A Soluble Receptor for Advanced Glycation End-Products Inhibits Hypoxia/Reoxygenation-Induced Apoptosis in Rat Cardiomyocytes via the Mitochondrial Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
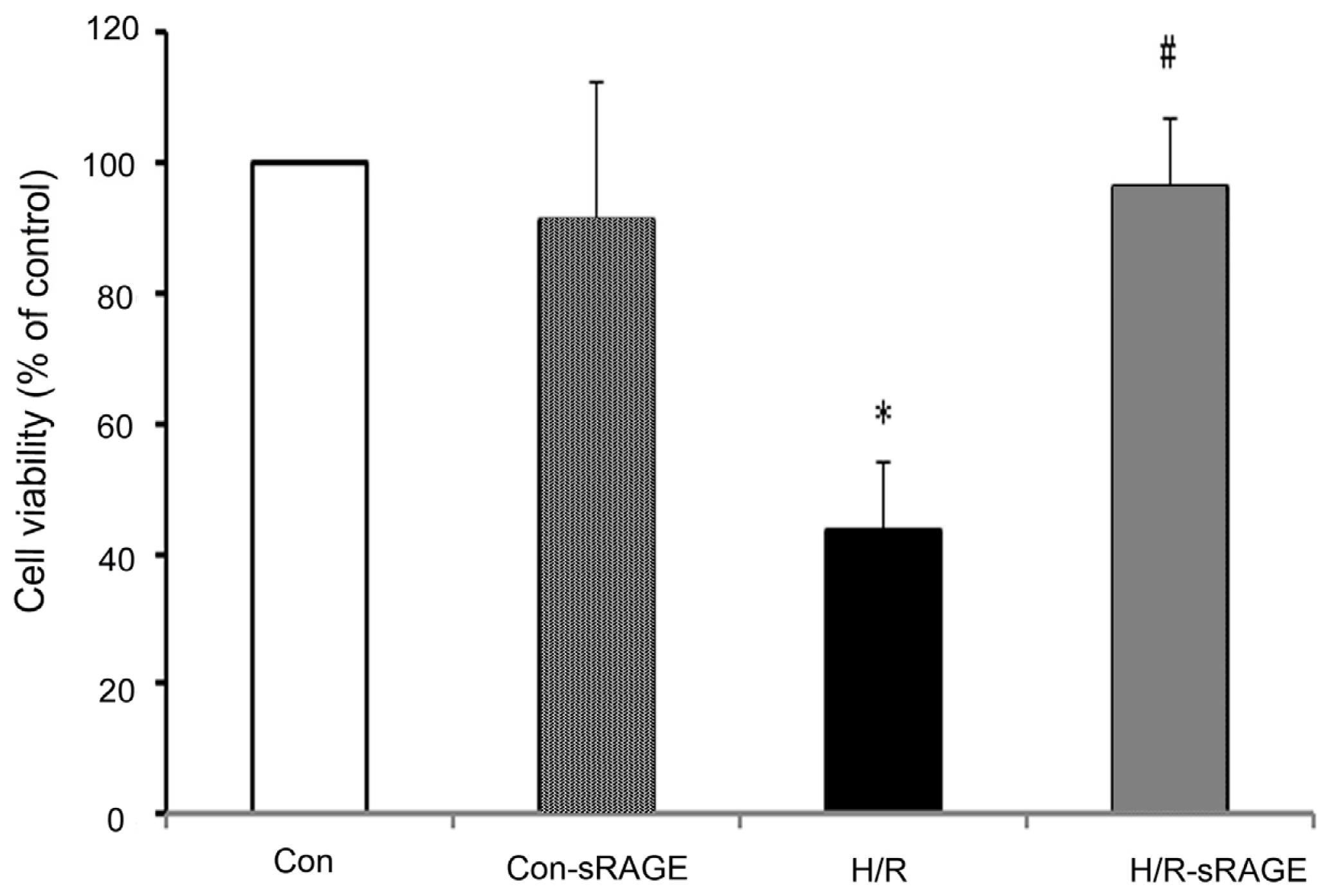
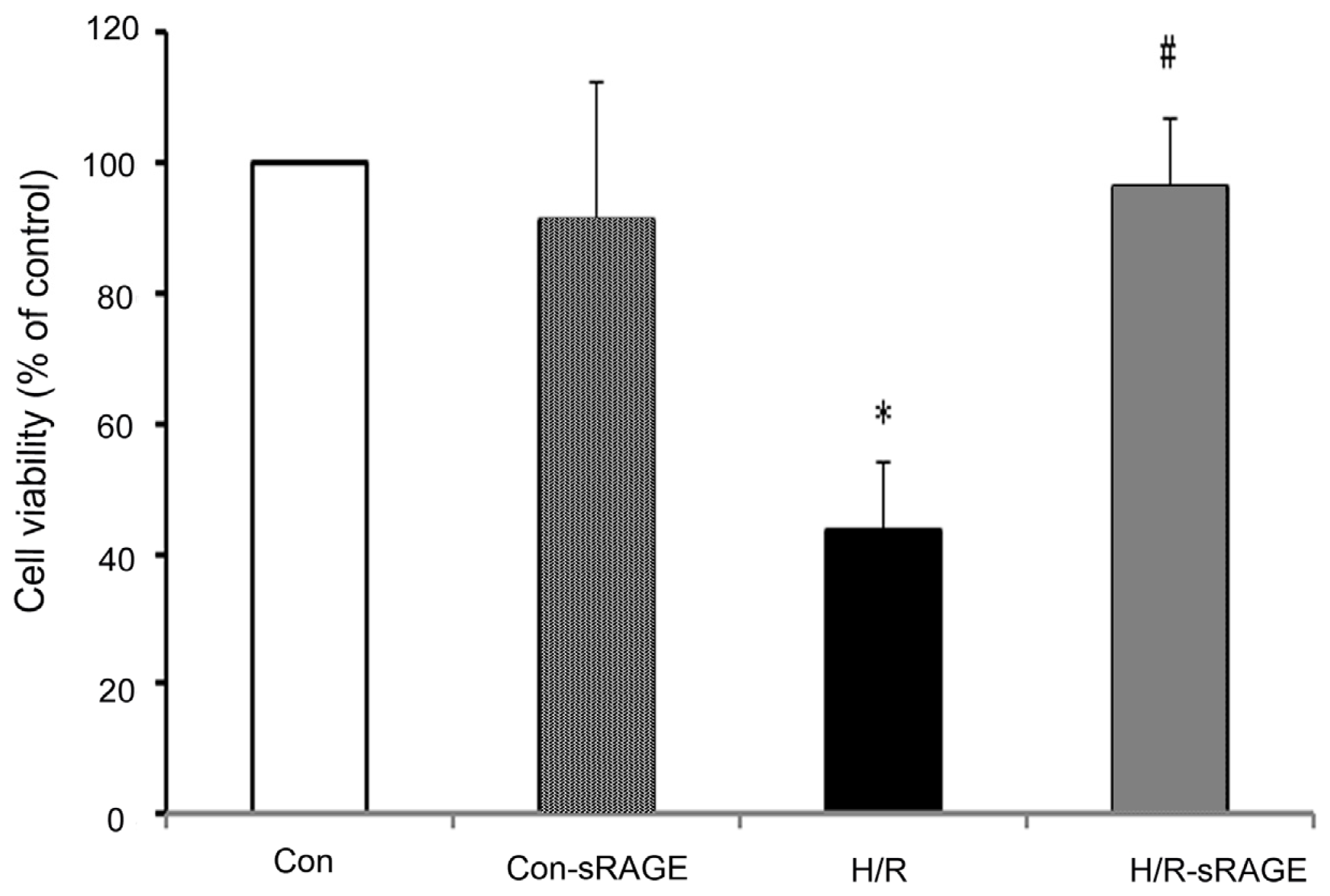
2.1. sRAGE Prevented the Reduction of Cell Viability Following H/R
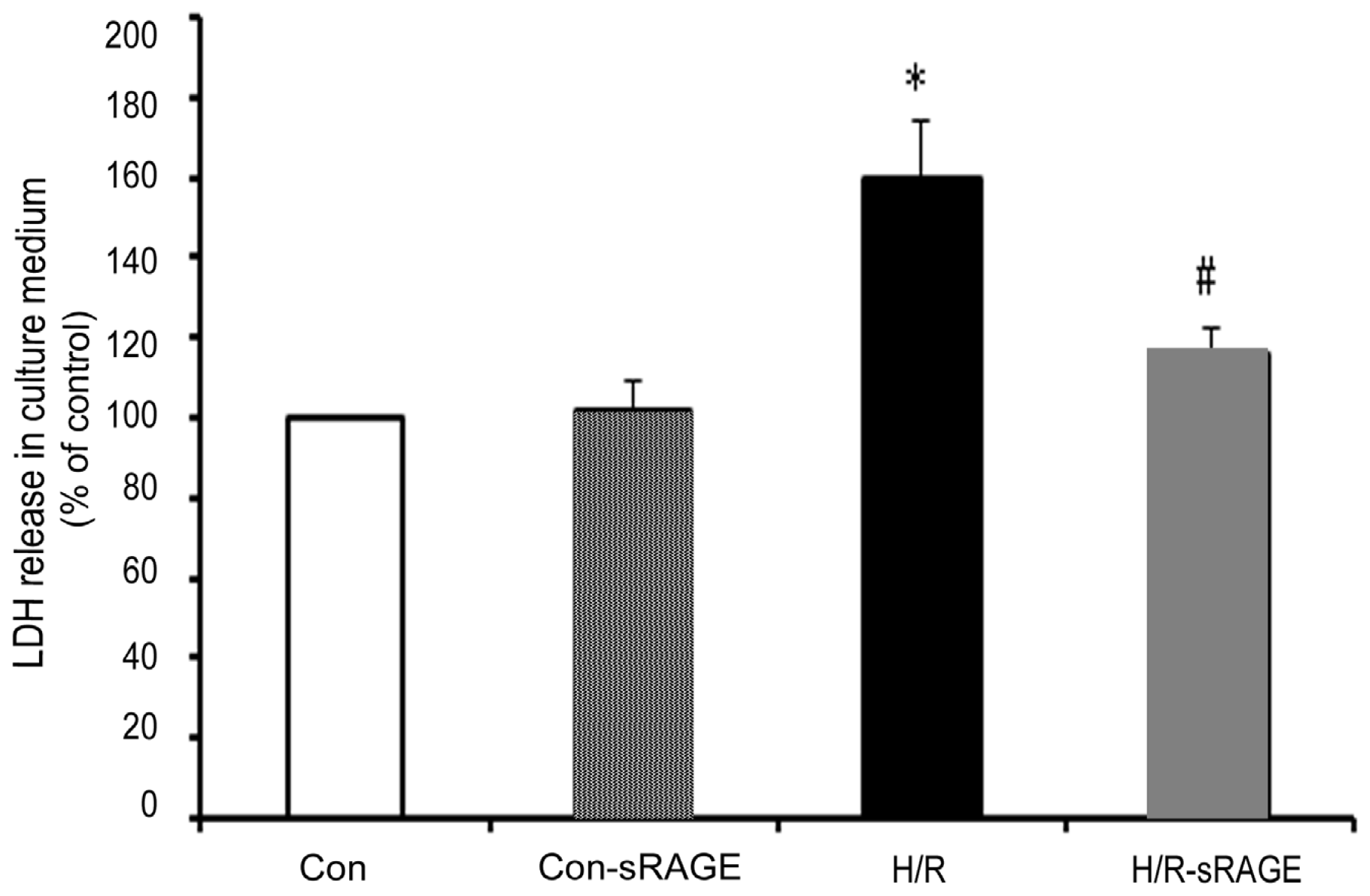
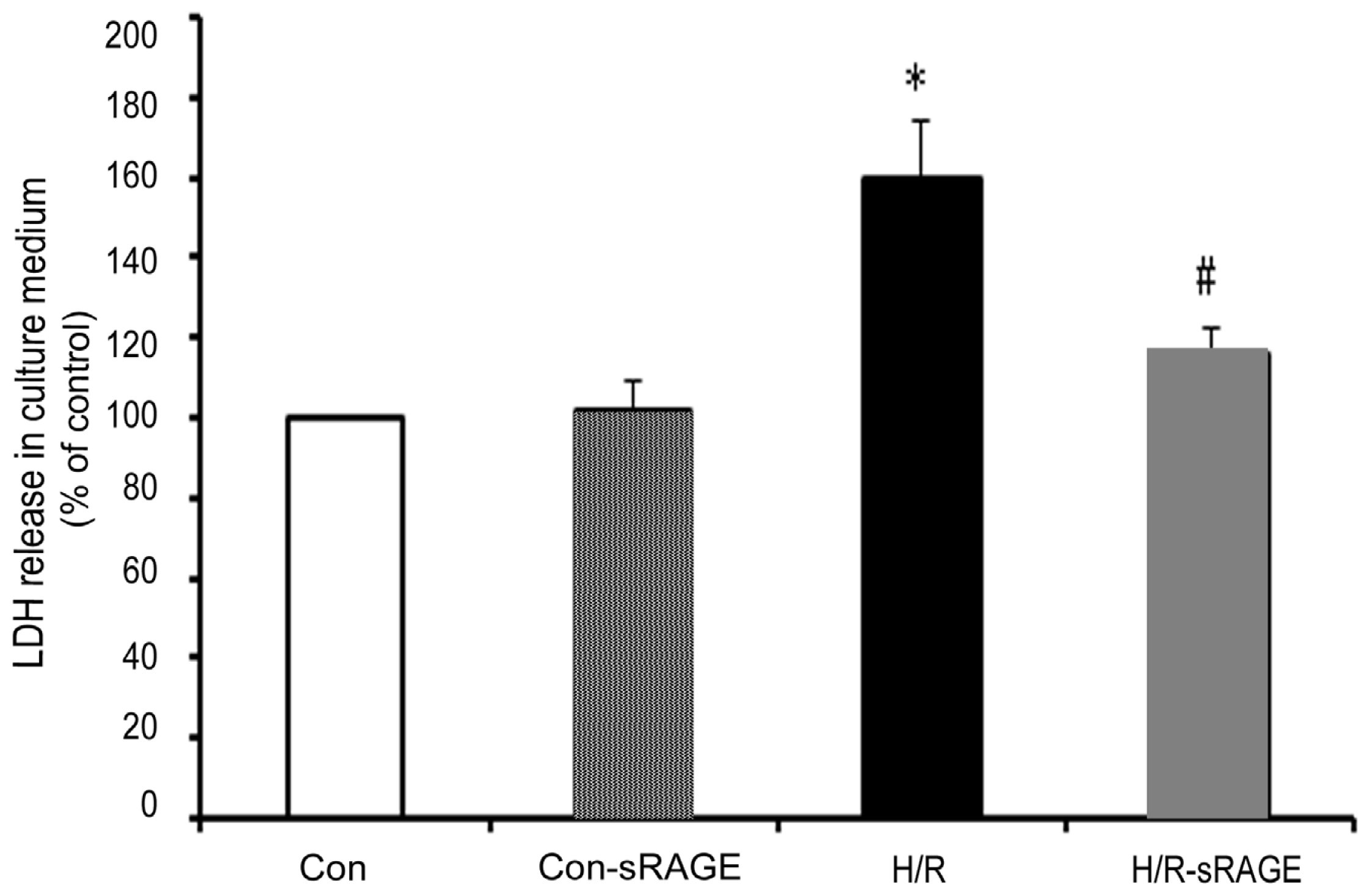
2.2. sRAGE Inhibited LDH Leakage Induced by H/R
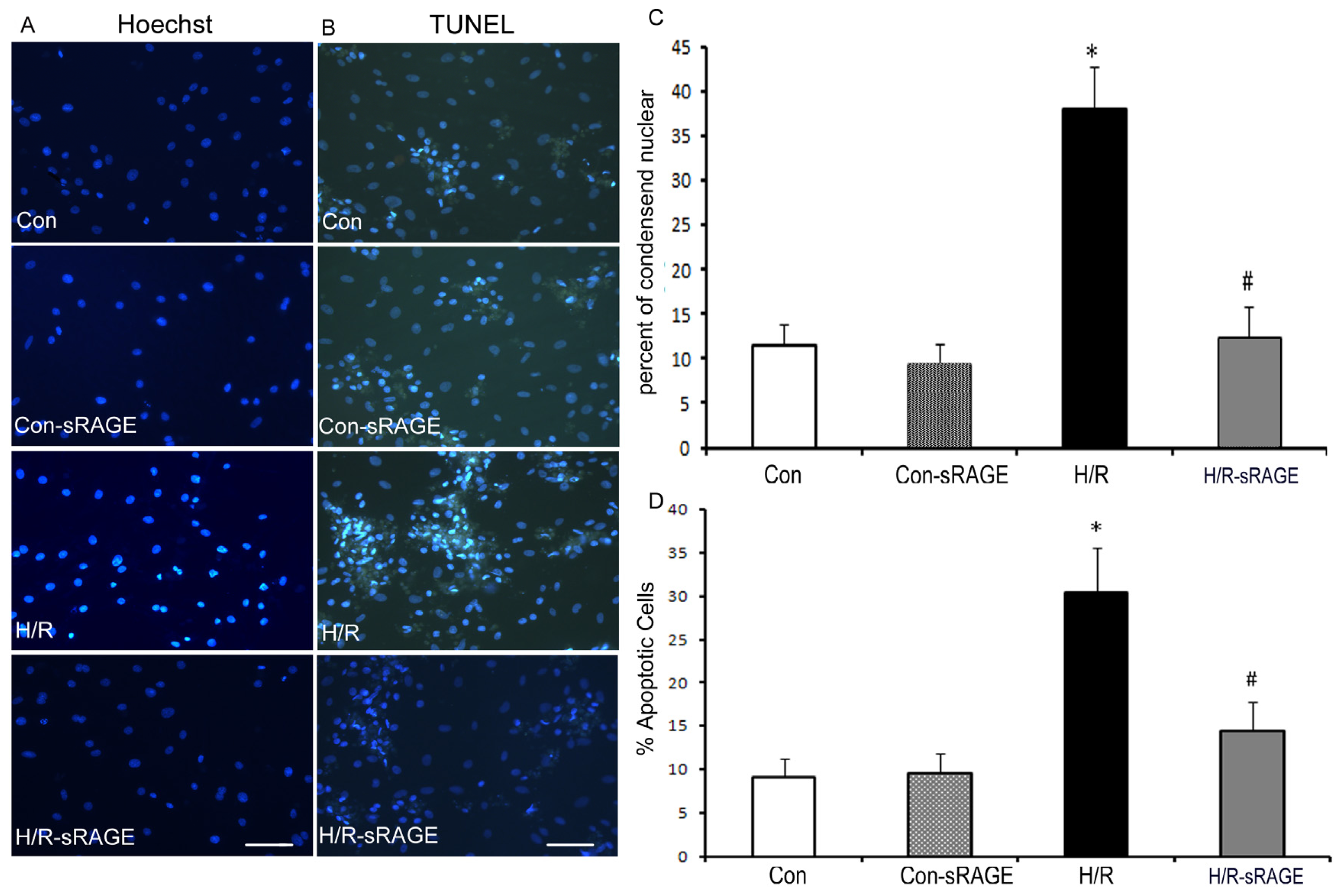
2.3. Exogenous Administration of sRAGE Inhibited H/R Induced Cardiomyocyte Apoptosis in vitro
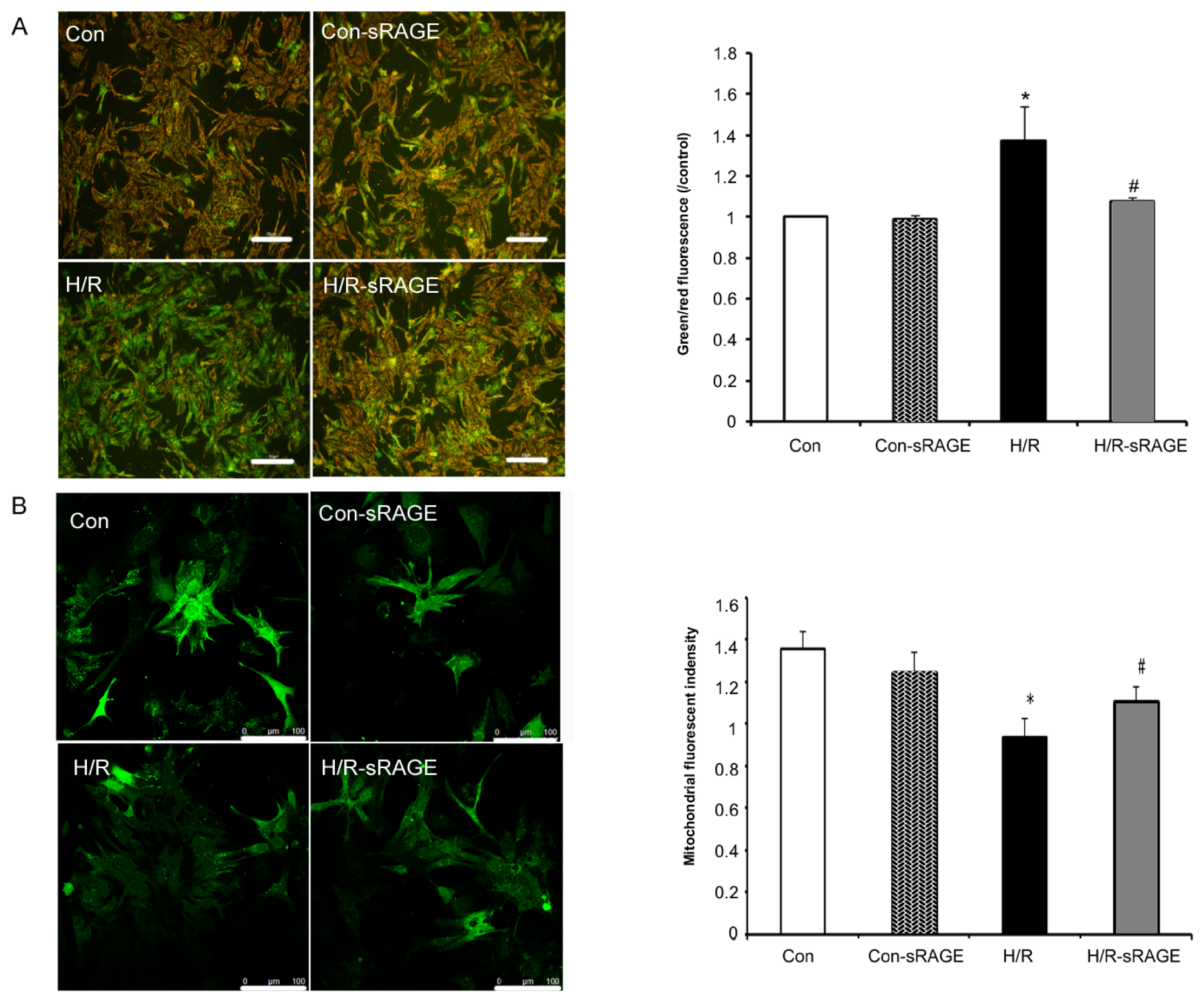
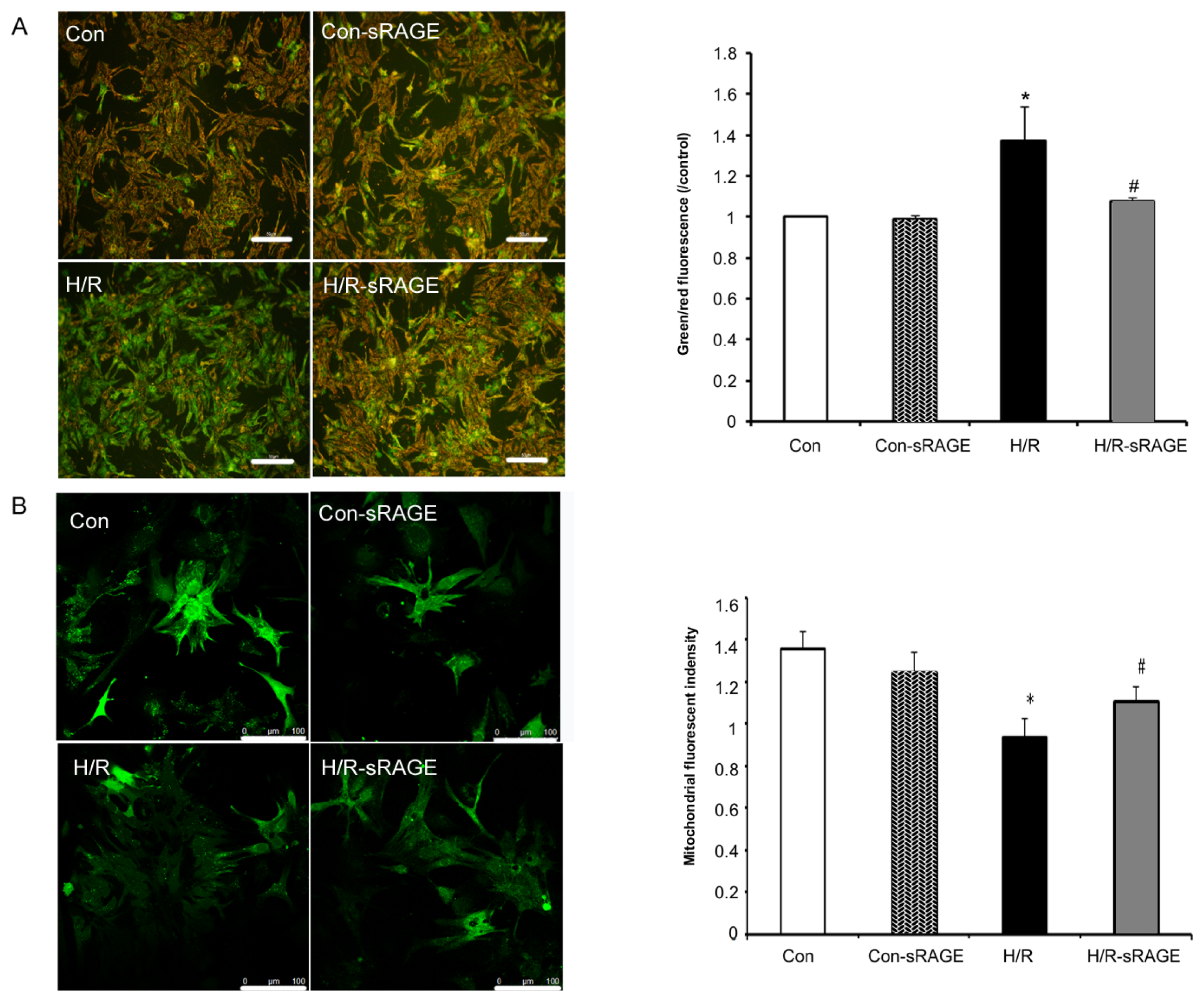
2.4. sRAGE Inhibited Mitochondrial Depolarization and mPTP Opening Induced by H/R
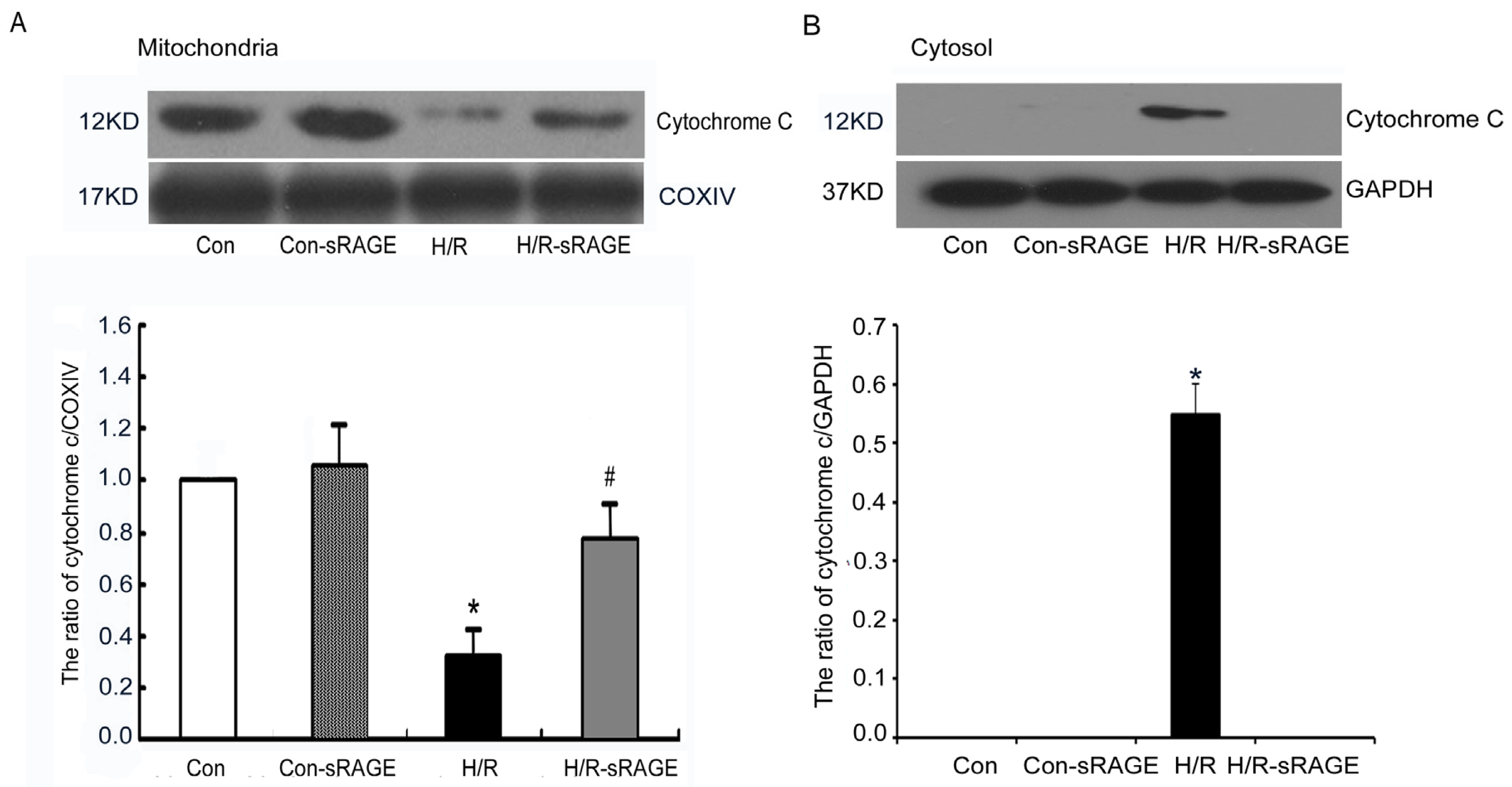
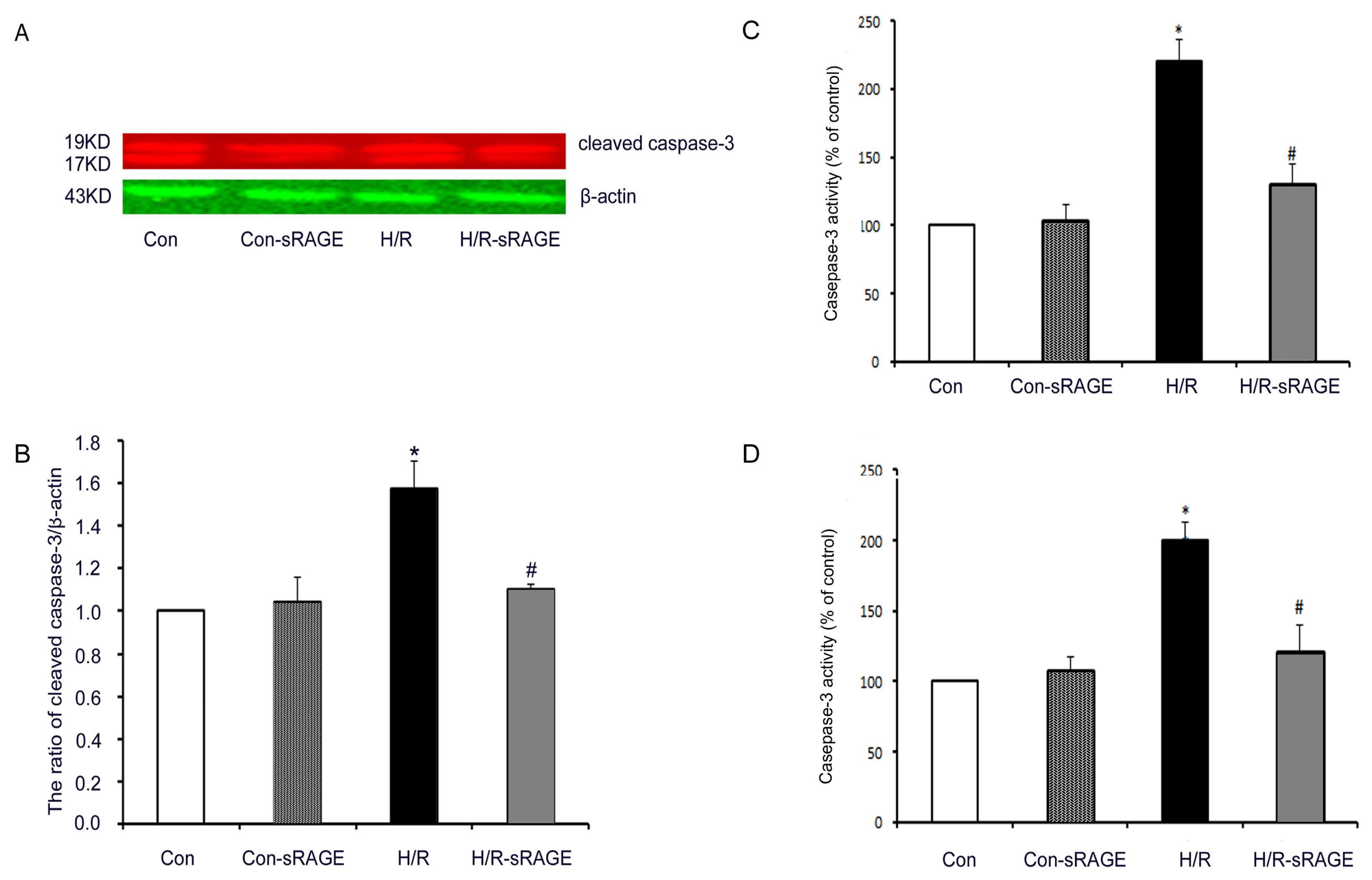
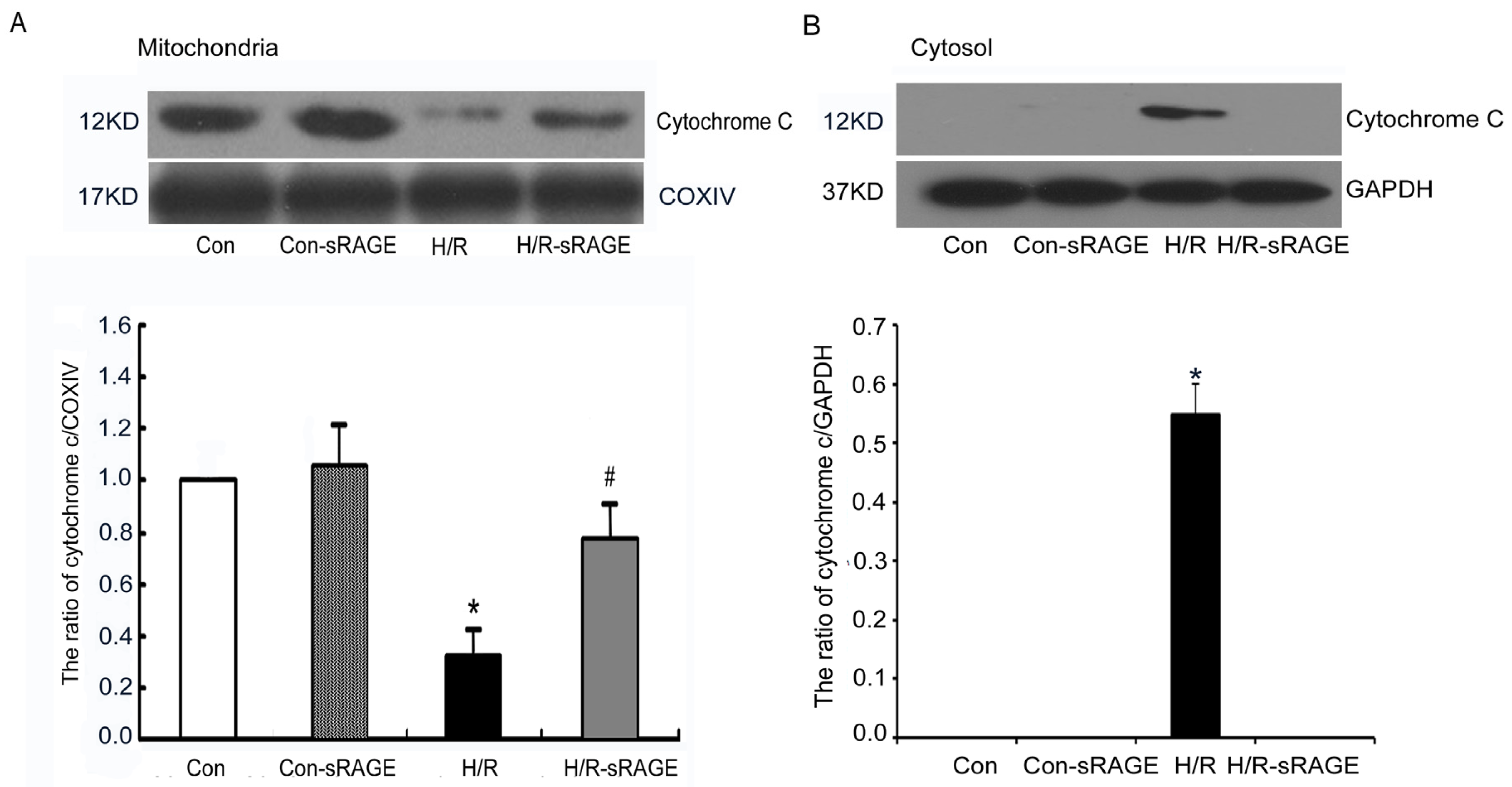
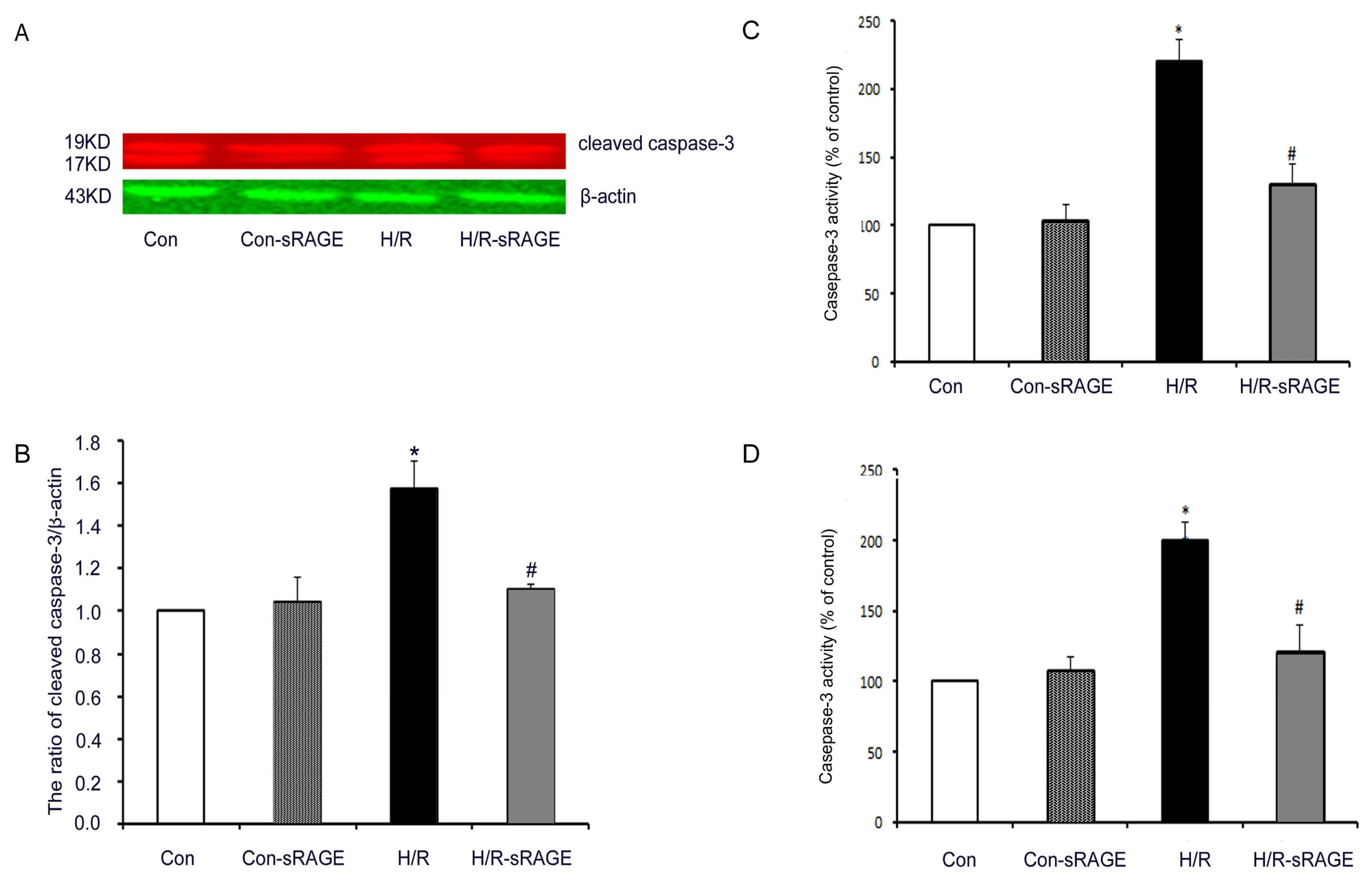
2.5. sRAGE Inhibited H/R-Induced Mitochondrial Cytochrome c Release and the Increase of Caspase-3 and Caspase-9 Activity during H/R
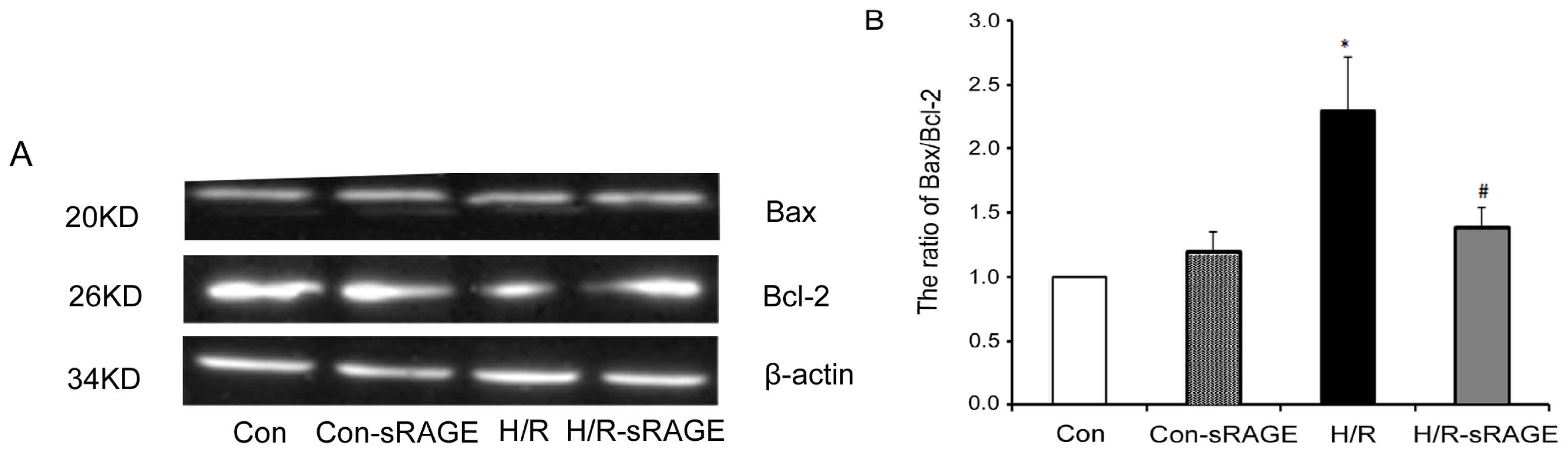
2.6. sRAGE Inhibited the Increase of the Ratio of Bax to Bcl-2 following H/R Injury
3. Materials and Methods
3.1. Materials
3.2. Isolation and Culture of Neonatal Rat Cardiomyocytes
3.3. Study Groups and Experimental Protocol
3.4. Cell Viability Assay
3.5. Assay of LDH Activity
3.6. Apoptosis Assessment
