Prediction of a New Ligand-Binding Site for Type 2 Motif based on the Crystal Structure of ALG-2 by Dry and Wet Approaches


Abstract
:
1. Introduction
2. Results and Discussion
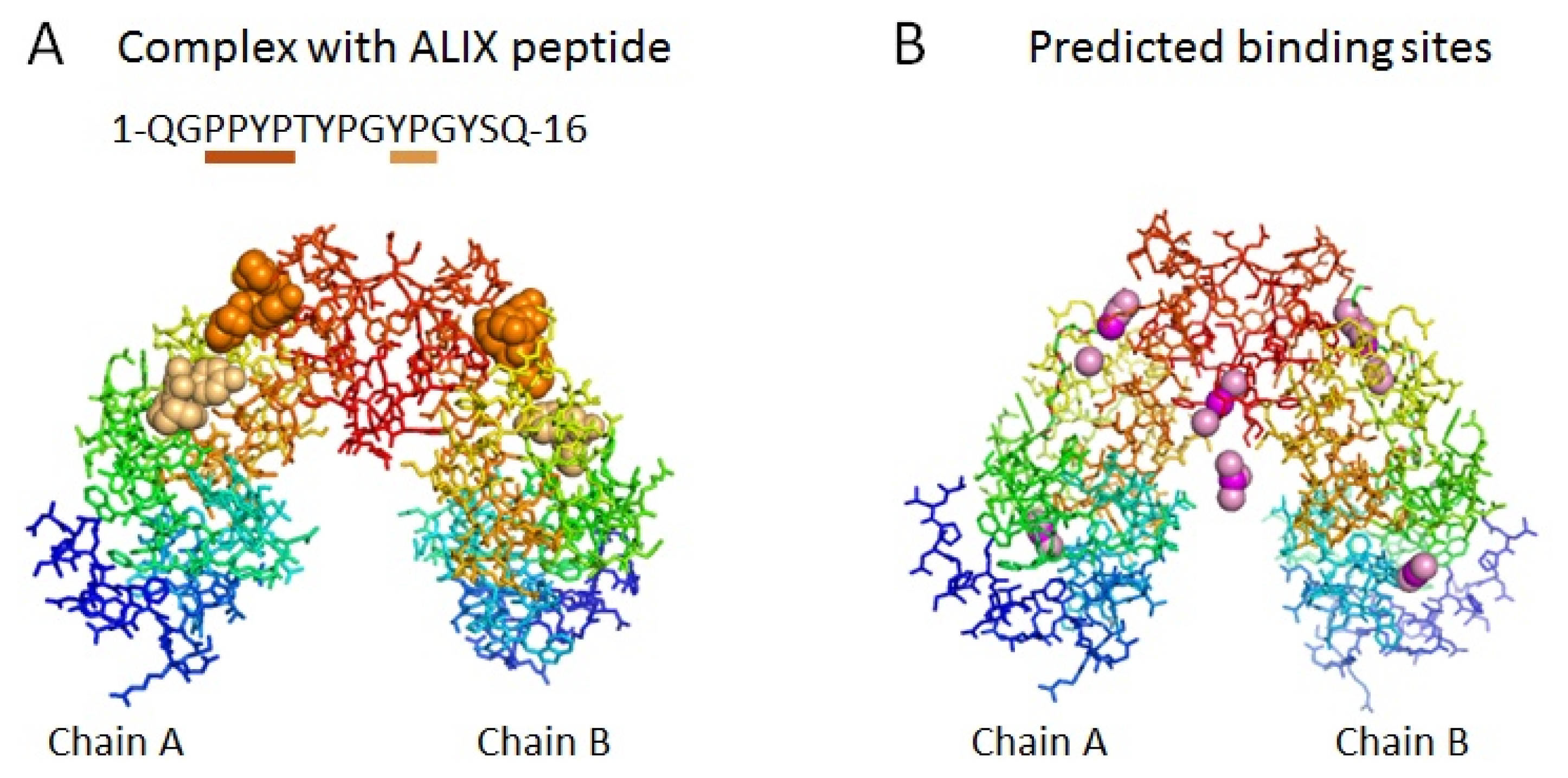
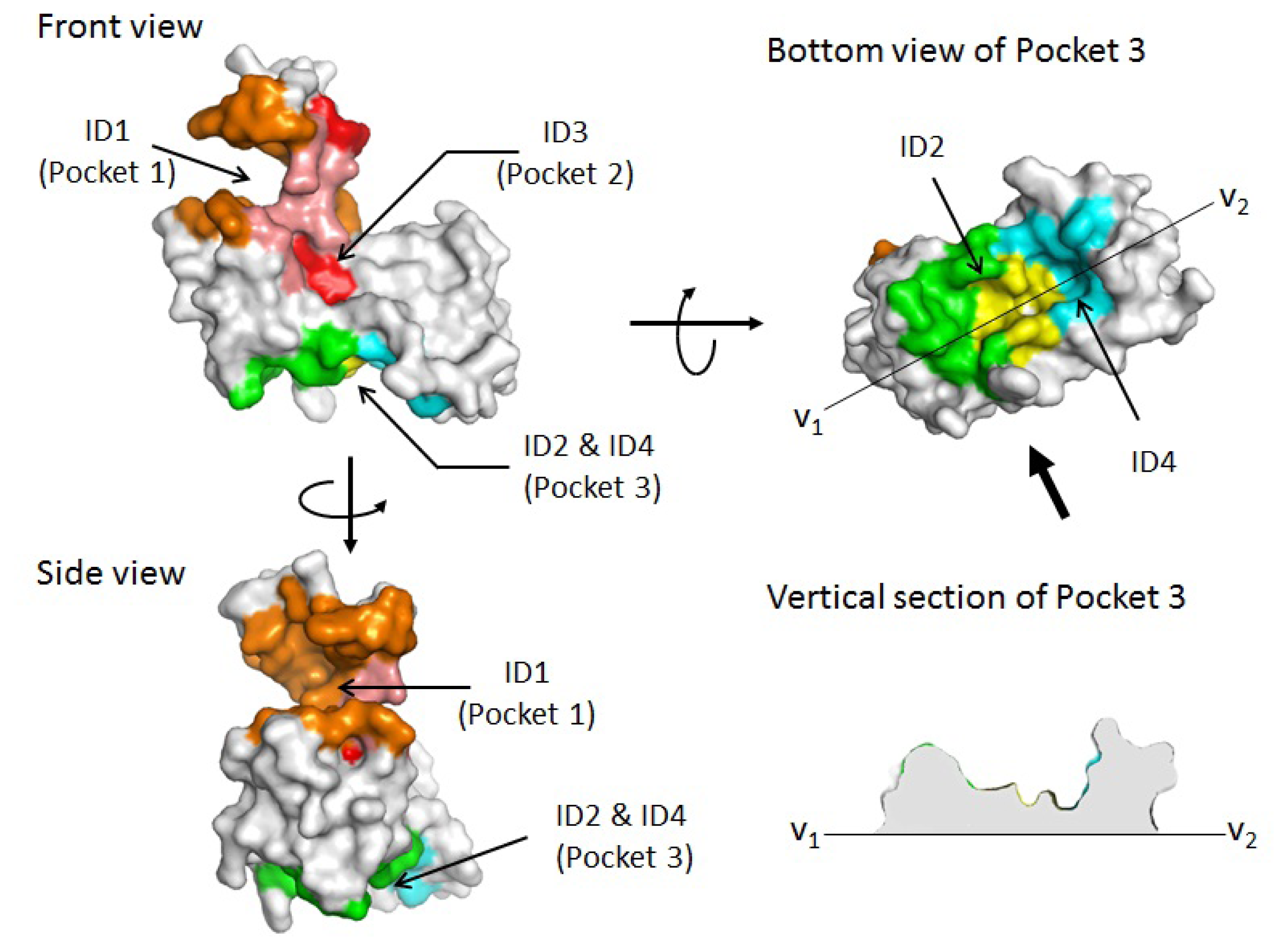
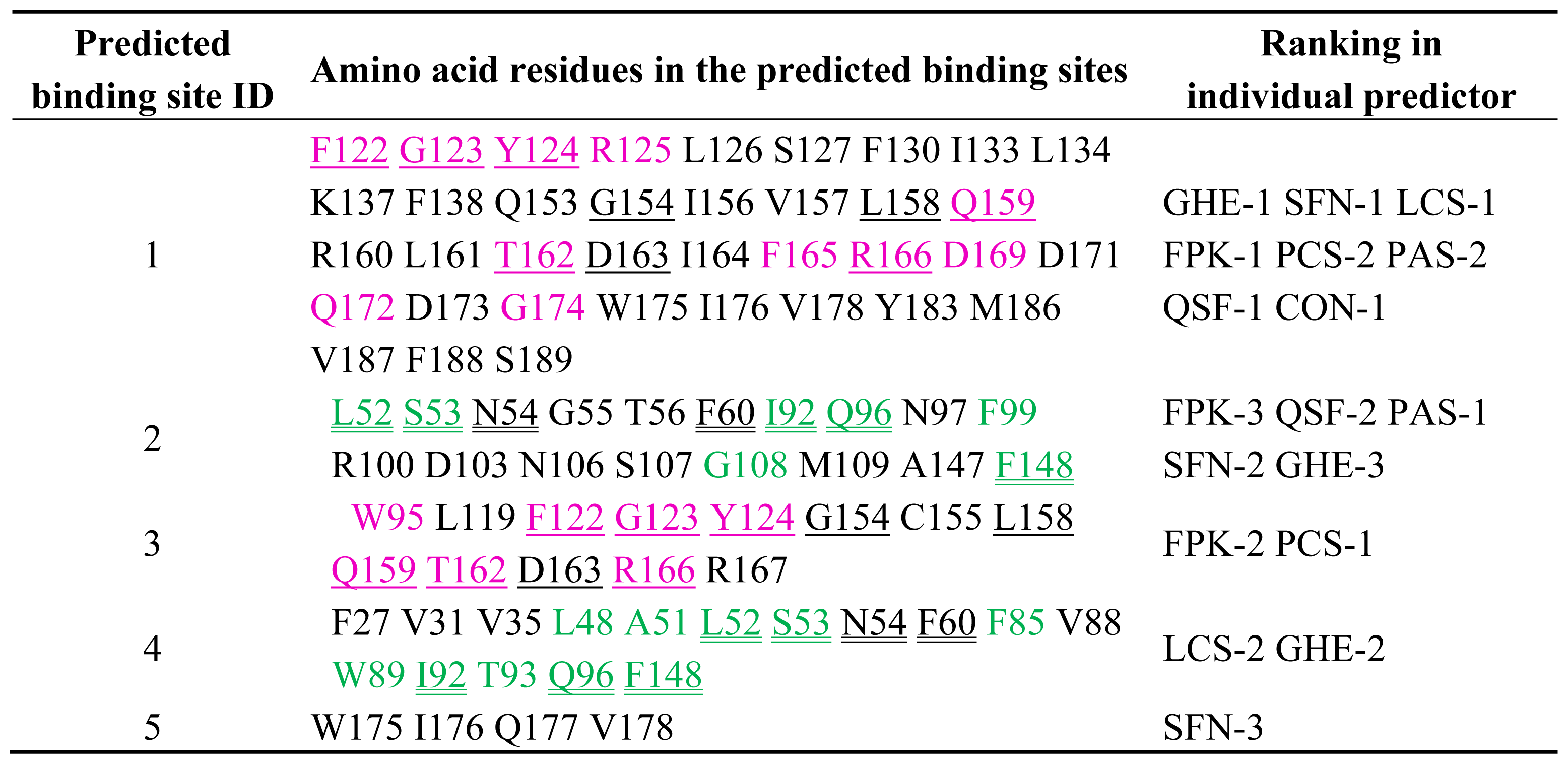
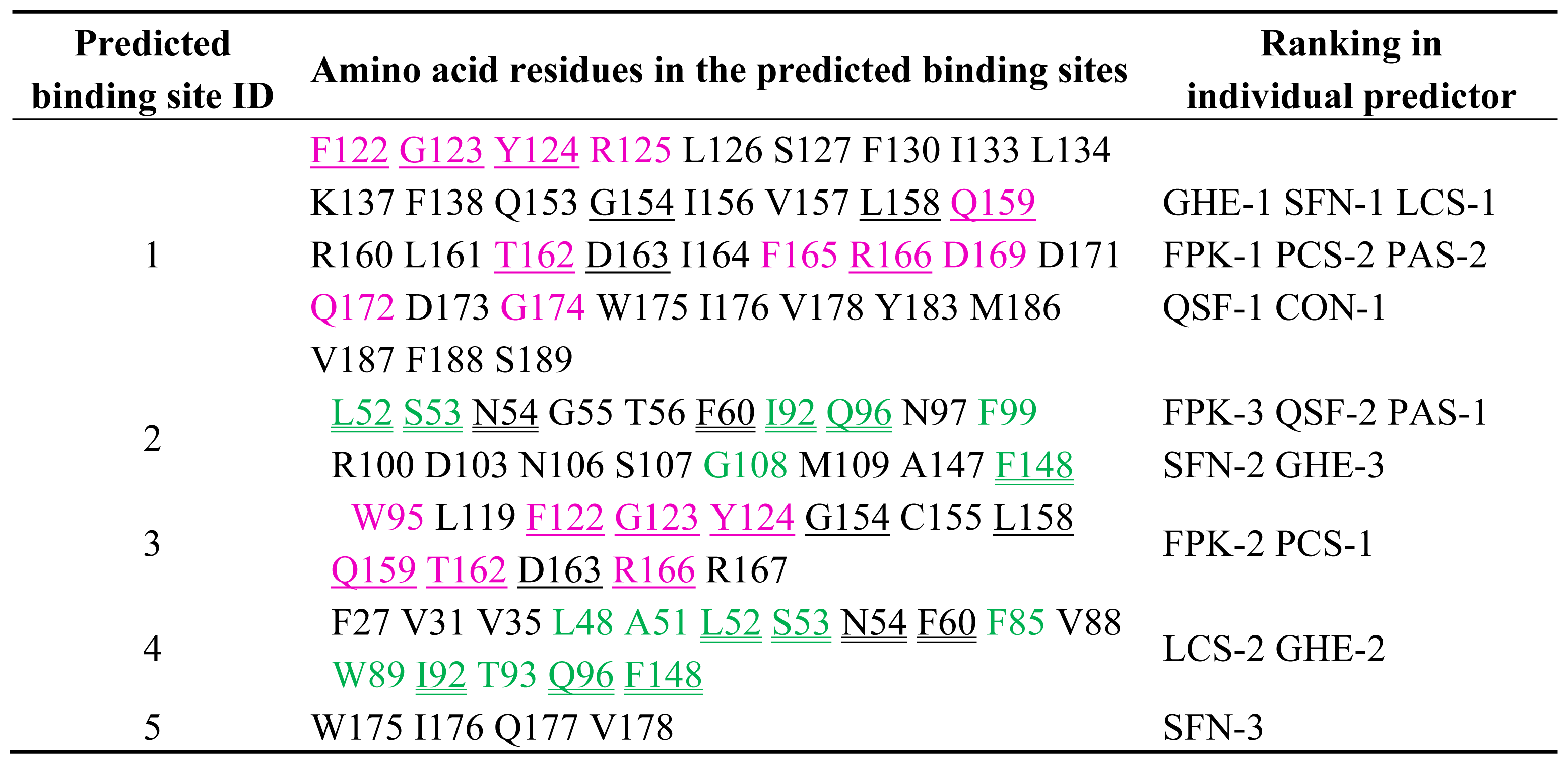
2.1. Prediction of Potential Binding Sites in the Dimer Molecule of ALG-2
