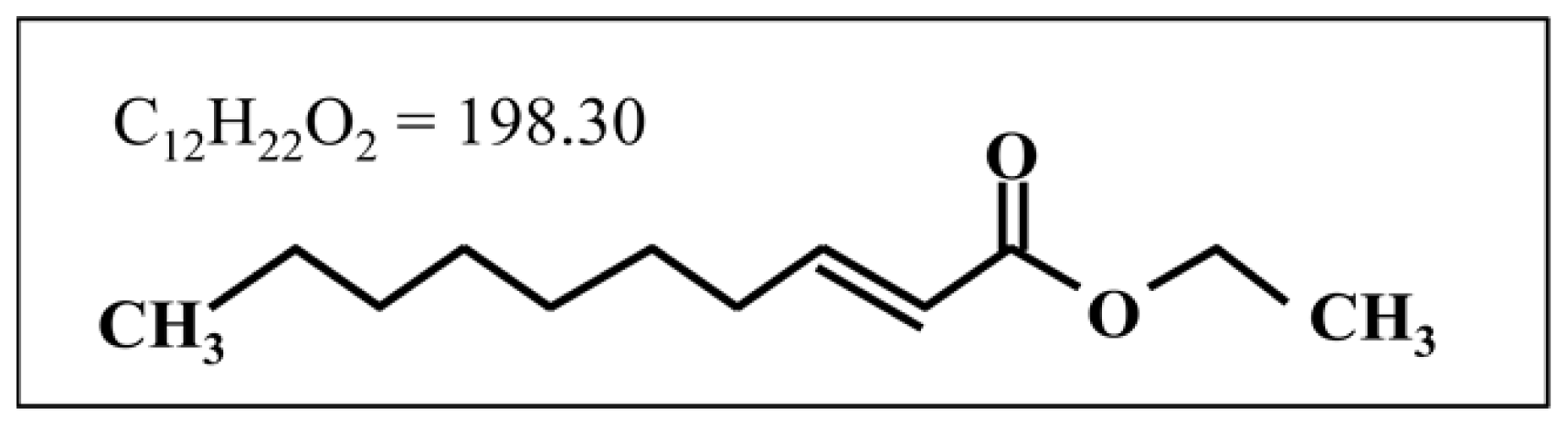
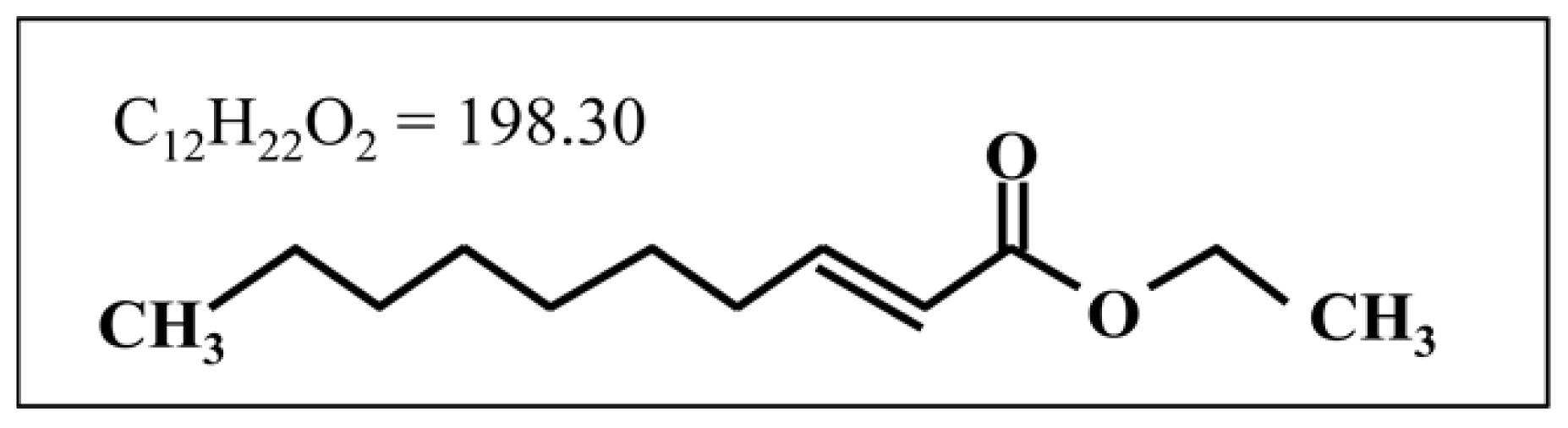
2-Decenoic Acid Ethyl Ester, a Compound That Elicits Neurotrophin-like Intracellular Signals, Facilitating Functional Recovery from Cerebral Infarction in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
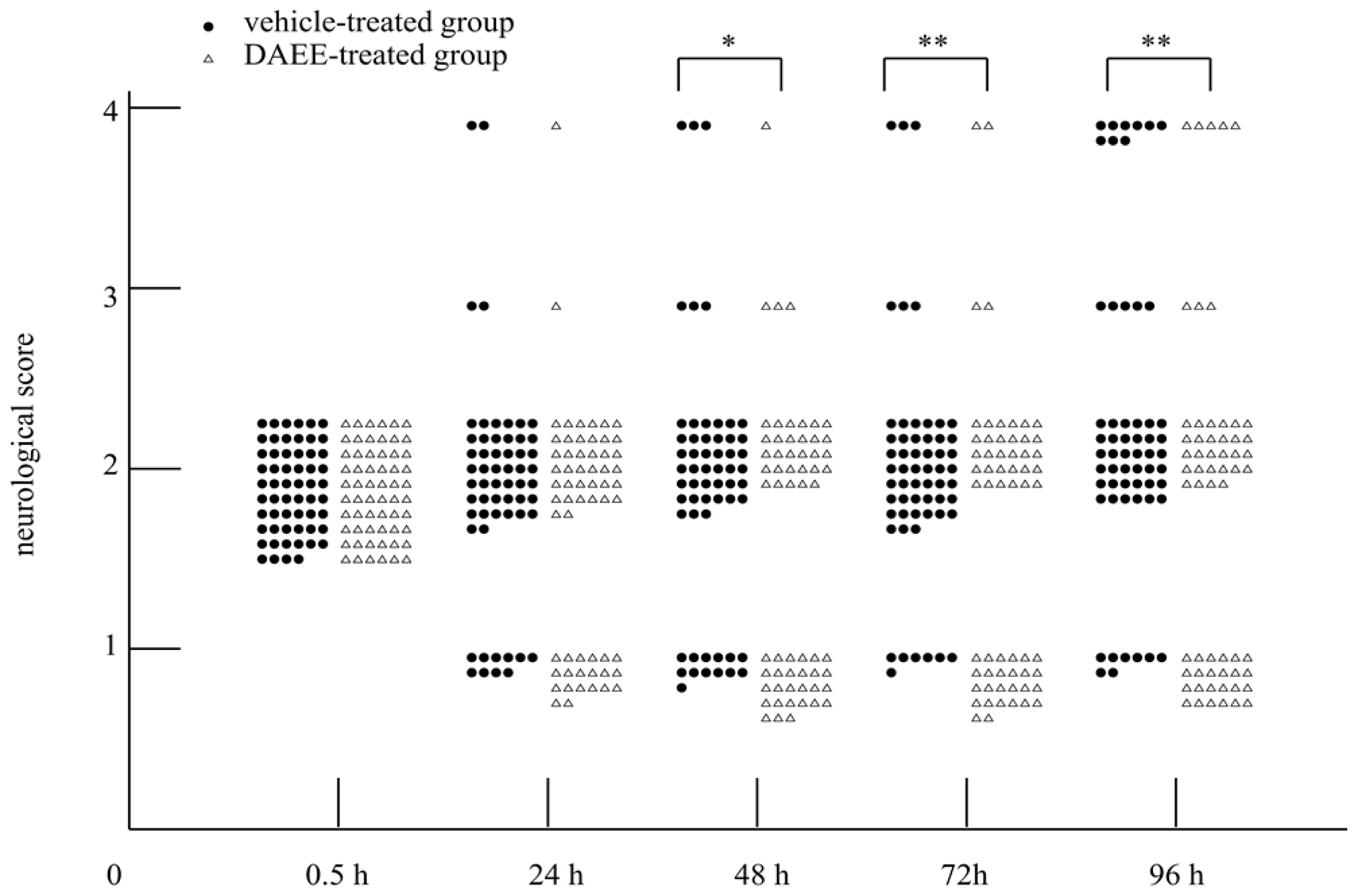
2.1. Evaluation of Functional Recovery after PMCAO
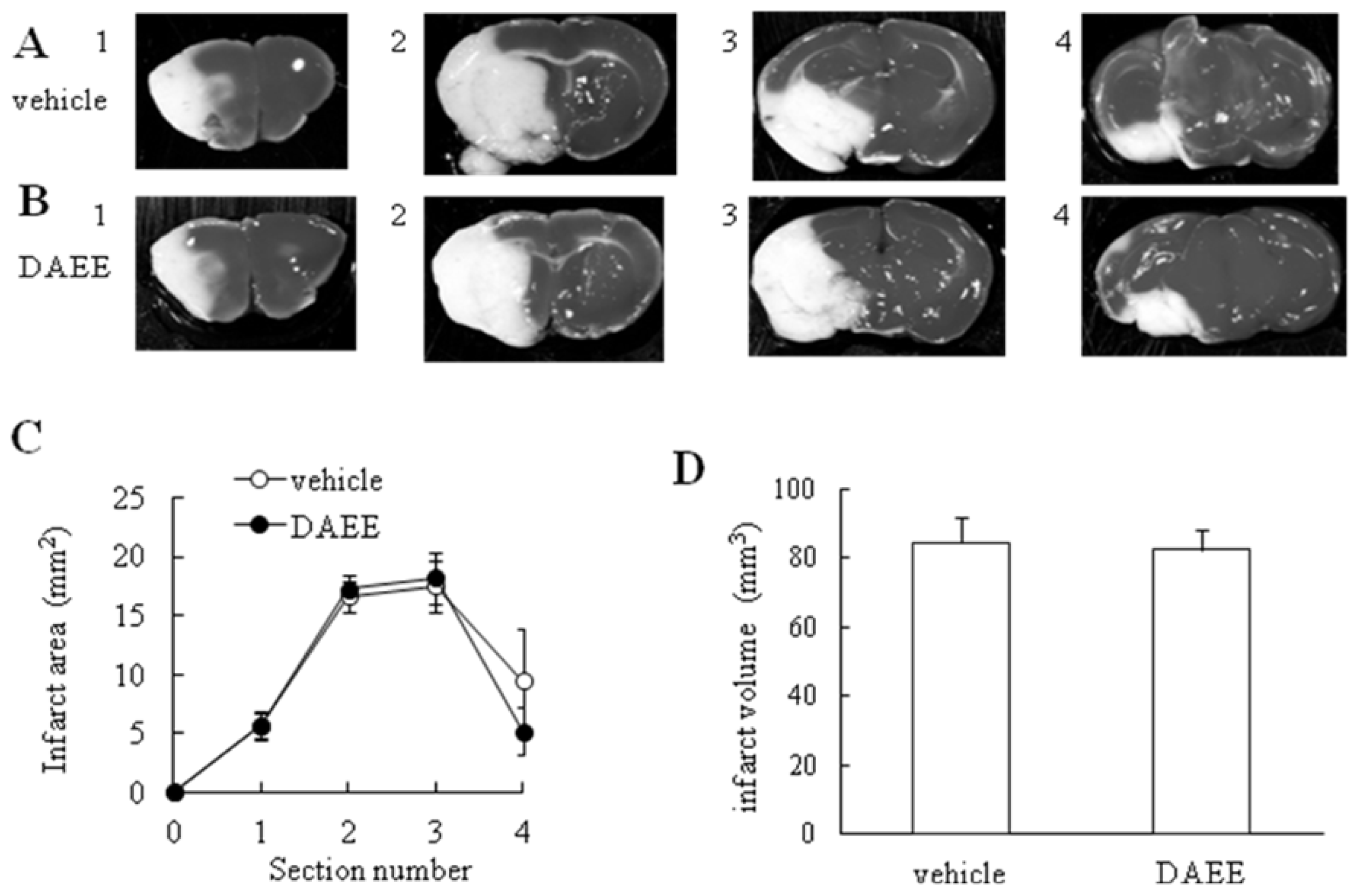
2.2. Measurement of Infarct Volume
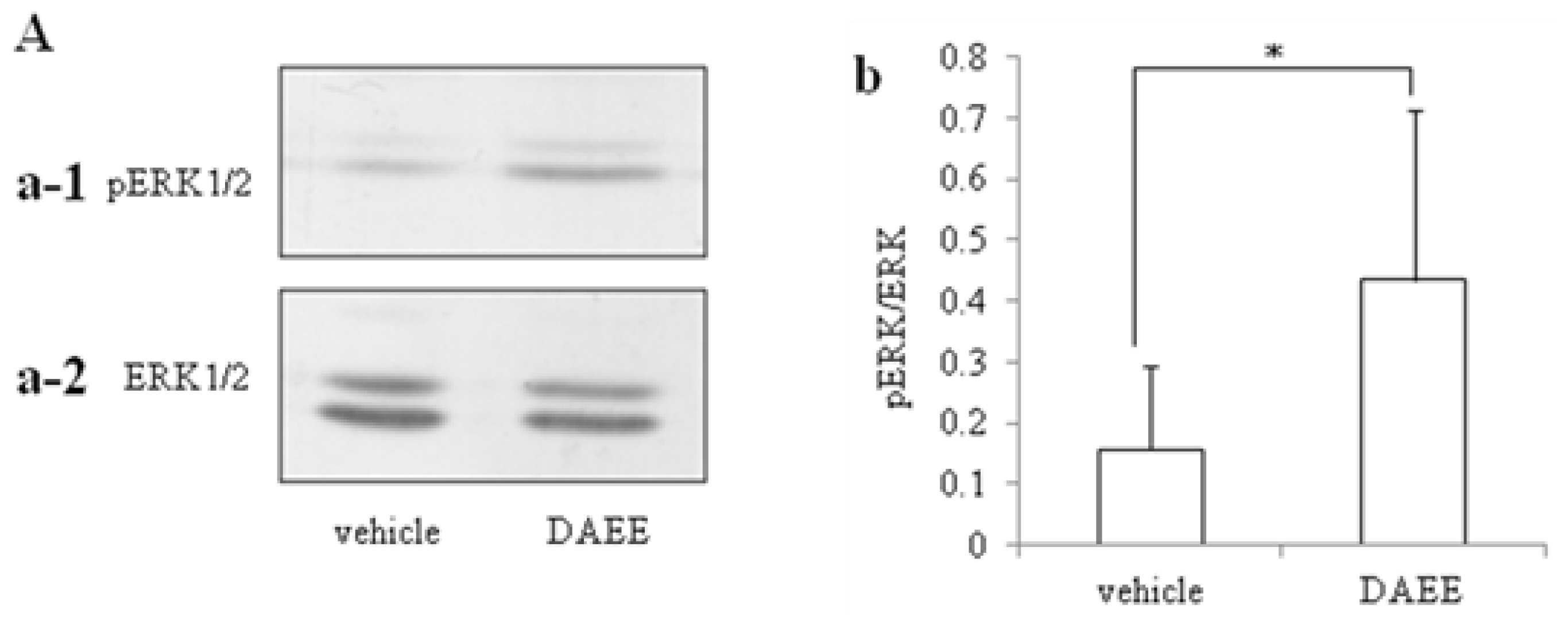
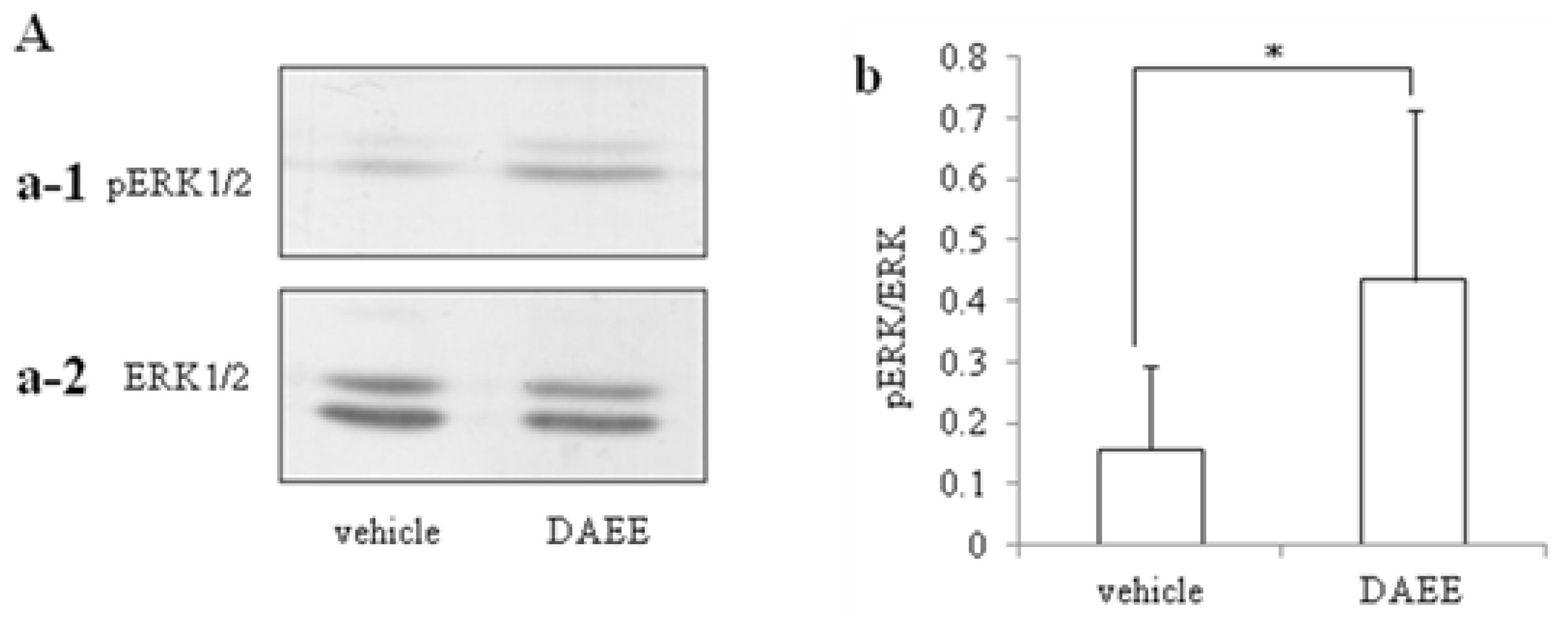
2.3. Activation of ERK1/2
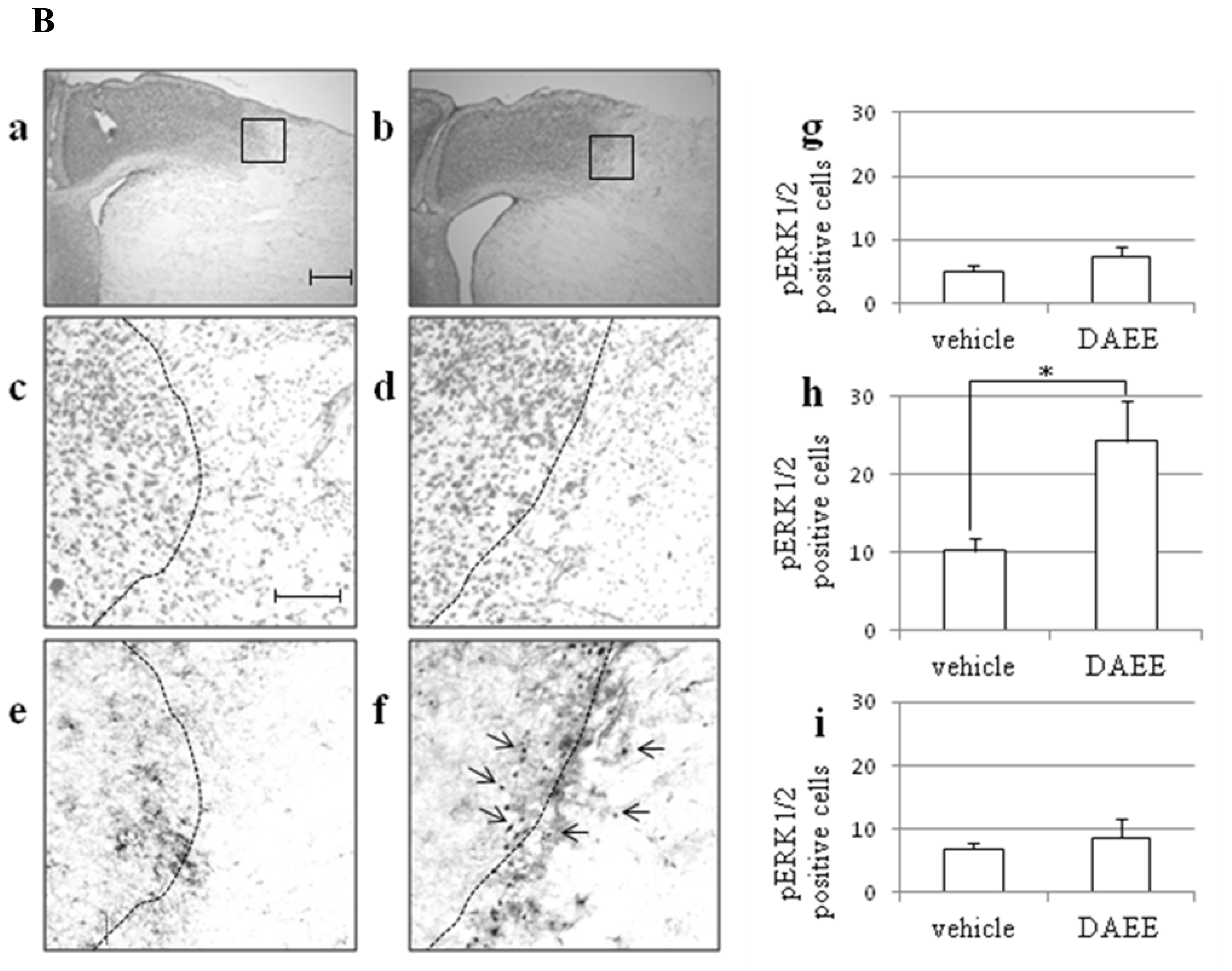
2.4. Expression of pERK1/2 in Ischemic Penumbra
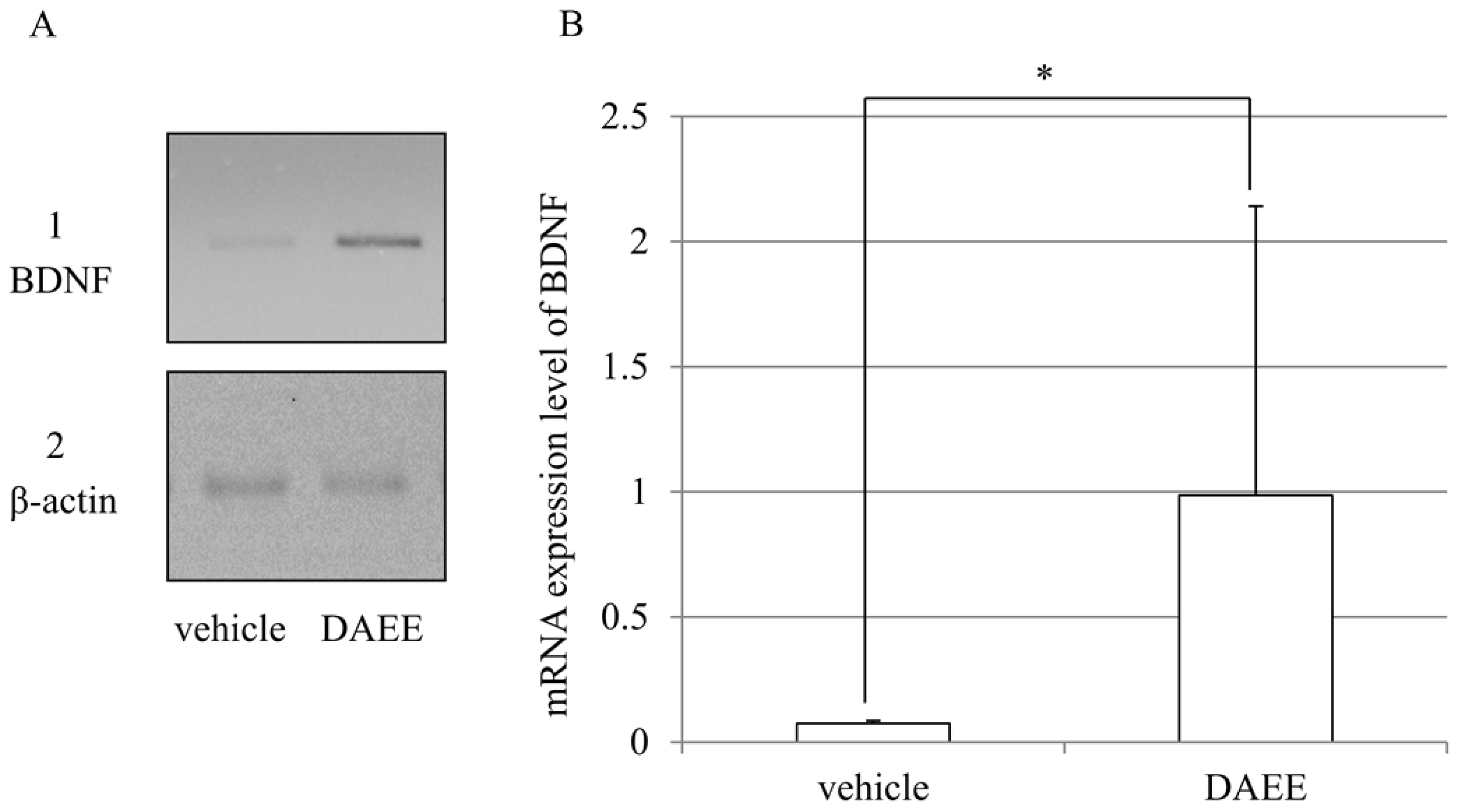
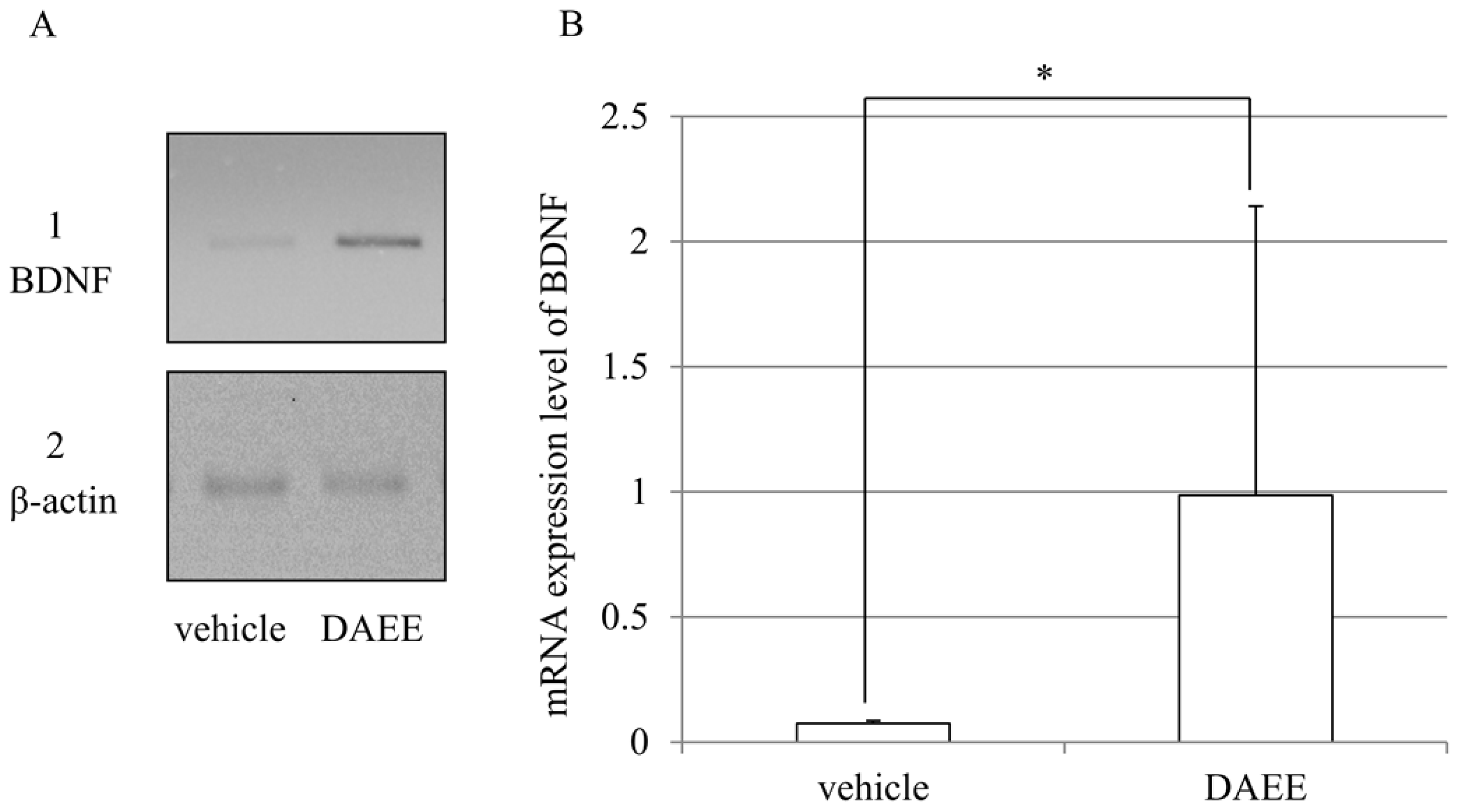
2.5. Expression of BDNF mRNAs after PMCAO
3. Discussion
4. Experimental Section
4.1. Animal Model of Cerebral Infarction
4.2. Administration of the Drug
4.3. Analysis of Animal Behavior
4.4. Measurement of Infarct Volume
4.5. Western Immunoblot Analysis
4.6. Histopathological and Immunohistchemical Study
4.7. RT-PCR
