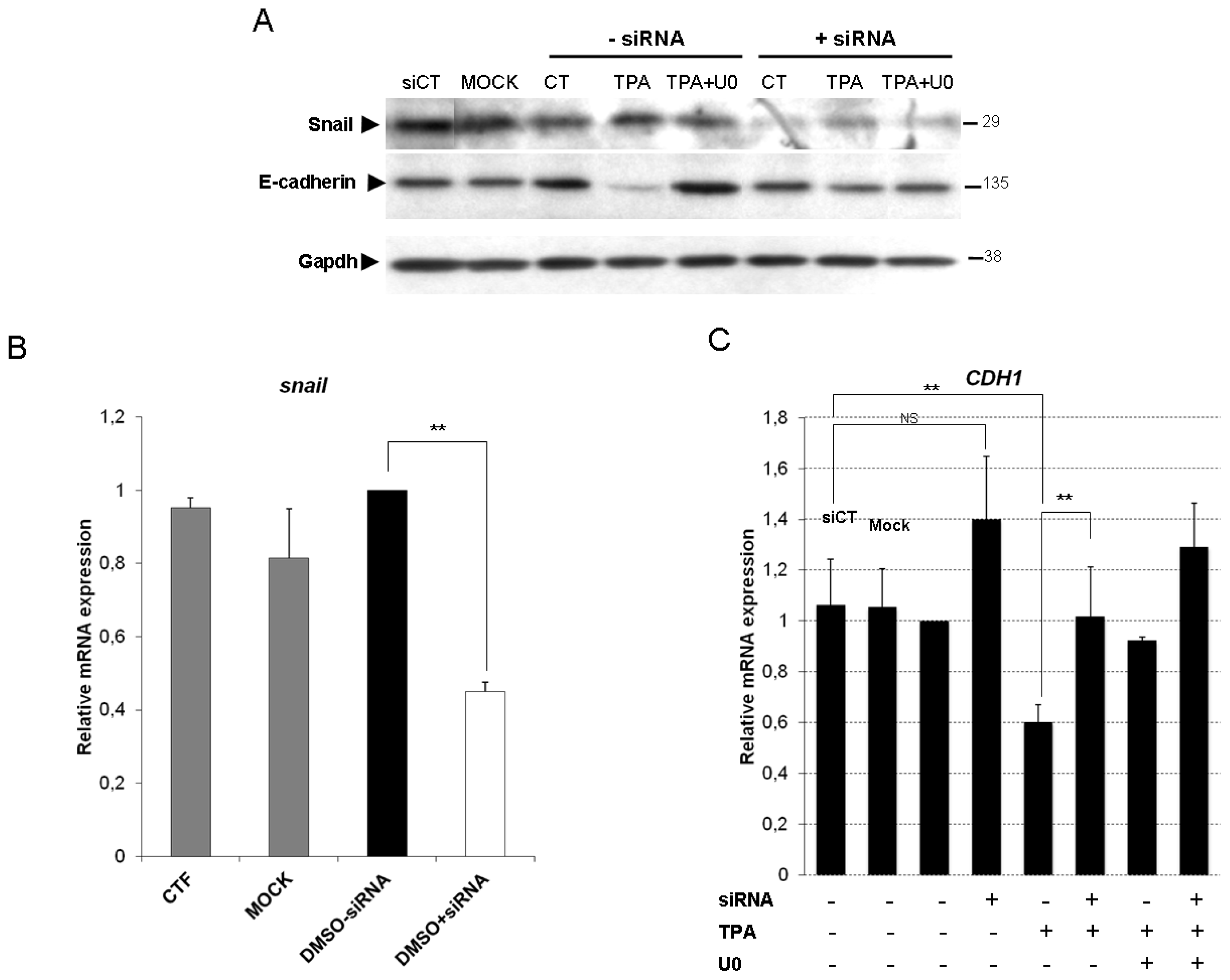{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Phosphorylation site | Source/type | Manufacturer | Dilution WB | Dilution IF |
|---|---|---|---|---|---|
| pErk1/2 | Thr202/Tyr204 | Rabbit mAb1 | Cell signaling | 1:2000 | |
| pErk1/2 | Thr202/Tyr204 | Mouse mAb | Sigma | 1:300 | |
| pMEK1/2 | Ser217/221 | Rabbit pAb2 | Cell signaling | 1:2000 | |
| Erk2 | Rabbit pAb | Cell signaling | 1:5000 | ||
| pP90RSK | Ser380 | Rabbit pAb | Cell signaling | 1:2000 | |
| pBad1 | Ser136 | Rabbit pAb | Cell signaling | 1:2000 | |
| Gapdh | Rabbit mAb | Cell signaling | 1:7500 | ||
| E-cadherin | Rabbit mAb | Epitomics | 1:5000 | ||
| b-catenin | Rabbit mAb | Santa Cruz | 1:2000 | 1:300 | |
| Fibronectin | Rabbit mAb | Epitomics | 1:1000 | 1:400 | |
| Snail | Rabbit pAb | Santa Cruz | 1:1000 |
Acknowledgments
Conflicts of Interest
References
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar]
- Kensler, T.W.; Qian, G.S.; Chen, J.G.; Groopman, J.D. Translational strategies for cancer prevention in liver. Nat. Rev. Cancer 2003, 3, 321–329. [Google Scholar]
- Bréchot, C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: Old and new paradigms. Gastroenterology 2004, 127, S56–S61. [Google Scholar]
- Coleman, W.B. Mechanisms of human hepatocarcinogenesis. Curr. Mol. Med 2003, 3, 573–588. [Google Scholar]
- Liang, T.J.; Heller, T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 2004, 127, S62–S71. [Google Scholar]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet 2002, 31, 339–346. [Google Scholar]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar]
- Firrincieli, D.; Boissan, M.; Chignard, N. Epithelial-mesenchymal transition in the liver. Gastroenterol. Clin. Biol 2010, 34, 523–528. [Google Scholar]
- Van Zijl, F.; Zulehner, G.; Petz, M.; Schneller, D.; Kornauth, C.; Hau, M.; Machat, G.; Grubinger, M.; Huber, H.; Mikulits, W. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol 2009, 5, 1169–1179. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig 2009, 119, 1420–1428. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol 2003, 15, 740–746. [Google Scholar]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol 2004, 48, 365–375. [Google Scholar] [Green Version]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol 2005, 17, 548–558. [Google Scholar]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig 2009, 119, 1429–1437. [Google Scholar]
- Cavallaro, U.; Christofori, G. Multitasking in tumor progression: Signaling functions of cell adhesion molecules. Ann. N. Y. Acad. Sci 2004, 1014, 58–66. [Google Scholar]
- Endo, K.; Ueda, T.; Ueyama, J.; Ohta, T.; Terada, T. Immunoreactive E-cadherin, alpha-catenin, beta-catenin, and gamma-catenin proteins in hepatocellular carcinoma: Relationships with tumor grade, clinicopathologic parameters, and patients’ survival. Hum. Pathol 2000, 31, 558–565. [Google Scholar]
- Peinado, H.; Olmeda, D.; Cano, A. Snail Zeb bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar]
- Fransvea, E.; Angelotti, U.; Antonaci, S.; Giannelli, G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008, 47, 1557–1566. [Google Scholar]