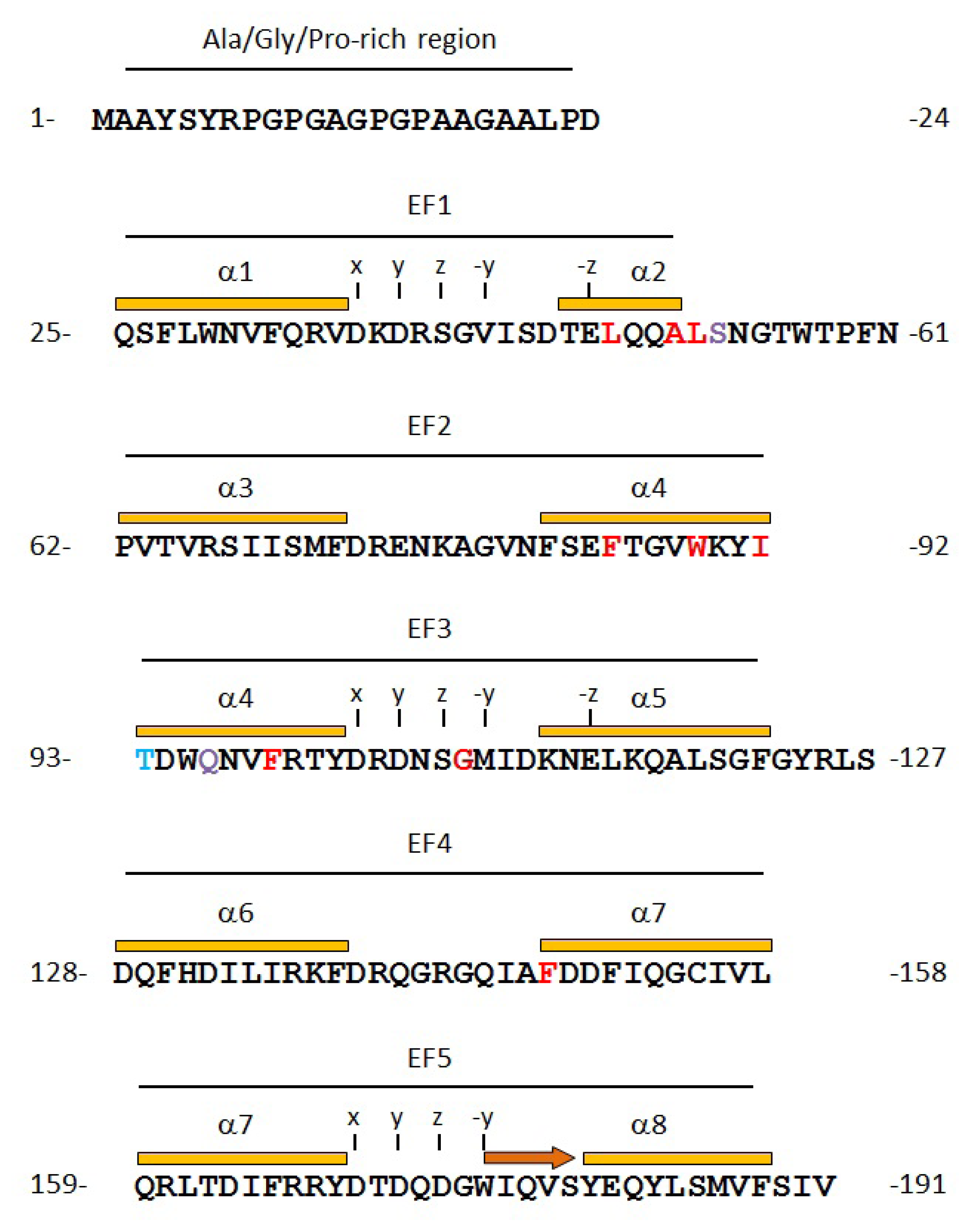
2.2. Prediction of Binding Sites for Type 2 Motif in the Monomer Molecule of ALG-2
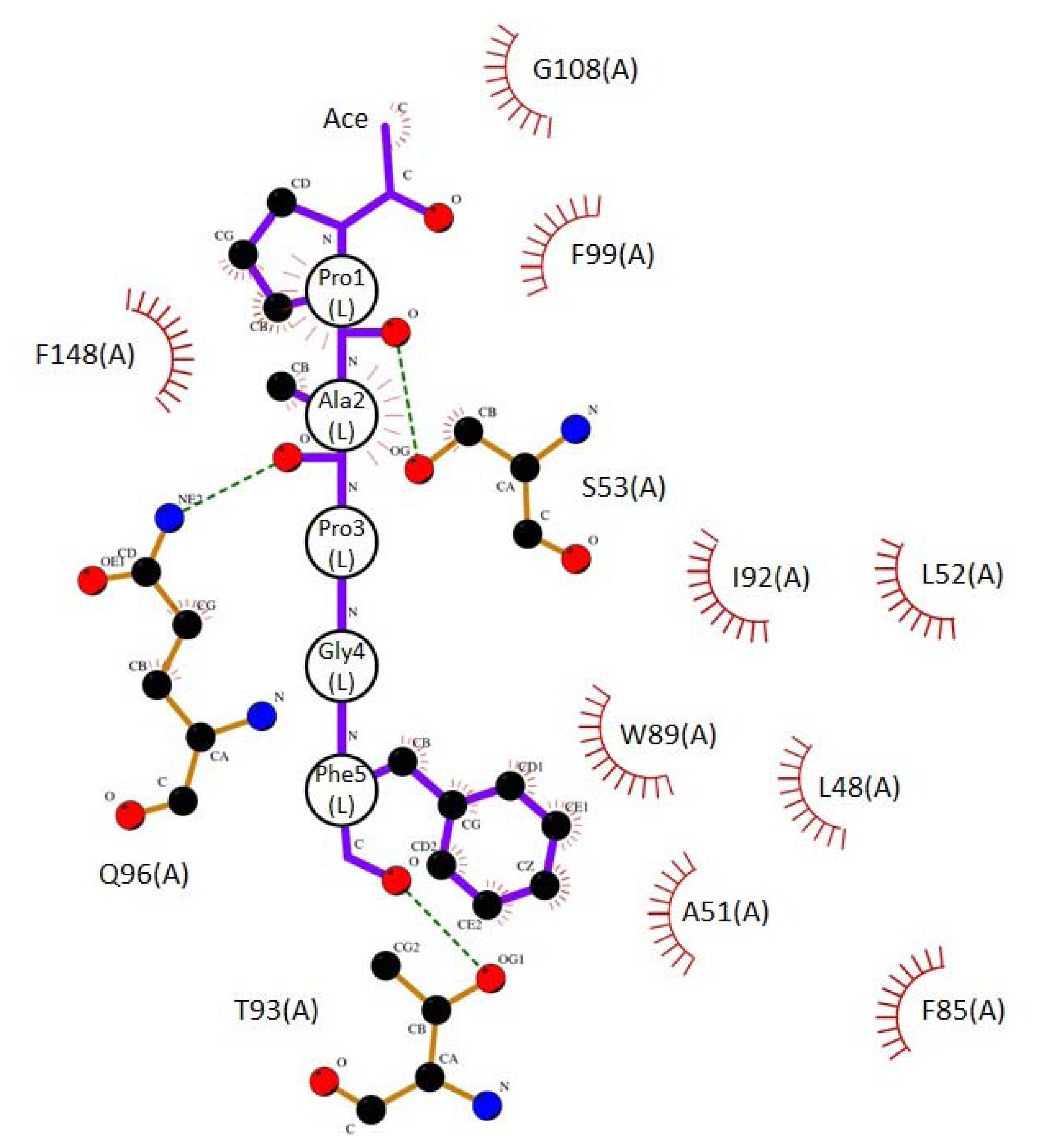
