Phages and HIV-1: From Display to Interplay

Abstract
:
1. Introduction
- Epitope mapping, which relies on the screening of random peptide libraries on immobilized monoclonal or polyclonal antibodies to determine the linear and/or conformational epitopes recognized by these antibodies (linear epitope: sequence of continuous amino acids recognized by the paratope of a given antibody; conformational/discontinuous epitope: group of amino acids scattered along a protein sequence which come together in the folded protein and are recognized by the paratope of a given antibody). Such screening usually results in the identification of sequences mimicking the natural epitope (mimotopes) and provides precise information about the location of residues forming the natural epitope. These mimotopes may in turn be used as valuable immunogens to elicit antibodies targeting the original epitope, an approach referred to as “reverse vaccinology”.
- Inhibitor discovery, based on screening of phage libraries displaying random peptides or antibody fragments against viral or host proteins critical for viral replication.
- “Phage Substrate” approach in which potential substrate sequences are displayed at the phage surface to enzymes such as proteases. This approach not only allows for proteolysis specificity profiling, but provides information and data to develop specific inhibitors.
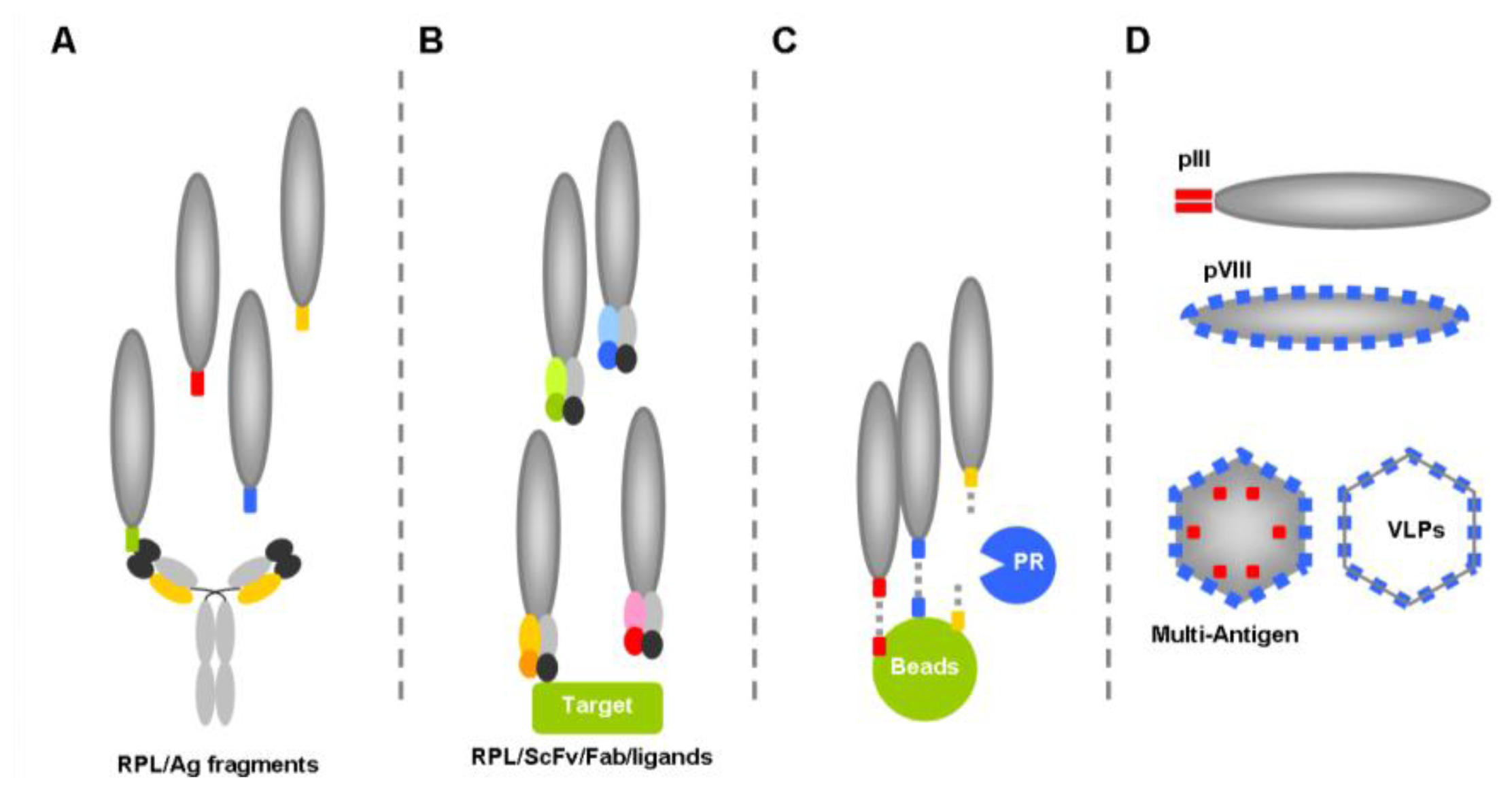
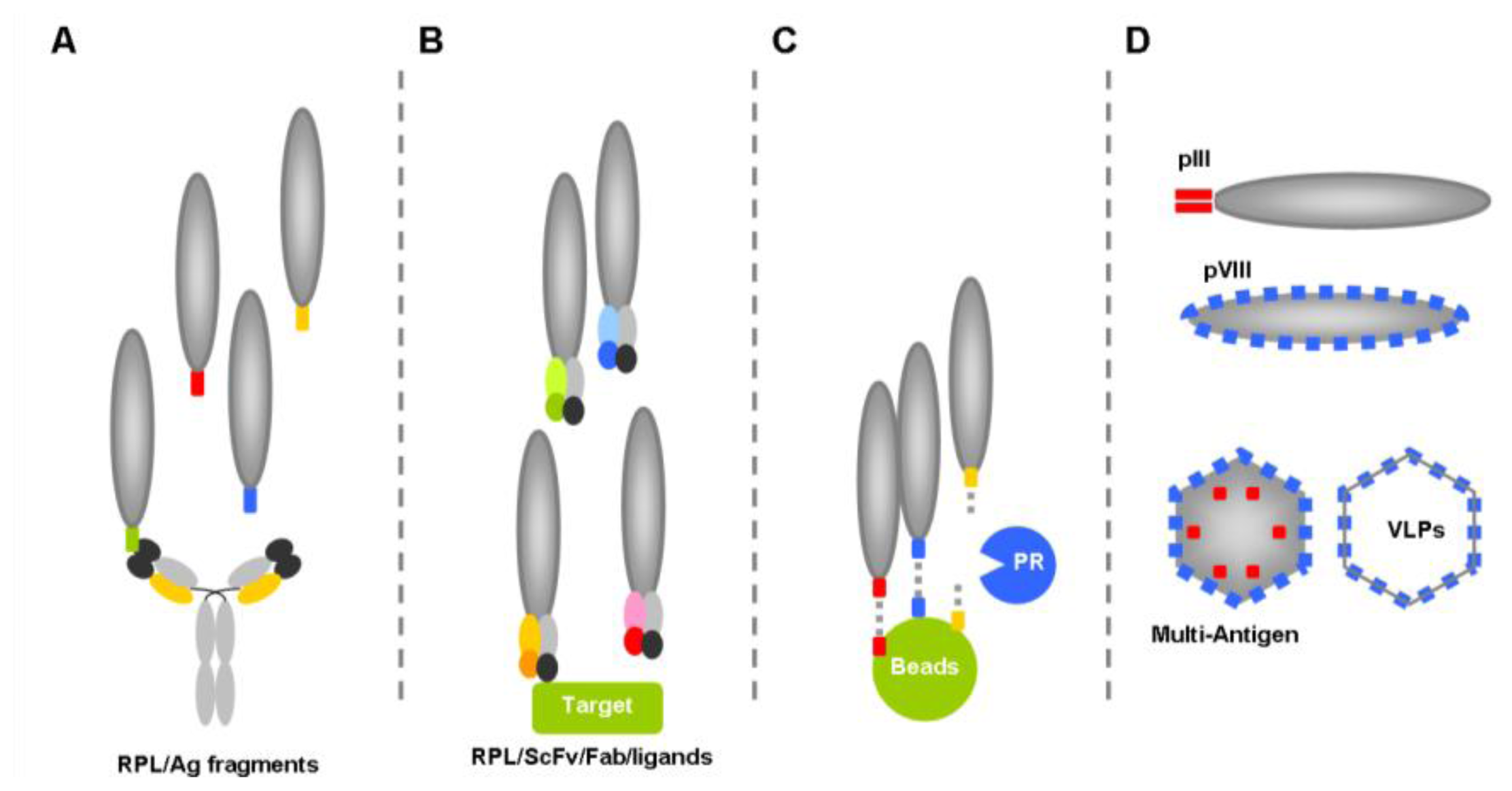
- Carrier phage, in which the phage functions as a “carrier”, “vehicle” or “virus-like particle” to display exogenous peptides such as mimotopes or even full size antigens to the immune system, to elicit specific humoral and/or cytotoxic T-cells responses.