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Makino, A.; Iinuma, M.; Fukumitsu, H.; Soumiya, H.; Furukawa, Y.; Furukawa, S. 2-Decenoic acid ethyl ester possesses neurotrophin-like activities to facilitate intracellular signals and increase synapse-specific proteins in neurons cultured from embryonic rat brain. Biomed. Res 2010, 31, 379–386. [Google Scholar]
- Hirakawa, A.; Shimizu, K.; Fukumitsu, H.; Soumiya, H.; Iinuma, M.; Furukawa, S. 2-Decenoic acid ethyl ester, a derivative of unsaturated medium-chain fatty acids, facilitates functional recovery of locomotor activity after spinal cord injury. Neuroscience 2010, 171, 1377–1385. [Google Scholar]
- Momose, Y.; Murata, M.; Kobayashi, K.; Tachikawa, M.; Nakabayashi, Y.; Kanazawa, I.; Toda, T. Association studies of multiple candidate genes for Parkinson’s disease using single nucleotide polymorphisms. Ann. Neurol 2002, 51, 133–136. [Google Scholar]
- Matsushita, S.; Arai, H.; Matsui, T.; Yuzuriha, T.; Urakami, K.; Masaki, T.; Higuchi, S. Brain-derived neurotrophic factor gene polymorphisms and Alzheimer’s disease. J. Neural. Transm 2005, 112, 703–711. [Google Scholar]
- Sen, S.; Nesse, R.M.; Stoltenberg, S.F.; Li, S.; Gleiberman, L.; Chakravarti, A.; Weder, A.B.; Burmeister, M. A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology 2003, 28, 397–401. [Google Scholar]
