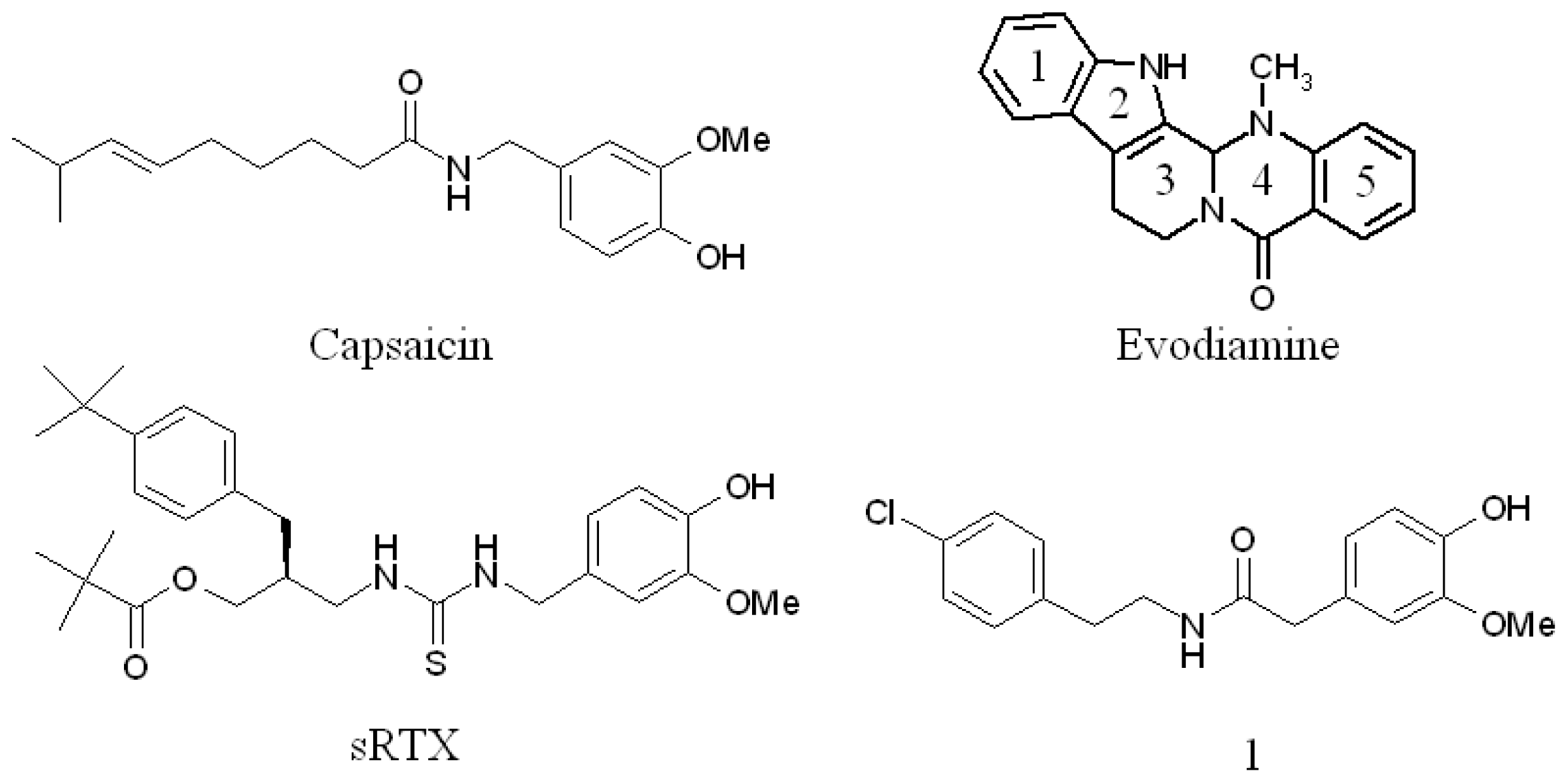
Binding Mode Prediction of Evodiamine within Vanilloid Receptor TRPV1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
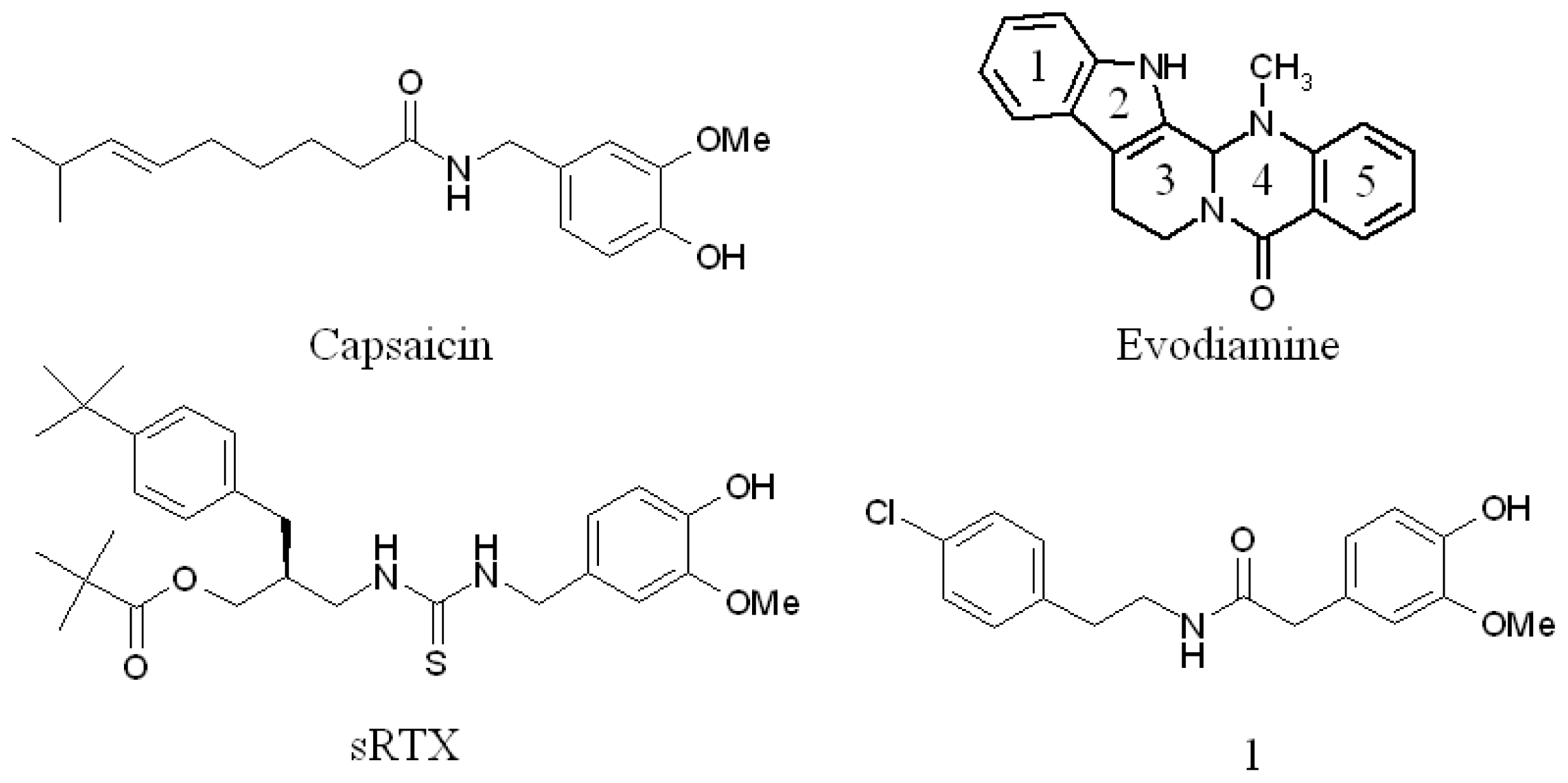
:1. Introduction
2. Results and Discussion
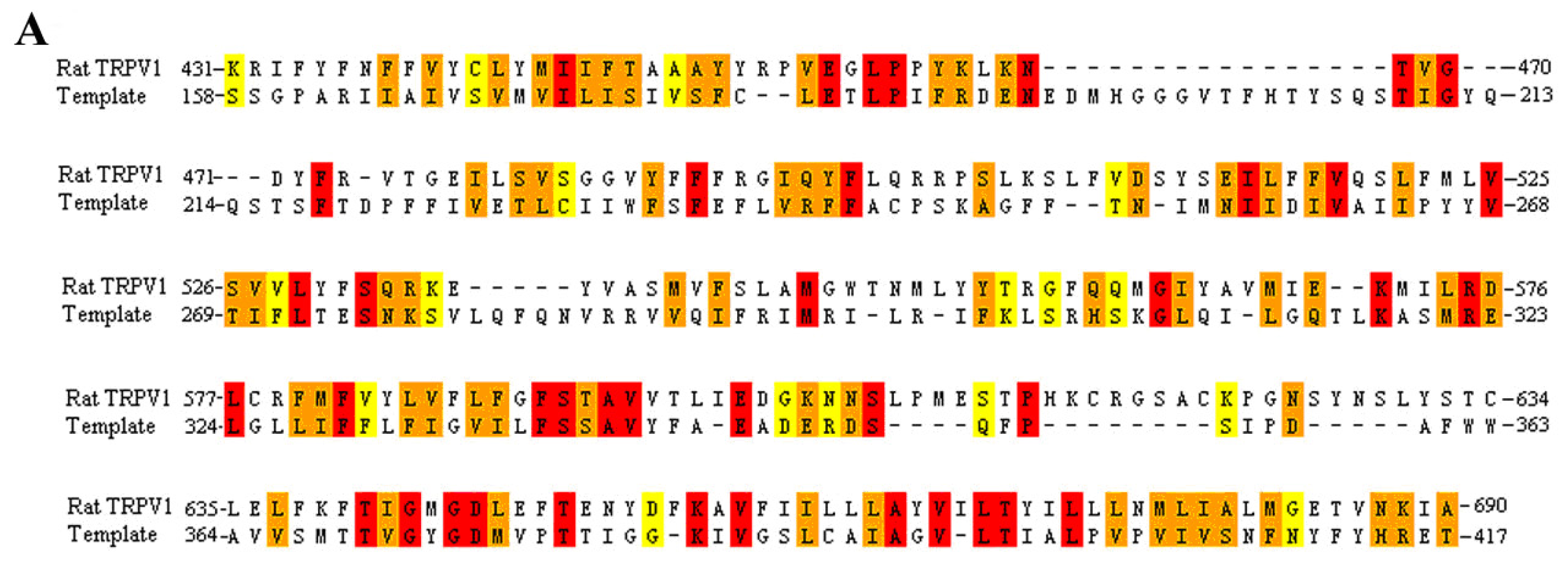
2.1. Building Rat, Human and Rabbit TRPV1 Homology Models
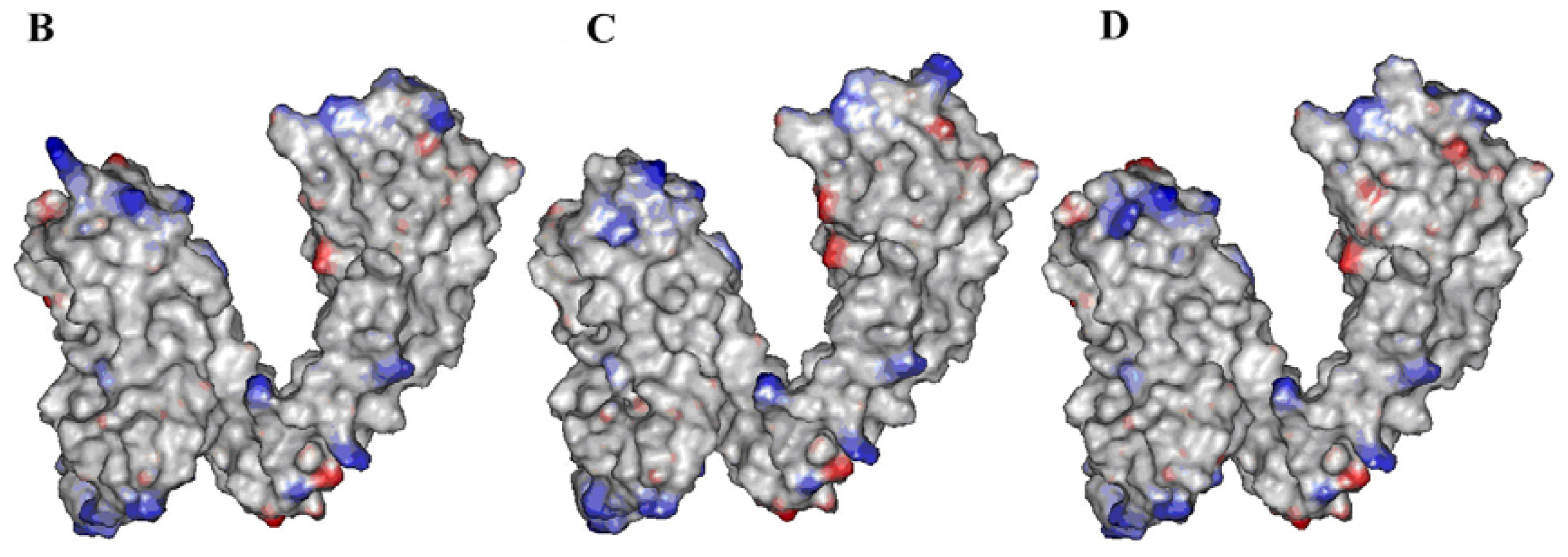
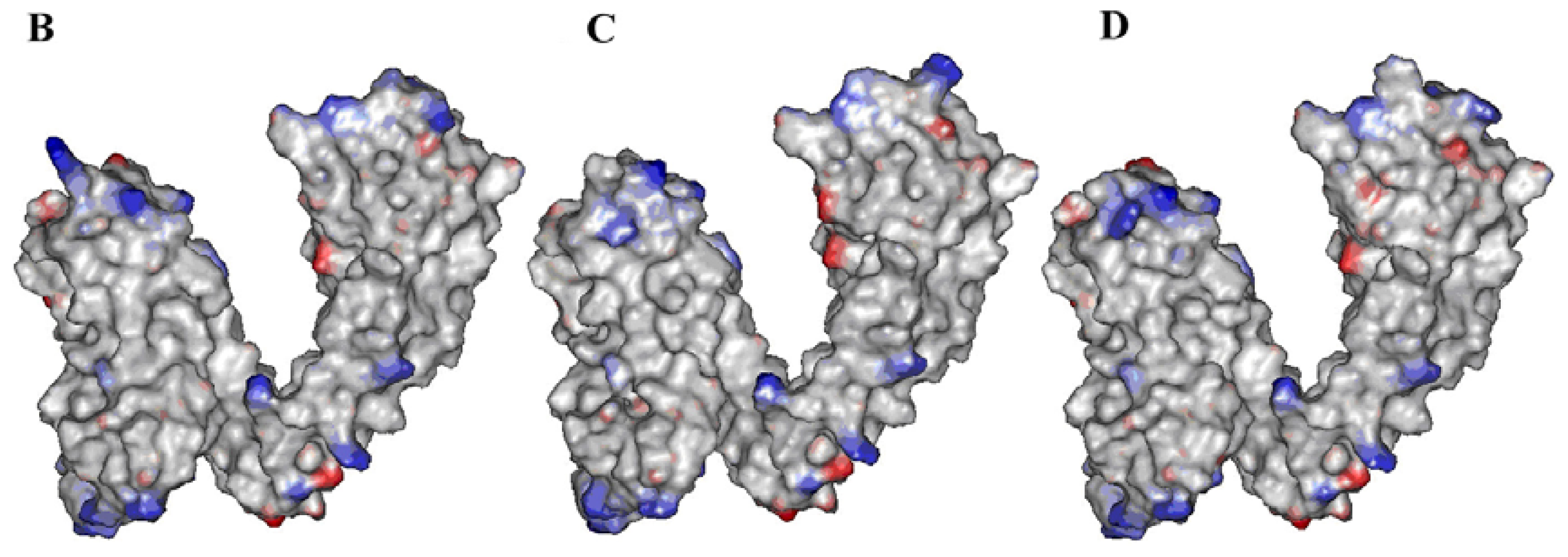
2.2. Identification of Ligand-Binding Region in TRPV1
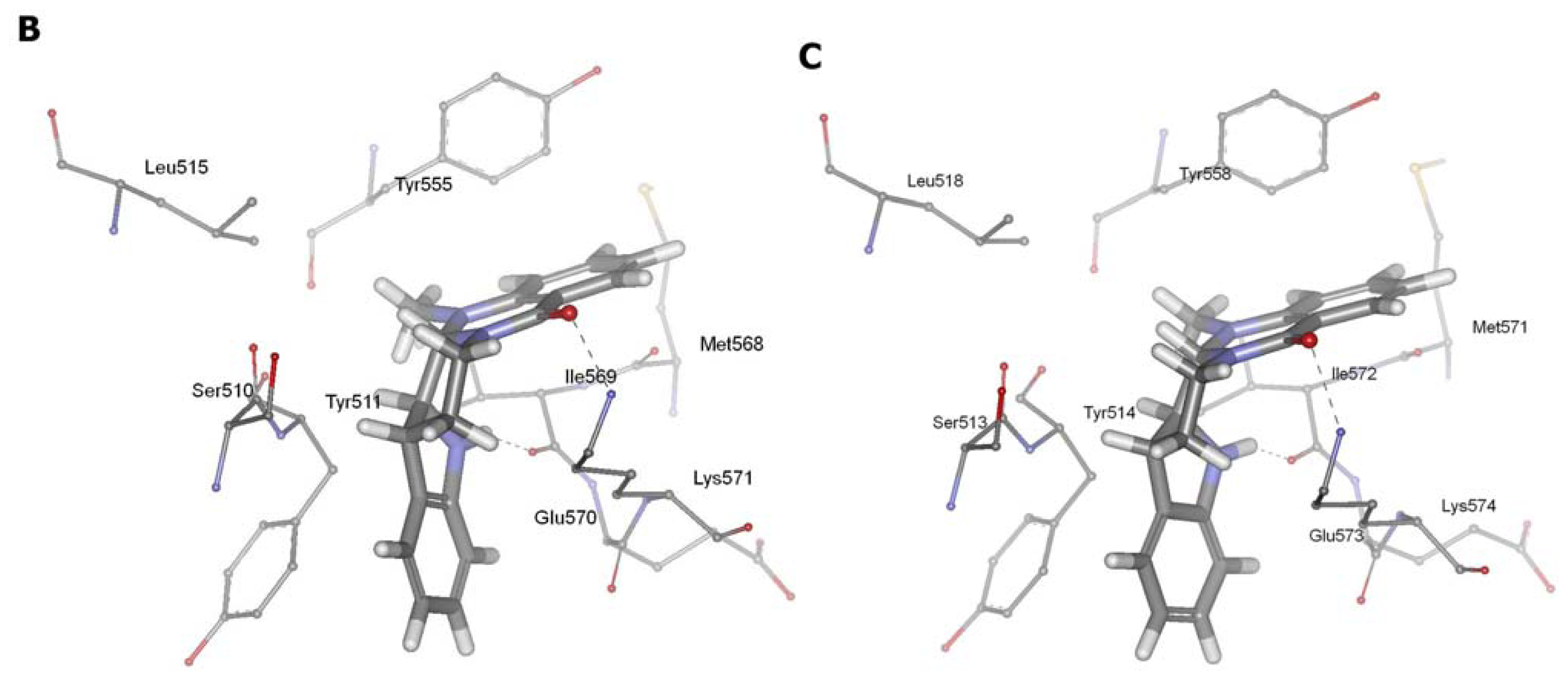
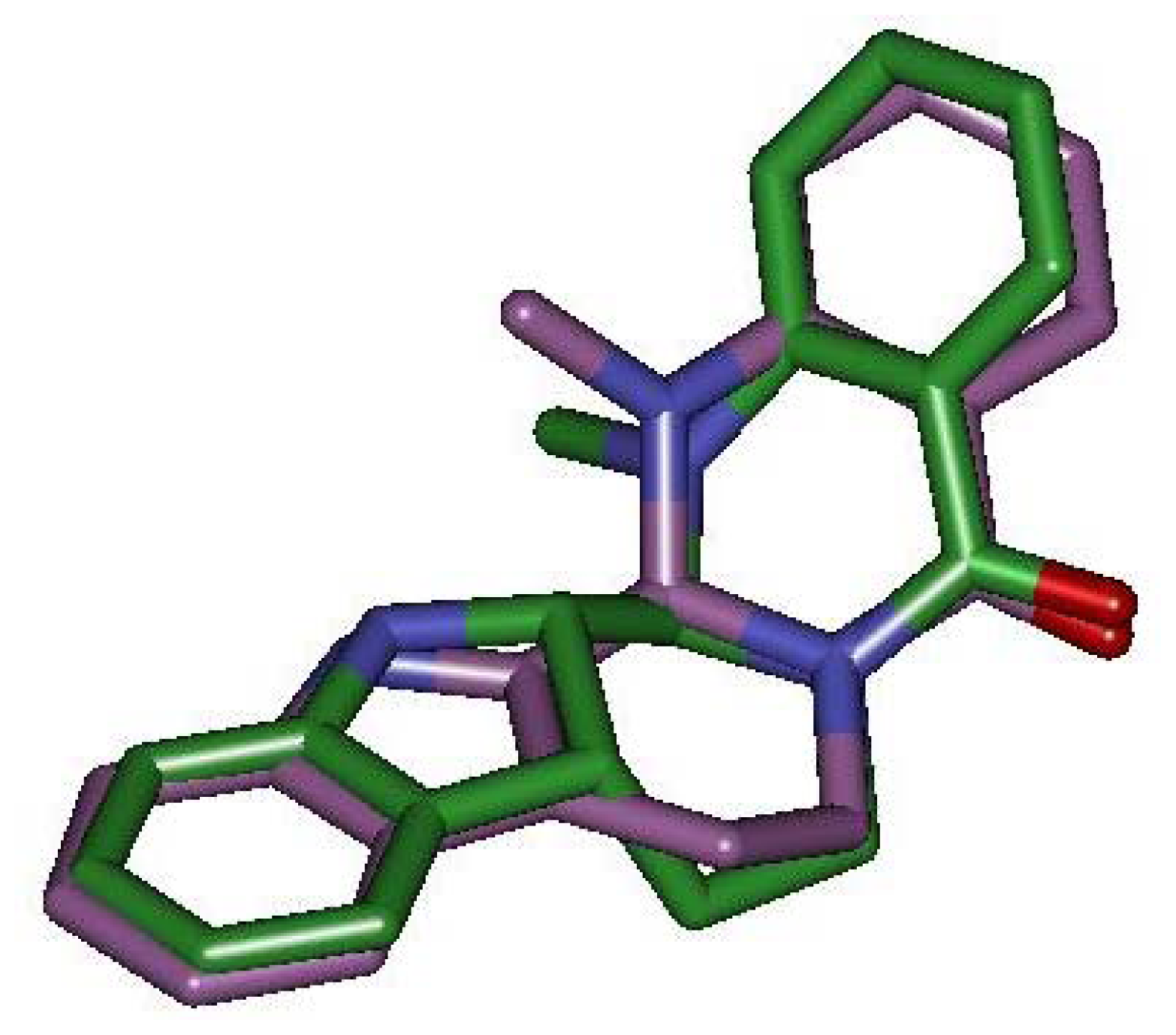
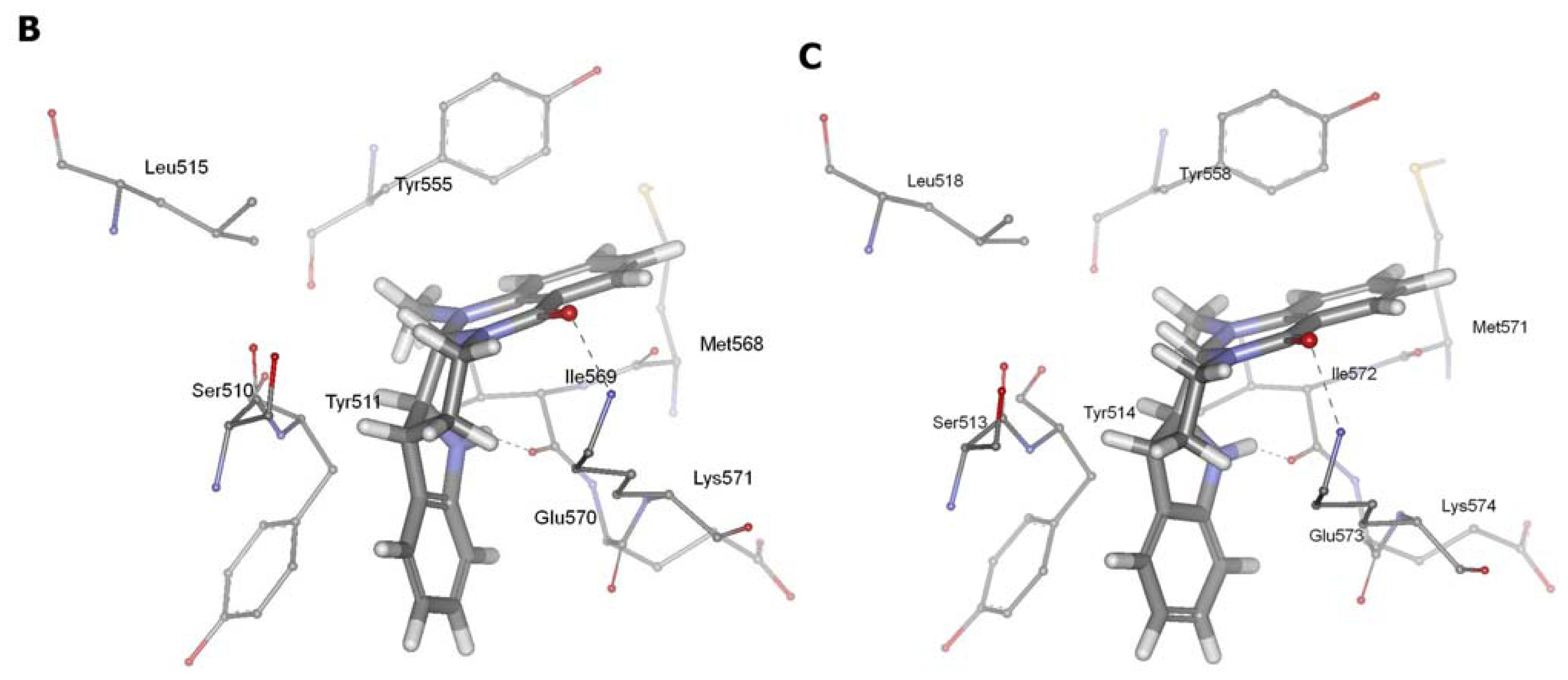
2.3. Detection of Binding between TRPV1 Homology Models and Evodiamine
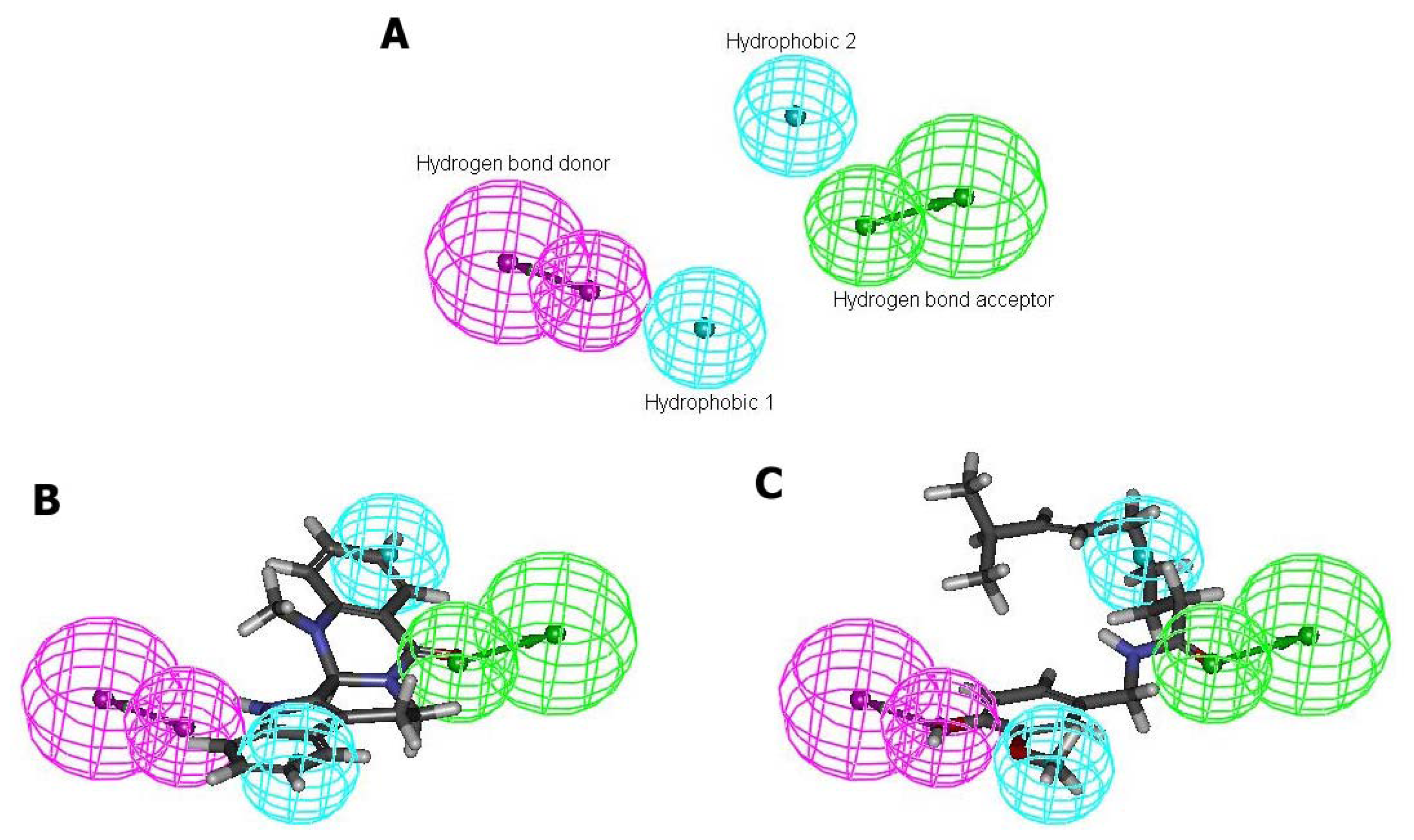
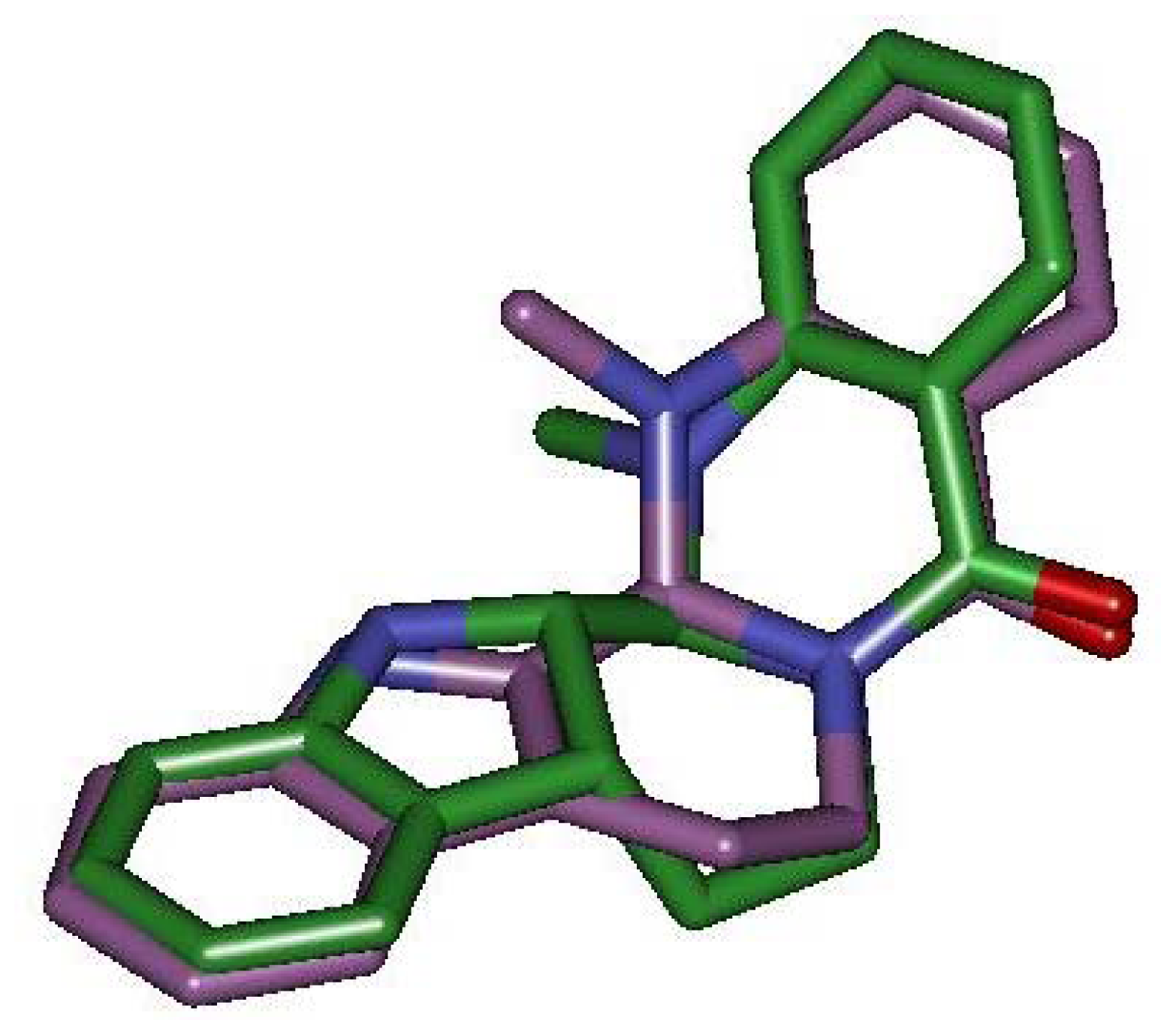
2.4. Pharmacophore Studies
3. Experimental Section
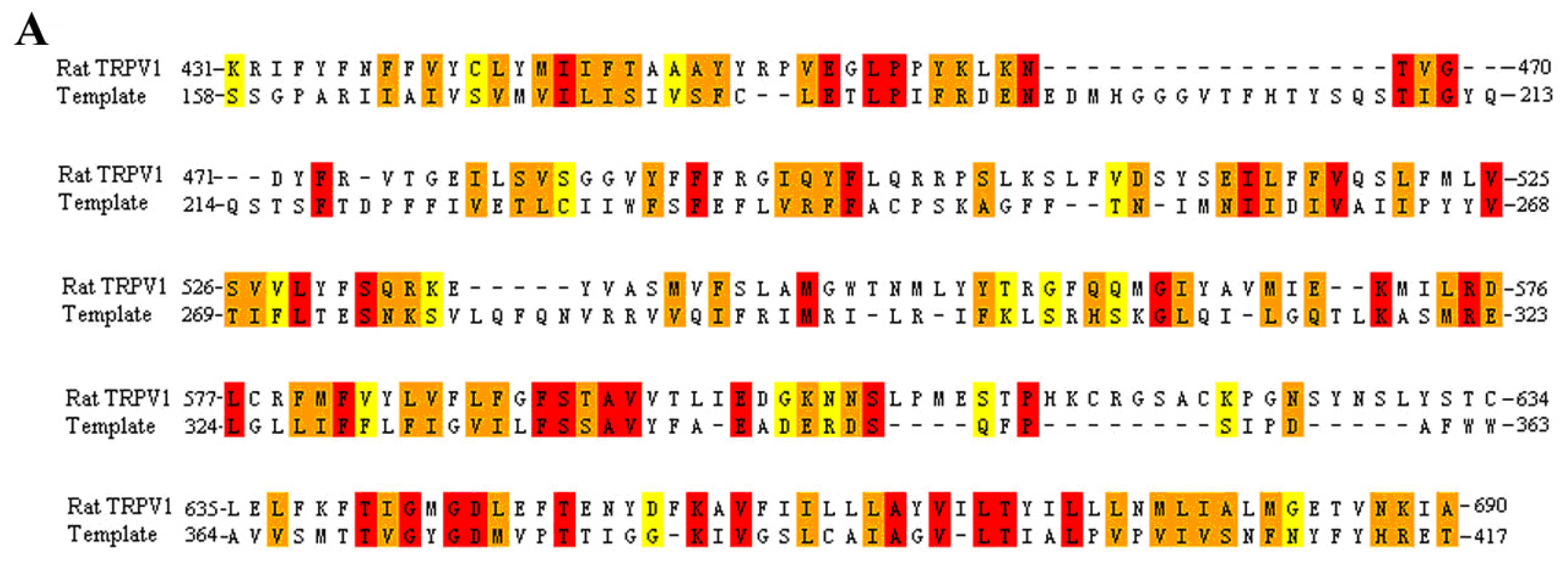
3.1. Multiple Sequence Alignment
3.2. Homology Modeling
