Streptozotocin-Induced Cytotoxicity, Oxidative Stress and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
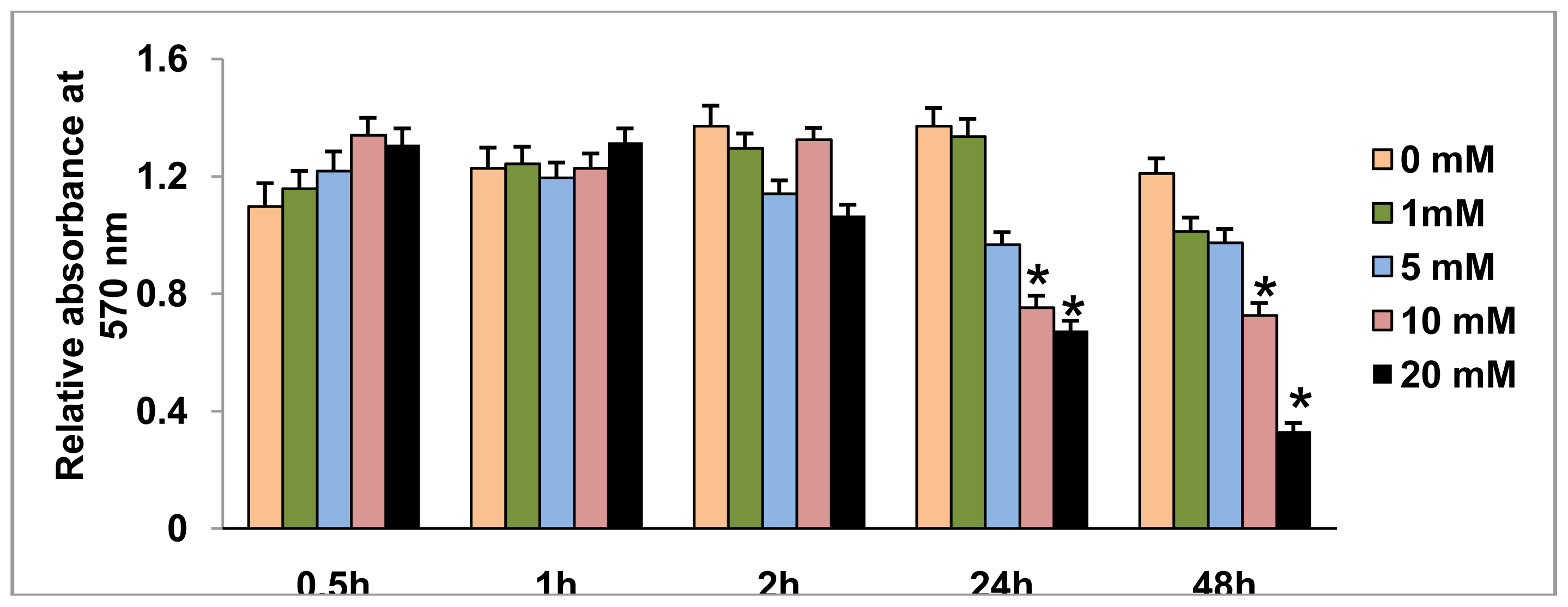
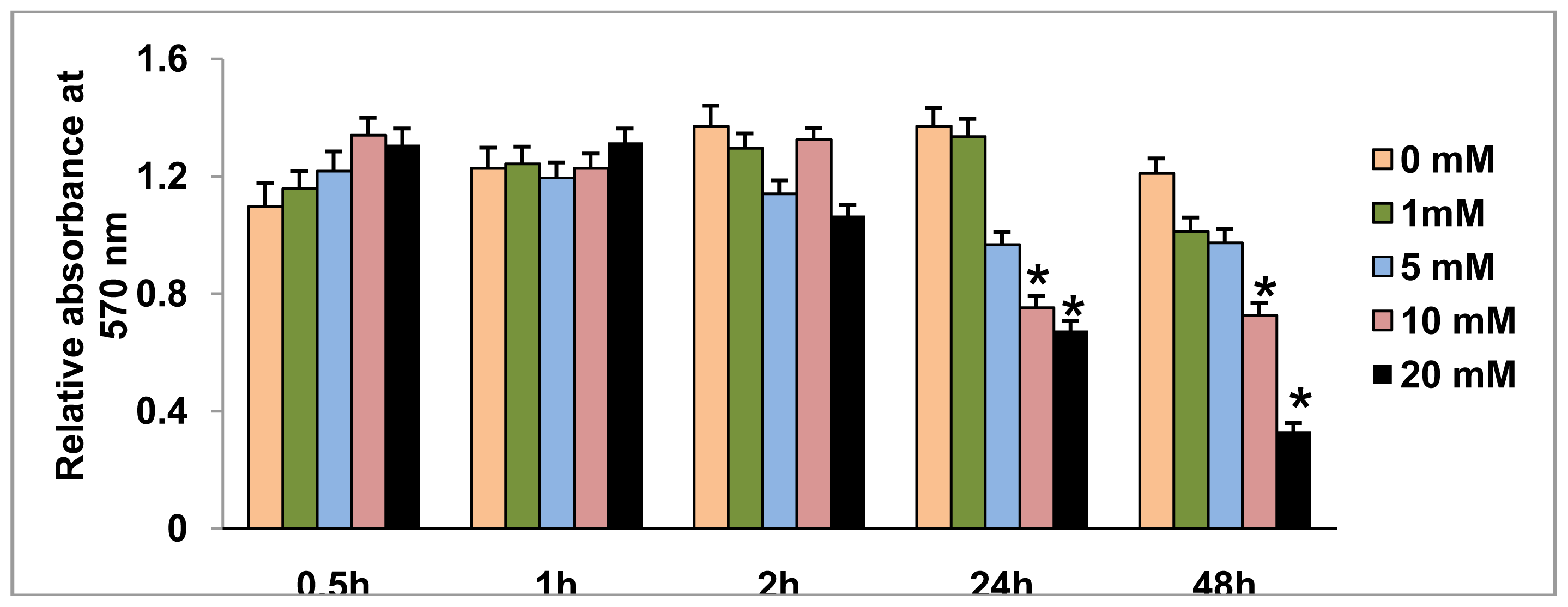
2.1. Effect of STZ on HepG2 Cell Viability
