Strengths and Weaknesses of Recently Engineered Red Fluorescent Proteins Evaluated in Live Cells Using Fluorescence Correlation Spectroscopy

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Autocorrelation Curves of Dyes and Purified Proteins in Solution
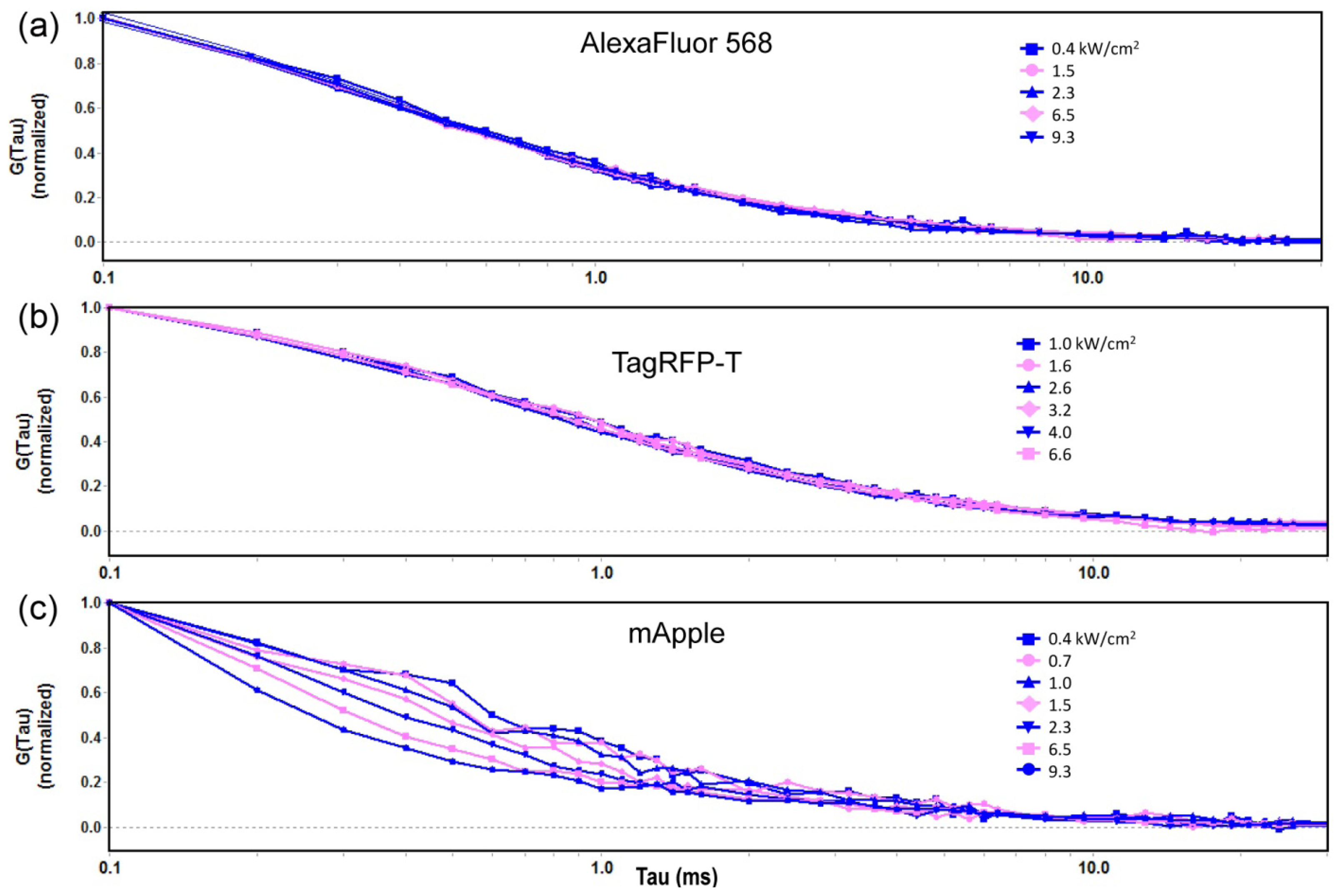
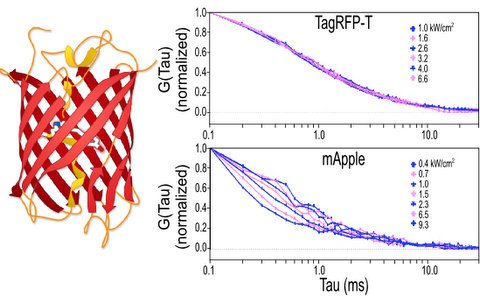
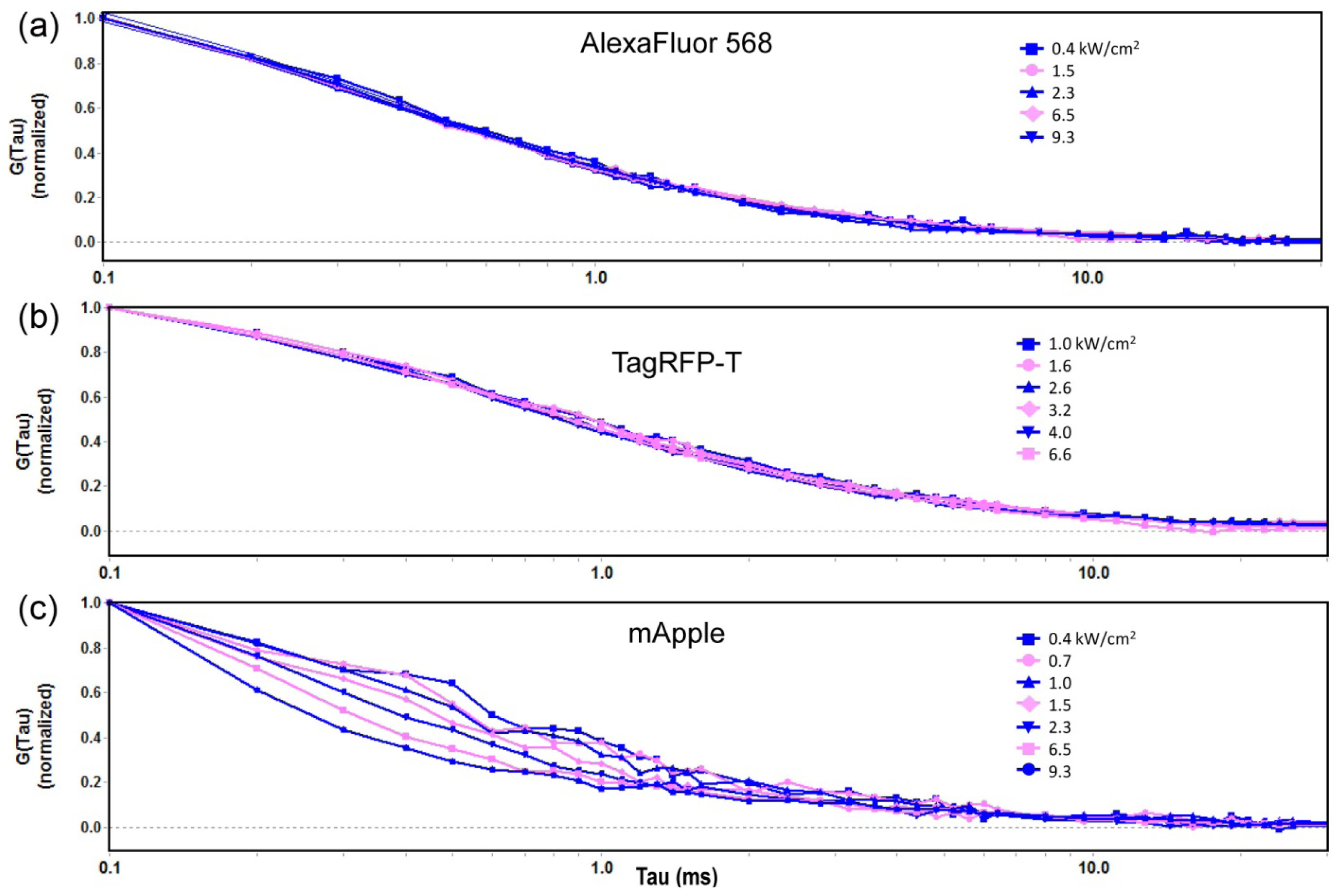
2.1.1. Autocorrelation Curves at a Range of Laser Powers
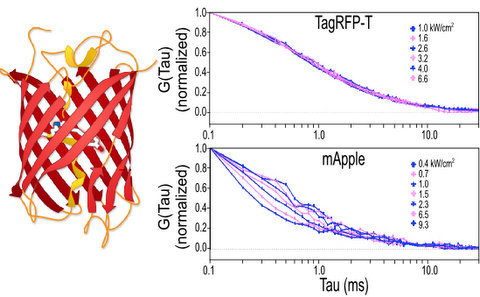
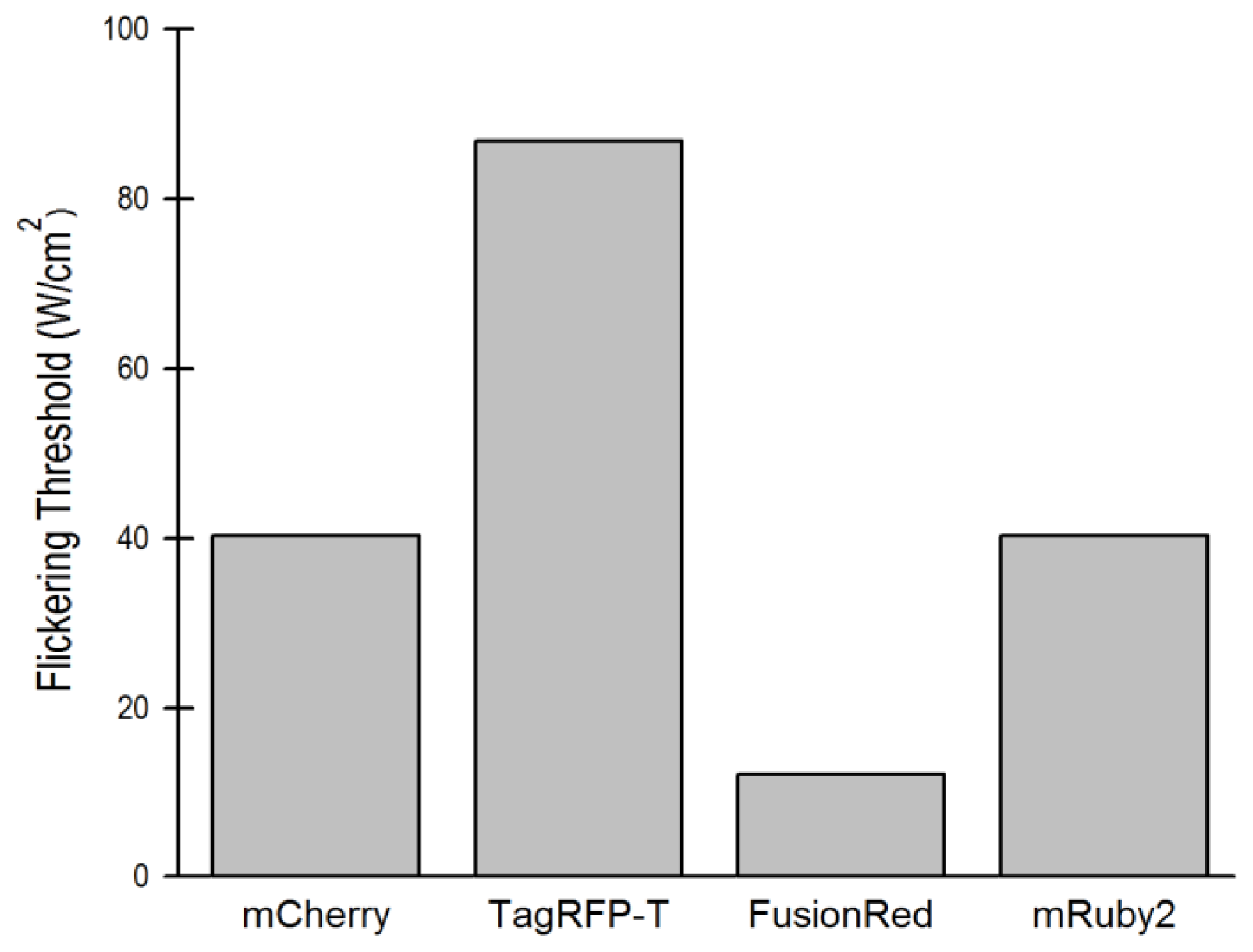
2.1.2. The Effect of Flickering on Red FP Autocorrelation Curves
2.2. Optimization of Excitation Intensity in Live Cells
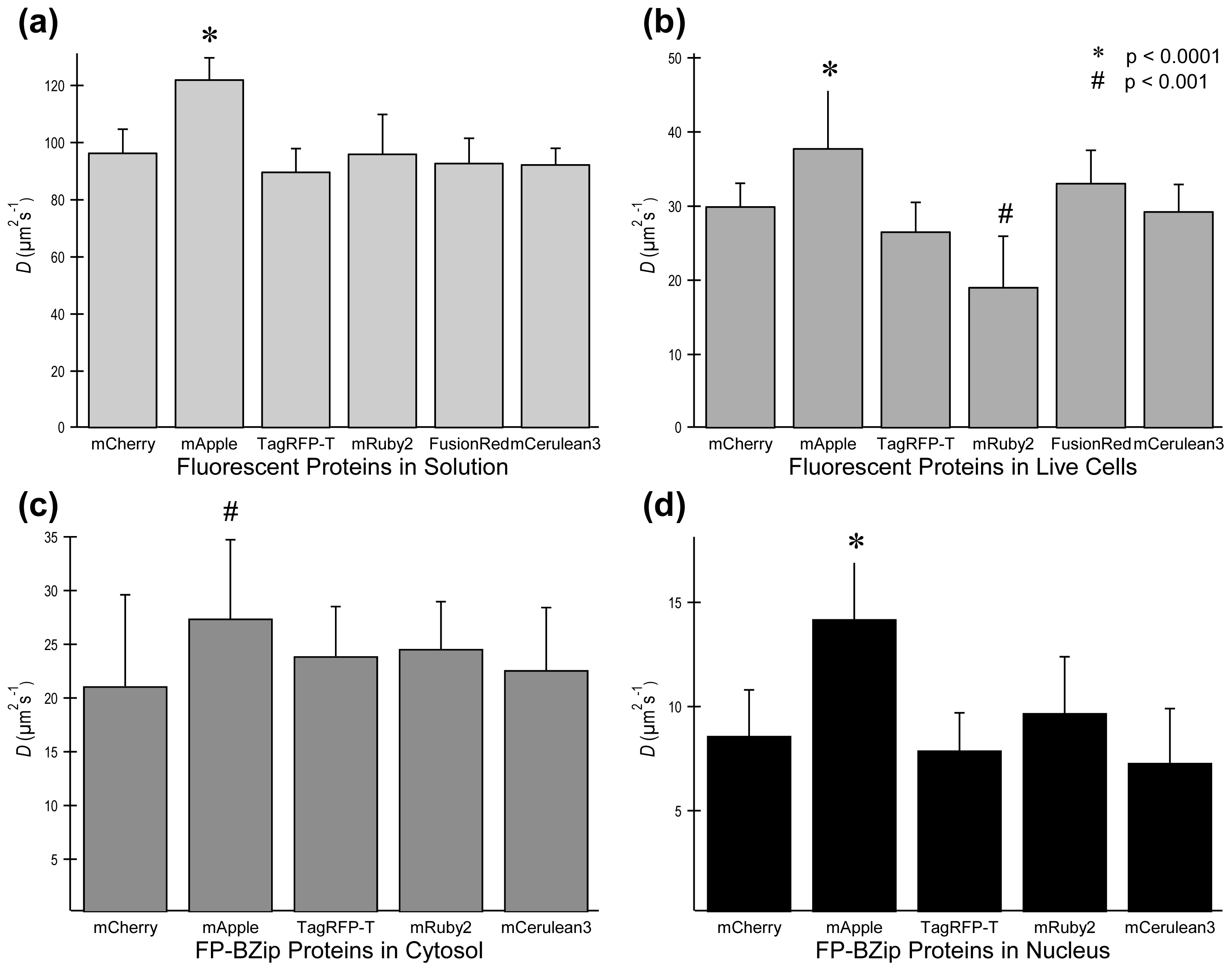
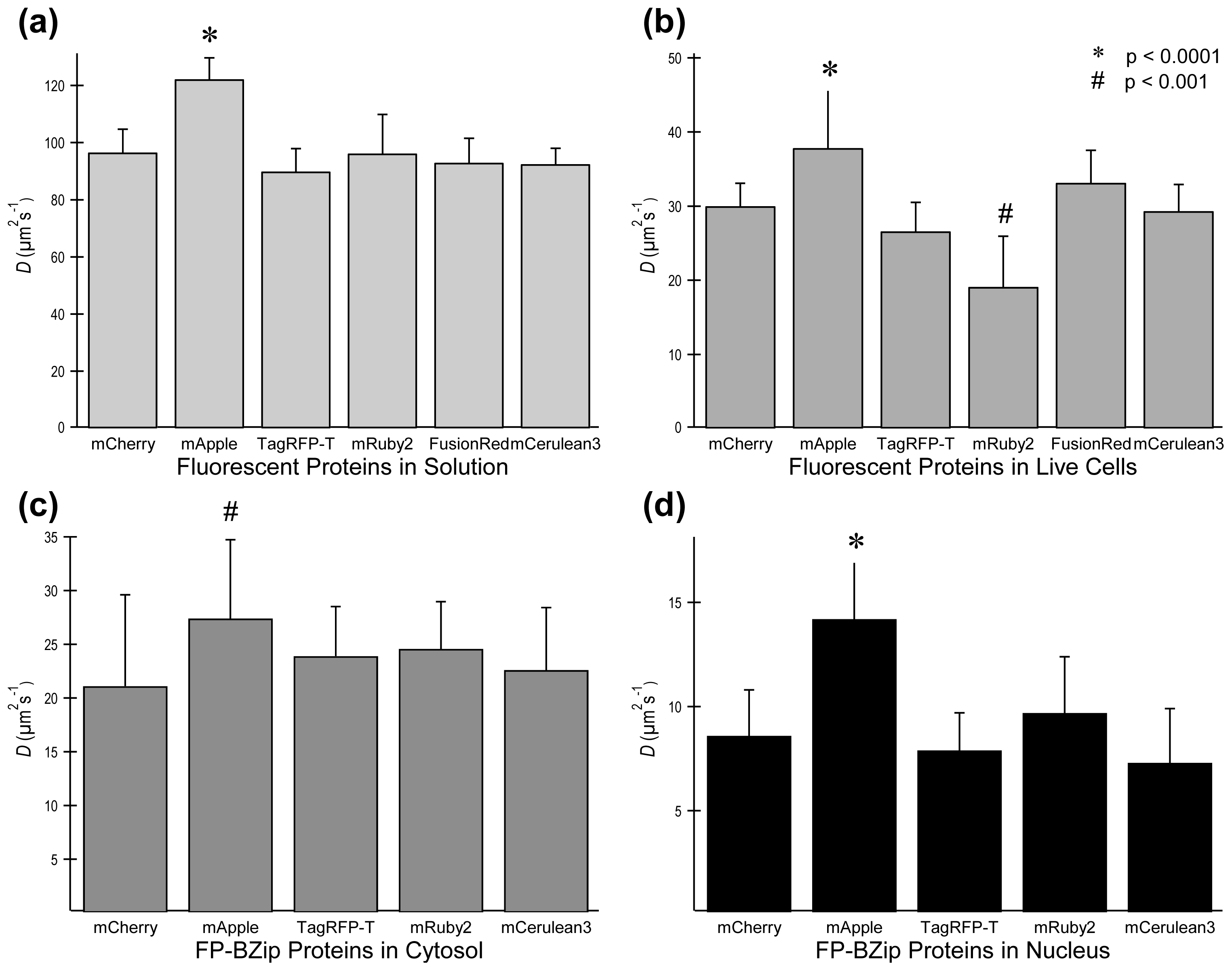
2.3. FP Diffusion Studies
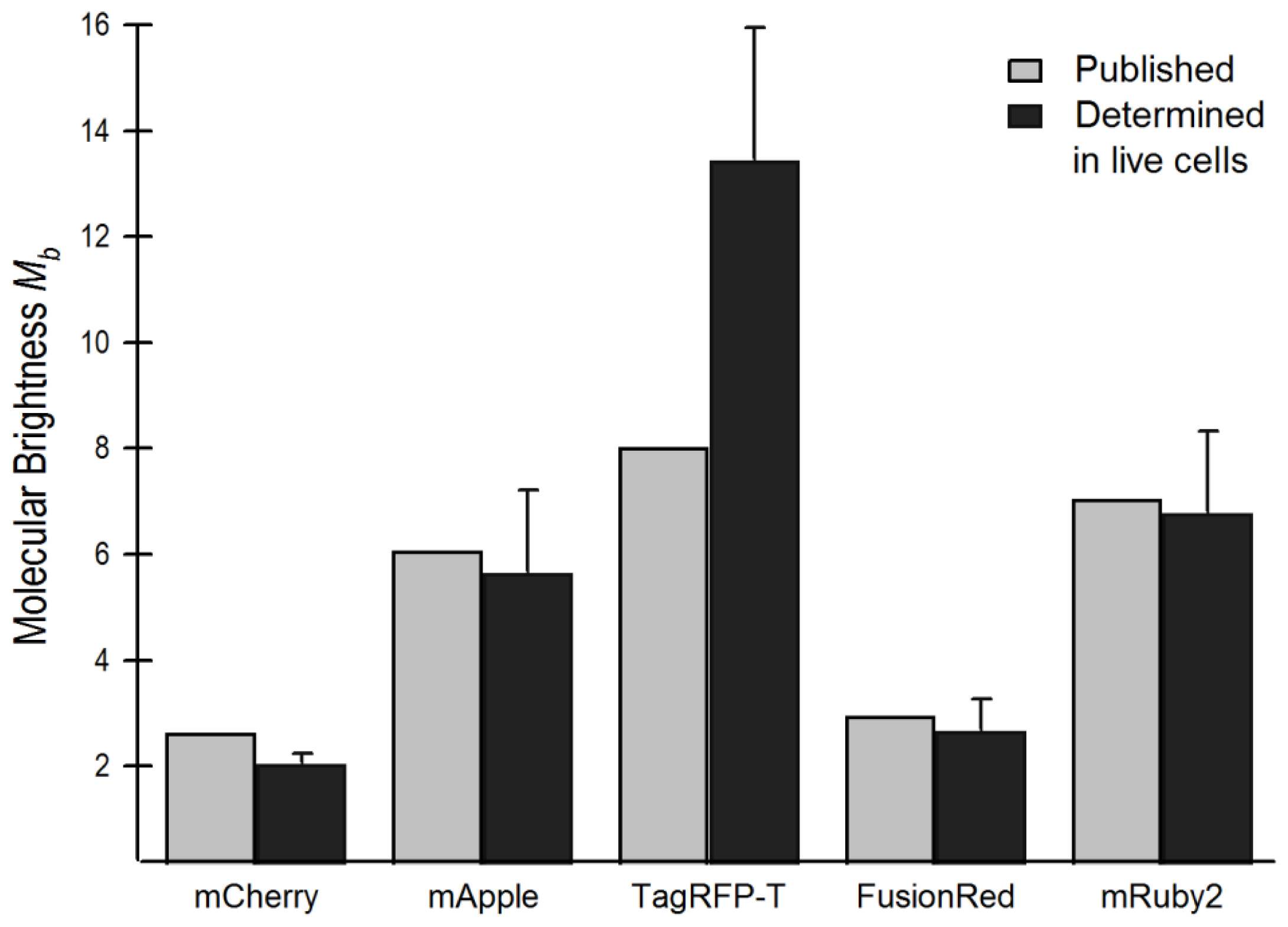
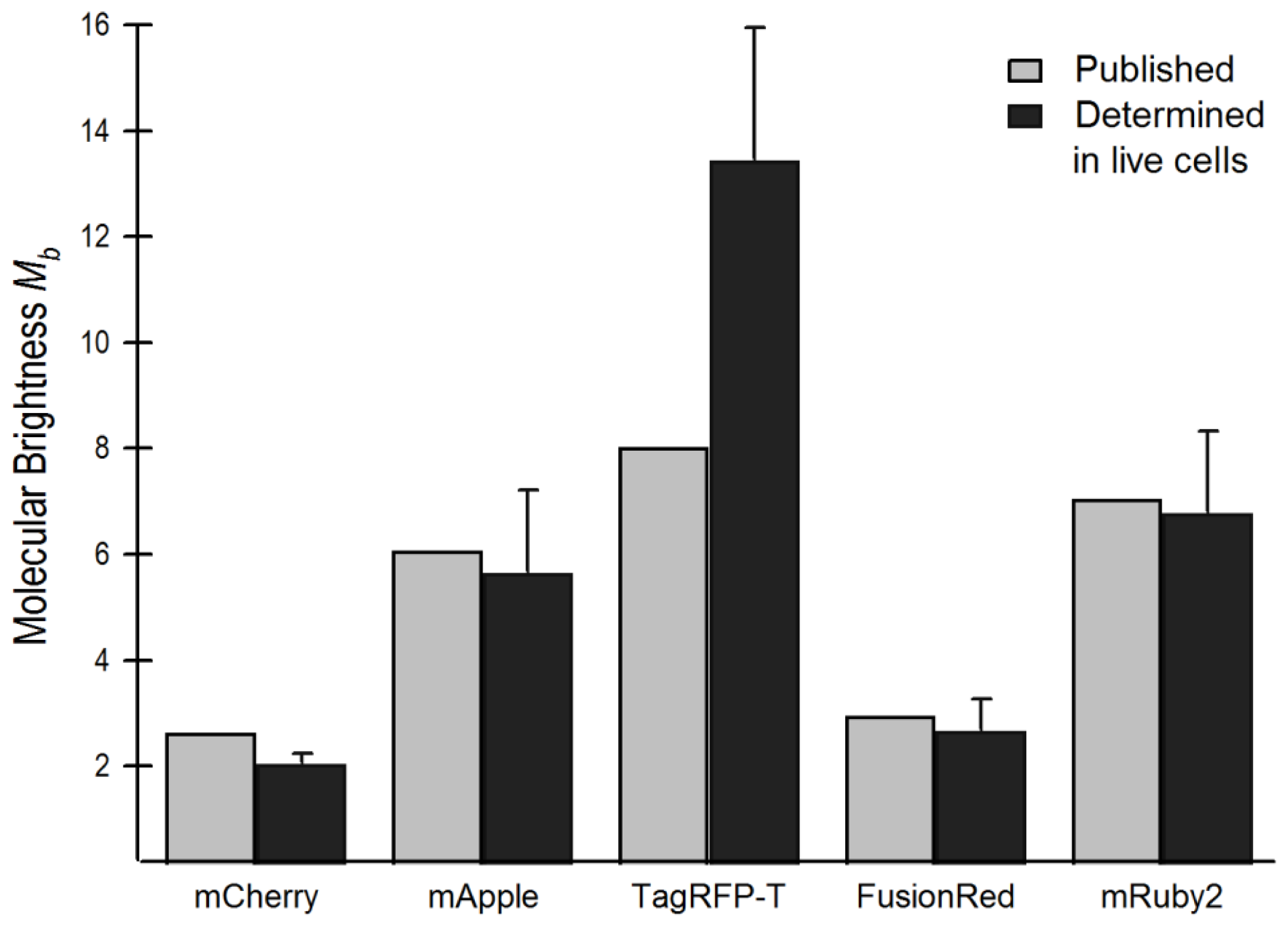
2.4. Brightness Analysis in Live Cells
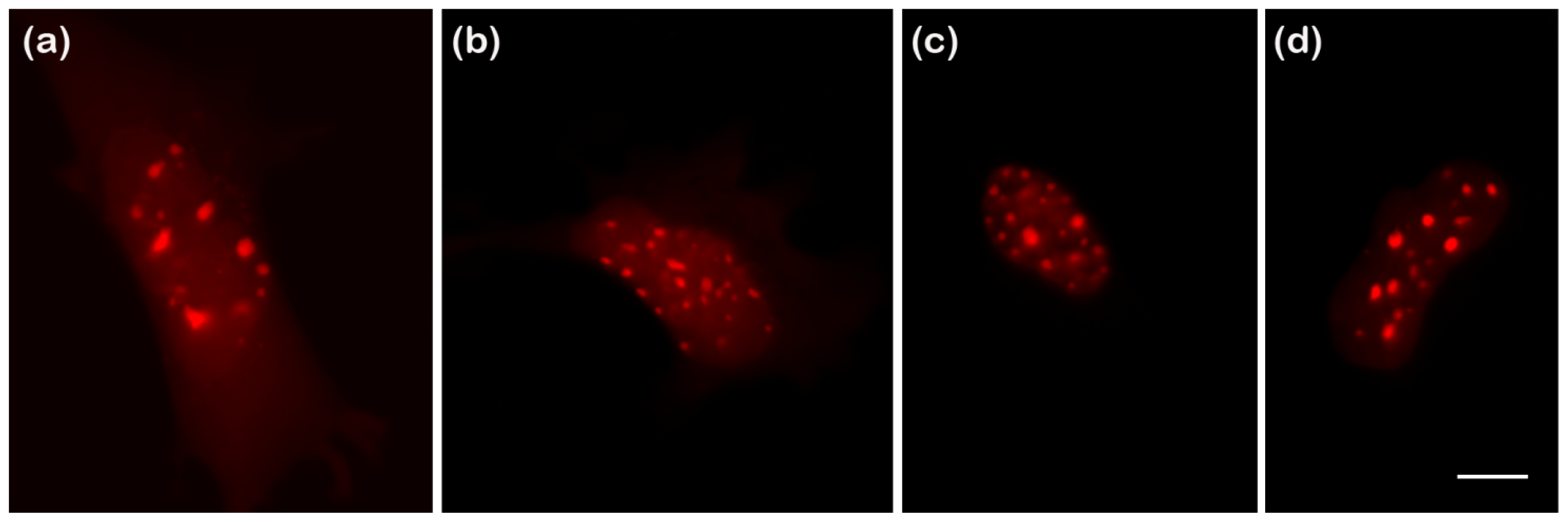
2.4.1. Using Brightness to Determine Oligomeric State
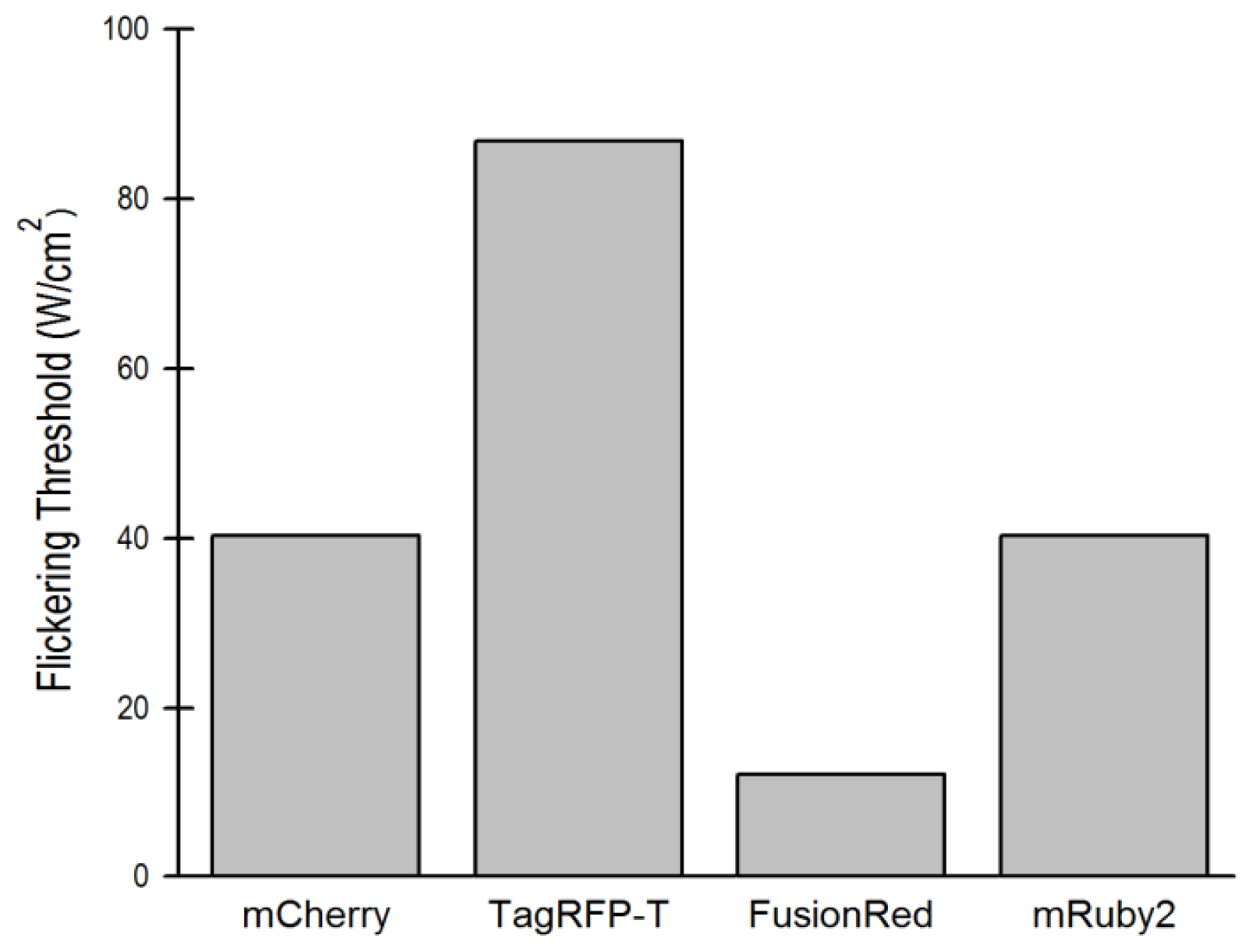
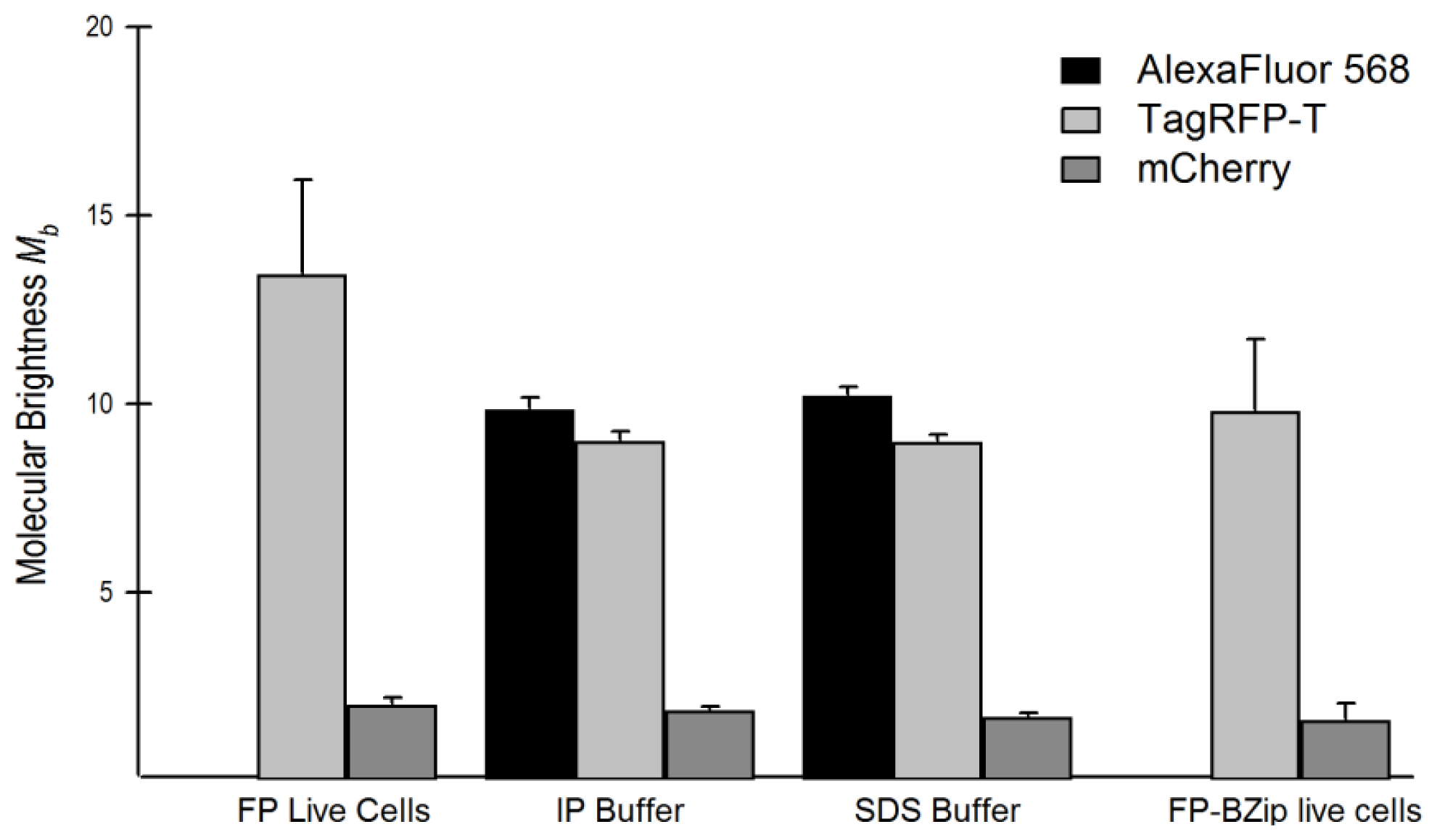
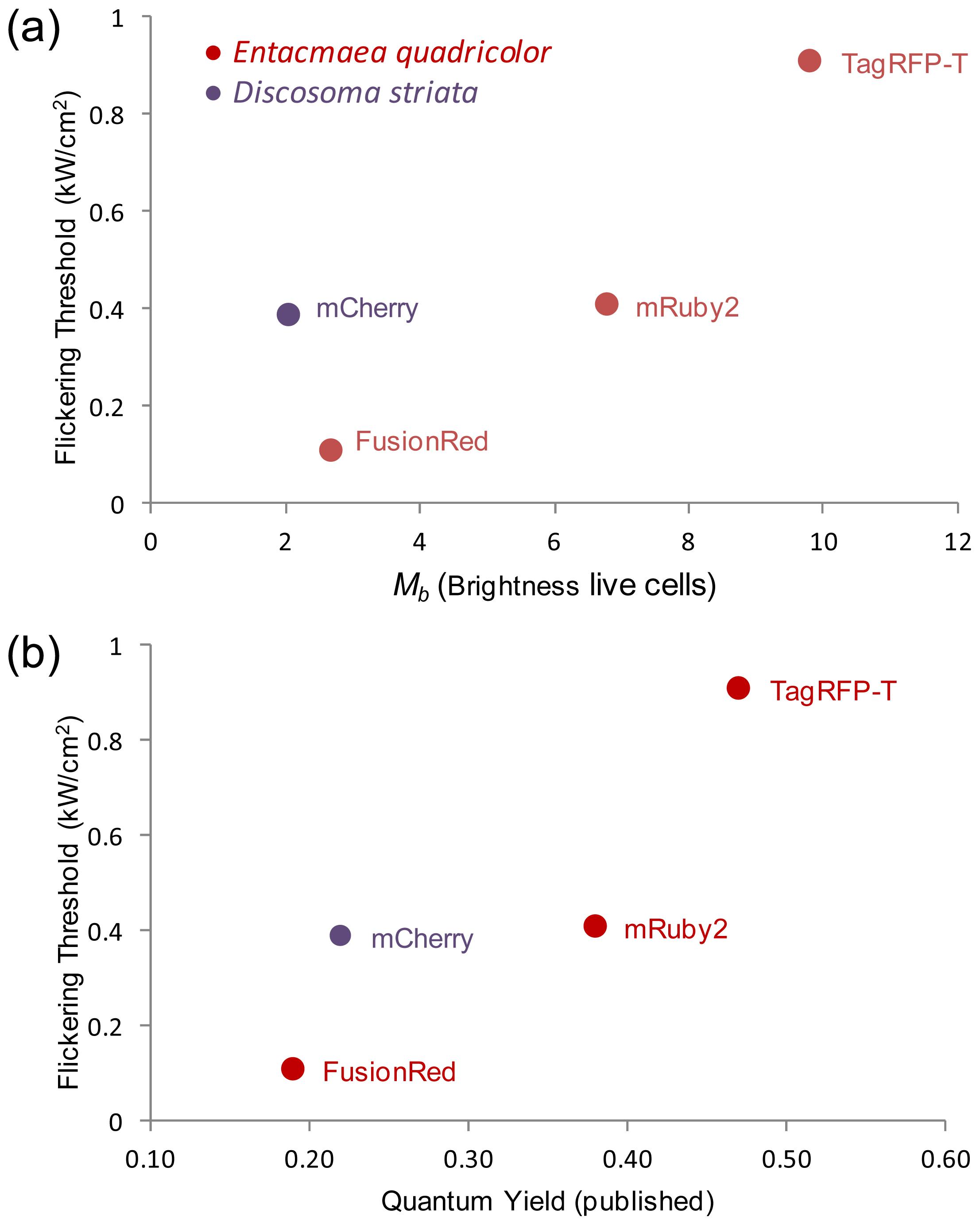