3.3. Binding Site Analysis
3.4. Molecular Docking
3.5. Binding Energy Calculations
3.6. Pharmacophore Modeling
4. Conclusions
Supplementary Information
ijms-13-08958-s001.pdfAcknowledgments
References
- Jara-Oseguera, A.; Simon, S.A.; Rosenbaum, T. TRPV1: On the road to pain relief. Curr. Mol. Pharmacol 2008, 1, 255–269. [Google Scholar]
- Kedei, N.; Szabo, T.; Lile, J.D.; Treanor, J.J.; Olah, Z.; Iadarola, M.J.; Blumberg, P.M. Analysis of the native quaternary structure of vanilloid receptor 1. J. Biol. Chem 2001, 276, 28613–28619. [Google Scholar]
- Szallasi, A.; Blumberg, P.M. Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 1989, 30, 515–520. [Google Scholar]
- Pearce, L.V.; Petukhov, P.A.; Szabo, T.; Kedei, N.; Bizik, F.; Kozikowski, A.P.; Blumberg, P.M. Evodiamine functions as an agonist for the vanilloid receptor TRPV1. Org. Biomol. Chem 2004, 2, 2281–2286. [Google Scholar]
- De Petrocellis, L.; Bisogno, T.; Davis, J.B.; Pertwee, R.G.; Di Marzo, V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: Inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett 2000, 483, 52–56. [Google Scholar]
- Ross, R.A. Anandamide and vanilloid TRPV1 receptors. Br. J. Pharmacol 2003, 140, 790–801. [Google Scholar]
- Picazo-Juárez, G.; Romero-Suárez, S.; Nieto-Posadas, A.; Llorente, I.; Jara-Oseguera, A.; Briggs, M.; McIntosh, T.J.; Simon, S.A.; Ladrón-de-Guevara, E.; Islas, L.D.; et al. Identification of a binding motif in the S5 helix that confers cholesterol sensitivity to the TRPV1 ion channel. J. Biol. Chem 2011, 286, 24966–24976. [Google Scholar]
- Jordt, S.E.; Julius, D. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell 2002, 108, 421–430. [Google Scholar]
- Gavva, N.R.; Klionsky, L.; Qu, Y.; Shi, L.; Tamir, R.; Edenson, S.; Zhang, T.J.; Viswanadhan, V.N.; Toth, A.; Pearce, L.V.; et al. Molecular determinants of vanilloid sensitivity in TRPV1. J. Biol. Chem 2004, 279, 20283–20295. [Google Scholar]
