Carcinogenesis of Pancreatic Adenocarcinoma: Precursor Lesions

 ,
,
Abstract
:1. Introduction
2. Precursor Lesions
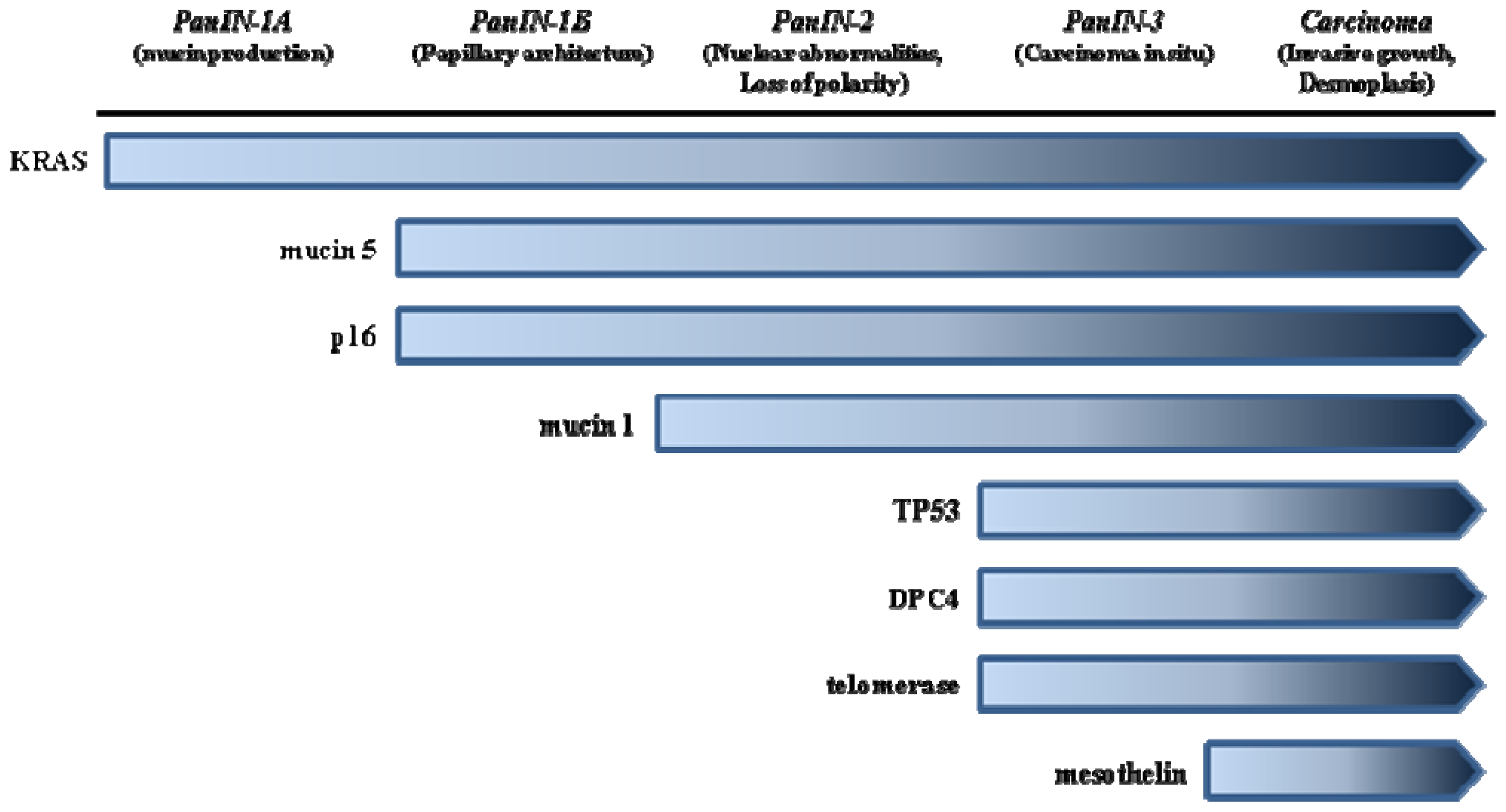
2.1. Pancreatic Intraephitelial Neoplasia (PanIN)
2.2. Intraductal Papillary Mucinous Neoplasms
2.3. Mucinous Cystic Neoplasms
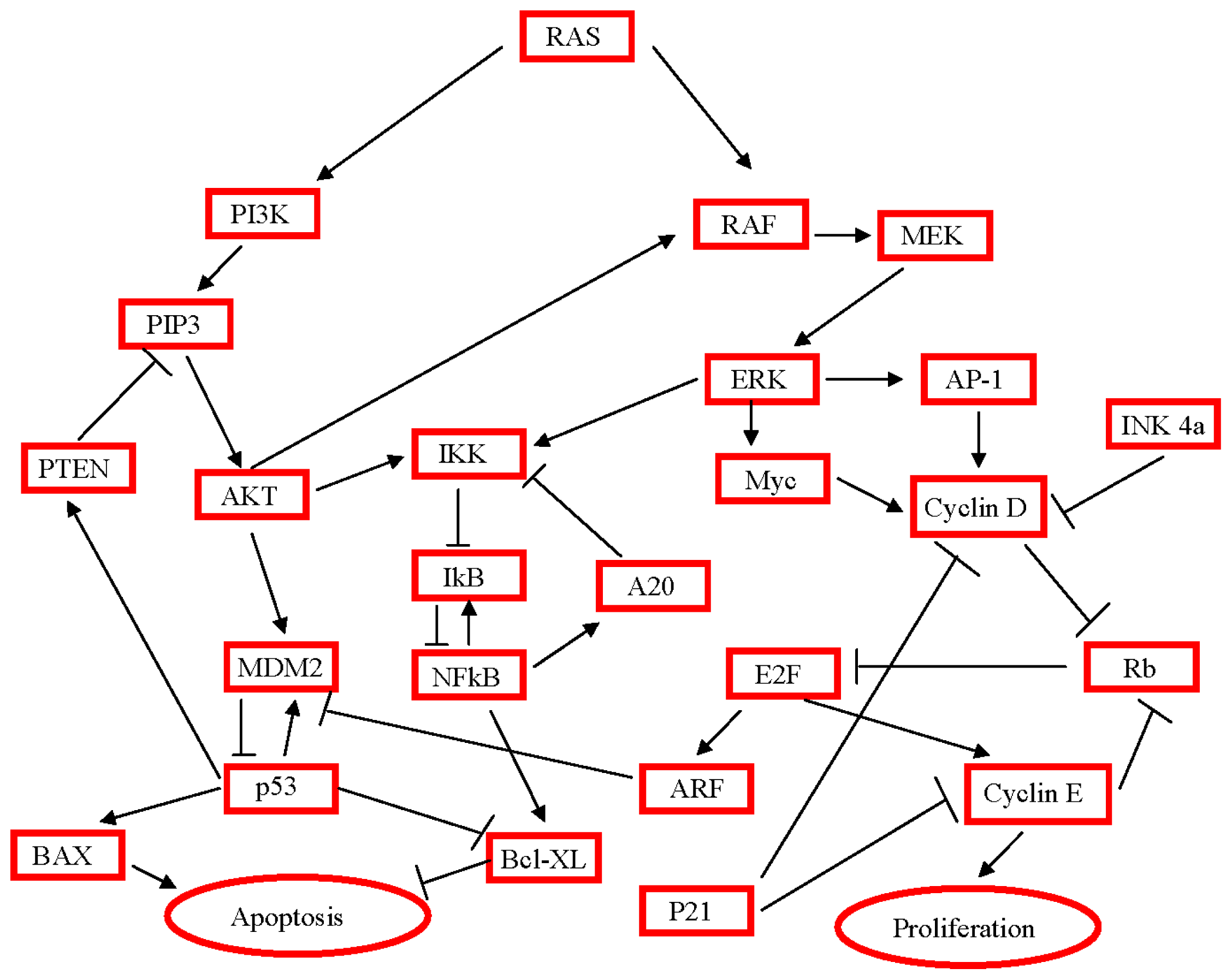
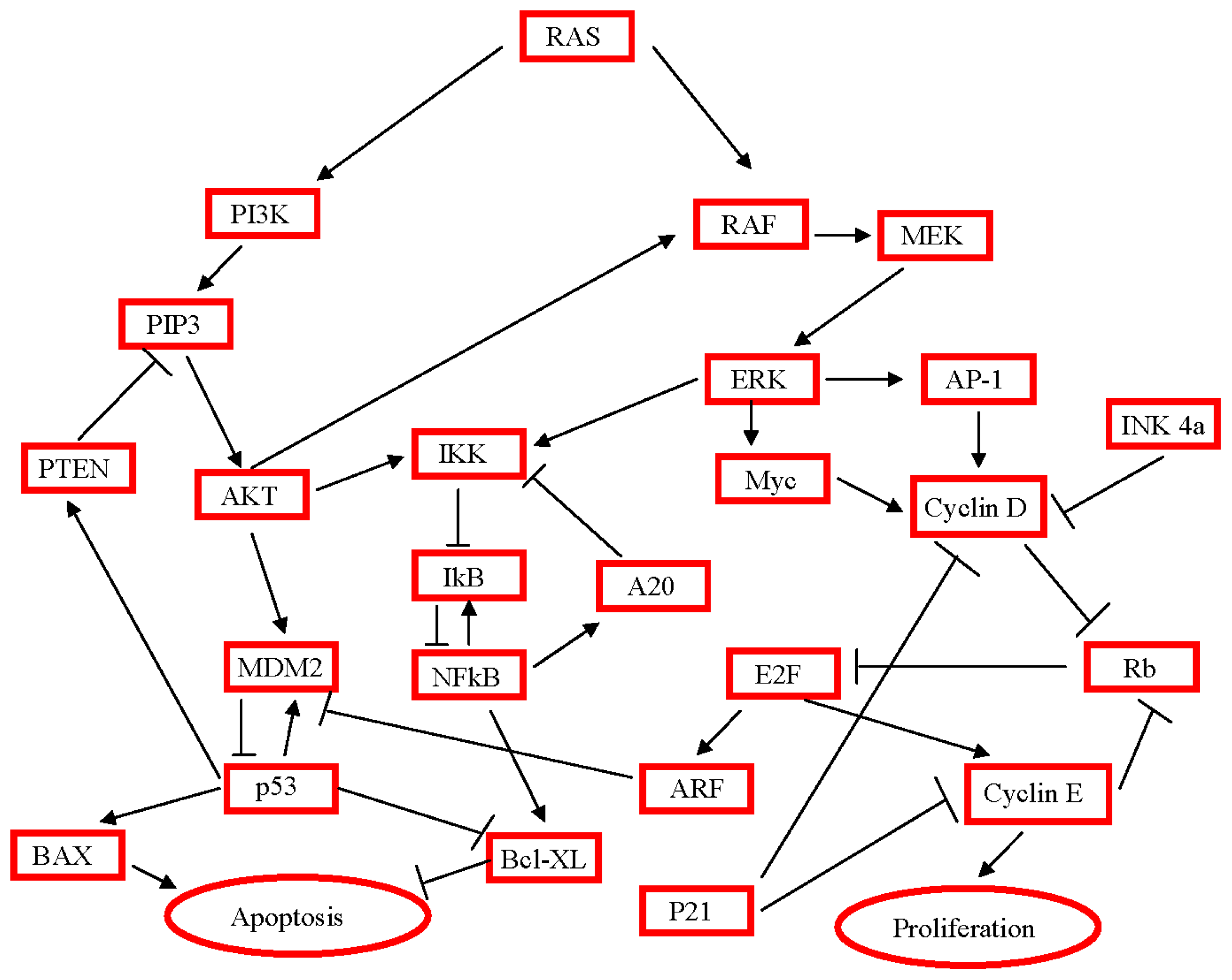
3. Activated Pathways and Altered Processes in Pancreatic Ductal Adenocarcinoma
4. Epithelial to Mesenchymal Transition in Invasion and Metastasis
5. Epigenetic
5.1. Chromatin-Based Epigenetics
5.2. MicroRNA
6. Tumor Microenvironment: Role in Carcinogenesis and Therapeutic Potential
7. Conclusions and Future Perspectives


{kind=link}
{kind=link}
| Molecular target | Genetic aberration (%) | Type aberration |
|---|---|---|
| EGFR | 43%–69% | overexpression |
| VEGF | 93% | overexpression |
| KRAS | 90% | point mutation |
| BRAF | 30% | point mutation |
| AKT2 | 15% | amplification |
| MYB | 10% | amplification |
| IGF-1R | 64% | amplification |
| MMPs | 85% | amplification |
| Hedgeogh | 70% | amplification |
| m-TOR | 65% | overexpression |
| MEK | 72% | point mutation |
| COX-2 | 67%–90% | point mutation |
| Molecular target | Genetic aberration (%) | Type aberration |
|---|---|---|
| p16INK4A/CDKN2A | 40% | deletion |
| P53 | 50%–75% | intragenic mutation |
| DPC4 (Smad4) | 30% | deletion |
Conflicts of Interest
References
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med 2010, 362, 1605–1617. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Parkin, D.M.; Bray, F.I.; Devesa, S.S. Cancer burden in the year 2000. The global picture. Eur. J. Cancer 2001, 37, S4–S66. [Google Scholar]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs. gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol 2006, 20, 197–209. [Google Scholar]
- Koorstra, J.B.; Hustinx, S.R.; Offerhaus, G.J.; Maitra, A. Pancreatic carcinogenesis. Pancreatology 2008, 8, 110–125. [Google Scholar]
- Brat, D.J.; Lillemoe, K.D.; Yeo, C.J.; Warfield, P.B.; Hruban, R.H. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol 1998, 22, 163–169. [Google Scholar]
- Hruban, R.H.; Maitra, A.; Goggins, M. Update on pancreatic intraepithelial neoplasia. Int. J. Clin. Exp. Pathol 2008, 1, 306–316. [Google Scholar]
- Sergeant, G.; Vankelecom, H.; Gremeaux, L.; Topal, B. Role of cancer stem cells in pancreatic ductal adenocarcinoma. Nat. Rev. Clin. Oncol 2009, 6, 580–586. [Google Scholar]
- Hermann, P.C.; Mueller, M.T.; Heeschen, C. Pancreatic cancer stem cells-insights and perspectives. Expert Opin. Biol. Ther 2009, 9, 1271–1278. [Google Scholar]
- Bhagwandin, V.J.; Shay, J.W. Pancreatic cancer stem cells: Fact or fiction? Biochim. Biophys. Acta 2009, 1792, 248–259. [Google Scholar]
- Simeone, D.M. Pancreatic cancer stem cells: Implications for the treatment of pancreatic cancer. Clin. Cancer Res 2008, 14, 5646–5648. [Google Scholar]
- Stelow, E.B.; Adams, R.B.; Moskaluk, C.A. The prevalence of pancreatic intraepithelial neoplasia in pancreata with uncommon types of primary neoplasms. Am. J. Surg. Pathol 2006, 30, 36–41. [Google Scholar]
- Iacobuzio-Donahue, C.A.; Wilentz, R.E.; Argani, P.; Yeo, C.J.; Cameron, J.L.; Kern, S.E. Dpc4 protein in mucinous cystic neoplasms of the pancreas: Frequent loss of expression in invasive carcinomas suggests a role in genetic progression. Am. J. Surg. Pathol 2000, 24, 1544–1548. [Google Scholar]
- Hruban, R.H.; Pitman, M.B.; Klimstra, D.S. Tumors of the Pancreas; Firminger, H.I., Ed.; Atlas of Tumor Pathology Series 4; American Registry of Pathology and Armed Forces Institute of Pathology: Washington, DC, USA, 2007. [Google Scholar]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Compton, C.; Garrett, E.; Goodman, S.N. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol 2001, 25, 579–586. [Google Scholar]
- Yamaguchi, K.; Yokohata, K.; Noshiro, H.; Chijiiwa, K.; Tanaka, M. Mucinous cystic neoplasm of the pancreas or intraductal papillary-mucinous tumour of the pancreas. Eur. J. Surg 2000, 166, 141–148. [Google Scholar]