2. Exploration of HIV-1 Epitope Landscape
2.1. Antibodies Directed against Viral Proteins
2.1.1. Monoclonal Antibodies Directed against Viral Epitopes
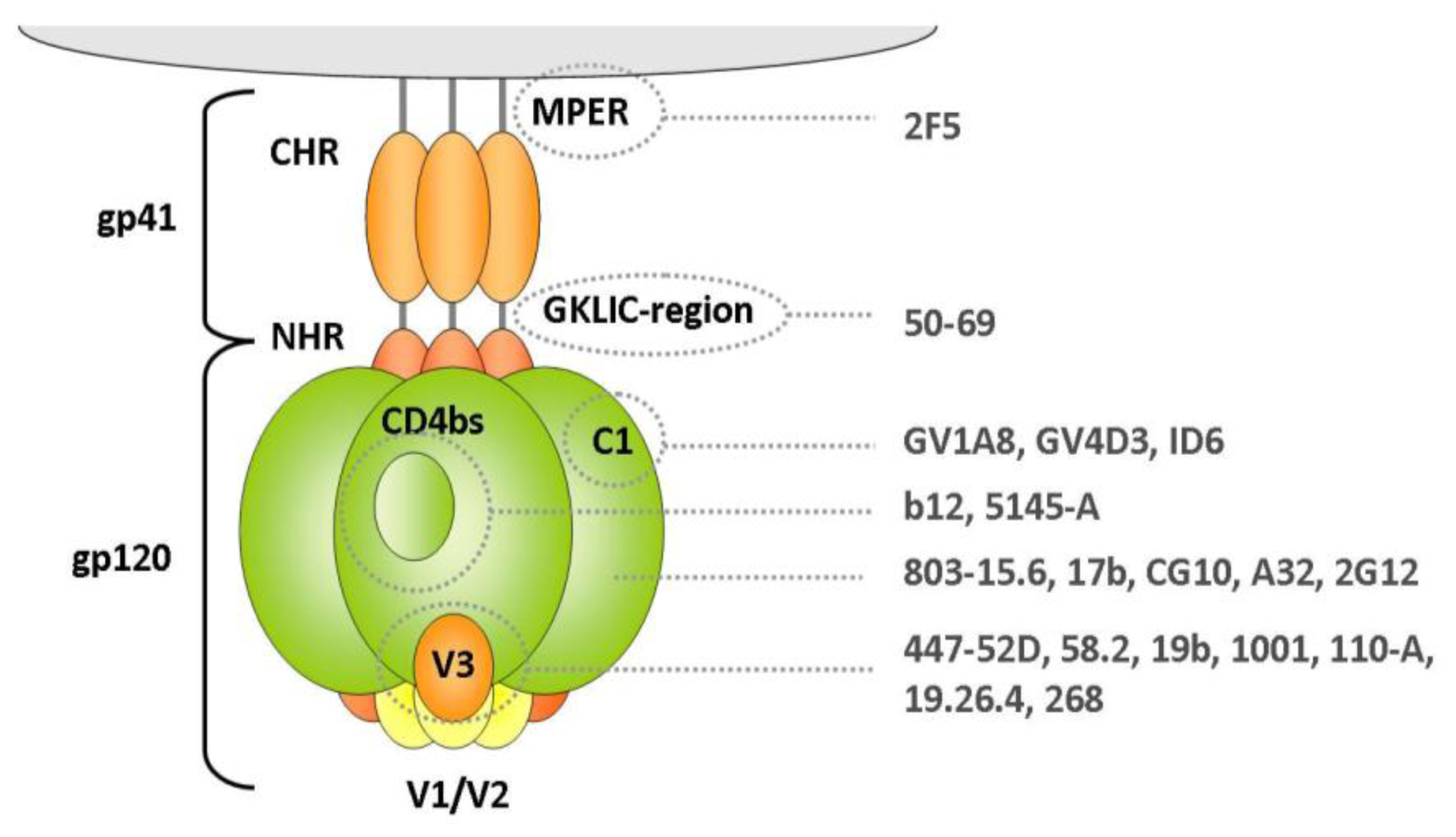
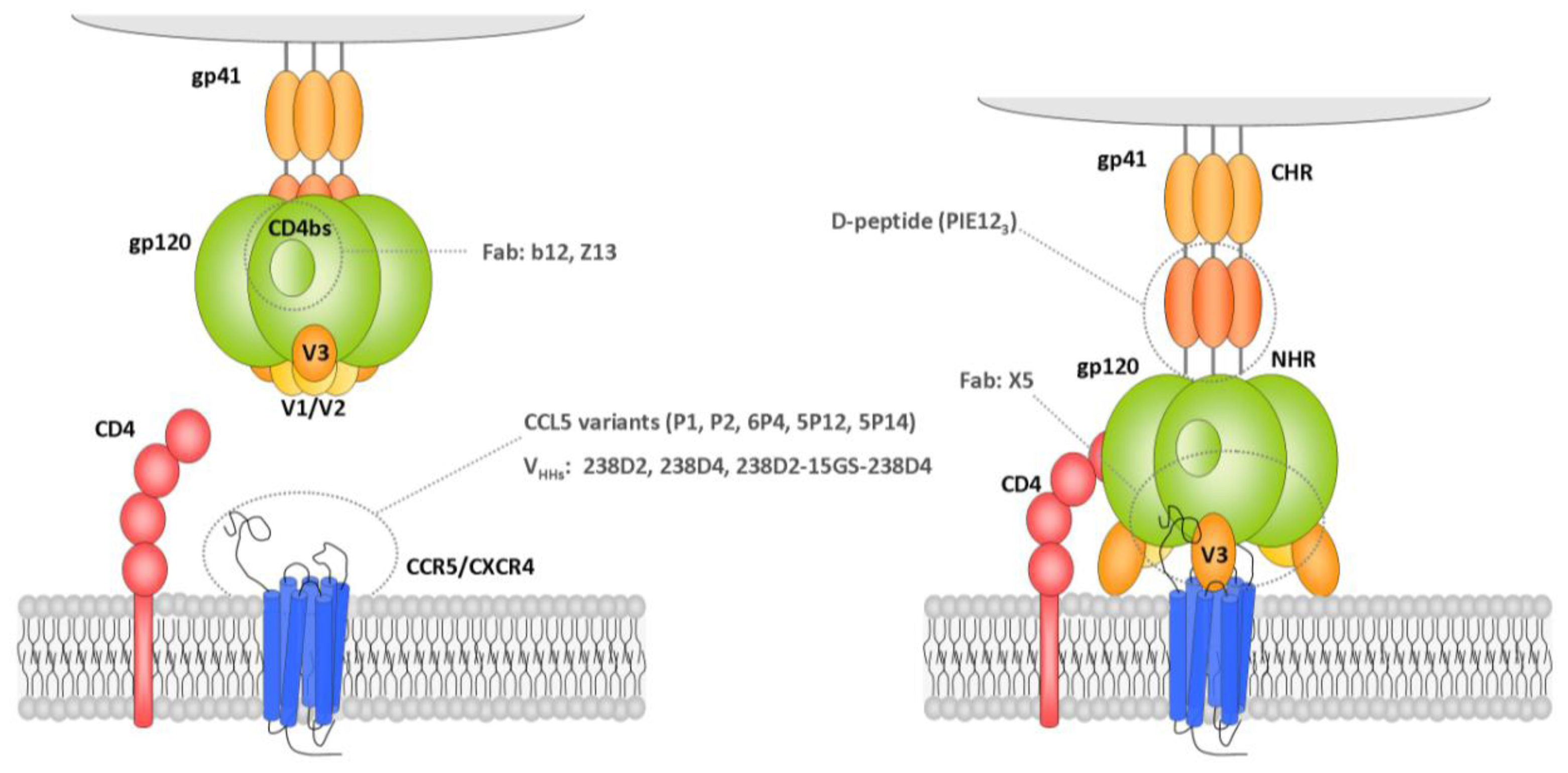
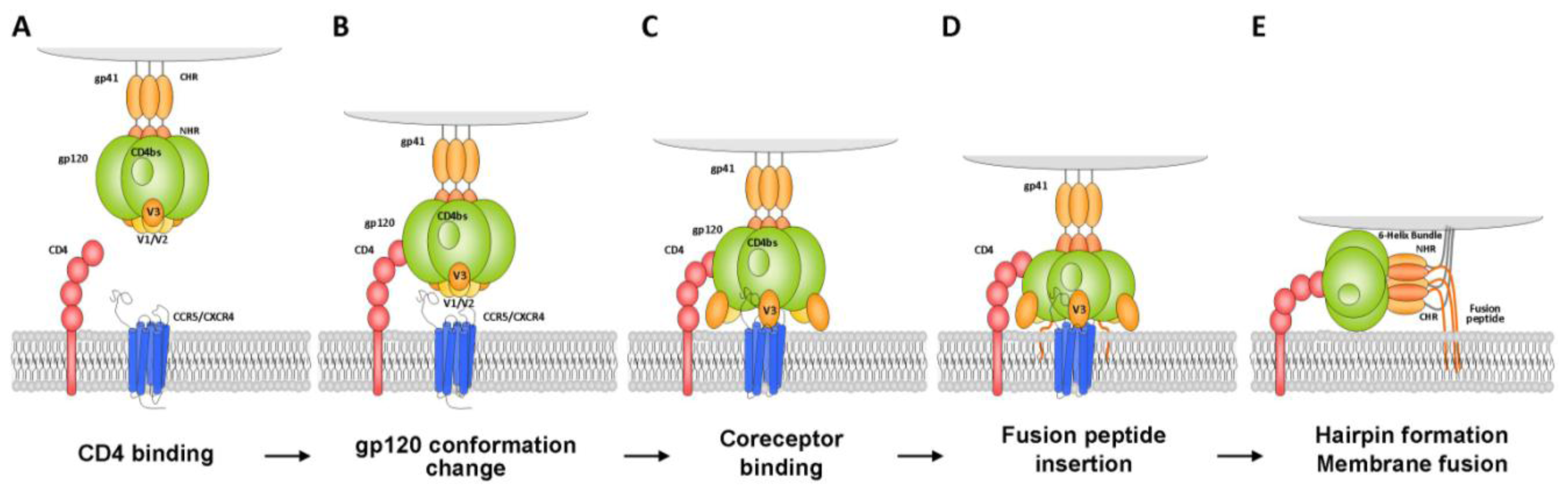
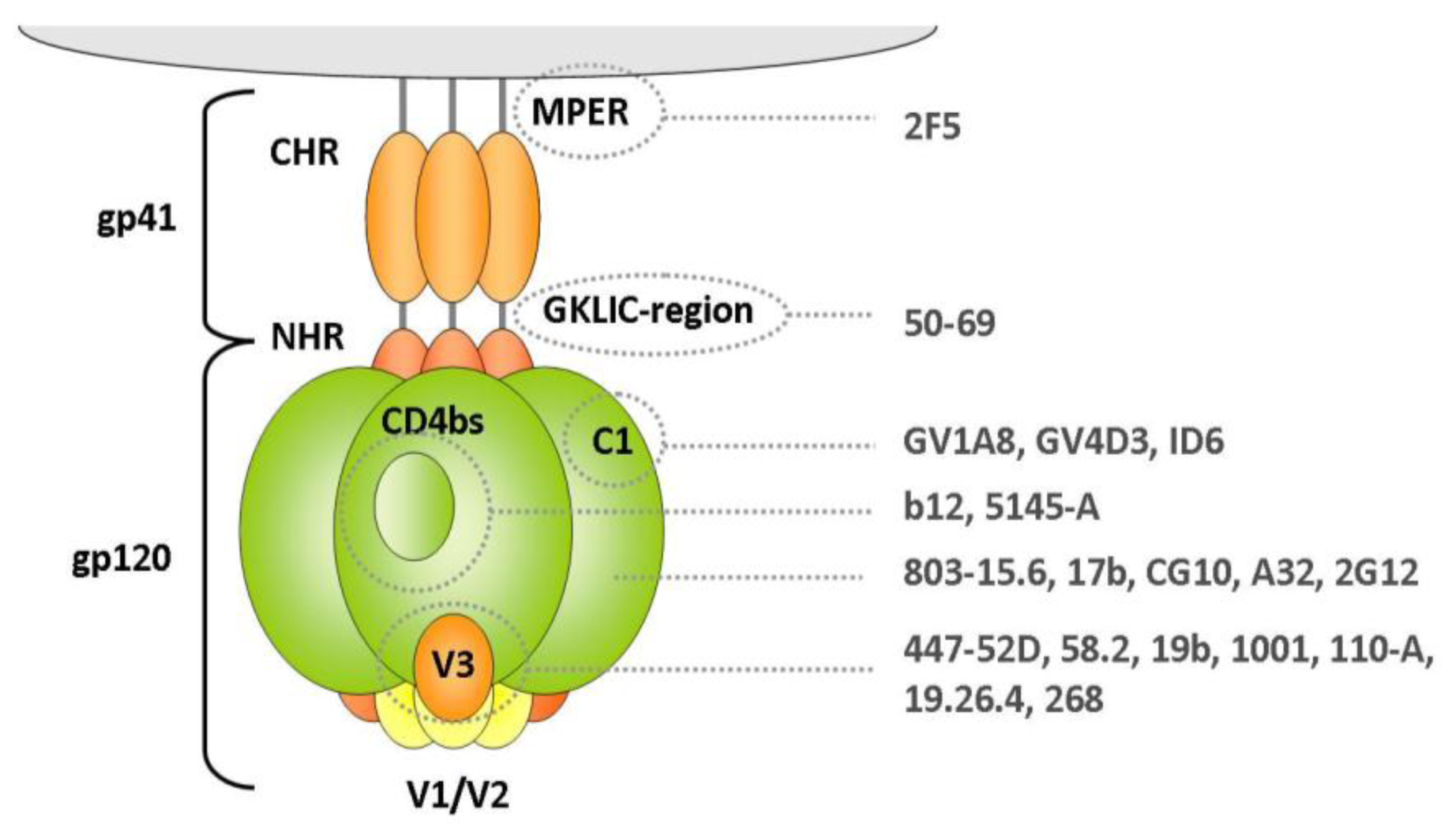
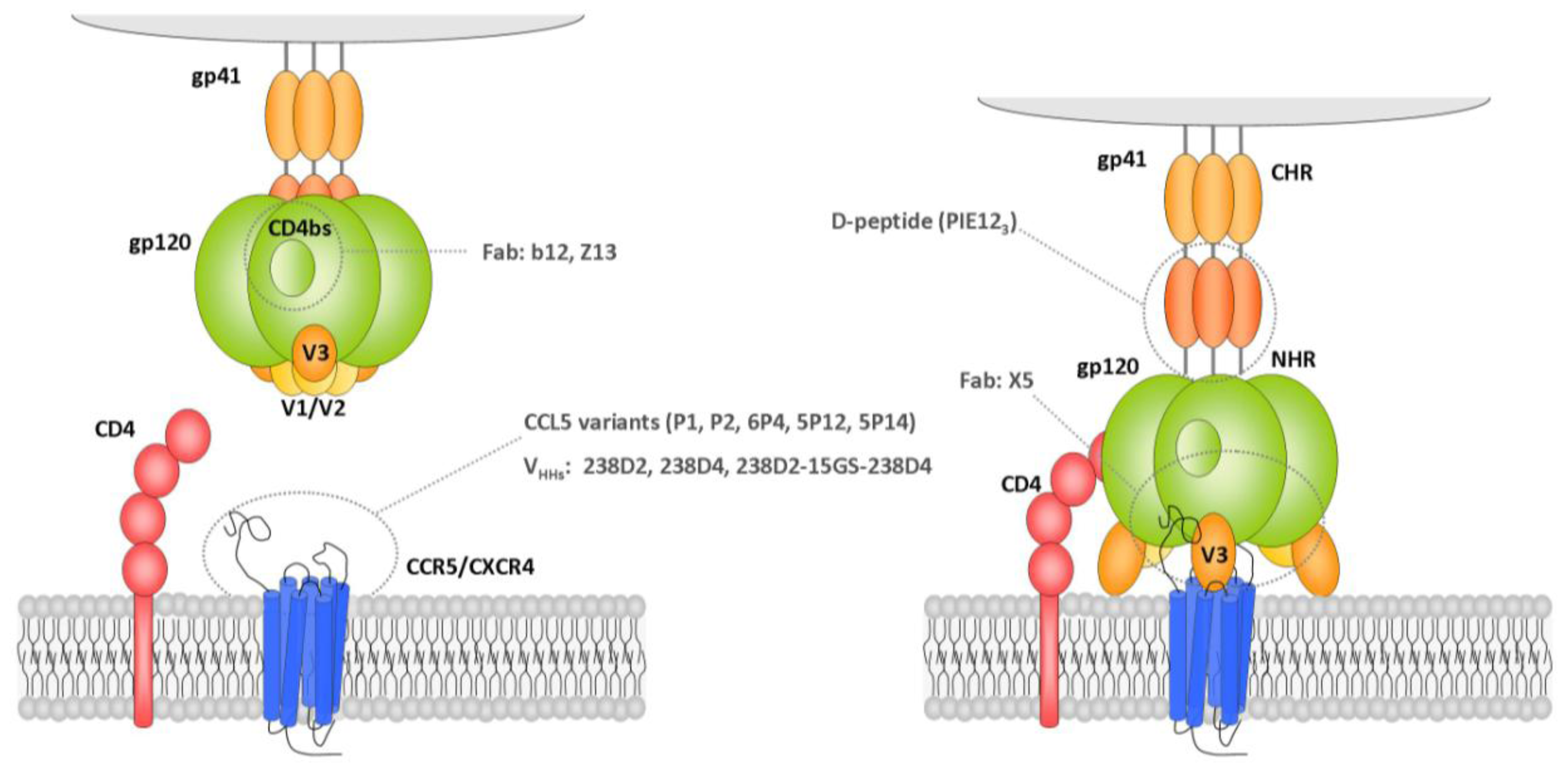
2.1.1.1. Gp120 V3 Loop
2.1.1.2. Gp41 Membrane Proximal External Region (MPER)
2.1.1.3. Gp120 C1 Domain
2.1.1.4. Gp120 CD4-Binding Site
2.1.1.5. Other Domains
2.1.2. Polyclonal Antibodies Directed Against Viral Epitopes
2.2. Antibodies Directed against Host Proteins
3. Identification of HIV-1 Inhibitors by Phage Display