- Chou, M.Z.; Mtui, T.; Gao, Y.D.; Kohler, M.; Middleton, R.E. Resiniferatoxin binds to the capsaicin receptor (TRPV1) near the extracellular side of the S4 transmembrane domain. Biochemistry 2004, 43, 2501–2511. [Google Scholar]
- Lee, J.H.; Lee, Y.; Ryu, H.; Kang, D.W.; Lee, J.; Lazar, J.; Pearce, L.V.; Pavlyukovets, V.A.; Blumberg, P.M.; Choi, S. Structural insights into transient receptor potential vanilloid type 1 (TRPV1) from homology modeling, flexible docking, and mutational studies. J. Comput. Aided Mol. Des 2011, 25, 317–327. [Google Scholar]
- Yu, H.; Tu, Y.; Zhang, C.; Fan, X.; Wang, X.; Wang, Z.; Liang, H. Evodiamine as a novel antagonist of aryl hydrocarbon receptor. Biochem. Biophys. Res. Commun 2010, 1, 94–98. [Google Scholar]
- Peng, L.; Li, Y.J. The vanilloid receptor TRPV-1: Role in cardiovascular and gastrointestinal protection. Eur. J. Pharmacol 2010, 627, 1–7. [Google Scholar]
- Kobayashi, Y.; Nakano, Y.; Hoshikuma, K.M.; Yokoo, Y.; Kamiya, T. The bronchoconstrictive action of evodiamine, an indoloquinazoline alkaloid isolated from the fruits of Evodia rutaecarpa, on guinea-pig isolated bronchus: Possible involvement on vanilloid receptors. Planta Med 2000, 66, 526–530. [Google Scholar]
- Kobayashi, Y.; Hoshikuma, K.; Nakano, Y.; Yokoo, Y.; Kamiya, T. The positive inotropic and chronotropic effects of evodiamine and rutaecarpine, indoloquinazoline alkaloids isolated from the fruits of Evodia rutaecarpa, on the guinea-pig isolated right atria: Possible involvement of vanilloid receptors. Planta Med 2001, 67, 244–248. [Google Scholar]
- Rang, W.Q.; Du, Y.H.; Hu, C.P.; Ye, F.; Xu, K.P.; Peng, J.; Deng, H.W.; Li, Y.J. Protective effects of evodiamine on myocardial ischemia-reperfusion injury in rats. Planta Med 2004, 70, 1140–1143. [Google Scholar]