- Cubilla, A.L.; Fitzgerald, P.J. Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res 1976, 36, 2690–2698. [Google Scholar]
- Hruban, R.H.; Wilentz, R.E.; Kern, S.E. Genetic progression in the pancreatic ducts. Am. J. Pathol 2000, 156, 1821–1825. [Google Scholar]
- Day, J.D.; Digiuseppe, J.A.; Yeo, C. Immunohistochemical evaluation of HER-2/neu expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasms. Hum. Pathol 1996, 27, 119–124. [Google Scholar]
- Wilentz, R.E.; Geradts, J.; Maynard, R. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: Loss of intranuclear expression. Cancer Res 1998, 58, 4740–4744. [Google Scholar]
- Rosty, C.; Geradts, J.; Sato, N.; Wilentz, R.E.; Roberts, H.; Sohn, T.; Cameron, J.L.; Yeo, C.J.; Hruban, R.H.; Goggins, M. p16 inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am. J. Surg. Pathol 2003, 27, 1495–1501. [Google Scholar]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet 1994, 8, 27–32. [Google Scholar]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res 2000, 60, 1835–1839. [Google Scholar]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000, 60, 2002–2006. [Google Scholar]
- Bassi, C.; Sarr, M.G.; Lillemoe, K.D.; Reber, H.A. Natural history of intraductal papillary mucinous neoplasms (IPMN): Current evidence and implications for management. J. Gastrointest. Surg 2008, 12, 645–650. [Google Scholar]
- Kloppel, G.; Kosmahl, M.; Luttges, J. Intraductal neoplasms of the pancreas: Cystic and common. Pathologe 2005, 26, 31–36. [Google Scholar]
- Zamboni, G.; Scarpa, A.; Bogina, G. Mucinous cystic tumors of the pancreas: Clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am. J. Surg. Pathol 1999, 23, 410–422. [Google Scholar]
- Fukushima, N.; Mukai, K. Pancreatic neoplasms with abundant mucus production: Emphasis on intraductal papillary-mucinous tumors and mucinous cystic tumors. Adv. Anat. Pathol 1999, 6, 65–77. [Google Scholar]
- Su, G.H.; Hruban, R.H.; Bansal, R.K. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am. J. Pathol 1999, 154, 1835–1840. [Google Scholar]
- Maire, F.; Hammel, P.; Terris, B. Intraductal papillary and mucinous pancreatic tumour: A new extracolonic tumour in familial adenomatous polyposis. Gut 2002, 51, 446–449. [Google Scholar]
- Tanaka, M.; Chari, S.; Adsay, V.; Fernandez-Del Castillo, C.; Falconi, M.; Shimizu, M.; Yamaguchi, K.; Yamao, K.; Matsuno, S. International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology 2006, 6, 17–32. [Google Scholar]
- Niedergethmann, M.; Grutzmann, R.; Hildebrand, R.; Dittert, D.; Aramin, N.; Franz, M.; Dobrowolsky, F.; Post, S.; Saeger, H.D. Outcome of invasive and noninvasive intraductal papillary-mucinous neoplasms of the pancreas (IPMN): A 10-year experience. World J. Surg 2008, 32, 2253–2260. [Google Scholar]
- Ban, S.; Naitoh, Y.; Ogawa, F. Intraductal papillary mucinous neoplasm (IPMN) of the gastric-type with focal nodular growth of the arborizing papillae: A case of high-grade transformation of the gastric-type IPMN. Virchows. Arch 2006, 449, 112–116. [Google Scholar]
- Adsay, N.V.; Pierson, C.; Sarkar, F. Colloid (mucinous non cystic) carcinoma of the pancreas. Am. J. Surg. Pathol 2001, 25, 26–42. [Google Scholar]
- Adsay, N.V.; Merati, K.; Basturk, O.; Iacpbuzio-Donahue, C.; Levi, E.; Cheng, J.D.; Sarkar, F.H.; Hruban, R.H.; Klimsta, D.S. Pathologically and biologically distinct types of epithelium in intraductal papillary mucinous neoplasms: Delineation of an “intestinal” pathway of carcinogenesis in the pancreas. Am. J. Surg. Pathol 2004, 28, 839–848. [Google Scholar]
- Adsay, N.V.; Adair, C.F.; Heffess, C.S. Intraductal oncocytic papillary neoplasms of the pancreas. Am. J. Surg. Pathol 1996, 20, 980–994. [Google Scholar]
- Ban, S.; Naitoh, Y.; Mino-Kenudson, M. Intraductal papillary mucinous neoplasm (IPMN) of the pancreas: Its histopathologic difference between 2 major types. Am. J. Surg. Pathol 2006, 30, 1561–1569. [Google Scholar]
- Crippa, S.; Partelli, S.; Tamburrino, D.; Falconi, M. The natural history of a branch-duct intraductal papillary mucinous neoplasm of the pancreas. Surgery 2012. [Google Scholar] [CrossRef]