3.1. Inhibitors of HIV-1 Proteins
3.1.1. Env Inhibitors
3.1.1.1. Gp120 CD4 Binding Site
3.1.1.1.1. Fab Libraries
3.1.1.1.2. ScFvs Hydrolyzing gp120
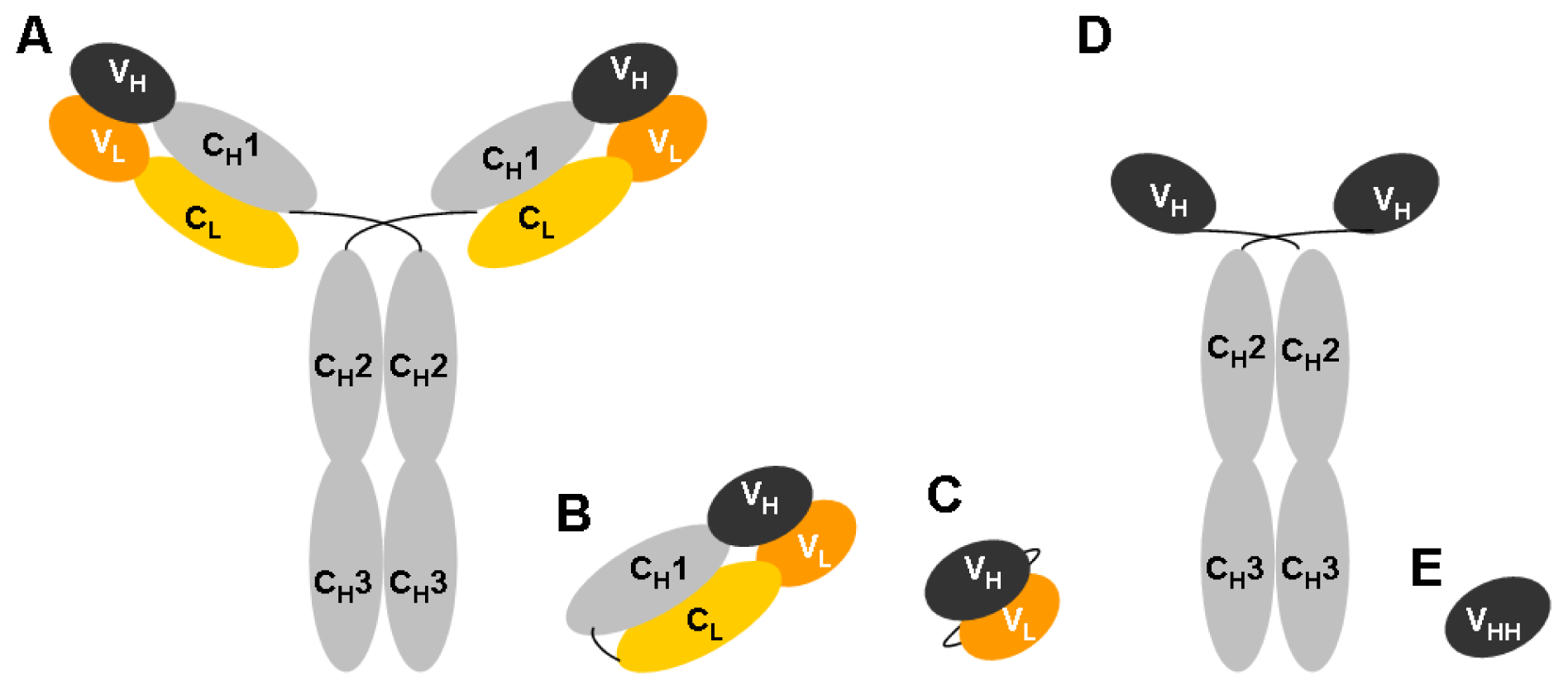
3.1.1.1.3. Nanobodies
3.1.1.1.4. CD4 Mimics
3.1.1.2. Gp120 V3 Loop
3.1.1.3. Gp120 CD4-Induced Epitope
3.1.1.4. Gp120 C1 Domain
3.1.1.5. Phage Display as a Tool to Unravel the HIV-1-Specific Humoral Response
3.1.1.6. Gp41 MPER Inhibitors
3.1.1.7. Gp41 Heptad Repeat Inhibitors
3.1.2. Other HIV-1 Proteins
3.1.2.1. Viral Protein of Regulation (Vpr)
3.1.2.2. Integrase (IN)
3.1.2.3. Transactivator of Transcription (Tat)/Transactivation Response element (TAR)
3.1.2.4. Nucleocapsid (NC)/Packaging Signal (psi) Sequence