3.7. Measurement of MMP by Florescent JC1
3.8. Determination of mPTP Opening
3.9. Isolation of Mitochondria
3.10. Western Blot Analysis
3.11. Caspase Activity Assay
3.12. Statistical Analysis
4. Discussion
5. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Murray, C.J.; Lopez, A.D. Alternative projections of mortality and disability by cause 1990–2020: Global burden of disease study. Lancet 1997, 349, 1498–1504. [Google Scholar]
- Verma, S.; Fedak, P.W.; Weisel, R.D.; Butany, J.; Rao, V.; Maitland, A.; Li, R.K.; Dhillon, B.; Yau, T.M. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation 2002, 105, 2332–2336. [Google Scholar]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med 2007, 357, 1121–1135. [Google Scholar]
- Kim, H.K.; Thu, V.T.; Heo, H.J.; Kim, N.; Han, J. Cardiac proteomic responses to ischemia-reperfusion injury and ischemic preconditioning. Expert Rev. Proteomics 2011, 8, 241–261. [Google Scholar]
- Miura, T.; Nishihara, M.; Miki, T. Drug development targeting the glycogen synthase kinase-3β (GSK-3β)-mediated signal transduction pathway: Role of GSK-3β in myocardial protection against ischemia/reperfusion injury. J. Pharmacol. Sci 2009, 109, 162–167. [Google Scholar]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res. Cardiol 2006, 101, 180–189. [Google Scholar]