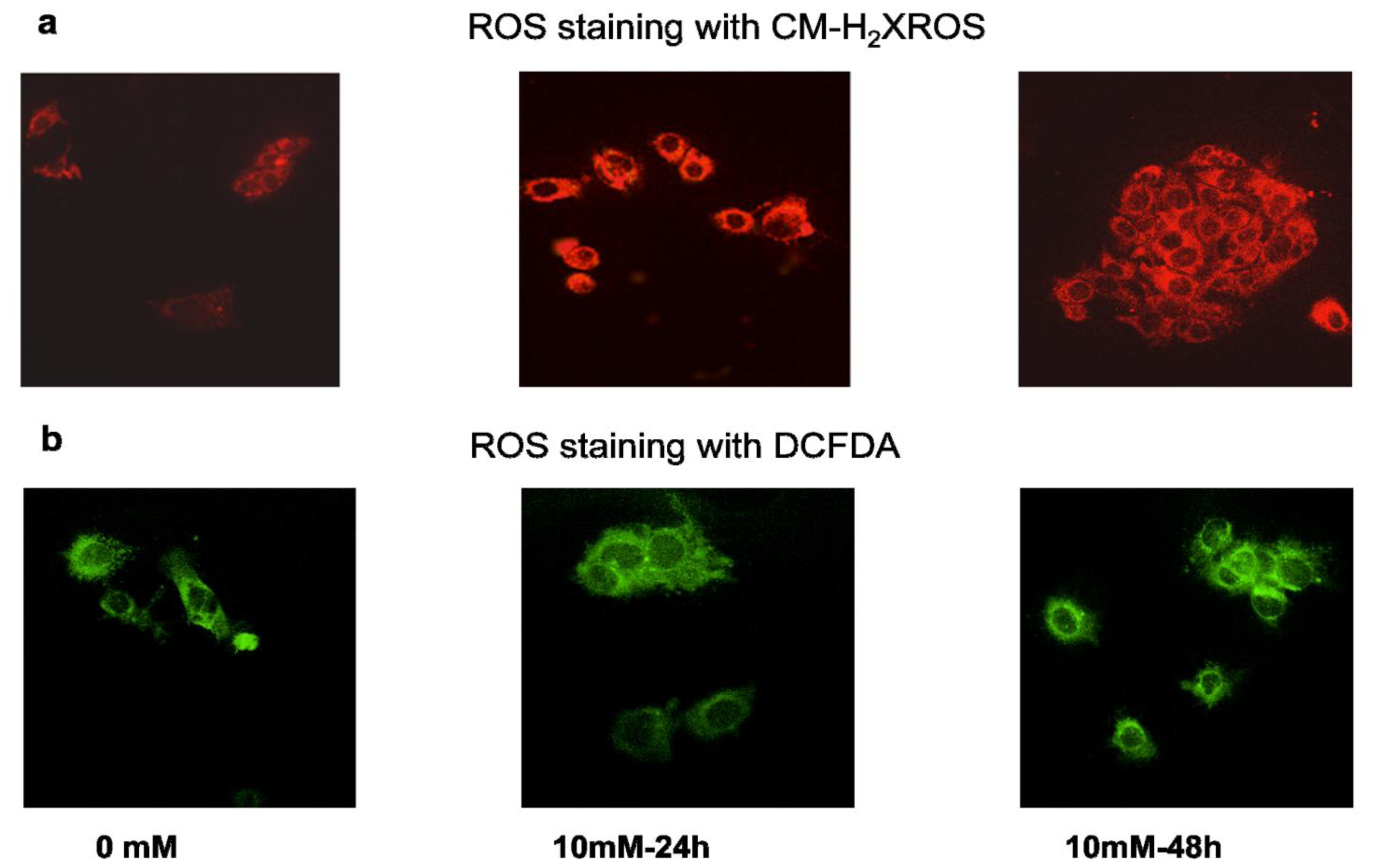
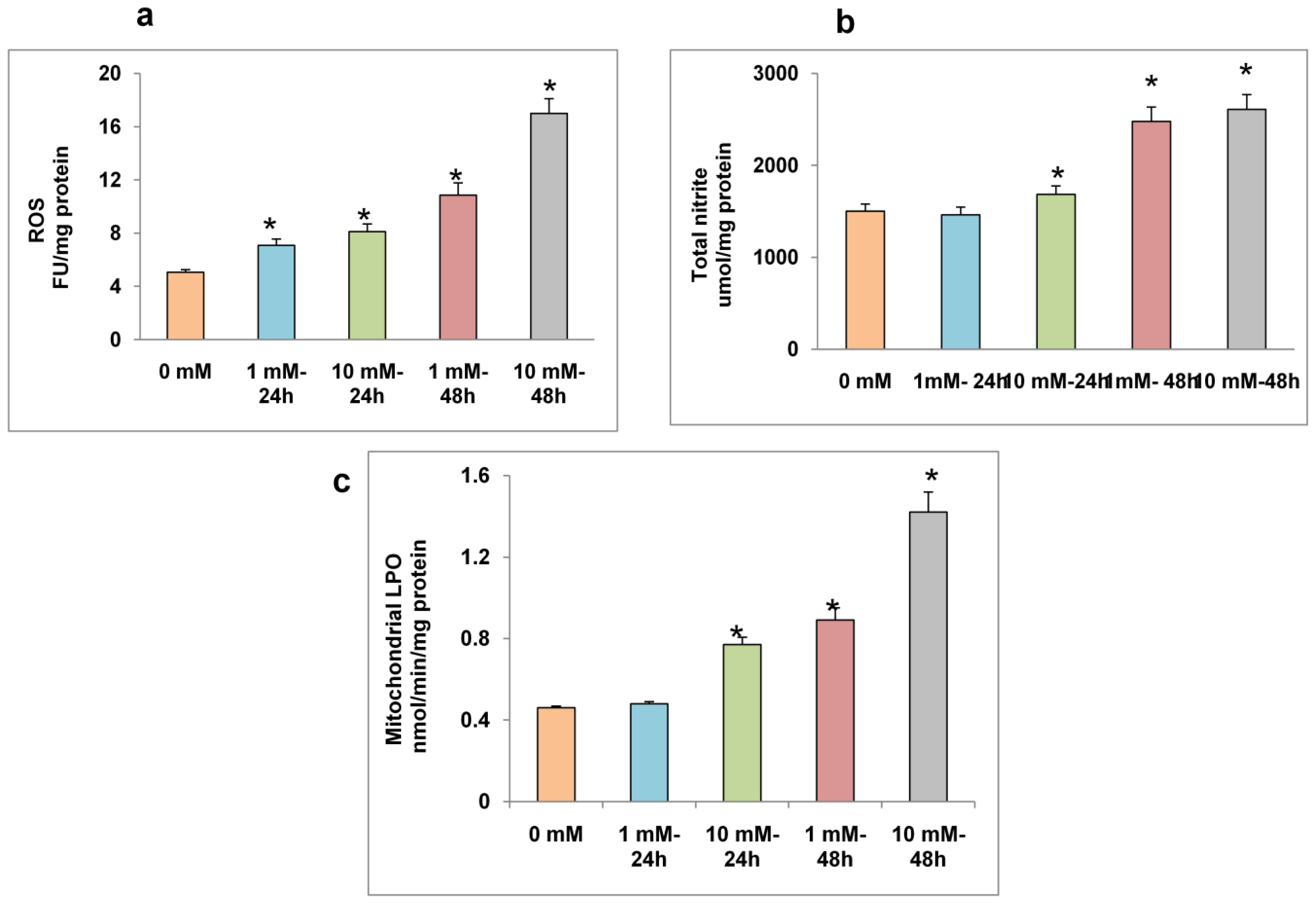
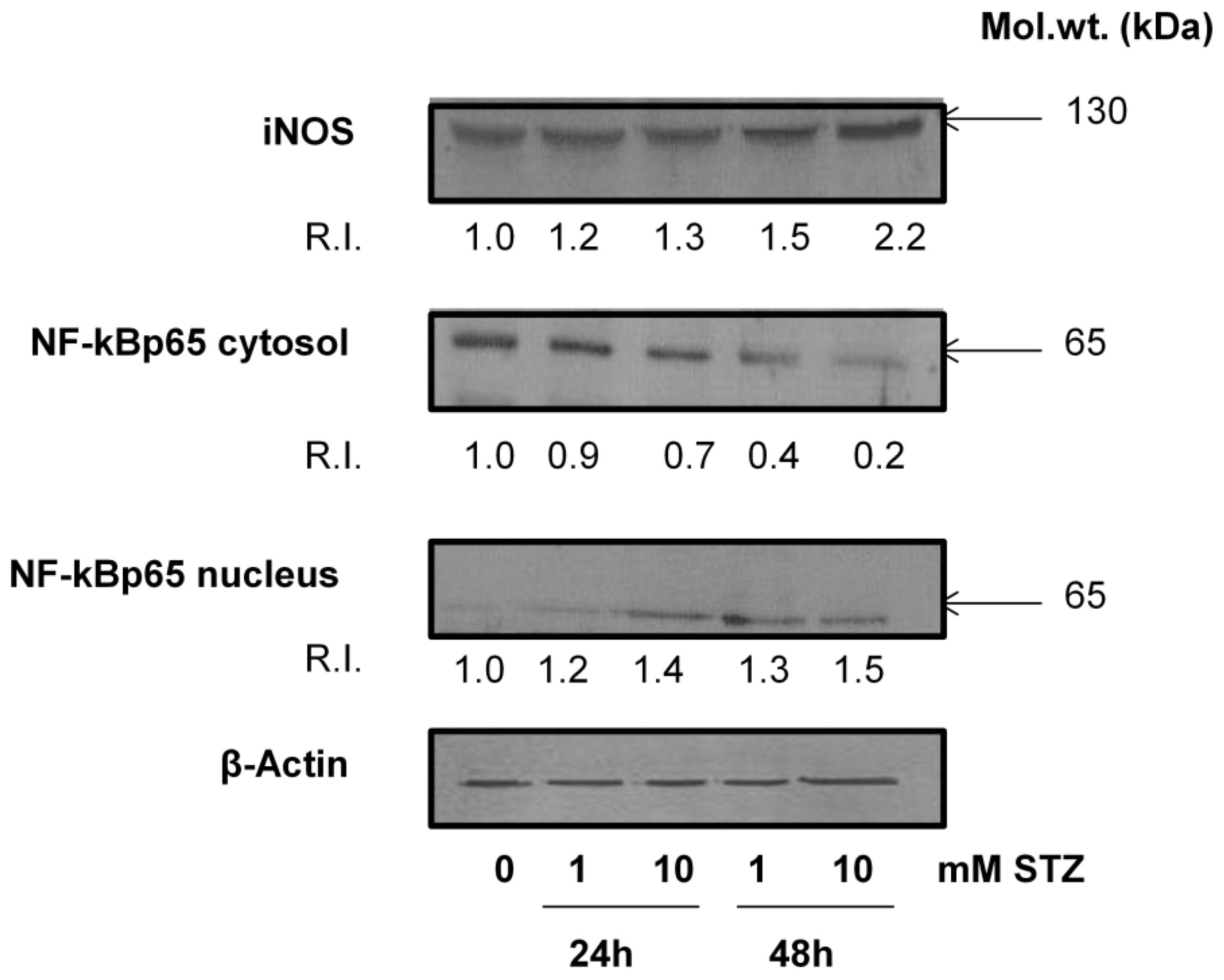
2.2. Effect of STZ on ROS, NO Production and LPO
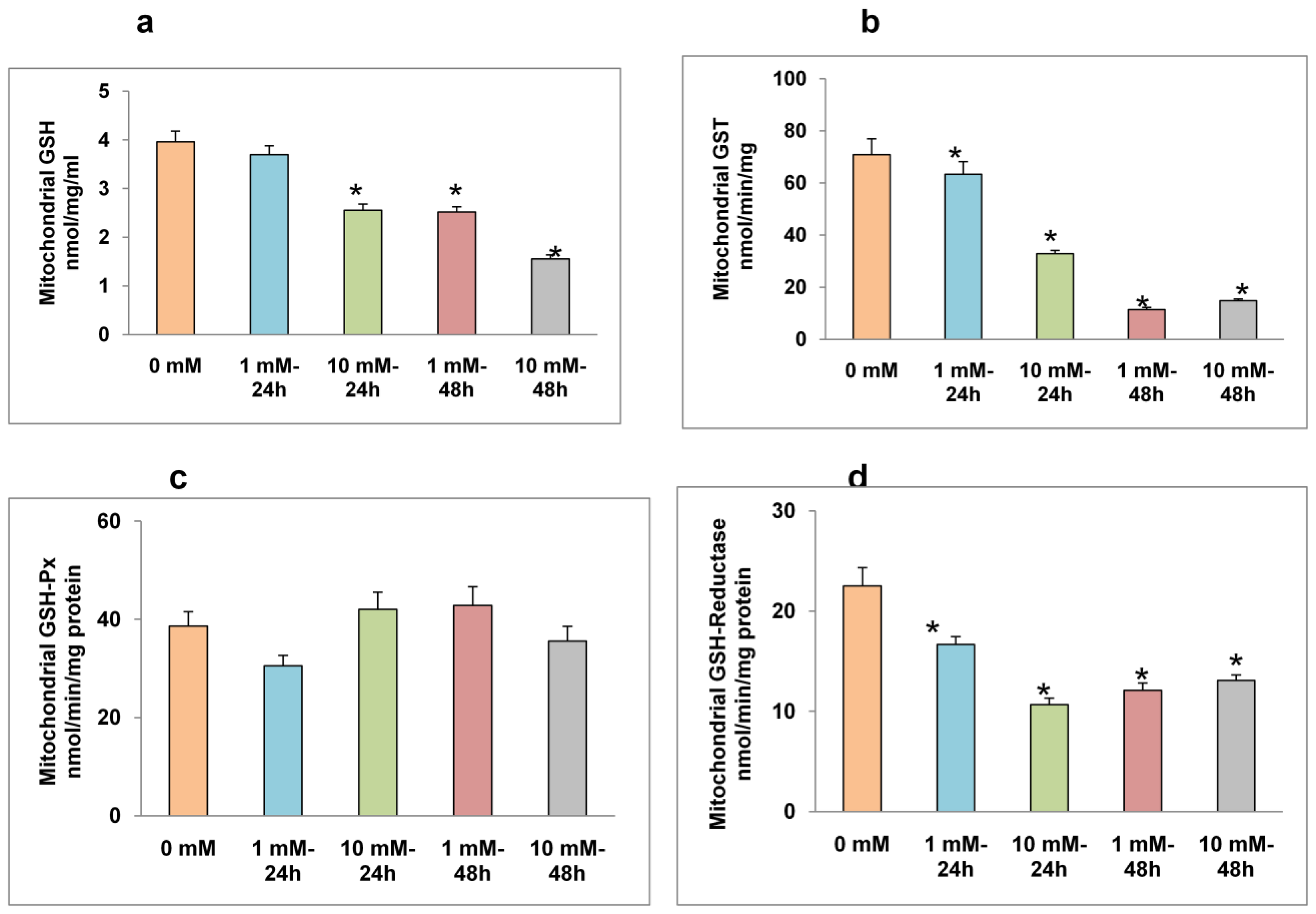
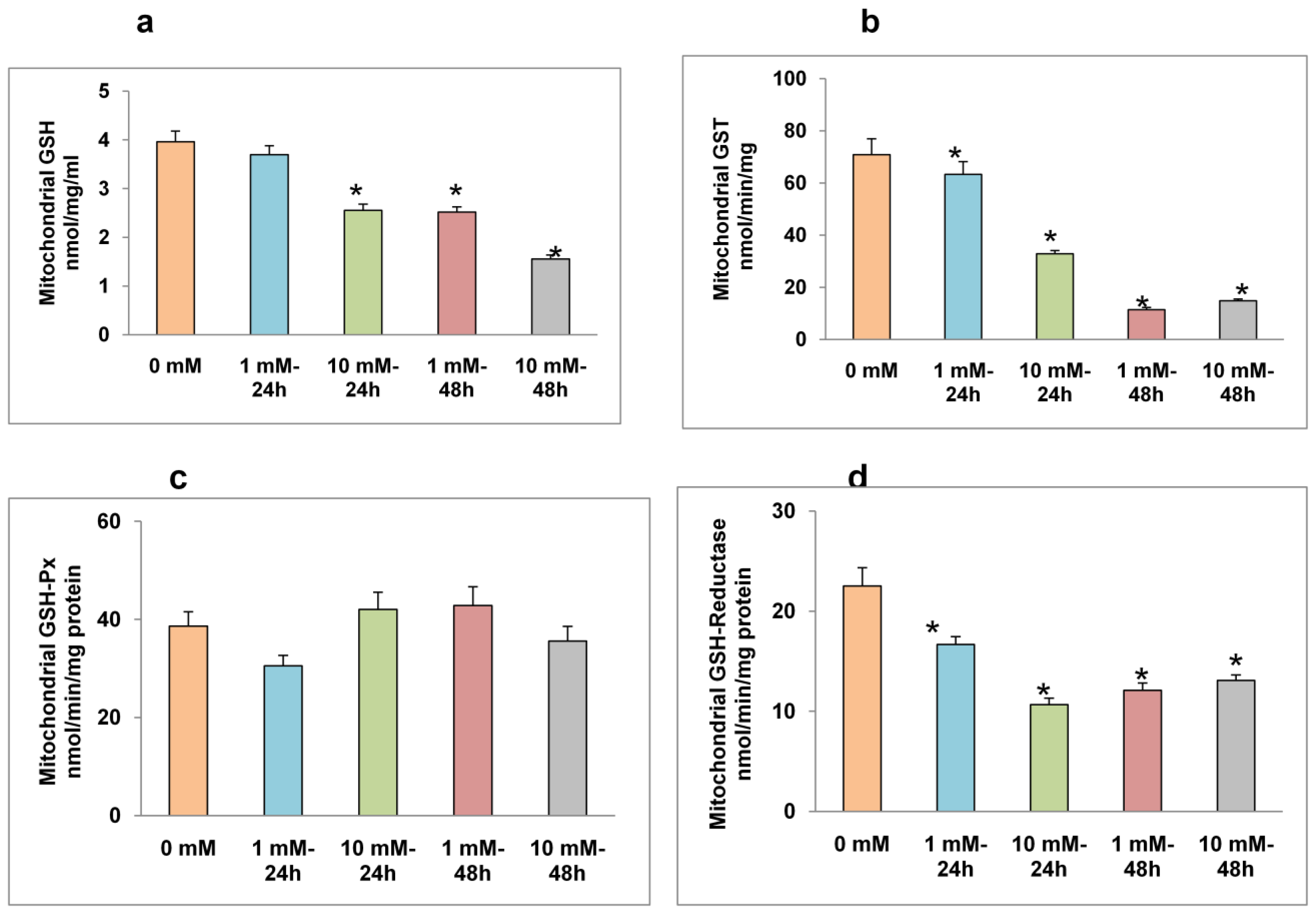