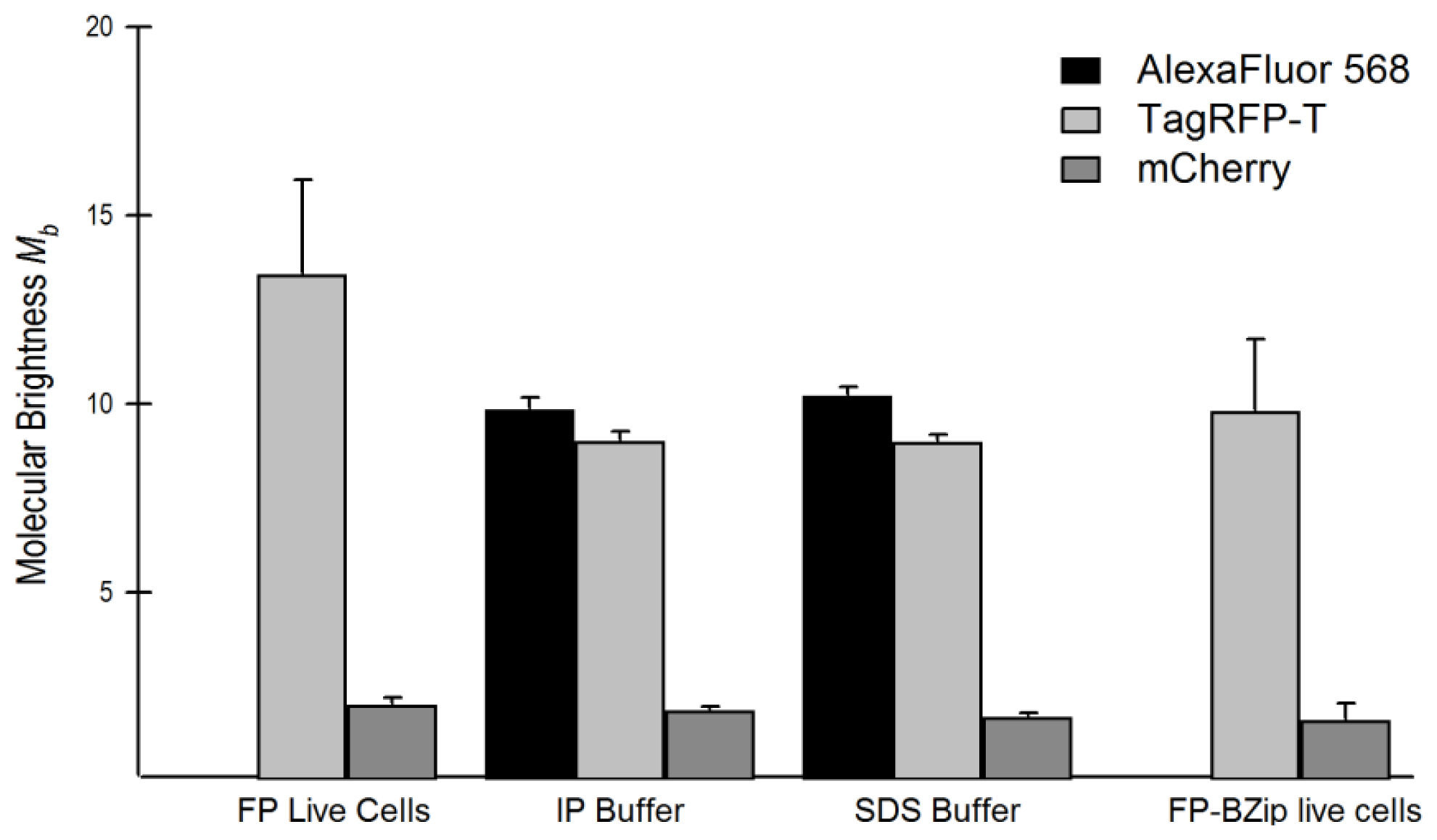
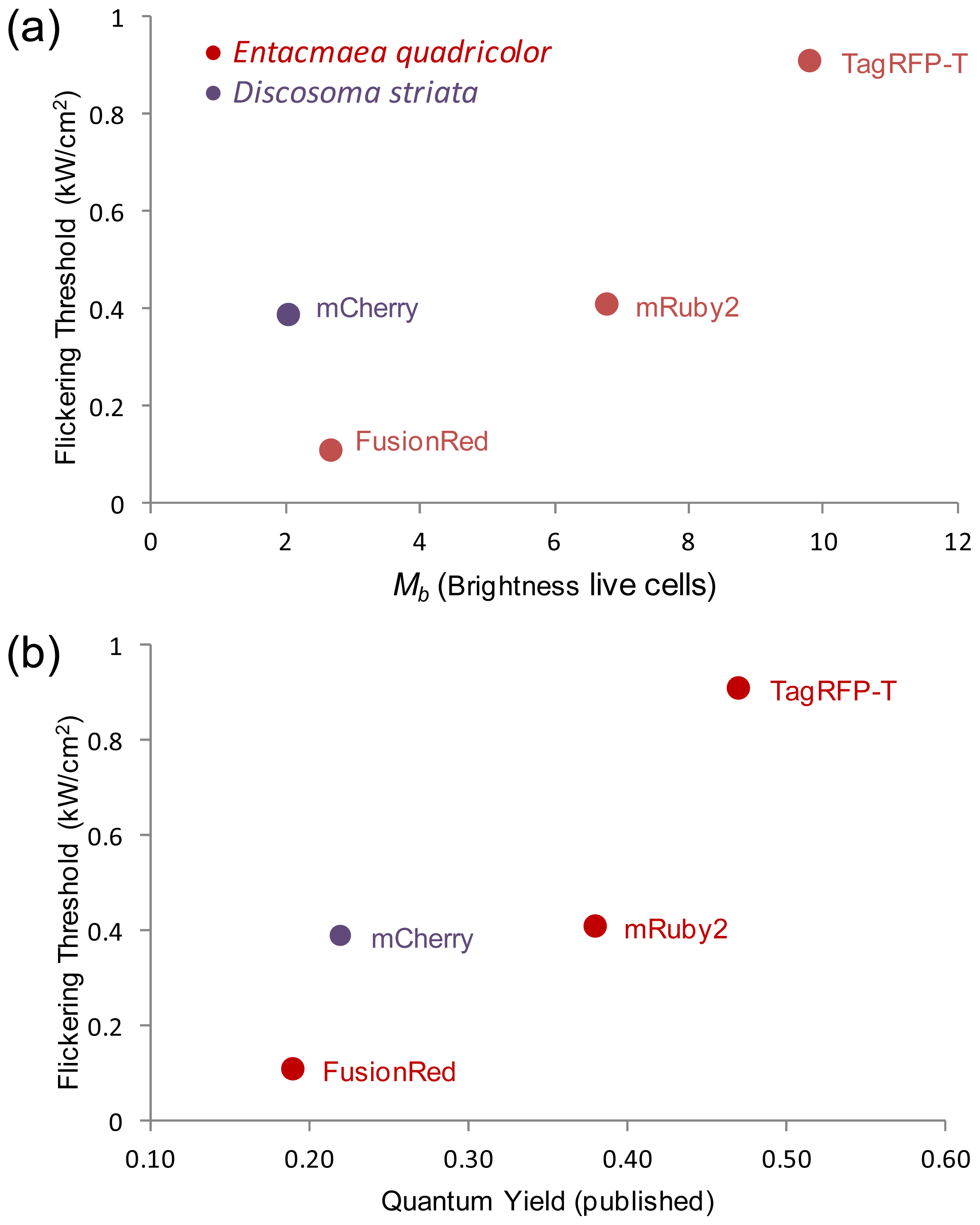
2.5. Relationship of Intrinsic Brightness to Flickering Threshold
3. Experimental Section
3.1. Protein Expression
3.2. Tissue Culture and Transfection
3.3. FCS Measurements and Image Acquisition
4. Conclusions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flickering threshold (kW/cm2) | Optimal power (kW/cm2) | D in soln (μm2/s) | D in live cells (μm2/s) | D FP-BZip in cytosol (μm2/s) | D FP-BZip in nucleus (μm2/s) | Brightness published (a.u.) [ref] | Mb (FPs in live cells) | Mb (FP-BZip live cells) | |
|---|---|---|---|---|---|---|---|---|---|
| mCherry | 0.39 | 0.11 | 96.6 ± 8.1 | 30.0 ± 3.1 | 21.1 ± 8.5 | 8.6 ± 2.2 | 2.62 [6] | 2.0 ± 0.2 | 1.65 |
| mApple | -- | 0.075 | 122.2 ± 7.5 | 37.8 ± 8.5 | 27.4 ± 7.3 | 14.2 ± 4.0 | 6.06 [11] | 5.6 ± 1.6a | n.d. |
| TagRFP-T | 0.91 | 0.10–0.13 | 89.9 ± 8.0 | 26.6 ± 3.9 | 23.9 ± 4.6 | 7.9 ± 1.8 | 8.02 [13] | 13.4 ± 2.5 | 9.8 ± 1.9 |
| mRuby2 | 0.41 | 0.17 | 96.2 ± 5.6 | 19.1 ± 3.6 | 24.6 ± 4.4 | 9.7 ± 2.7 | 7.04 [9] | 6.8 ± 1.6 | 7.0 ± 0.9 |