- Dal Molin, M.; Matthaei, H.; Wu, J.; Blackford, A.; Debeljak, M.; Rezaee, N.; Wolfgang, C.L.; Butturini, G.; Salvia, R.; Bassi, C.; et al. Clinicopathological correlates of activating GNAS mutations in Intraductal Papillary Mucinous Neoplasm (IPMN) of the pancreas. Ann. Surg. Oncol 2013, in press. [Google Scholar]
- Sessa, F.; Solcia, E.; Capella, C.; Bonato, M.; Scarpa, A.; Zamboni, G.; Pellegata, N.S.; Ranzani, G.N.; Rickaert, F.; Kloppel, G. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: An investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows. Arch 1994, 425, 357–367. [Google Scholar]
- Biankin, A.V.; Biankin, S.A.; Kench, J.G. Aberrant p16 (INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut 2002, 50, 861–868. [Google Scholar]
- Sato, N.; Ueki, T.; Fukushima, N.; Lacobuzio-Donahue, C.A.; Yeo, C.J.; Cameron, J.L.; Hruban, R.H.; Goggins, M. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 2002, 123, 365–372. [Google Scholar]
- House, M.G.; Guo, M.; Iacobuzio-Donahue, C.; Herman, J.G. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis 2003, 24, 193–198. [Google Scholar]
- Pitman, M.B.; Michaels, P.J.; Deshpande, V. Cytological and cyst fluid analysis of small (< or = 3 cm) branch duct intraductal papillary mucinous neoplasms adds value to patient management decisions. Pancreatology 2008, 8, 277–284. [Google Scholar]
- Sakorafas, G.H.; Sarr, M.G.; van de Velde, C.J. Intraductal papillary mucinous neoplasms of the pancreas: A surgical perspective. Surg. Oncol 2005, 14, 155–178. [Google Scholar]
- Sho, M.; Nakajima, Y.; Kanehiro, H.; Hisanaga, M.; Nishio, K.; Nagao, M.; Ikeda, N.; Kanokogi, H.; Yamada, T.; Nakano, H. Pattern of recurrence after resection for intraductal papillary mucinous tumors of the pancreas. World J. Surg 1998, 22, 874–878. [Google Scholar]
- Raut, C.P.; Cleary, K.R.; Staerkel, G.A.; Abruzzese, J.L.; Wolff, R.A.; Lee, J.H.; Vauthey, J.N.; Lee, J.E.; Pisters, P.W.; Evans, D.B. Intraductal papillary mucinous neoplasms of the pancreas: Effect of invasion and pancreatic margin status on recurrence and survival. Ann. Surg. Oncol 2006, 13, 582–594. [Google Scholar]
- Reddy, R.P.; Smyrk, T.C.; Zapiach, M.; Levy, M.J.; Pearson, R.K.; Clain, J.E.; Farnell, M.B.; Sarr, M.G.; Chari, S.T. Pancreatic mucinous cystic neoplasm defined by ovarian stroma: Demographics, clinical features, and prevalence of cancer. Clin. Gastroenterol. Hepatol 2004, 2, 1026–1031. [Google Scholar]
- Campbell, F.; Azadeh, B. Cystic neoplasms of the exocrine pancreas. Histopathology 2008, 52, 539–551. [Google Scholar]
- Zamboni, G.; Capelli, P.; Pesci, A.; Brighenti, A. Pathology of Cystic Tumor. In Imaging of the Pancreas: Cystic and Rare Tumors; Procacci, C., Megibow, A.J., Eds.; Springer-Verlag: Berlin, Germany, 2003; pp. 9–30. [Google Scholar]
- Erdogan, D.; Lamers, W.H.; Offerhaus, G.J.; Busch, O.R.; Gouma, D.J.; van Gulik, T.M. Cystadenomas with ovarian stroma in liver and pancreas: An evolving concept. Dig. Surg 2006, 23, 186–191. [Google Scholar]
- Flejou, J.F.; Boulange, B.; Bernades, P.; Belghiti, J.; Henin, D. p53 protein expression and DNA ploidy in cystic tumors of the pancreas. Pancreas 1996, 13, 247–252. [Google Scholar]
- Bartsch, D.; Bastian, D.; Barth, P. K-ras oncogene mutations indicate malignancy in cystic tumors of the pancreas. Ann. Surg 1998, 228, 79–86. [Google Scholar]
- Wenig, B.M.; Albores-Saavedra, J.; Buetow, P.C.; Heffess, C.S. Pancreatic mucinous cystic neoplasm with sarcomatous stroma: A report of three cases. Am. J. Surg. Pathol 1997, 21, 70–80. [Google Scholar]
- Longnecker, D.S.; Adler, G.; Hruban, R.H.; Kloppel, G. Intraductal Papillary-Mucinous Neoplasms of the Pancreas. In Pathology and Genetics of Tumours of the Digestive System. WHO Classification of Tumours; Hamilton, S.R., Aaltonen, L.A., Eds.; IARC Press: Lyon, France, 2000; pp. 237–240. [Google Scholar]