2.3. Effects of STZ on Mitochondrial GSH Concentration and GSH Metabolism
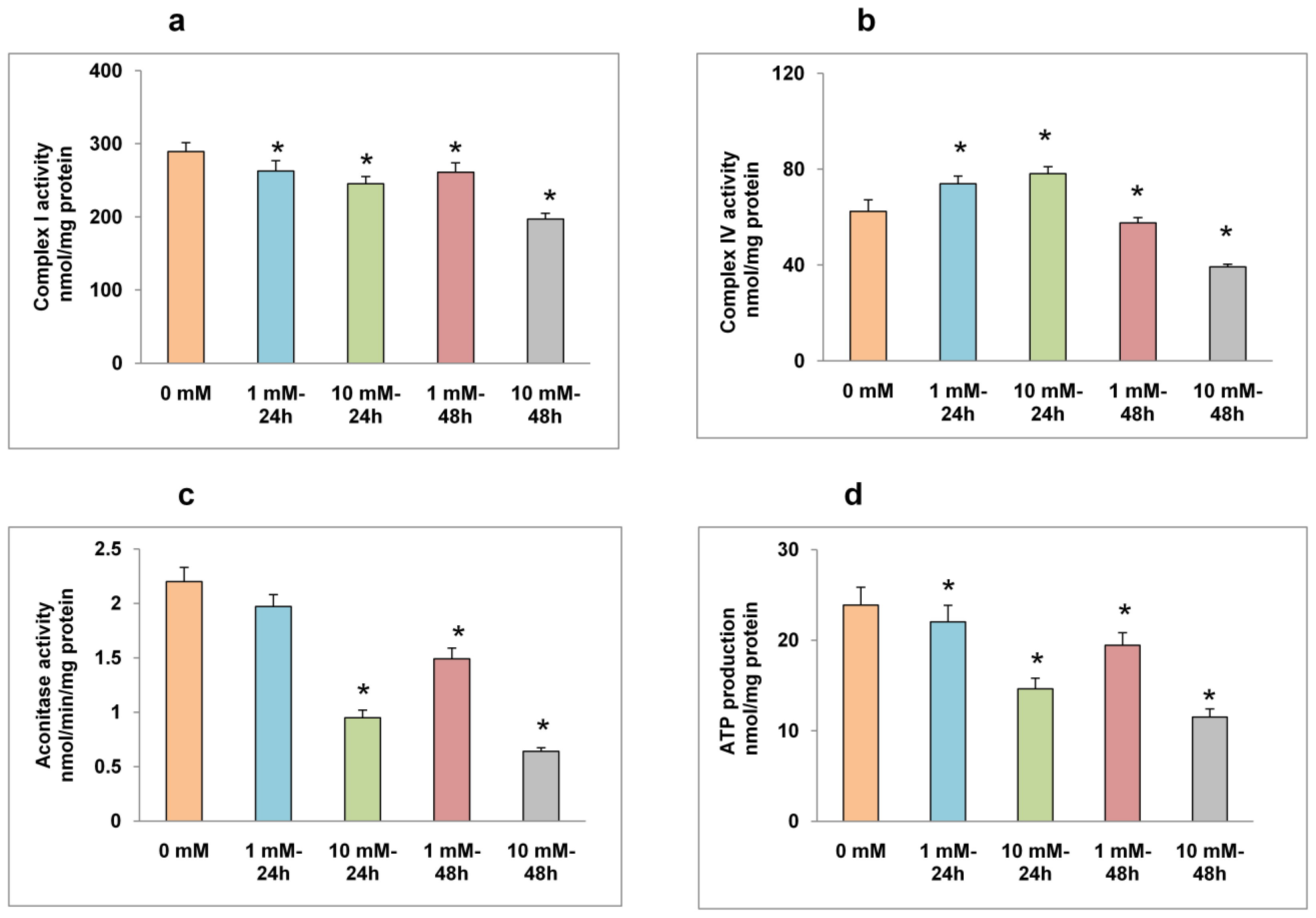
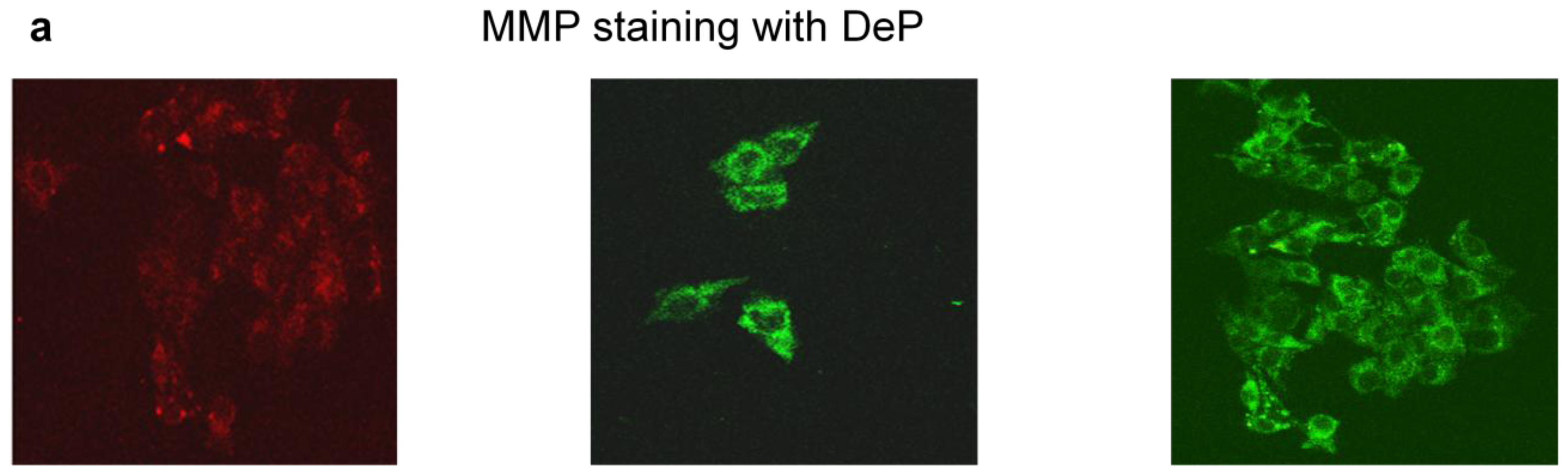
2.4. Effects of STZ on Mitochondrial Respiratory Function and ATP Production