| FusionRed | 0.11 | 0.11 | 93.0 ± 13.7 | 33.2 ± 6.8 | n.d. | n.d. | 2.95 [8] | 2.7 ± 0.6 | n.d. |
| mCerulean3 | n.d. | n.d. | 92.5 ± 8.5 | 29.3 ± 4.4 | 22.6 ± 5.8 | 7.3 ± 2.6 | n.d. | n.d. | n.d. |
Acknowledgments
Conflicts of Interest
References
- Day, R.N.; Davidson, M.W. The fluorescent protein palette: Tools for cellular imaging. Chem. Soc. Rev 2009, 38, 2887–2921. [Google Scholar]
- Matz, M.V.; Fradkov, A.F.; Labas, Y.A.; Savitsky, A.P.; Zaraisky, A.G.; Markelov, M.L.; Lukyanov, S.A. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotech 1999, 17, 969–973. [Google Scholar]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol 2004, 22, 1567–1572. [Google Scholar]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Meth 2005, 2, 905–909. [Google Scholar]
- Hendrix, J.; Flors, C.; Dedecker, P.; Hofkens, J.; Engelborghs, Y. Dark states in monomeric red fluorescent proteins studied by fluorescence correlation and single molecule spectroscopy. Biophys. J 2008, 94, 4103–4113. [Google Scholar]