- Lang, U.E.; Hellweg, R.; Kalus, P.; Bajbouj, M.; Lenzen, K.P.; Sander, T.; Kunz, D.; Gallinat, J. Association of a functional BDNF polymorphism and anxiety-related personality traits. Psychopharmacology 2005, 180, 5–9. [Google Scholar]
- Apfel, S.C. Neurotrophic factor therapy-prospects and problems. Clin. Chem. Lab. Med 2001, 39, 351–355. [Google Scholar]
- Kotani, Y.; Morimoto, N.; Oida, Y.; Tamura, Y.; Tamura, S.; Inoue, T.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Prevention of in vitro and in vivo acute ischemic neuronal damage by (2S)-1-(4-amino-2,3,5-trimethylphenoxy)-3-{4-[4-(4-fluorobenzyl) phenyl]-1-piparazinyl}-2-propanol dimethanesulfonate (SUN N8075), a novel neuroprotective agent with antioxidant propaties. Neuroscience 2007, 149, 779–788. [Google Scholar]
- Ferrer, I.; Krupinski, J.; Goutan, E.; Marti, E.; Ambrosio, S.; Arenas, E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol 2001, 101, 229–238. [Google Scholar]
- Higashi, Y. Edaravone for treatment of acute cerebral infarction: role of endothelium-derived nitric oxide and oxidative stress. Expert. Opin. Pharmacother 2009, 10, 323–331. [Google Scholar]