- Argaud, L.; Gateau-Roesch, O.; Augeul, L.; Couture-Lepetit, E.; Loufouat, J.; Gomez, L.; Robert, D.; Ovize, M. Increased mitochondrial calcium coexists with decreased reperfusion injury in postconditioned (but not preconditioned) hearts. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H386–H391. [Google Scholar]
- Borutaite, V.; Budriunaite, A.; Morkuniene, R.; Brown, G.C. Release of mitochondrial cytochrome c and activation of cytosolic caspases induced by myocardial ischaemia. Biochim. Biophys. Acta 2001, 1537, 101–109. [Google Scholar]
- Bopassa, J.C.; Ferrera, R.; Gateau-Roesch, O.; Couture-Lepetit, E.; Ovize, M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc. Res 2006, 69, 178–185. [Google Scholar]
- Infanger, M.; Faramarzi, S.; Grosse, J.; Kurth, E.; Ulbrich, C.; Bauer, J.; Wehland, M.; Kreutz, R.; Kossmehl, P.; Paul, M.; et al. Expression of vascular endothelial growth factor and receptor tyrosine kinases in cardiac ischemia/reperfusion injury. Cardiovasc Pathol 2007, 16, 291–299. [Google Scholar]
- Bucciarelli, L.G.; Kaneko, M.; Ananthakrishnan, R.; Harja, E.; Lee, L.K.; Hwang, Y.C.; Lerner, S.; Bakr, S.; Li, Q.; Lu, Y.; et al. Receptor for advanced-glycation end products: Key modulator of myocardial ischemic injury. Circulation 2006, 113, 1226–1234. [Google Scholar]
- Lindsey, J.B.; Cipollone, F.; Abdullah, S.M.; McGuire, D.K. Receptor for advanced glycation end-products (RAGE) and soluble RAGE (sRAGE): Cardiovascular implications. Diab. Vasc. Dis. Res 2009, 6, 7–14. [Google Scholar]