- Shu, X.; Shaner, N.C.; Yarbrough, C.A.; Tsien, R.Y.; Remington, S.J. Novel chromophores and buried charges control color in mFruits. Biochemistry 2006, 45, 9639–9647. [Google Scholar]
- Subach, O.M.; Malashkevich, V.N.; Zencheck, W.D.; Morozova, K.S.; Piatkevich, K.D.; Almo, S.C.; Verkhusha, V.V. Structural characterization of acylimine-containing blue and red chromophores in mTagBFP and TagRFP fluorescent proteins. Chem. Biol 2010, 17, 333–341. [Google Scholar]
- Shemiakina, I.; Ermakova, G.; Cranfill, P.; Baird, M.; Evans, R.; Souslova, E.; Staroverov, D.; Gorokhovatsky, A.; Putintseva, E.; Gorodnicheva, T. A monomeric red fluorescent protein with low cytotoxicity. Nat. Commun 2012, 3. [Google Scholar] [CrossRef]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar]
- Kredel, S.; Oswald, F.; Nienhaus, K.; Deuschle, K.; Röcker, C.; Wolff, M.; Heilker, R.; Nienhaus, G.U.; Wiedenmann, J. mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS One 2009, 4, e4391. [Google Scholar]
- Shaner, N.C.; Lin, M.Z.; McKeown, M.R.; Steinbach, P.A.; Hazelwood, K.L.; Davidson, M.W.; Tsien, R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 2008, 5, 545–551. [Google Scholar]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 2007, 4, 555–557. [Google Scholar]
- Dean, K.M.; Lubbeck, J.L.; Binder, J.K.; Schwall, L.R.; Jimenez, R.; Palmer, A.E. Analysis of red-fluorescent proteins provides insight into dark-state conversion and photodegradation. Biophys. J 2011, 101, 961–969. [Google Scholar]
- Siegel, A.P.; Hays, N.M.; Day, R.N. Unraveling transcription factor interactions with heterochromatin protein 1 using fluorescence lifetime imaging microscopy and fluorescence correlation spectroscopy. J. Biomed. Opt 2013, 18. [Google Scholar] [CrossRef]
- Schenk, A.; Ivanchenko, S.; Röcker, C.; Wiedenmann, J.; Nienhaus, G.U. Photodynamics of red fluorescent proteins studied by fluorescence correlation spectroscopy. Biophys. J 2004, 86, 384–394. [Google Scholar]
- Burnette, D.T.; Sengupta, P.; Dai, Y.; Lippincott-Schwartz, J.; Kachar, B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 21081–21086. [Google Scholar]