- Nonaka, Y.; Koumura, A.; Hyakkoku, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Combination treatment with normobaric hyperoxia and cilostazol protects mice against focal cerebral ischemia-induced neuronal damage better than each treatment alone. J. Pharmacol. Exp. Ther 2009, 330, 13–22. [Google Scholar]
- Bendersen, J.B.; Pittu, L.H.; Cermano, S.M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H.M. Evaluation of 2,3,5-triphenyltetrazolium chloride as a strain for detection and quantification of experimental cerebral infarction in rats. Stroke 1986, 17, 1304–1308. [Google Scholar]
- Popp, A.; Jaenisch, N.; Witte, O.W.; Frahm, C. Identification of ischemic regions in a rat model of stroke. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Hetman, M.; Gozdz, A. Role of extracellular signal regulated kinase 1 and 2 in neuronal survival. Eur. J. Biochem 2004, 271, 2050–2055. [Google Scholar]
- Jiang, Y.; Wei, N.; Lu, J.; Zhu, J.; Xu, G.; Liu, X. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience 2011, 172, 398–405. [Google Scholar]
- Lauritzen, I.; Blondeau, N.; Heurteaux, C.; Widmann, C.; Romey, G.; Lazdunski, M. Polyunsaturated fatty acids are potent neuroprotectors. EMBO J 2000, 19, 1784–1793. [Google Scholar]
- Blondeau, N.; Widmann, C.; Lazdunski, M.; Heurteaux, C. Polyunsaturated fatty acids induce ischemic and epileptic tolerance. Neuroscience 2002, 109, 231–241. [Google Scholar]
- Emsley, R.; Oosthuizen, P.; van Rensburg, S.J. Clinical potential of omega-3 fatty acids in the treatment of schizophrenia. CNS. Drugs 2003, 17, 1081–1091. [Google Scholar]
- Kamata, Y.; Shiraga, H.; Tai, A.; Kawamoto, Y.; Gohda, E. Induction of neurite outgrowth in PC12 cells by the medium-chain fatty acid octanoic acid. Neuroscience 2007, 146, 1073–1081. [Google Scholar]
- Hattori, N.; Nomoto, H.; Fukumitsu, H.; Mishima, S.; Furukawa, S. Royal jelly-induced neurite outgrowth from rat pheochromocytoma PC12 cells requires integrin signal independent of activation of extracellular signal-regulated kinases. Biomed. Res 2007, 28, 139–146. [Google Scholar]
- Barbacid, M. Structural and functional properties of the TRK family of neurotrophin receptors. Ann. N. Y. Acad. Sci 1995, 766, 442–458. [Google Scholar]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol 2000, 10, 381–391. [Google Scholar]