3.1.2.5. Negative Factor (Nef)/Virion Infectivity Factor (Vif)
3.1.2.6. Reverse Transcriptase (RT)
3.1.2.7. Regulator of Virion Expression (Rev)
3.1.2.8. Group-Specific Antigen (Gag)
3.1.3. Diagnostic Applications
3.2. Inhibitors of Host Proteins
3.2.1. Host receptors inhibitors
3.2.1.1. CCR5 Coreceptor
3.2.1.2. CXCR4 Coreceptor
3.2.2. Other Host Protein Inhibitors
4. Phage Substrate
5. Phage particles as HIV-1 Antigen Carriers
6. Conclusions and Future Challenges
Acknowledgements
Abbreviations
| AA | amino acids |
| Ab | Antibody |
| ADCC | Antibody-Dependent Cellular Cytotoxicity |
| AIDS | Acquired ImmunoDeficiency Syndrome |
| ARM | Arginine Rich Motif |
| BNtAb | Broadly Neutralizing Antibody |
| BS | Binding Site |
| BSA | Bovine Serum Albumin |
| CCR5 | CC chemokine Receptor 5 |
| CD | Cluster of Differentiation |
| CDR | Complementarity Determining Region |
| CH | Constant Heavy chain domain |
| CHR | C-terminal Heptad Repeats |
| CTL | Cytotoxic T-Lymphocytes |
| CXCR4 | CXC chemokine Receptor 4 |
| DDDP | DNA-Dependent DNA Polymerase |
| ECL | ExtraCellular Loop |
| Fab | Fragment antigen-binding |
| FIV | Feline ImmunoDeficiency Virus |
| FR | Frameworks |
| Gag | group-specific antigen |
| gp | glycoprotein |
| HAART | Highly Active Anti-Retroviral Therapy |
| HB | Helix Bundle |
| HcAb | Heavy-chain only Antibody |
| HCDR | Heavy-chain Complementarity Determining Region |