- Drobizhev, M.; Hughes, T.E.; Stepanenko, Y.; Wnuk, P.; O’Donnell, K.; Scott, J.N.; Callis, P.R.; Mikhaylov, A.; Dokken, L.; Rebane, A. Primary role of the chromophore bond length Alternation in reversible photoconversion of red fluorescence proteins. Sci. Rep 2012, 2. [Google Scholar] [CrossRef]
- Enderlein, J.; Gregor, I. Using fluorescence lifetime for discriminating detector afterpulsing in fluorescence-correlation spectroscopy. Rev. Sci. Instrum 2005, 76, 033102:1–033102:5. [Google Scholar]
- Markwardt, M.L.; Kremers, G.J.; Kraft, C.A.; Ray, K.; Cranfill, P.J.C.; Wilson, K.A.; Day, R.N.; Wachter, R.M.; Davidson, M.W.; Rizzo, M.A. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One 2011, 6, e17896. [Google Scholar]
- Landschulz, W.H.; Johnson, P.F.; McKnight, S.L. The leucine zipper: A hypothetical structure common to a new class of DNA binding proteins. Science 1988, 240, 1759–1764. [Google Scholar]
- Bancaud, A.; Huet, S.; Daigle, N.; Mozziconacci, J.; Beaudouin, J.; Ellenberg, J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J 2009, 28, 3785–3798. [Google Scholar]