- Lu, B. BDNF and activity-dependent synaptic modulation. Learn. Mem 2003, 10, 86–98. [Google Scholar]
- Shi, Q.; Zhang, P.; Zhang, J.; Chen, X.; Lu, H.; Tian, Y.; Parker, T.L.; Liu, Y. Adenovirus-mediated brain derived neurotrophic factor expression regulated by hypoxia response element protects brain from injury of transient middle cerebral artery occlusion in mice. Neurosci. Lett 2009, 465, 220–225. [Google Scholar]
- Schabitz, W.R.; Steigleder, T.; Cooper-Kuhn, C.M.; Schwab, S.; Sommer, C.; Schneider, A.; Kuhn, H.G. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke 2007, 38, 2165–2172. [Google Scholar]
- Hefti, F. Pharmacology of neurotrophic factors. Annu. Rev. Pharmacol. Toxicol 1997, 37, 239–267. [Google Scholar]
- Kotani, Y.; Nakajima, Y.; Hasegawa, T.; Satoh, M.; Nagase, H.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Propofol exerts greater neuroprotection with disodium edetate than without it. J. Cereb. Blood. Flow. Metab 2008, 28, 354–366. [Google Scholar]
- Nonaka, Y.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Combination effects of normobaric hyperoxia and edaravone on focal cerebral ischemia-induced neuronal damage in mice. Neurosci. Lett 2008, 22, 224–228. [Google Scholar]
- Kikuchi, K.; Kawahara, K.; Tancharoen, S.; Matsuda, F.; Morimoto, Y.; Ito, T.; Biswas, K.K.; Takenouchi, K.; Miura, N.; Oyama, Y.; et al. The free radical scavenger edaravone rescues rats from cerebral infarction by attenuating the release of high-mobility group box-1 in neuronal cells. J. Pharmacol. Exp. Ther 2009, 329, 865–874. [Google Scholar]
- Liu, N.; Shang, J.; Tian, F.; Nishi, H.; Abe, K. In vivo optical imaging for evaluating the efficacy of edaravone after transient cerebral ischemia in mice. Brain Res 2011, 23, 66–75. [Google Scholar]
- Hara, H.; Huang, P.L.; Panahian, N.; Fishman, M.C.; Moskowitz, M.A. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J. Cereb. Blood. Flow. Metab 1996, 16, 605–611. [Google Scholar]
- Hara, H.; Friedlander, R.M.; Gagliardini, V.; Ayata, C.; Fink, K.; Huang, Z.; Shimizu-Sasamata, M.; Yuan, J.; Moskowitz, M.A. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl. Acad. Sci. USA 1997, 94, 2007–2012. [Google Scholar]