- Fernandez-del Castillo, C.; Targarona, J.; Thayer, S.P.; Rattner, D.W.; Brugge, W.R.; Warshaw, A.L. Incidental pancreatic cysts: Clinicopathologic characteristics and comparison with symptomatic patients. Arch. Surg 2003, 138, 427–433. [Google Scholar]
- Sahani, D.V.; Kadavigere, R.; Saokar, A.; Fernandez-Del Castillo, C.; Brugge, W.R.; Hahn, P.F. Cystic pancreatic lesions: A simple imaging-based classification system for guiding management. Radiographics 2005, 25, 1471–1484. [Google Scholar]
- Takaori, K.; Tanigawa, N. Laparoscopic pancreatic resection: The past, present, and future. Surg. Today 2007, 37, 535–545. [Google Scholar]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar]
- Hruban, R.H.; van Mansfeld, A.D.; Offerhaus, G.J.; van Weering, D.H.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bos, J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol 1993, 143, 545–554. [Google Scholar]
- Lohr, M.; Kloppel, G.; Maisonneuve, P. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 2005, 7, 17–23. [Google Scholar]
- Schneider, G.; Schmid, R.M. Genetic alterations in pancreatic carcinoma. Mol. Cancer 2003, 2, 15. [Google Scholar]
- Tada, M.; Omata, M.; Kawai, S.; Saisho, H.; Ohto, M.; Saiki, R.K.; Sninsky, J.J. Detection of Ras gene mutations in pancreatic juice and peripheral blood of patients with pancreatic adenocarcinoma. Cancer Res 1993, 53, 2472–2474. [Google Scholar]
- Jimeno, A.; Hidalgo, M. Molecular biomarkers: Their increasing role in the diagnosis, characterization, and therapy guidance in pancreatic cancer. Mol. Cancer Ther 2006, 5, 787–796. [Google Scholar]
- End, D.W.; Smets, G.; Todd, A.V. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001, 61, 131–137. [Google Scholar]
- Van Cutsem, E.; van de Velde, H.; Karasek, P. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol 2004, 22, 1430–1438. [Google Scholar]
- Laheru, D.; Shah, P.; Rajeshkumar, N.V. Integrated preclinical and clinical development of S-trans,trans-farnesylthiosalicylic acid (FTS), Salirasib in pancreatic cancer. Invest. New Drugs 2012, 30, 2391–2399. [Google Scholar]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar]
- Schonleben, F.; Qiu, W.L.; Ciau, N.T.; Ho, D.J.; Li, X.J.; Allendorf, J.D.; Remotti, H.E.; Su, G.H. PIK-3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clin. Cancer Res 2006, 12, 3851–3855. [Google Scholar]
- Asano, T.; Yao, Y.; Zhu, J.; Li, D.; Abbruzzese, J.L.; Reddy, S.A. The PI 3-kinase/Akt signalling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene 2004, 23, 8571–8580. [Google Scholar]
- Feig, L.A. Ral GTPases: Approaching their 15 min of fame. Trends Cell Biol 2003, 13, 419–425. [Google Scholar]
- Lim, K.H.; O’Hayer, K.; Adam, S.J.; Kendall, S.D.; Campbell, P.M.; Der, C.J.; Counter, C.M. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol 2006, 16, 2385–2394. [Google Scholar]
- Elghazi, L.; Weiss, A.J.; Barker, D.J.; Callaghan, J.; Staloch, L.; Sandgren, E.P. Regulation of pancreas plasticity and malignant transformation by Akt signaling. Gastroenterology 2009, 136, 1091–1103. [Google Scholar]
- Stanger, B.Z.; Stiles, B.; Lauwers, G.Y.; Bardeesy, N.; Mendoza, M.; Wang, Y. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 2005, 8, 185–195. [Google Scholar]
- Ying, H.; Elpekc, K.G.; Vinjamoori, A.; Zimmermanh, S.M.; Chua, G.C.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. Pten is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB-cytokine network. Cancer Discov 2011, 1, 158–169. [Google Scholar]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res 2008, 14, 5995–6004. [Google Scholar]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res 2007, 67, 187–219. [Google Scholar]
- Hidalgo, M.; Maitra, A. The hedgehog pathway and pancreatic cancer. N. Engl. J. Med 2009, 361, 2094–2096. [Google Scholar]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev 2009, 23, 24–36. [Google Scholar]
- Onishi, H.; Morifuji, Y.; Kai, M.; Suyama, K.; Iwasaki, H.; Katano, M. Hedgehog inhibitor decreases chemosensitivity to 5-fluorouracil and gemcitabine under hypoxic conditions in pancreatic cancer. Cancer Sci 2012, 103, 1272–1279. [Google Scholar]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem 2009, 52, 4400–4418. [Google Scholar]