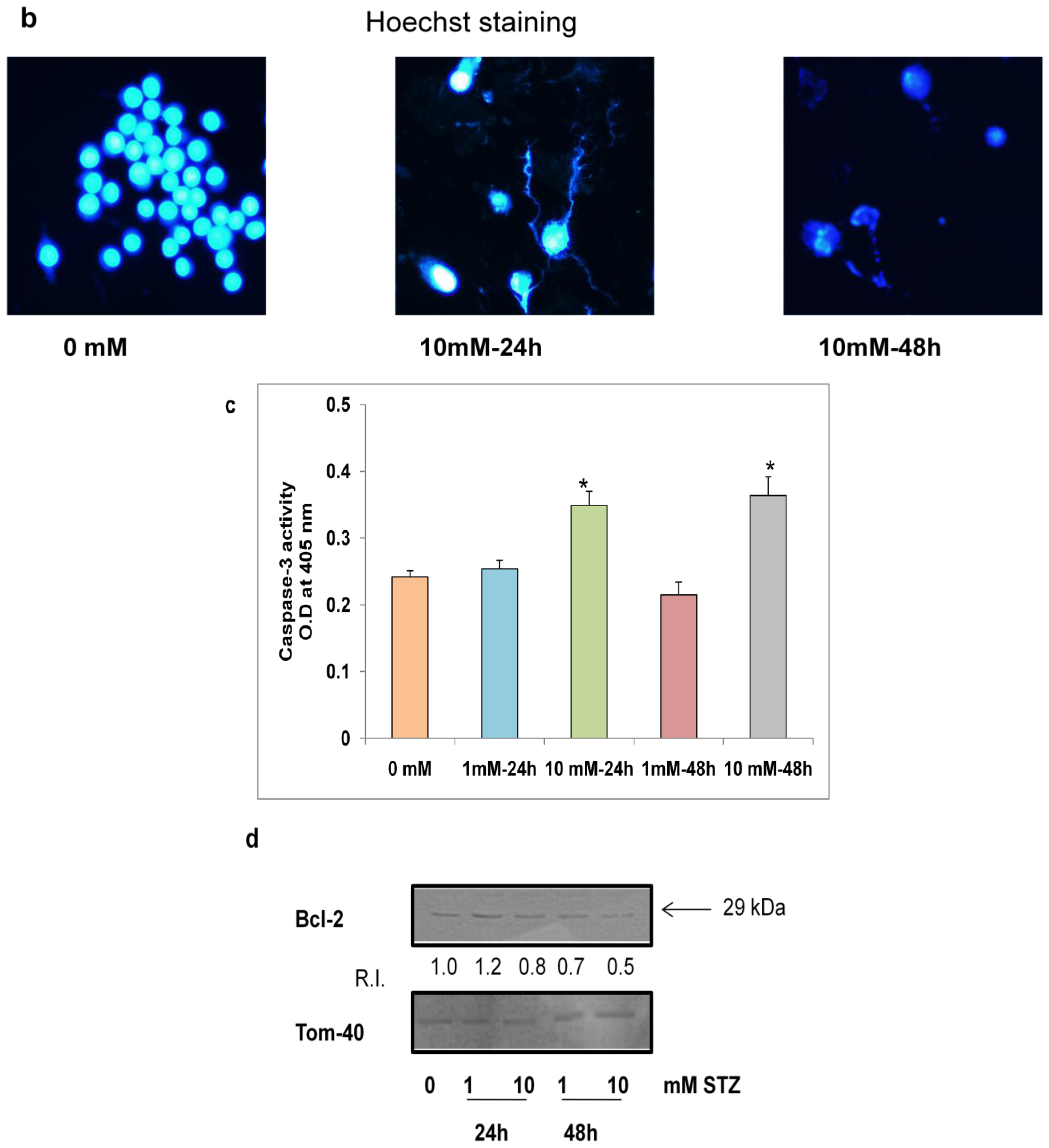
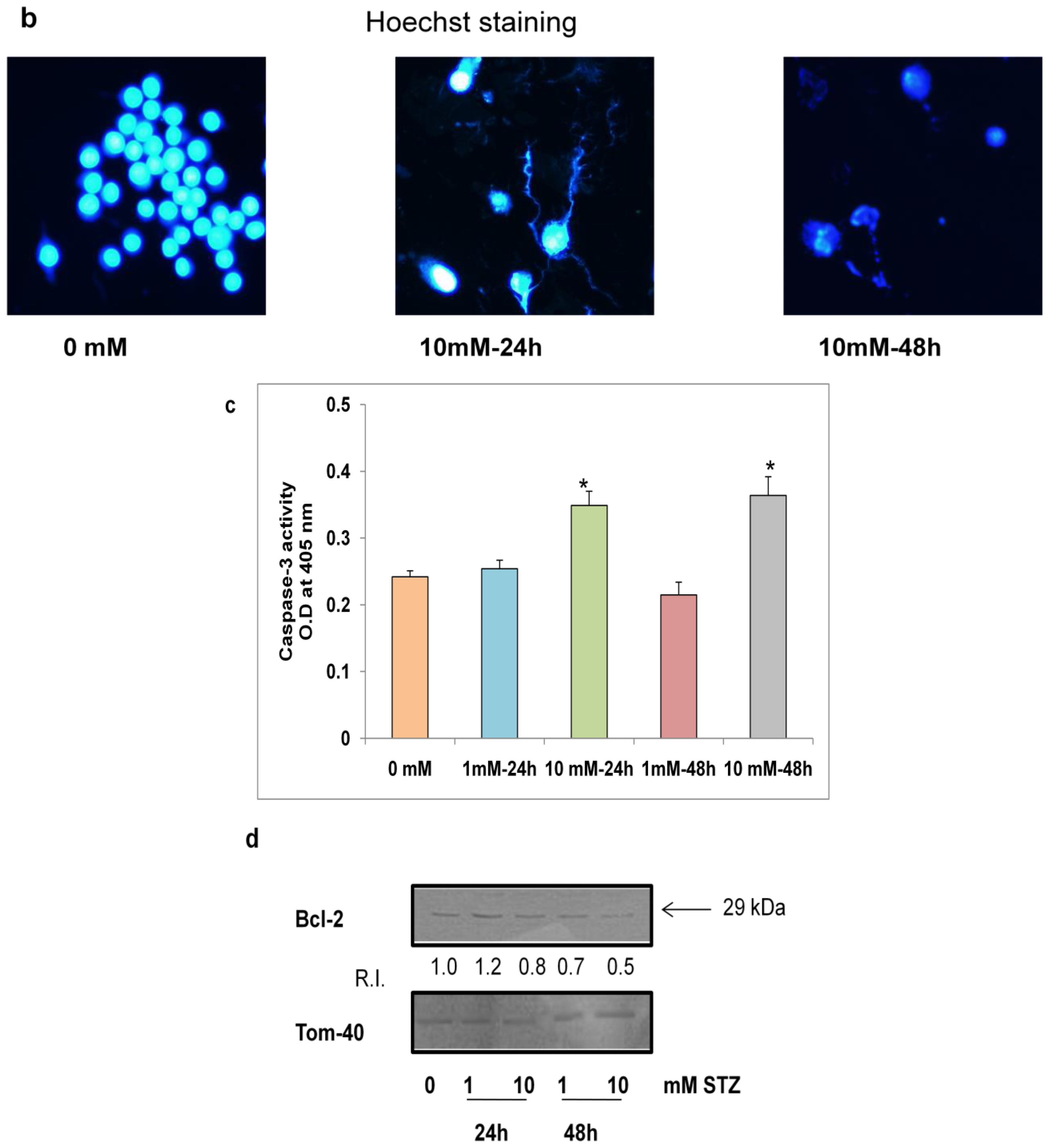
2.5. Effects of STZ on Cell Survival and Apoptosis
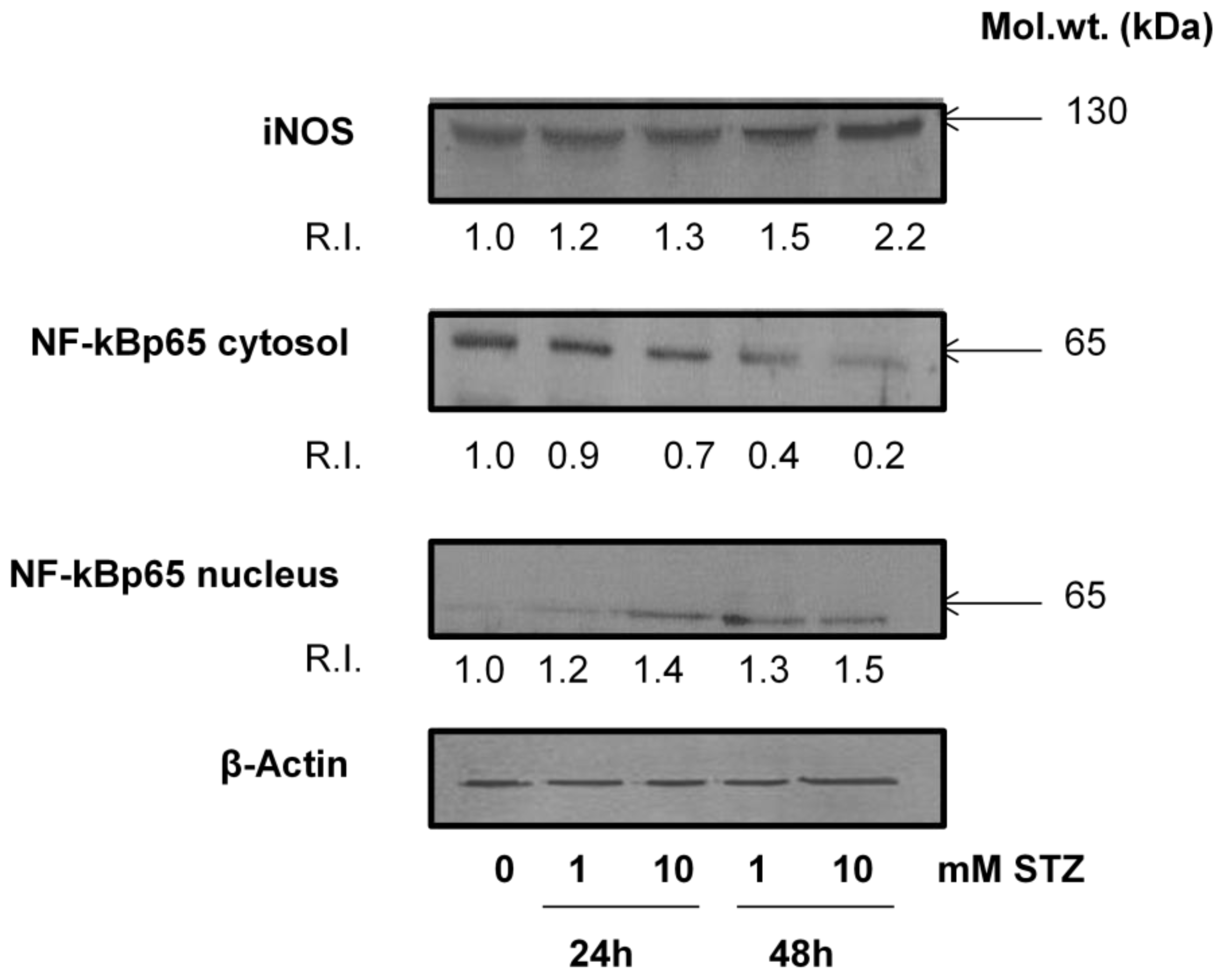
2.6. Effect of STZ on the Expression of Apoptotic Protein Markers