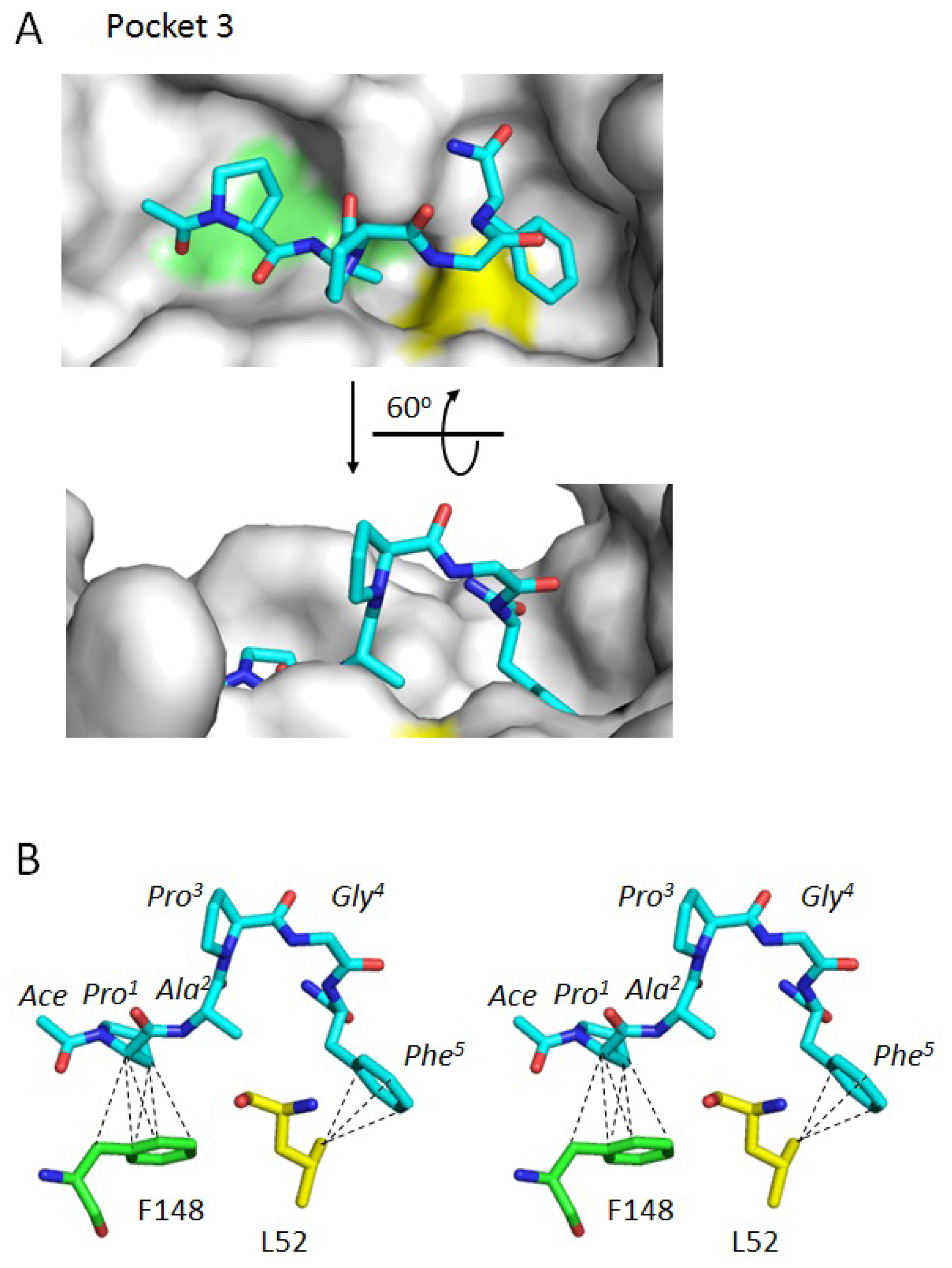
2.3. Docking Model of Type 2 Motif Peptide Binding to Hydrophobic Pocket 3
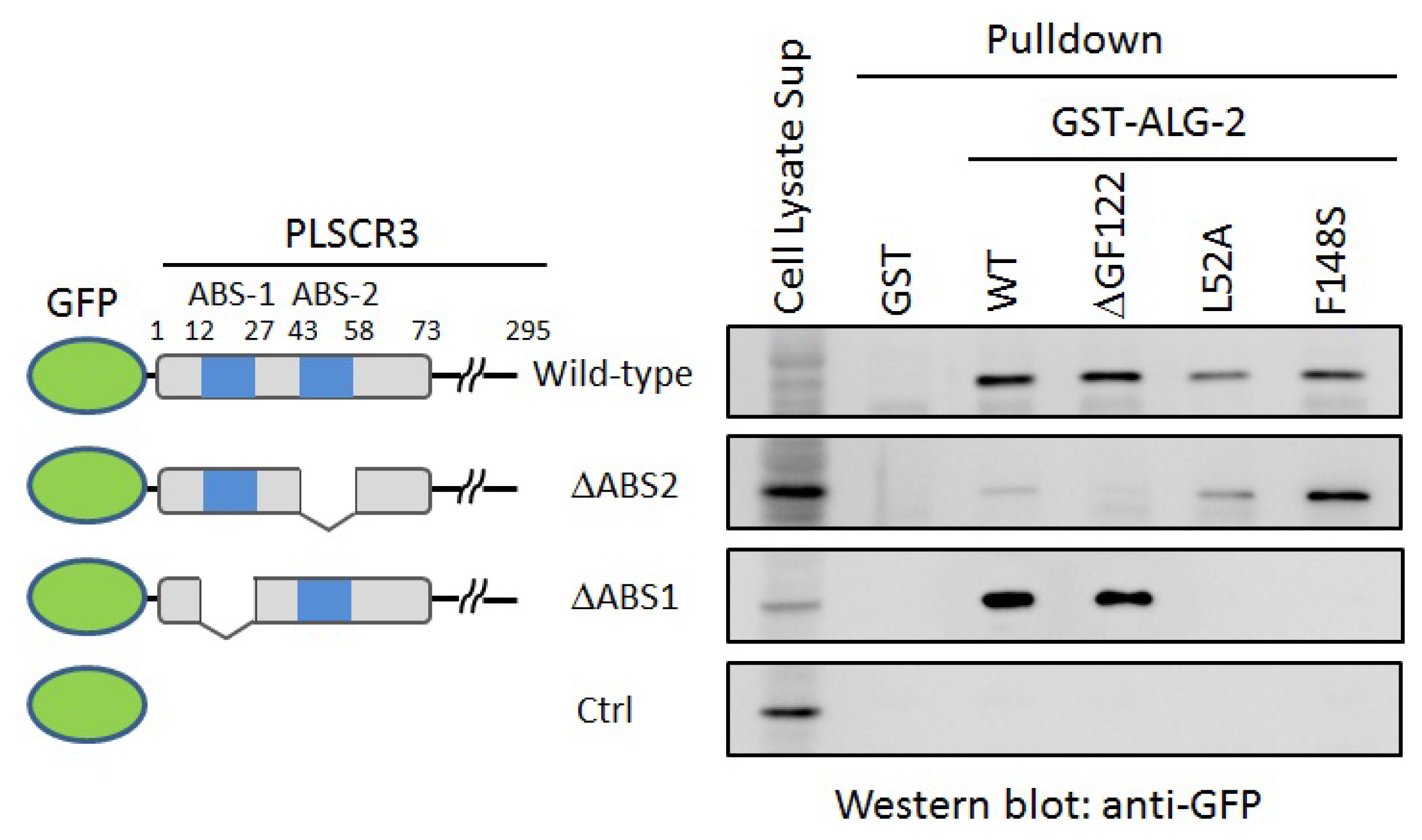
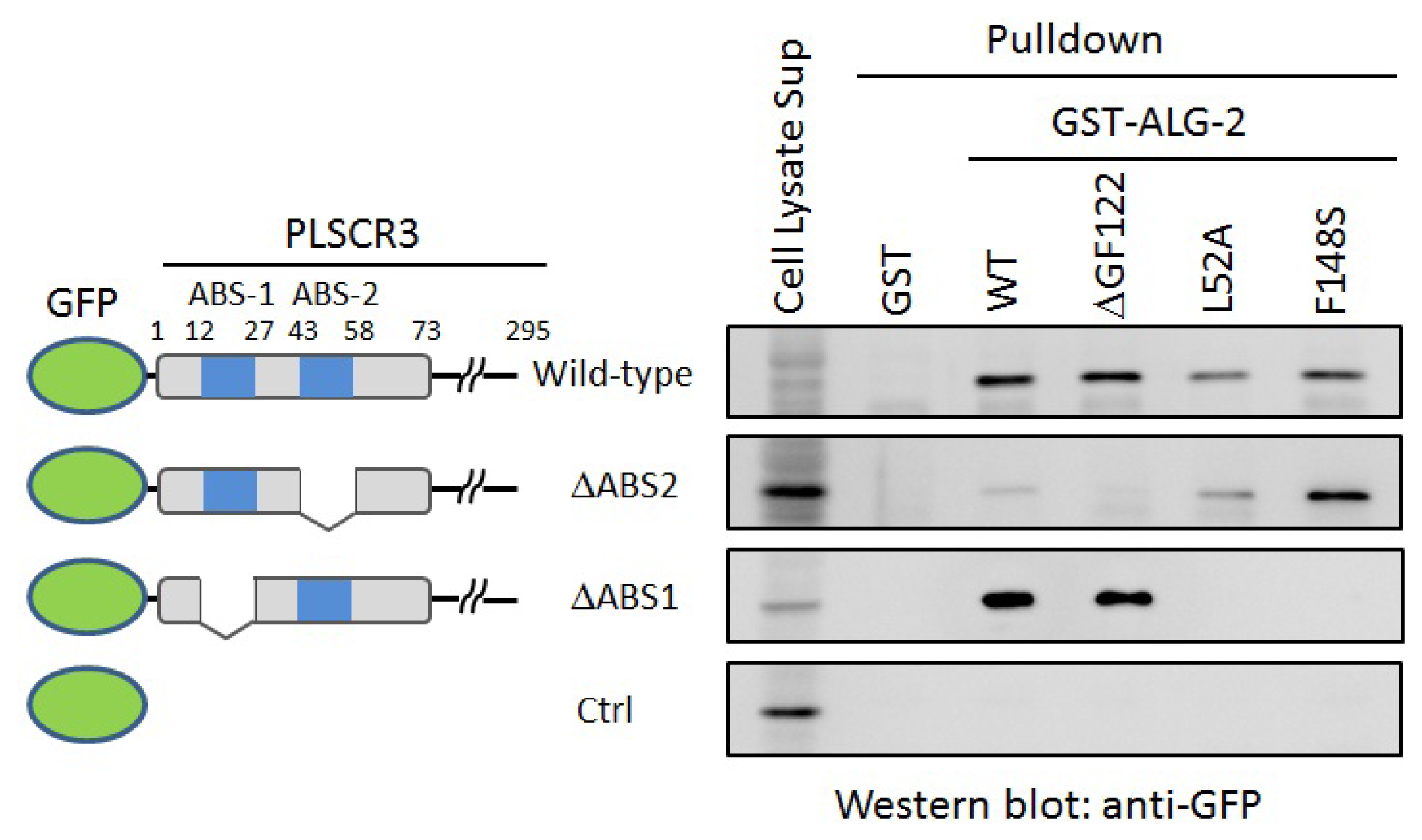
2.4. In Vitro Binding Assay
2.5. Ca2+-Dependent Interaction
3. Experimental Section
3.1. Prediction of Potential Binding Sites and Docking Simulation
3.2. Expression and Purification of Mutant ALG-2 Proteins
3.3. GST-Pulldown Assays
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
- Supplementary MaterialsSupplementary Figures S1, S2 and Supplementary files in PDB formats are available at IJMS online.