- Santilli, F.; Vazzana, N.; Bucciarelli, L.G.; Davi, G. Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Curr. Med. Chem 2009, 16, 940–952. [Google Scholar]
- Dong, H.W.; Guo, C.X.; Du, F.H.; Wang, H.X.; Zeng, X.J.; Zhang, L.K.; Tian, J.P. Effects of myocardial ischemia/reperfusion injury on the level of endogenous soluble receptor for advanced glycation end-products (sRAGE). J. Cap. Med. Univ 2011, 32, 640–644. [Google Scholar]
- Aleshin, A.; Ananthakrishnan, R.; Li, Q.; Rosario, R.; Lu, Y.; Qu, W.; Song, F.; Bakr, S.; Szabolcs, M.; D’Agati, V.; et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H1823–H1832. [Google Scholar]
- Andrassy, M.; Volz, H.C.; Igwe, J.C.; Funke, B.; Eichberger, S.N.; Kaya, Z.; Buss, S.; Autschbach, F.; Pleger, S.T.; Lukic, I.K.; Bea, F.; et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 2008, 117, 3216–3226. [Google Scholar]
- Feeney, C.J.; Pennefather, P.S.; Gyulkhandanyan, A.V. A cuvette-based fluorometric analysis of mitochondrial membrane potential measured in cultured astrocyte monolayers. J. Neurosci. Methods 2003, 125, 13–25. [Google Scholar]
- Han, L.; Xu, C.; Jiang, C.; Li, H.; Zhang, W.; Zhao, Y.; Zhang, L.; Zhang, Y.; Zhao, W.; Yang, B. Effects of polyamines on apoptosis induced by simulated ischemia/reperfusion injury in cultured neonatal rat cardiomyocytes. Cell Biol. Int 2007, 31, 1345–1352. [Google Scholar]