- Orford, K.; Crockett, C.; Jensen, J.P.; Weissman, A.M.; Byers, S.W. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J. Biol. Chem 1997, 272, 24735–24738. [Google Scholar]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar]
- Roose, J.; Huls, G.; van Beest, M.; Moerer, P.; van der Horn, K.; Goldschmeding, R.; Logtenberg, T.; Clevers, H. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 1999, 285, 1923–1926. [Google Scholar]
- Wielenga, V.J.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol 1999, 154, 515–523. [Google Scholar]
- Brabletz, T.; Jung, A.; Dag, S.; Hlubek, F.; Kirchner, T. Beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am. J. Pathol 1999, 155, 1033–1038. [Google Scholar]
- Tetsu, O.; McCormick, F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar]
- Hu, C.T.; Wu, J.R.; Chang, T.Y.; Cheng, C.C.; Wu, W.S. The transcriptional factor Snail simultaneously triggers cell cycle arrest and migration of human hepatoma HepG2. J. Biomed. Sci 2008, 15, 343–355. [Google Scholar]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 2007, 67, 10123–10128. [Google Scholar]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar]
- Boyer, B.; Tucker, G.C.; Vallés, A.M.; Gavrilovic, J.; Thiery, J.P. Reversible transition towards a fibroblastic phenotype in a rat carcinoma cell line. Int. J. Cancer Suppl 1989, 4, 69–75. [Google Scholar]
- Schwarzbauer, J.E.; Sechler, J.L. Fibronectin fibrillogenesis: A paradigm for extracellular matrix assembly. Curr. Opin. Cell Biol 1999, 11, 622–627. [Google Scholar]
- Mauro, A.; Ciccarelli, C.; de Cesaris, P.; Scoglio, A.; Bouché, M.; Molinaro, M.; Aquino, A.; Zani, B.M. PKCalpha-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J. Cell Sci 2002, 115, 3587–3599. [Google Scholar]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol 2004, 6, 931–940. [Google Scholar]
- Barberà, M.J.; Puig, I.; Domínguez, D.; Julien-Grille, S.; Guaita-Esteruelas, S.; Peiró, S.; Baulida, J.; Francí, C.; Dedhar, S.; Larue, L.; et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 2004, 23, 7345–7354. [Google Scholar]
- Shin, S.Y.; Rath, O.; Zebisch, A.; Choo, S.M.; Kolch, W.; Cho, K.H. Functional roles of multiple feedback loops in extracellular signal-regulated kinase and Wnt signaling pathways that regulate epithelial-mesenchymal transition. Cancer Res 2010, 70, 6715–6724. [Google Scholar]
- Christen, B.; Slack, J.M. Spatial response to fibroblast growth factor signalling in Xenopus embryos. Development 1999, 126, 119–125. [Google Scholar]
- Yao, Y.; Li, W.; Wu, J.; Germann, U.A.; Su, M.S.; Kuida, K.; Boucher, D.M. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 12759–12764. [Google Scholar]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res 2001, 61, 4222–4228. [Google Scholar]
- Grände, M.; Franzen, A.; Karlsson, J.O.; Ericson, L.E.; Heldin, N.E.; Nilsson, M. Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci 2002, 115, 4227–4236. [Google Scholar]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem 2003, 278, 21113–21123. [Google Scholar]
- McKillop, I.H.; Schmidt, C.M.; Cahill, P.A.; Sitzmann, J.V. Altered expression of mitogen-activated protein kinases in a rat model of experimental hepatocellular carcinoma. Hepatology 1997, 26, 1484–1491. [Google Scholar]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol 2008, 48, 83–90. [Google Scholar]