- Sjolund, J.; Manetopoulos, C.; Stockhausen, M.T.; Axelson, H. The Notch pathway in cancer: Differentiation gone awry. Eur. J. Cancer 2005, 41, 2620–2629. [Google Scholar]
- Wang, Z.; Zhang, Y.; Li, Y.; Banerjee, S.; Liao, J.; Sarkar, F.H. Down-regulation of Notch -1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol. Cancer Ther 2006, 5, 483–493. [Google Scholar]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar]
- Miwa, W.; Yasuda, J.; Murakami, Y.; Yashima, K.; Sugano, K.; Sekine, T.; Kono, A.; Egawa, S.; Yamaguchi, K.; Hayashizaki, Y.; et al. Isolation of DNA sequences amplified at chromosome 19q13.1-q13.2 including the AKT2 locus in human pancreatic cancer. Biochem. Biophys. Res. Commun 1996, 225, 968–974. [Google Scholar]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar]
- Wallrapp, C.; Muller-Pillasch, F.; Solinas-Toldo, S.; Lichter, P.; Friess, H.; Buchler, M.; Fink, T.; Adler, G.; Gress, T.M. Characterization of a high copy number amplification at 6q24 in pancreatic cancer identifies c-myb as a candidate oncogene. Cancer Res 1997, 57, 3135–3139. [Google Scholar]
- Hezel, A.F. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006, 13, 1218–1249. [Google Scholar]
- Gerdes, B. Tumor-suppressing pathways in cystic pancreatic tumors. Pancreas 2003, 13, 42–48. [Google Scholar]
- Russo, A.A.; Tong, L.; Lee, J.O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 1998, 395, 237–243. [Google Scholar]
- De vos Nederveen Cappel, W.H.; Lagendijk, M.A.; Lamers, C.B.; Morreau, H.; Vasen, H.F. Surveillance for familial pancreatic cancer. Scand. J. Gastroenterol. Suppl 2003, 329, 94–99. [Google Scholar]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell. Biol 2013, 15, 2–8. [Google Scholar]
- Redston, M.S.; Caldas, C.; Seymour, A.B.; Hruban, R.H.; da Costa, L.; Yeo, C.J.; Kern, S.E. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54, 3025–3033. [Google Scholar]
- Weiss, R.H.; Marshall, D.; Howard, L.; Corbacho, A.M.; Cheung, A.T.; Sawai, E.T. Suppression of breast cancer growth and angiogenesis by an antisense oligodeoxynucleotide to p21 (Waf1/Cip1). Cancer Lett 2003, 189, 39–48. [Google Scholar]
- Garcea, G.; Neal, C.P.; Pattenden, C.J.; Steward, W.P.; Berry, D.P. Molecular prognostic markers in pancreatic cancer: A systematic review. Eur. J. Cancer 2005, 41, 2213–2236. [Google Scholar]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar]
- Gaspar, N.J.; Li, L.Y.; Kapoun, A.M.; Medicherla, S.; Reddy, M.; Li, G.; O’Young, G.; Quon, D.; Henson, M.; Damm, D.L.; et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol. Pharmacol 2007, 72, 152–161. [Google Scholar]
- Maitra, A.; Kern, S.E.; Hruban, R.H. Molecular pathogenesis of pancreatic cancer. Best Pract. Res. Clin. Gastroenterol 2006, 20, 211–226. [Google Scholar]
- Han, H.J.; Yanagisawa, A.; Kato, Y.; Park, J.G.; Nakamura, Y. Genetic instability in pancreatic cancer and poorly differentiated type of gastric cancer. Cancer Res 1993, 53, 5087–5089. [Google Scholar]
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol 1998, 152, 1501–1507. [Google Scholar]
- Marcus, V.A.; Madlensky, L.; Gryfe, R. Immunohistochemistry for hMLH1 and hMSH2: A practical test for DNA mismatch repairdeficient tumors. Am. J. Surg. Pathol 1999, 23, 1248–1255. [Google Scholar]
- Tomaszewska, R.; Okon, K.; Stachura, J. Expression of the DNA mismatch repair proteins (hMLH1 and hMSH2) in infiltrating pancreatic cancer and its relation to some phenotypic features. Pol. J. Pathol 2003, 54, 31–37. [Google Scholar]
- Couch, F.J.; Johnson, M.R.; Rabe, K.; Boardman, L.; McWilliams, R.; de Andrade, M.; Petersen, G. Germ line fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res 2005, 65, 383–386. [Google Scholar]
- Van der Heijden, M.S.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res 2003, 63, 2585–2588. [Google Scholar]
- Hahn, S.A.; Greenhalf, B.; Ellis, I.; Sina-Frey, M.; Rieder, H.; Korte, B.; Gerdes, B.; Kress, R.; Ziegler, A.; Raeburn, J.A.; et al. BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst 2003, 95, 214–221. [Google Scholar]