2.7. Discussion
3. Experimental Section
3.1. Materials
3.2. HepG2 Cell Culture and STZ Treatment
3.3. MTT Cell Viability Test
3.4. Measurement of ROS, NO and LPO
3.5. Measurement of GSH Metabolism
3.6. Measurement of Mitochondrial Functions
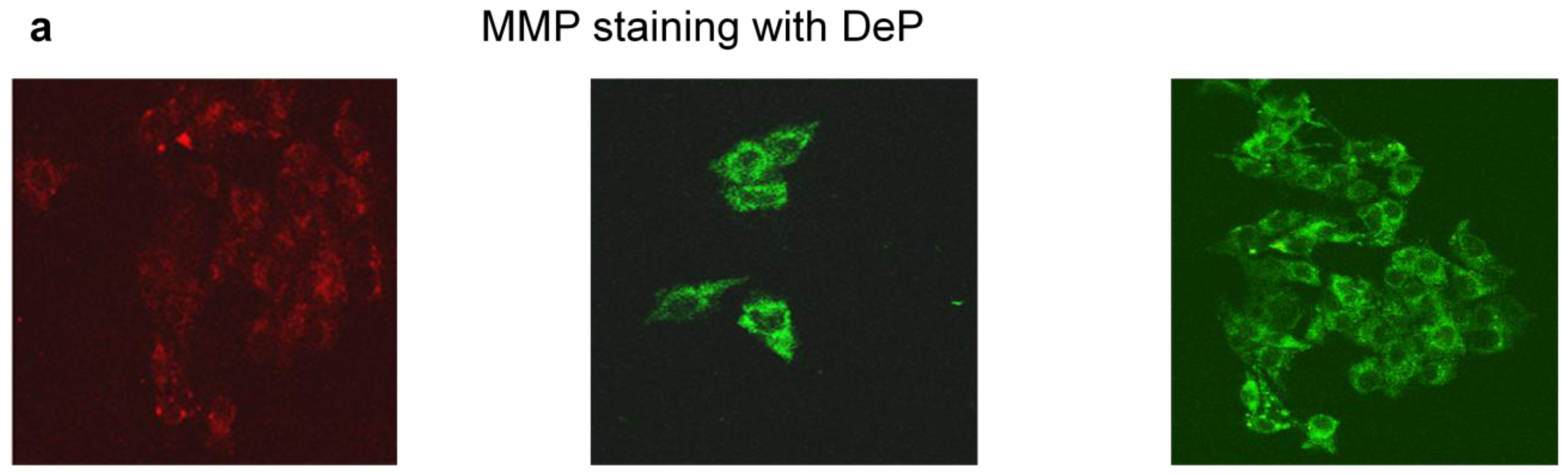
3.6.1. Measurement of Mitochondrial Membrane Potential (MMP)
3.6.2. Measurement of Mitochondrial Respiratory Functions and ATP Content
3.7. Measurement of Apoptosis
3.7.1. Nuclear Staining with Hoechst 33342
3.7.2. Assay of Caspase-3 Activity
3.8. SDS-PAGE and Western Blot Analysis
3.9. Statistical Analysis
4. Conclusions
Acknowledgements
- Conflict of InterestAuthors declare that there is no conflict of interest in publishing this study.