- Domínguez, D.; Montserrat-Sentís, B.; Virgós-Soler, A.; Guaita, S.; Grueso, J.; Porta, M.; Puig, I.; Baulida, J.; Francí, C.; García de Herreros, A. Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol. Cell. Biol 2003, 23, 5078–5089. [Google Scholar]
- Delfini, M.C.; de La Celle, M.; Gros, J.; Serralbo, O.; Marics, I.; Seux, M.; Scaal, M.; Marcelle, C. The timing of emergence of muscle progenitors is controlled by an FGF/ERK/SNAIL1 pathway. Dev. Biol 2009, 333, 229–237. [Google Scholar]
- Jiao, W.; Miyazaki, K.; Kitajima, Y. Inverse correlation between E-cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo. Br. J. Cancer 2002, 86, 98–101. [Google Scholar]
- Giroldi, L.A.; Bringuier, P.P.; de Weijert, M.; Jansen, C.; van Bokhoven, A.; Schalken, J.A. Role of E boxes in the repression of E-cadherin expression. Biochem. Biophys. Res. Commun 1997, 241, 453–458. [Google Scholar]
- Behrens, J.; Frixen, U.; Schipper, J.; Weidner, M.; Birchmeier, W. Cell adhesion in invasion and metastasis. Semin. Cell Biol 1992, 3, 169–178. [Google Scholar]
- Takeichi, M. Cadherins in cancer: Implications for invasion and metastasis. Curr. Opin. Cell Biol 1993, 5, 806–811. [Google Scholar]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci 1999, 24, 73–76. [Google Scholar]
- Kozyraki, R.; Scoazec, J.Y.; Flejou, J.F.; D’Errico, A.; Bedossa, P.; Terris, B.; Fiorentino, M.; Bringuier, A.F.; Grigioni, W.F.; Feldmann, G. Expression of cadherins and alpha-catenin in primary epithelial tumors of the liver. Gastroenterology 1996, 110, 1137–1149. [Google Scholar]
- Zhai, B.; Yan, H.X.; Liu, S.Q.; Chen, L.; Wu, M.C.; Wang, H.Y. Reduced expression of E-cadherin/catenin complex in hepatocellular carcinomas. World J. Gastroenterol 2008, 14, 5665–5673. [Google Scholar]
- Miyoshi, A.; Kitajima, Y.; Kido, S.; Shimonishi, T.; Matsuyama, S.; Kitahara, K.; Miyazaki, K. Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br. J. Cancer 2005, 92, 252–258. [Google Scholar]
- Sugimachi, K.; Tanaka, S.; Kameyama, T.; Taguchi, K.; Aishima, S.; Shimada, M.; Tsuneyoshi, M. Transcriptional repressor snail and progression of human hepatocellular carcinoma. Clin. Cancer Res 2003, 9, 2657–2664. [Google Scholar]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol 2002, 3, 155–166. [Google Scholar]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol 2001, 21, 8184–8188. [Google Scholar]
- Lundgren, K.; Nordenskjöld, B.; Landberg, G. Hypoxia Snail incomplete epithelial-mesenchymal transition in breast cancer. Br. J. Cancer 2009, 101, 1769–1781. [Google Scholar]
- Zavadil, J.; Böttinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar]
- Uematsu, K.; Kanazawa, S.; You, L.; He, B.; Xu, Z.; Li, K.; Peterlin, B.M.; McCormick, F.; Jablons, D.M. Wnt pathway activation in mesothelioma: Evidence of Dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res 2003, 63, 4547–4551. [Google Scholar]
- Yook, J.I.; Li, X.Y.; Ota, I.; Fearon, E.R.; Weiss, S.J. Wnt-dependent regulation of the E-cadherin repressor snail. J. Biol. Chem 2005, 280, 11740–11748. [Google Scholar]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar]
- Conacci-Sorrell, M.; Zhurinsky, J.; Ben-Ze’ev, A. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Investig 2002, 109, 987–991. [Google Scholar]