- Gallmeier, E.; Calhoun, E.S.; Rago, C.; Brody, J.R.; Cunningham, S.C.; Hucl, T.; Gorospe, M.; Kohli, M.; Lengauer, C.; Kern, S.E. Targeted disruption of FANCC and FANCG in human cancer provides a preclinical model for specific therapeutic options. Gastroenterology 2006, 130, 2145–2154. [Google Scholar]
- Micalizzi, D.S.; Ford, H.L. Epithelial-mesenchymal transition in development and cancer. Future Oncol 2009, 5, 1129–1143. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest 2009, 119, 1420–1428. [Google Scholar]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res 2006, 66, 46–51. [Google Scholar]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; Depinho, R.A.; Jacks, T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar]
- Maier, H.J.; Schmidt-Strassburger, U.; Huber, M.A.; Wiedemann, E.M.; Beug, H.; Wirth, T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett 2010, 295, 214–228. [Google Scholar]
- Baumgart, S.; Ellenrieder, V.; Fernandez-Zapico, M.E. Oncogenic transcription factors: Cornerstones of inflammation-linked pancreatic carcinogenesis. Gut 2013, 62, 310–316. [Google Scholar]
- Singh, S.; Sadanandam, A.; Singh, R.K. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev 2007, 26, 453–467. [Google Scholar]
- Denz, A.; Pilarsky, C.; Muth, D.; Ruckert, F.; Saeger, H.D.; Grutzmann, R. Inhibition of MIF leads to cell cycle arrest and apoptosis in pancreatic cancer cells. J. Surg. Res 2010, 160, 29–34. [Google Scholar]
- Jin, Z.Q.; Zhi, F.C.; Chen, X.Q.; Wang, Y.D. Expression of macrophage migration inhibition factor in pancreatic carcinoma tissue. Di Yi Jun Yi Da Xue Xue Bao 2004, 24, 1301–1303. [Google Scholar]
- Funamizu, N.; Hu, C.; Lacy, C.; Schetter, A.; Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, M.B.; Ried, T.; Yfantis, H.G.; et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2012. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res 2006, 12, 4147–4153. [Google Scholar]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009, 69, 2400–2407. [Google Scholar]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.; Parikh, N.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol 2007, 14, 3629–3637. [Google Scholar]
- Buck, E.; Eyzaguirre, A.; Barr, S.; Thompson, S.; Sennello, R.; Young, D.; Iwata, K.K.; Gibson, N.W.; Cagnoni, P.; Haley, J.D. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol. Cancer Ther 2007, 6, 532–541. [Google Scholar]
- Menke, A.; Adler, G. TGFbeta-induced fibrogenesis of the pancreas. Int. J. Gastrointest. Cancer 2002, 31, 41–46. [Google Scholar]
- Wu, K.; Zeng, J.; Li, L.; Fan, J.; Zhang, D.; Xue, Y.; Zhu, G.; Yang, L.; Wang, X.; He, D. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol. Rep 2010, 23, 1545–1552. [Google Scholar]
- Nguyen, C.; Liang, G.; Nguyen, T.T.; Tsao-Wei, D.; Groshen, S.; Lubbert, M.; Zhou, J.H.; Benedict, W.F.; Jones, P.A. Susceptibility of nonpromoter CpG islands to de novo methylation in normal and neoplastic cells. J. Natl. Cancer Inst 2001, 93, 1465–1472. [Google Scholar]
- Jones, P.A. The DNA methylation paradox. Trends Genet 1999, 15, 34–37. [Google Scholar]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar]
- Baylin, S.; Bestor, T.H. Altered methylation patterns in cancer cell genomes: Cause or consequence? Cancer Cell 2002, 1, 299–305. [Google Scholar]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar]
- Squazzo, S.L.; O’Geen, H.; Komashko, V.M.; Krig, S.R.; Jin, V.X.; Jang, S.W.; Margueron, R.; Reinberg, D.; Green, R.; Farnham, P.J. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res 2006, 16, 890–900. [Google Scholar]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 2008, 4, e1000242. [Google Scholar]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of Eve. Cell 2006, 124, 1111–1115. [Google Scholar]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar]