References
- Maki, M.; Narayana, S.V.; Hitomi, K. A growing family of the Ca2+-binding proteins with five EF-hand motifs. Biochem. J 1997, 328, 718–720. [Google Scholar]
- Maki, M.; Kitaura, Y.; Satoh, H.; Ohkouchi, S.; Shibata, H. Structures, functions and molecular evolution of the penta-EF-hand Ca2+-binding proteins. Biochim. Biophys. Acta 2002, 1600, 51–60. [Google Scholar]
- Maki, M.; Maemoto, Y.; Osako, Y.; Shibata, H. Evolutionary and physical linkage between calpains and penta-EF-hand Ca2+-binding proteins. FEBS J 2012, 279, 1414–1421. [Google Scholar]
- Maki, M.; Suzuki, H.; Shibata, H. Structure and function of ALG-2, a penta-EF-hand calcium-dependent adaptor protein. Sci. China Life Sci 2011, 54, 770–779. [Google Scholar]
- Vito, P.; Lacana, E.; D’Adamio, L. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science 1996, 271, 521–525. [Google Scholar]
- Missotten, M.; Nichols, A.; Rieger, K.; Sadoul, R. Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell. Death Differ 1999, 6, 124–129. [Google Scholar]
- Vito, P.; Pellegrini, L.; Guiet, C.; D’Adamio, L. Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J. Biol. Chem 1999, 274, 1533–1540. [Google Scholar]
- Satoh, H.; Nakano, Y.; Shibata, H.; Maki, M. The penta-EF-hand domain of ALG-2 interacts with amino-terminal domains of both annexin VII and annexin XI in a Ca2+-dependent manner. Biochim. Biophys. Acta 2002, 1600, 61–67. [Google Scholar]
- Satoh, H.; Shibata, H.; Nakano, Y.; Kitaura, Y.; Maki, M. ALG-2 interacts with the amino-terminal domain of annexin XI in a Ca2+-dependent manner. Biochem. Biophys. Res. Commun 2002, 291, 1166–1172. [Google Scholar]
- Katoh, K.; Suzuki, H.; Terasawa, Y.; Mizuno, T.; Yasuda, J.; Shibata, H.; Maki, M. The penta-EF-hand protein ALG-2 interacts directly with the ESCRT-I component TSG101, and Ca2+-dependently co-localizes to aberrant endosomes with dominant-negative AAA ATPase SKD1/Vps4B. Biochem. J 2005, 391, 677–685. [Google Scholar]
- Yamasaki, A.; Tani, K.; Yamamoto, A.; Kitamura, N.; Komada, M. The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol. Biol. Cell 2006, 17, 4876–4887. [Google Scholar]
- Shibata, H.; Suzuki, H.; Yoshida, H.; Maki, M. ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem. Biophys. Res. Commun 2007, 353, 756–763. [Google Scholar]
- Montaville, P.; Dai, Y.; Cheung, C.Y.; Giller, K.; Becker, S.; Michalak, M.; Webb, S.E.; Miller, A.L.; Krebs, J. Nuclear translocation of the calcium-binding protein ALG-2 induced by the RNA-binding protein RBM22. Biochim. Biophys. Acta 2006, 1763, 1335–1343. [Google Scholar]
- Draeby, I.; Woods, Y.L.; la Cour, J.M.; Mollerup, J.; Bourdon, J.C.; Berchtold, M.W. The calcium binding protein ALG-2 binds and stabilizes Scotin, a p53-inducible gene product localized at the endoplasmic reticulum membrane. Arch. Biochem. Biophys 2007, 467, 87–94. [Google Scholar]
- Shibata, H.; Suzuki, H.; Kakiuchi, T.; Inuzuka, T.; Yoshida, H.; Mizuno, T.; Maki, M. Identification of Alix-type and Non-Alix-type ALG-2-binding sites in human phospholipid scramblase 3: Differential binding to an alternatively spliced isoform and amino acid-substituted mutants. J. Biol. Chem 2008, 283, 9623–9632. [Google Scholar]
- Osugi, K.; Suzuki, H.; Nomura, T.; Ariumi, Y.; Shibata, H.; Maki, M. Identification of the P-body component PATL1 as a novel ALG-2-interacting protein by in silico and Far-Western screening of proline-rich proteins. J. Biochem 2012. [Google Scholar] [CrossRef]
- Hwang, I.S.; Jung, Y.S.; Kim, E. Interaction of ALG-2 with ASK1 influences ASK1 localization and subsequent JNK activation. FEBS Lett 2002, 529, 183–187. [Google Scholar]
- Chen, C.; Sytkowski, A.J. Apoptosis-linked gene-2 connects the Raf-1 and ASK1 signalings. Biochem. Biophys. Res. Comm 2005, 333, 51–57. [Google Scholar]
- Vergarajauregui, S.; Martina, J.A.; Puertollano, R. Identification of the penta-EF-hand protein ALG-2 as a Ca2+-dependent interactor of mucolipin-1. J. Biol. Chem 2009, 284, 36357–36366. [Google Scholar]
- Shibata, H.; Inuzuka, T.; Yoshida, H.; Sugiura, H.; Wada, I.; Maki, M. The ALG-2 binding site in Sec31A influences the retention kinetics of Sec31A at the endoplasmic reticulum exit sites as revealed by live-cell time-lapse imaging. Biosci. Biotechnol. Biochem 2010, 74, 1819–1826. [Google Scholar]
- Suzuki, H.; Kawasaki, M.; Inuzuka, T.; Okumura, M.; Kakiuchi, T.; Shibata, H.; Wakatsuki, S.; Maki, M. Structural basis for Ca2+-dependent formation of ALG-2/Alix peptide complex: Ca2+/EF3-driven arginine switch mechanism. Structure 2008, 16, 1562–1573. [Google Scholar]
- Ikura, M.; Ames, J.B. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: Two ways to promote multifunctionality. Proc. Natl. Acad. Sci. USA 2006, 103, 1159–1164. [Google Scholar]
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 2011, 27, 2083–2088. [Google Scholar]
- Huang, B.; Schroeder, M. LIGSITEcsc: Predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol 2006, 6. [Google Scholar] [CrossRef]
- Brady, G.P., Jr; Stouten, P.F. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des 2000, 14, 383–401. [Google Scholar]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar]
- Laskowski, R.A. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph 1995, 13, 323–330. [Google Scholar]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 2009, 10. [Google Scholar] [CrossRef]
- Kawabata, T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins 2010, 78, 1195–1211. [Google Scholar]
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol 2009, 5. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar]
- Okumura, M.; Ichioka, F.; Kobayashi, R.; Suzuki, H.; Yoshida, H.; Shibata, H.; Maki, M. Penta-EF-hand protein ALG-2 functions as a Ca2+-dependent adaptor that bridges Alix and TSG101. Biochem. Biophys. Res. Commun 2009, 386, 237–241. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng 1995, 8, 127–134. [Google Scholar]
- Collaborative Computational Project. Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr 1994, 50, 760–763.