- Liu, Y.; Ma, Y.Z.; Wang, R.T.; Xia, C.H.; Zhang, R.Q.; Lian, K.; Luan, R.H.; Sun, L.; Yang, L.; Lau, W.B.; Wang, H.C.; et al. Advanced glycation end products accelerate ischemia/reperfusion injury through receptor of advanced end product/nitrative thioredoxin inactivation in cardiac microvascular endothelial cells. Antioxid. Redox Signal 2011, 15, 1769–1778. [Google Scholar]
- Moore, T.C.; Moore, J.E.; Kaji, Y.; Frizzell, N.; Usui, T.; Poulaki, V.; Campbell, I.L.; Stitt, A.W.; Gardiner, T.A.; Archer, D.B.; et al. The role of advanced glycation end products in retinal microvascular leukostasis. Invest. Ophthalmol. Visual Sci 2003, 44, 4457–4464. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar]
- Gurevich, R.M.; Regula, K.M.; Kirshenbaum, L.A. Serpin protein CrmA suppresses hypoxia-mediated apoptosis of ventricular myocytes. Circulation 2001, 103, 1984–1991. [Google Scholar]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D.J.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3671–3675. [Google Scholar]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys 1999, 76, 725–734. [Google Scholar]
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W.; Clauss, M.; Wang, F.; et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem 1992, 267, 14987–14997. [Google Scholar]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar]
- Donato, R. RAGE: A single receptor for several ligands and different cellular responses: The case of certain S100 proteins. Curr. Mol. Med 2007, 7, 711–724. [Google Scholar]