- Trumpp, A.; Wiestler, O.D. Mechanisms of disease: Cancer stem cells—Targeting the evil twin. Nat. Clin. Pract. Oncol 2008, 5, 337–347. [Google Scholar]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A. A chromatinmediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar]
- Easwaran, H.; Johnstone, S.E.; van Neste, L.; Ohm, J.; Mosbruger, T.; Wang, Q.; Aryee, M.J.; Joyce, P.; Ahuja, N.; Weisenberger, D.; et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res 2012, 22, 837–849. [Google Scholar]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 2010, 20, 440–446. [Google Scholar] [Green Version]
- Zhang, L.; Gao, J.; Li, Z.; Gong, Y. Neuronal pentraxin II (NPTX2) is frequently down-regulated by promoter hypermethylation in pancreatic cancers. Dig. Dis. Sci 2012, 57, 2608–2614. [Google Scholar]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar]
- Cai, H.H.; Yue-Ming, S.; Yi, M.; Wen-Tao, G.; Quan, P.; Jie, Y.; Han-Lin, Z. Aberrant methylation frequency of TNFRSF10C promoter in pancreatic cancer cell lines. Hepatobiliary Pancreat. Dis. Int 2011, 10, 95–100. [Google Scholar]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350. [Google Scholar]
- Wang, J.; Yi, X.; Tang, H. Direct quantification of microRNA at low picomolar level in sera of glioma patients using a competitive hybridization followed by amplified voltammetric detection. Anal. Chem 2012, 84, 6400. [Google Scholar]
- Baker, M.B.; Bao, G.; Searles, C.D. In vitro quantification of specific microRNA using molecular beacons. Nucleic Acids Res 2012, 40, e13. [Google Scholar]
- Lee, E.J.; Gusev, Y.; Jiang, J. Expression profiling identifies microRNA signature in pancreatic cancer. Int. J. Cancer 2007, 120, 1046. [Google Scholar]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol 2006, 24, 4677–4684. [Google Scholar]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, M. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar]
- Wang, J.; Chen, J.; Chang, P. MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev. Res 2009, 2, 807. [Google Scholar]
- Zhang, Y.; Li, M.; Wang, H.; Fisher, W.E.; Lin, P.H.; Yao, Q.; Chen, C. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by realtime PCR analysis. World J. Surg 2009, 33, 698–709. [Google Scholar]
- Habbe, N.; Koorstra, J.B.; Mendell, J.T. MicroRNA miR-155 is a biomarker of early pancreatic neoplasia. Cancer Biol. Ther 2009, 8, 340. [Google Scholar]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189. [Google Scholar]
- Yu, S.; Lu, Z.; Liu, C.; Meng, Y.; Ma, Y.; Zhao, W.; Liu, J.; Chen, J. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res 2010, 70, 6015. [Google Scholar]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet 2009, 10, 704. [Google Scholar]
- Lin, F.; Wang, X.; Jie, Z. Inhibitory effects of miR-146b-5p on cell migration and invasion of pancreatic cancer by targeting MMP16. J. Huazhong Univ. Sci. Technol. Med. Sci 2011, 31, 509. [Google Scholar]
- Laurila, E.M.; Sandstrom, S.; Rantanen, L.M.; Autio, R.; Kallioniemi, A. Both inhibition and enhanced expression of miR-31 lead to reduced migration and invasion of pancreatic cancer cells. Genes Chromosomes Cancer 2012, 51, 557. [Google Scholar]
- Dillhoff, M.; Liu, J.; Frankel, W. MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J. Gastrointest. Surg 2008, 12, 2171. [Google Scholar]
- Tsuda, N.; Ishiyama, S.; Li, Y.; Yoannides, C.G.; Abruzzese, J.L.; Chang, D.Z. Synthetic microRNA designed to target glioma-associated antigen 1 transcription factor inhibits division and induces late apoptosis in pancreatic tumor cells. Clin. Cancer Res 2006, 12, 6557. [Google Scholar]
- Ma, Y.; Yu, S.; Zhao, W. miR-27a regulates the growth, colony formation and migration of pancreatic cancer cells by targeting Sprouty2. Cancer Lett 2010, 298, 150. [Google Scholar]
- Paterson, E.L.; Kolesnikoff, N.; Gregory, P.A.; Bert, A.G.; Khew-Goodall, Y.; Goodall, G.J. The micro-RNA-200 family regulates epithelial to mesenchymal transition. Sci. World J 2008, 8, 901. [Google Scholar]
- Park, J.K.; Lee, E.J.; Esau, C. Antisense inhibition of microRNA-21 or -221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas 2009, 38, e190. [Google Scholar]