- Maki, M.; Yamaguchi, K; Kitaura, Y; Satoh, H; Hitomi, K. Calcium-induced exposure of a hydrophobic surface of mouse ALG-2, which is a member of the penta-EF-hand protein family. J. Biochem 1998, 124, 1170–1177. [Google Scholar]
- Jia, J.; Tarabykina, S.; Hansen, C.; Berchtold, M.; Cygler, M. Structure of apoptosis-linked protein ALG-2: Insights into Ca2+-induced changes in penta-EF-hand proteins. Structure 2001, 9, 267–275. [Google Scholar]
- MetaPocket 2.0. Available online: http://projects.biotec.tu-dresden.de/metapocket accessed on 24 February 2012.
- PyMol. Available online: http://www.pymol.org/ accessed on 26 January 2012.
- Kristian, R. PyMOL plugins. 2007. Available online: http://www.rubor.de/pymol_extensions_en.html accessed on 27 February 2012.
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 2126–2132. [Google Scholar]
- AutoDock Vina. Available online: http://autodock.scripps.edu/ accessed on 3 March 2012.
- LIGPLOT. Available online: http://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/ accessed on 3 March 2012.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Color | Predicted binding site ID number | Pocket number | |
|---|---|---|---|
| orange | 1 | Pocket 1 | |
| red | 3 | Pocket 2 | |
| salmon | 1 & 3 | Pockets 1 & 2 | |
| green | 2 | Pocket 3 | |
| cyan | 4 | Pocket 3 | |
| yellow | 2 & 4 | Pocket 3 |
| Interacting atoms in peptide | Interacting atoms in ALG-2 | Distance (Å) | |
|---|---|---|---|
| Hydrophobic | |||
| ACE | C a | CE2F99 | 3.7 |
| CAG108 | 3.6 | ||
| Pro1 | CG | CBQ96 | 3.9 |
| CB | CGQ96 | 3.7 | |
| CG | CGQ96 | 3.9 | |
| CA | CD2F148 | 3.9 | |
| CB | CD2F148 | 3.5 | |
| CB | CE2F148 | 3.8 | |
| CB | CGF148 | 3.7 | |
| Ala2 | CA | CBS53 | 3.5 |
| CB | CBS53 | 3.5 | |
| Phe5 | CZ | CD1L48 | 3.8 |
| CE2 | CBA51 | 3.6 | |
| CD2 | CD2L52 | 3.8 | |
| CE2 | CD2L52 | 3.7 | |
| CZ | CD2L52 | 3.9 | |
| CE1 | CE1F85 | 3.8 | |
| CZ | CE1F85 | 3.5 | |
| CZ | CZF85 | 3.7 | |
| CD1 | CD1W89 | 3.8 | |
| CE1 | CAW89 | 3.8 | |
| CB | CG2I92 | 3.7 | |
| CD1 | CG2I92 | 3.6 | |
| CG | CG2I92 | 3.8 | |
| Hydrogen bonds | |||
| Pro1 | O | OGS53 | 3.1 |
| Ala2 | O | NE2Q96 | 3.1 |
| Phe5 | O | OG1T93 | 3.0 |
| NH2 | N | OE1Q96 | 3.0 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takahashi, T.; Suzuki, H.; Inuzuka, T.; Shibata, H.; Maki, M. Prediction of a New Ligand-Binding Site for Type 2 Motif based on the Crystal Structure of ALG-2 by Dry and Wet Approaches. Int. J. Mol. Sci. 2012, 13, 7532-7549. https://doi.org/10.3390/ijms13067532
Takahashi T, Suzuki H, Inuzuka T, Shibata H, Maki M. Prediction of a New Ligand-Binding Site for Type 2 Motif based on the Crystal Structure of ALG-2 by Dry and Wet Approaches. International Journal of Molecular Sciences. 2012; 13(6):7532-7549. https://doi.org/10.3390/ijms13067532
Chicago/Turabian StyleTakahashi, Takeshi, Hironori Suzuki, Tatsutoshi Inuzuka, Hideki Shibata, and Masatoshi Maki. 2012. "Prediction of a New Ligand-Binding Site for Type 2 Motif based on the Crystal Structure of ALG-2 by Dry and Wet Approaches" International Journal of Molecular Sciences 13, no. 6: 7532-7549. https://doi.org/10.3390/ijms13067532