Abbreviations
| DCFDA | 2′,7′-dichlorofluorescein diacetate |
| GSH | glutathione |
| GST | glutathione S-transferase |
| GSH-Px | glutathione peroxidase |
| LPO | lipid peroxidation |
| MMP | mitochondrial membrane potential |
| NO | nitric oxide |
| ROS | reactive oxygen species |
| STZ | streptozotocin |
| SDS-PAGE | sodium dodecylsulphate polyacrylamide gel electrophoresis |
References
- Weiss, R.B. Streptozotocin: A review of its pharmacology, efficacy, and toxicity. Cancer Treat. Rep 1982, 66, 427–438. [Google Scholar]
- Dolan, M.E. Inhibition of DNA repair as a means of increasing the antitumor activity of DNA active agents. Adv. Drug Del. Rev 1997, 26, 105–118. [Google Scholar]
- Lewis, C.; Barbiers, A.R. Streptozotocin, a new antibiotic. In vitro and in vivo evaluation. Antibiot. Ann 1959, 7, 247–254. [Google Scholar]
- Schroy, P.C., III; Cohen, A.; Winawer, S.J.; Friedman, E.A. New chemotherapeutic drug sensitivity assay for colon carcinomas in monolayer culture. Cancer Res 1988, 48, 3236–3244. [Google Scholar]
- Van Dyke, K.; Jabbour, N.; Hoeldtke, R.; van dyke, C.; van dyke, M. Oxidative/nitrosative stresses trigger type I diabetes: Preventable in streptozotocin rats and detectable in human disease. Ann. N. Y. Acad. Sci 2010, 1203, 138–145. [Google Scholar]
- LeDoux, S.P.; Woodley, S.E.; Patton, N.J.; Wilson, G.L. Mechanisms of nitrosourea-induced beta-cell damage. Alterations in DNA. Diabetes 1986, 35, 866–872. [Google Scholar]
- Capucci, M.S.; Hoffmann, M.E.; Natarajan, A.T. Streptozotocin-induced genotoxic effects in Chinese hamster cells: The resistant phenotype of V79 cells. Mutat. Res 1995, 347, 79–85. [Google Scholar]
- Ridolfi, R.; Amaducci, L.; Derni, S.; Fabbri, L.; Innocenti, M.P.; Vignutelli, P. Chemotherapy with 5-fluorouracil and streptozotocin in carcinoid tumors of gastrointestinal origin: Experience with 13 patients. J. Chemother 1991, 3, 328–331. [Google Scholar]
- Islam, M.S.; Loots du, T. Experimental rodent models of type 2 diabetes: A review. Methods Find Exp. Clin. Pharmacol 2009, 31, 249–261. [Google Scholar]
- Wei, L.; Lu, Y.; He, S.; Jin, X.; Zeng, L.; Zhang, S.; Chen, Y.; Tian, B.; Mai, G.; Yang, G.; et al. Induction of diabetes with signs of autoimmunity in primates by the injection of multiple-low-dose streptozotocin. Biochem. Biophys. Res. Commun 2011, 412, 373–378. [Google Scholar]
- Hosokawa, M.; Dolci, W.; Thorens, B. Differential sensitivity of GLUT1 and GLUT2-expressing beta cells to streptozotocin. Biochem. Biophys. Res. Commun 2001, 289, 1114–1117. [Google Scholar]
- Friederich, M.; Hansell, P.; Palm, F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Curr. Diabetes Rev 2009, 5, 120–144. [Google Scholar]
- Simmons, R.A. Developmental origins of diabetes: The role of oxidative stress. Free Radic. Biol. Med 2006, 40, 917–922. [Google Scholar]
- Raza, H.; Prabu, S.K.; Robin, M.A.; Avadhani, N.G. Elevated Mitochondrial cytochrome P450 2E1 and Glutathione S-transferase A4–4 in streptozotocin-induced diabetic rats: Tissue-specific variations and role in oxidative stress. Diabetes 2004, 53, 185–194. [Google Scholar]
- Raza, H.; Subbuswamy, K.P.; John, A.; Avadhani, N.G. Impaired mitochondrial respiratory functions and oxidative stress in streptozotocin-induced diabetic rats. Int. J. Mol. Sci 2011, 12, 3133–3147. [Google Scholar]
- Van Dyke, K.; Ghareeb, E.; van Dyke, M.; Sosa, A.; Hoeldtke, R.D.; van Thiel, D.H. Luminescence experiments involved in the mechanism of streptozotocin diabetes and cataract formation. Luminescence 2008, 23, 386–391. [Google Scholar]
- Liu, H.K.; McCluskey, J.T.; Mc Clenghan, N.H.; Flatt, P.R. Streptozotocin-resistant BRIN-BD11 cells possess wide spectrum of toxin tolerance and enhanced insulin-secretory capacity. Endocrine 2007, 32, 20–29. [Google Scholar]
- Tuch, B.E.; Beynon, S.; Tabiin, M.T.; Sassoon, R.; Goodman, R.J.; Simpson, A.M. Effect of beta-cell toxins on genetically engineered insulin-secreting cells. J. Autoimmun 1997, 10, 239–244. [Google Scholar]
- Saini, K.S.; Thompson, C.; Winterford, C.M.; Walker, N.I.; Cameron, D.P. Streptozotocin at low doses induces apoptosis and at high doses causes necrosis in a murine pancreatic β-cells line, INS-1. Biochem. Mol. Biol. Int 1996, 39, 1229–1236. [Google Scholar]
- Harel, A.; Bloch, O.; Vardi, P.; Bloch, K. Sensitivity of HaCat keratinocytes to diabetogenic toxins. Biochem. Pharmacol 2002, 63, 171–178. [Google Scholar]
- Koo, K.B.; Suh, H.J.; Ra, K.S.; Choi, J.W. Protective effect of cyclo(his-pro) on streptozotocin-induced cytotoxicity and apoptosis in vitro. J. Microbiol. Biotechnol 2011, 21, 218–227. [Google Scholar]
- Latha, M.; Pari, L.; Sitasawad, S.; Bhonde, R. Scoparia dulcis, a traditional antidiabetic plant, protects against streptozotocin induced oxidative stress and apoptosis in vitro and in vivo. J. Biochem. Mol. Toxicol 2004, 18, 261–272. [Google Scholar]
- Locke, M.; Anderson, J. NF-κB activation in organs from STZ-treated rats. Appl. Physiol. Nutr. Metab 2011, 36, 121–127. [Google Scholar]
- Kim, E.K.; Song, M.Y.; Kim, I.S.; Moon, W.S.; Ryu, D.G.; So, H.S.; Park, R.; Park, J.W.; Kwon, K.B.; Park, B.H. Beneficial effect of Flos magnoliae extract on multiple low dose streptozotocin-induced type 1 diabetes development and cytokine-induced β-cell damage. Int. J. Mol. Med 2008, 22, 481–488. [Google Scholar]
- Bathaie, S.Z.; Sedghgoo, F.; Jafarnejad, A.; Farzami, B.; Khayatian, M. Spectroscopic studies of STZ-induced methylated-DNA in both in vivo and in vitro conditions. Spectrochim. Acta A Mol. Biomol. Spectrosc 2008, 71, 803–808. [Google Scholar]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptozotocin. Mutat. Res 2002, 512, 121–134. [Google Scholar]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur. J. Pharmacol 2011, 668, 15–24. [Google Scholar]
- Raza, H.; John, A.; Brown, E.M.; Benedict, S.; Kambal, A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol. Appl. Pharmacol 2008, 226, 161–168. [Google Scholar]
- Birch-Machin, M.A.; Turnbull, D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol 2001, 65, 97–117. [Google Scholar]
- Spear, J.F.; Prabu, S.K.; Galati, N.; Raza, H.; Anandatheerthavarada, H.K.; Avadhani, N.G. β1-adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H2459–H2466. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoresis transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raza, H.; John, A. Streptozotocin-Induced Cytotoxicity, Oxidative Stress and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells. Int. J. Mol. Sci. 2012, 13, 5751-5767. https://doi.org/10.3390/ijms13055751
Raza H, John A. Streptozotocin-Induced Cytotoxicity, Oxidative Stress and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells. International Journal of Molecular Sciences. 2012; 13(5):5751-5767. https://doi.org/10.3390/ijms13055751
Chicago/Turabian StyleRaza, Haider, and Annie John. 2012. "Streptozotocin-Induced Cytotoxicity, Oxidative Stress and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells" International Journal of Molecular Sciences 13, no. 5: 5751-5767. https://doi.org/10.3390/ijms13055751