- Iwagami, Y.; Eguchi, H.; Nagano, H.; Akita, H.; Hama, N.; Wada, H.; Kawamoto, K.; Kobayashi, S.; Tomokuni, A.; Tomimaru, Y.; et al. miR-320c regulates gemcitabine-resistance in pancreatic cancer via SMARCC1. Br. J. Cancer 2013, 10, 1038. [Google Scholar]
- Mace, T.A.; Collins, A.L.; Wojcik, S.E.; Croce, C.M.; Lesinski, G.B.; Bloomston, M. Hypoxia induces the overexpression of microRNA-21 in pancreatic cancer cells. J. Surg Res 2013, 18, S0022–S4804. [Google Scholar]
- Torrisani, J.; Bournet, B.; du Rieu, M.C.; Bouisson, M.; Souque, A.; Escourrou, J.; Buscail, L.; Cordelier, P. let-7 MicroRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum. Gene Ther 2009, 20, 831. [Google Scholar]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res 2012, 18, 4266–4276. [Google Scholar]
- Singh, M.; Lima, A.; Molina, R.; Hamilton, P.; Clermont, A.C.; Devasthali, V. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat. Biotechnol 2010, 28, 585–593. [Google Scholar]
- Gopinathan, A.; Tuveson, D.A. The use of GEM models for experimental cancer therapeutics. Dis. Model. Mech 2008, 1, 83–86. [Google Scholar]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Invest 2007, 117, 50–59. [Google Scholar]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res 2008, 68, 2085–2093. [Google Scholar]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol 2010, 177, 2585–2596. [Google Scholar]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar]
- Hamada, S.; Masamune, A; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun 2012, 421, 349–354. [Google Scholar]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar]
- Vismodegib and Gemcitabine Hydrochloride in Treating Patients with Advanced Pancreatic Cancer. Available online: http://clinicaltrials.gov/show/NCT01195415 (accessed on 13 September 2013).
- Gemcitabine Hydrochloride with or without Vismodegib in Treating Patients with Recurrent or Metastatic Pancreatic Cancer. Available online: http://clinicaltrials.gov/ct2/show/study/NCT01064622 (accessed on 13 September 2013).
- A Study Evaluating IPI-926 in Combination with Gemcitabine in Patients with Metastatic Pancreatic Cancer. Available online: http://clinicaltrials.gov/show/NCT01130142 (accessed on 13 September 2013).
- Hedgehog Inhibition for Pancreatic Ductal Adenocarcinoma (PDAC) in the Preoperative Setting (HIPPoS). Available online: http://clinicaltrials.gov/show/NCT01096732 (accessed on 13 September 2013).
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol 2007, 25, 319–325. [Google Scholar]
- Desai, N.; Trieu, V.; Yao, Z.; Louie, L.; Ci, S.; Yang, A. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res 2006, 12, 1317–1324. [Google Scholar]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol 2011, 29, 4548–4554. [Google Scholar]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med 1993, 328, 1433–1437. [Google Scholar]
- Dodson, L.F.; Hawkins, W.G.; Goedegebuure, P. Potential targets for pancreatic cancer immunotherapeutics. Immunother 2011, 3, 517–537. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gnoni, A.; Licchetta, A.; Scarpa, A.; Azzariti, A.; Brunetti, A.E.; Simone, G.; Nardulli, P.; Santini, D.; Aieta, M.; Delcuratolo, S.; et al. Carcinogenesis of Pancreatic Adenocarcinoma: Precursor Lesions. Int. J. Mol. Sci. 2013, 14, 19731-19762. https://doi.org/10.3390/ijms141019731
Gnoni A, Licchetta A, Scarpa A, Azzariti A, Brunetti AE, Simone G, Nardulli P, Santini D, Aieta M, Delcuratolo S, et al. Carcinogenesis of Pancreatic Adenocarcinoma: Precursor Lesions. International Journal of Molecular Sciences. 2013; 14(10):19731-19762. https://doi.org/10.3390/ijms141019731
Chicago/Turabian StyleGnoni, Antonio, Antonella Licchetta, Aldo Scarpa, Amalia Azzariti, Anna Elisabetta Brunetti, Gianni Simone, Patrizia Nardulli, Daniele Santini, Michele Aieta, Sabina Delcuratolo, and et al. 2013. "Carcinogenesis of Pancreatic Adenocarcinoma: Precursor Lesions" International Journal of Molecular Sciences 14, no. 10: 19731-19762. https://doi.org/10.3390/ijms141019731