- Riuzzi, F.; Sorci, G.; Donato, R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J. Biol. Chem 2006, 281, 8242–8253. [Google Scholar]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med 2003, 9, 907–913. [Google Scholar]
- Monteiro, F.A.; Cardoso, I.; Sousa, M.M.; Saraiva, M.J. In vitro inhibition of transthyretin aggregate-induced cytotoxicity by full and peptide derived forms of the soluble receptor for advanced glycation end products (RAGE). FEBS Lett 2006, 580, 3451–3456. [Google Scholar]
- Pullerits, R.; Brisslert, M.; Jonsson, I.M.; Tarkowski, A. Soluble receptor for advanced glycation end products triggers a proinflammatory cytokine cascade via β2 integrin Mac-1. Arthritis Rheum 2006, 54, 3898–3907. [Google Scholar]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 2007, 148, 548–558. [Google Scholar]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J. Clin. Invest 2008, 118, 183–194. [Google Scholar]
- Basta, G.; Lazzerini, G.; Del Turco, S.; Ratto, G.M.; Schmidt, A.M.; de Caterina, R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler. Thromb. Vasc. Biol 2005, 25, 1401–1407. [Google Scholar]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 2008, 22, 3716–3727. [Google Scholar]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J 2008, 22, 1572–1580. [Google Scholar]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J 2003, 370, 1097–1109. [Google Scholar]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest 1994, 94, 1621–1628. [Google Scholar]