- Siegel, A.P.; Hays, N.M.; Day, R.N. Dynamic nuclear protein interactions investigated using fluorescence lifetime and fluorescence fluctuation spectroscopy. Proc. SPIE 2012, 8226. [Google Scholar] [CrossRef]
- Wiedenmann, J.; Schenk, A.; Röcker, C.; Girod, A.; Spindler, K.-D.; Nienhaus, G.U. A far-red fluorescent protein with fast maturation and reduced oligomerization tendency from Entacmaea quadricolor (Anthozoa, Actinaria). Proc. Natl. Acad. Sci.USA 2002, 99, 11646–11651. [Google Scholar]
- Subach, F.V.; Patterson, G.H.; Renz, M.; Lippincott-Schwartz, J.; Verkhusha, V.V. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J. Am. Chem. Soc 2010, 132, 6481–6491. [Google Scholar]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Meth 2013, 10, 407–409. [Google Scholar]
- Gebhardt, J.C.M.; Suter, D.M.; Roy, R.; Zhao, Z.W.; Chapman, A.R.; Basu, S.; Maniatis, T.; Xie, X.S. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat. Methods 2013, 10, 421–426. [Google Scholar]
- Hanson, C.A.; Drake, K.R.; Baird, M.A.; Han, B.; Kraft, L.J.; Davidson, M.W.; Kenworthy, A.K. Overexpression of caveolin-1 is sufficient to phenocopy the behavior of a disease-associated mutant. Traffic 2013, 14, 663–677. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Siegel, A.P.; Baird, M.A.; Davidson, M.W.; Day, R.N. Strengths and Weaknesses of Recently Engineered Red Fluorescent Proteins Evaluated in Live Cells Using Fluorescence Correlation Spectroscopy. Int. J. Mol. Sci. 2013, 14, 20340-20358. https://doi.org/10.3390/ijms141020340
Siegel AP, Baird MA, Davidson MW, Day RN. Strengths and Weaknesses of Recently Engineered Red Fluorescent Proteins Evaluated in Live Cells Using Fluorescence Correlation Spectroscopy. International Journal of Molecular Sciences. 2013; 14(10):20340-20358. https://doi.org/10.3390/ijms141020340
Chicago/Turabian StyleSiegel, Amanda P., Michelle A. Baird, Michael W. Davidson, and Richard N. Day. 2013. "Strengths and Weaknesses of Recently Engineered Red Fluorescent Proteins Evaluated in Live Cells Using Fluorescence Correlation Spectroscopy" International Journal of Molecular Sciences 14, no. 10: 20340-20358. https://doi.org/10.3390/ijms141020340