- Stemmer, V.; de Craene, B.; Berx, G.; Behrens, J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene 2008, 27, 5075–5080. [Google Scholar]
- Thompson, M.D.; Monga, S.P. WNT/beta-catenin signaling in liver health and disease. Hepatology 2007, 45, 1298–1305. [Google Scholar]
- Takigawa, Y.; Brown, A.M. Wnt signaling in liver cancer. Curr. Drug Targets 2008, 9, 1013–1024. [Google Scholar]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Monges, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar]
- Merle, P.; de la Monte, S.; Kim, M.; Herrmann, M.; Tanaka, S.; von Dem Bussche, A.; Kew, M.C.; Trepo, C.; Wands, J.R. Functional consequences of frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology 2004, 127, 1110–1122. [Google Scholar]
- Ban, K.C.; Singh, H.; Krishnan, R.; Seow, H.F. GSK-3beta phosphorylation and alteration of beta-catenin in hepatocellular carcinoma. Cancer Lett 2003, 199, 201–208. [Google Scholar]
- Schmitt-Graeff, A.; Ertelt-Heitzmann, V.; Allgaier, H.P.; Olschewski, M.; Nitschke, R.; Haxelmans, S.; Koelble, K.; Behrens, J.; Blum, H.E. Coordinated expression of cyclin D1 and LEF-1/TCF transcription factor is restricted to a subset of hepatocellular carcinoma. Liver Int 2005, 25, 839–847. [Google Scholar]
- De La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar]
- Zeng, G.; Apte, U.; Cieply, B.; Singh, S.; Monga, S.P. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia 2007, 9, 951–959. [Google Scholar]
- Mori, M.; Barnard, G.F.; Mimori, K.; Ueo, H.; Akiyoshi, T.; Sugimachi, K. Overexpression of matrix metalloproteinase-7 mRNA in human colon carcinomas. Cancer 1995, 75, 1516–1519. [Google Scholar]
- Günthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zöller, M.; Haussmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar]
- Zucchini-Pascal, N.; de Sousa, G.; Rahmani, R. Lindane and cell death: At the crossroads between apoptosis, necrosis and autophagy. Toxicology 2009, 256, 32–41. [Google Scholar]
- Gebäck, T.; Schulz, M.M.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 2009, 46, 265–274. [Google Scholar]
- Saito, T.; Nagai, M.; Ladanyi, M. SYT-SSX1 and SYT-SSX2 interfere with repression of E-cadherin by snail and slug: A potential mechanism for aberrant mesenchymal to epithelial transition in human synovial sarcoma. Cancer Res 2006, 66, 6919–6927. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zucchini-Pascal, N.; Peyre, L.; Rahmani, R. Crosstalk between Beta-Catenin and Snail in the Induction of Epithelial to Mesenchymal Transition in Hepatocarcinoma: Role of the ERK1/2 Pathway. Int. J. Mol. Sci. 2013, 14, 20768-20792. https://doi.org/10.3390/ijms141020768
Zucchini-Pascal N, Peyre L, Rahmani R. Crosstalk between Beta-Catenin and Snail in the Induction of Epithelial to Mesenchymal Transition in Hepatocarcinoma: Role of the ERK1/2 Pathway. International Journal of Molecular Sciences. 2013; 14(10):20768-20792. https://doi.org/10.3390/ijms141020768
Chicago/Turabian StyleZucchini-Pascal, Nathalie, Ludovic Peyre, and Roger Rahmani. 2013. "Crosstalk between Beta-Catenin and Snail in the Induction of Epithelial to Mesenchymal Transition in Hepatocarcinoma: Role of the ERK1/2 Pathway" International Journal of Molecular Sciences 14, no. 10: 20768-20792. https://doi.org/10.3390/ijms141020768