- Soumiya, H.; Fukumitsu, H.; Furukawa, S. Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. J. Neurosci. Res 2011, 89, 1575–1585. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tanaka, Y.; Fukumitsu, H.; Soumiya, H.; Yoshimura, S.; Iwama, T.; Furukawa, S. 2-Decenoic Acid Ethyl Ester, a Compound That Elicits Neurotrophin-like Intracellular Signals, Facilitating Functional Recovery from Cerebral Infarction in Mice. Int. J. Mol. Sci. 2012, 13, 4968-4981. https://doi.org/10.3390/ijms13044968
Tanaka Y, Fukumitsu H, Soumiya H, Yoshimura S, Iwama T, Furukawa S. 2-Decenoic Acid Ethyl Ester, a Compound That Elicits Neurotrophin-like Intracellular Signals, Facilitating Functional Recovery from Cerebral Infarction in Mice. International Journal of Molecular Sciences. 2012; 13(4):4968-4981. https://doi.org/10.3390/ijms13044968
Chicago/Turabian StyleTanaka, Yoshitaka, Hidefumi Fukumitsu, Hitomi Soumiya, Shinichi Yoshimura, Toru Iwama, and Shoei Furukawa. 2012. "2-Decenoic Acid Ethyl Ester, a Compound That Elicits Neurotrophin-like Intracellular Signals, Facilitating Functional Recovery from Cerebral Infarction in Mice" International Journal of Molecular Sciences 13, no. 4: 4968-4981. https://doi.org/10.3390/ijms13044968