- Shang, L.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Abdillahi, M.; Zhu, Z.; Qu, W.; Rosario, R.; Toure, F.; Yan, S.F.; et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS One 2010, 5, e10092. [Google Scholar]
- Falcone, C.; Emanuele, E.; D’Angelo, A.; Buzzi, M.P.; Belvito, C.; Cuccia, M.; Geroldi, D. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler. Thromb. Vasc. Biol 2005, 25, 1032–1037. [Google Scholar]
- Song, J.Q.; Teng, X.; Cai, Y.; Tang, C.S.; Qi, Y.F. Activation of Akt/GSK-3β signaling pathway is involved in intermedin(1-53) protection against myocardial apoptosis induced by ischemia/reperfusion. Apoptosis 2009, 14, 1299–1307. [Google Scholar]
- Fu, J.; Huang, H.; Liu, J.; Pi, R.; Chen, J.; Liu, P. Tanshinone IIA protects cardiac myocytes against oxidative stress-triggered damage and apoptosis. Eur. J. Pharmacol 2007, 568, 213–221. [Google Scholar]
- Ussher, J.R.; Lopaschuk, G.D. The malonyl CoA axis as a potential target for treating ischaemic heart disease. Cardiovasc. Res 2008, 79, 259–268. [Google Scholar]
- Stanley, W.C.; Morgan, E.E.; Huang, H.; McElfresh, T.A.; Sterk, J.P.; Okere, I.C.; Chandler, M.P.; Cheng, J.; Dyck, J.R.; Lopaschuk, G.D. Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am. J. Physiol. Heart Circ. Physiol 2005, 289, H2304–H2309. [Google Scholar]