- Chiou, W.F.; Chen, C.F. Pharmacological profile of evodiamine in isolated rabbit corpus cavernosum. Eur. J. Pharmacol 2002, 446, 151–159. [Google Scholar]
- Ghasemi, M.; Sadeghipour, H.; Dehpour, A.R. Anandamide improves the impaired nitric oxide-mediated neurogenic relaxation of the corpus cavernosum in diabetic rats: Involvement of cannabinoid CB1 and vanilloid VR1 receptors. BJU Int 2007, 100, 1385–1390. [Google Scholar]
- Yu, F.H.; Yarov-Yarovoi, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev 2005, 57, 387–395. [Google Scholar]
- Brauchi, S.; Orta, G.; Mascayano, C.; Salazar, M.; Raddatz, N.; Urbina, H.; Rosenmann, E.; Gonzalez-Nilo, F.; Latorre, R. Dissection of the components for PIP2 activation and thermosensation in TRP channels. Proc. Natl. Acad. Sci. USA 2007, 104, 10246–10251. [Google Scholar]
- UniProtKB. UniProtKB/Swiss-Prot. Available online: http://www.uniprot.org/uniprot/ accessed on 29 January 2012.
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic. Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Laskoswki, R.A.; MacArthur, M.W.; Moss, D.S.; Thorton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst 1993, 26, 283–291. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci 1993, 2, 1511–1519. [Google Scholar]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model 2003, 21, 289–307. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [Google Scholar]
- Schapira, M.; Totrov, M.; Abagyan, R. Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recognit 1999, 12, 177–190. [Google Scholar]
- Brooks, B.R.; Brucolleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy minimization, and dynamic calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- Smellie, A.; Teig, S.L.; Towbin, P. Poling: Promoting conformational variation. J. Comp. Chem 1995, 16, 171–187. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Z.; Sun, L.; Yu, H.; Zhang, Y.; Gong, W.; Jin, H.; Zhang, L.; Liang, H. Binding Mode Prediction of Evodiamine within Vanilloid Receptor TRPV1. Int. J. Mol. Sci. 2012, 13, 8958-8969. https://doi.org/10.3390/ijms13078958
Wang Z, Sun L, Yu H, Zhang Y, Gong W, Jin H, Zhang L, Liang H. Binding Mode Prediction of Evodiamine within Vanilloid Receptor TRPV1. International Journal of Molecular Sciences. 2012; 13(7):8958-8969. https://doi.org/10.3390/ijms13078958
Chicago/Turabian StyleWang, Zhanli, Lidan Sun, Hui Yu, Yanhui Zhang, Wuzhuang Gong, Hongwei Jin, Liangren Zhang, and Huaping Liang. 2012. "Binding Mode Prediction of Evodiamine within Vanilloid Receptor TRPV1" International Journal of Molecular Sciences 13, no. 7: 8958-8969. https://doi.org/10.3390/ijms13078958