- Cohen, M.V.; Downey, J.M. Ischemic postconditioning: from receptor to end-effector. Antioxid. Redox Signal 2011, 14, 821–831. [Google Scholar]
- Paillard, M.; Gomez, L.; Augeul, L.; Loufouat, J.; Lesnefsky, E.J.; Ovize, M. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J. Mol. Cell Cardiol 2009, 46, 902–909. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, C.; Zeng, X.; Song, J.; Zhang, M.; Wang, H.; Xu, X.; Du, F.; Chen, B. A Soluble Receptor for Advanced Glycation End-Products Inhibits Hypoxia/Reoxygenation-Induced Apoptosis in Rat Cardiomyocytes via the Mitochondrial Pathway. Int. J. Mol. Sci. 2012, 13, 11923-11940. https://doi.org/10.3390/ijms130911923
Guo C, Zeng X, Song J, Zhang M, Wang H, Xu X, Du F, Chen B. A Soluble Receptor for Advanced Glycation End-Products Inhibits Hypoxia/Reoxygenation-Induced Apoptosis in Rat Cardiomyocytes via the Mitochondrial Pathway. International Journal of Molecular Sciences. 2012; 13(9):11923-11940. https://doi.org/10.3390/ijms130911923
Chicago/Turabian StyleGuo, Caixia, Xiangjun Zeng, Juanjuan Song, Min Zhang, Hongxia Wang, Xiaowei Xu, Fenghe Du, and Buxing Chen. 2012. "A Soluble Receptor for Advanced Glycation End-Products Inhibits Hypoxia/Reoxygenation-Induced Apoptosis in Rat Cardiomyocytes via the Mitochondrial Pathway" International Journal of Molecular Sciences 13, no. 9: 11923-11940. https://doi.org/10.3390/ijms130911923