| HIV-1 | Human ImmunoDeficiency Virus |
| HLA | Human Leukocyte Antigen |
| HOC | Highly antigenic Outer Capsid protein |
| IC50 | Inhibitory Concentration 50 |
| ID | Immuno Dominant |
| ID50 | Inhibitory Dose 50 |
| Ig | Immunoglobulin |
| IN | Integrase |
| KD | dissociation constant |
| KLH | Keyhole Limpet Hemocyanin |
| LTNP | Long-Term Non-Progressor |
| MAb | Monoclonal Antibody |
| MHC | Major Histocompatibility Complex |
| MPER | Membrane Proximal External Region |
| NC | NucleoCapsid |
| NCp | Nucleocapsid protein |
| Nef | Negative factor |
| NHR | N-terminal Heptad Repeats; NLS, Nuclear Localization Signal |
| NNY | n-nonanoyl |
| NtAb | Neutralizing Antibody |
| PAb | Polyclonal Antibody |
| PBL | Peripheral Blood Lymphocytes |
| PBMC | peripheral blood mononuclear cell |
| PHI | PreHairpin Intermediate |
| PIC | PreIntegration Complex |
| PIE | Pocket-specific Inhibitor of Entry |
| PR | Protease |
| psi | packaging signal |
| RDDP | RNA-Dependent DNA Polymerase |
| Rev | Regulator of virion expression |
| RPL | Randomized Peptide Library |
| RT | Reverse Transcriptase |
| ScFv | Single-chain variable fragment |
| SOC | Small Outer Capsid protein |
| SPR | Surface Plasmon Resonance |
| TAR | Transactivation Response element |
| TCR | T-Cells Receptor |
| TRM | Tat/TAR Recognition Motif |
| UDG | Uracyl DNA Glycosylase |
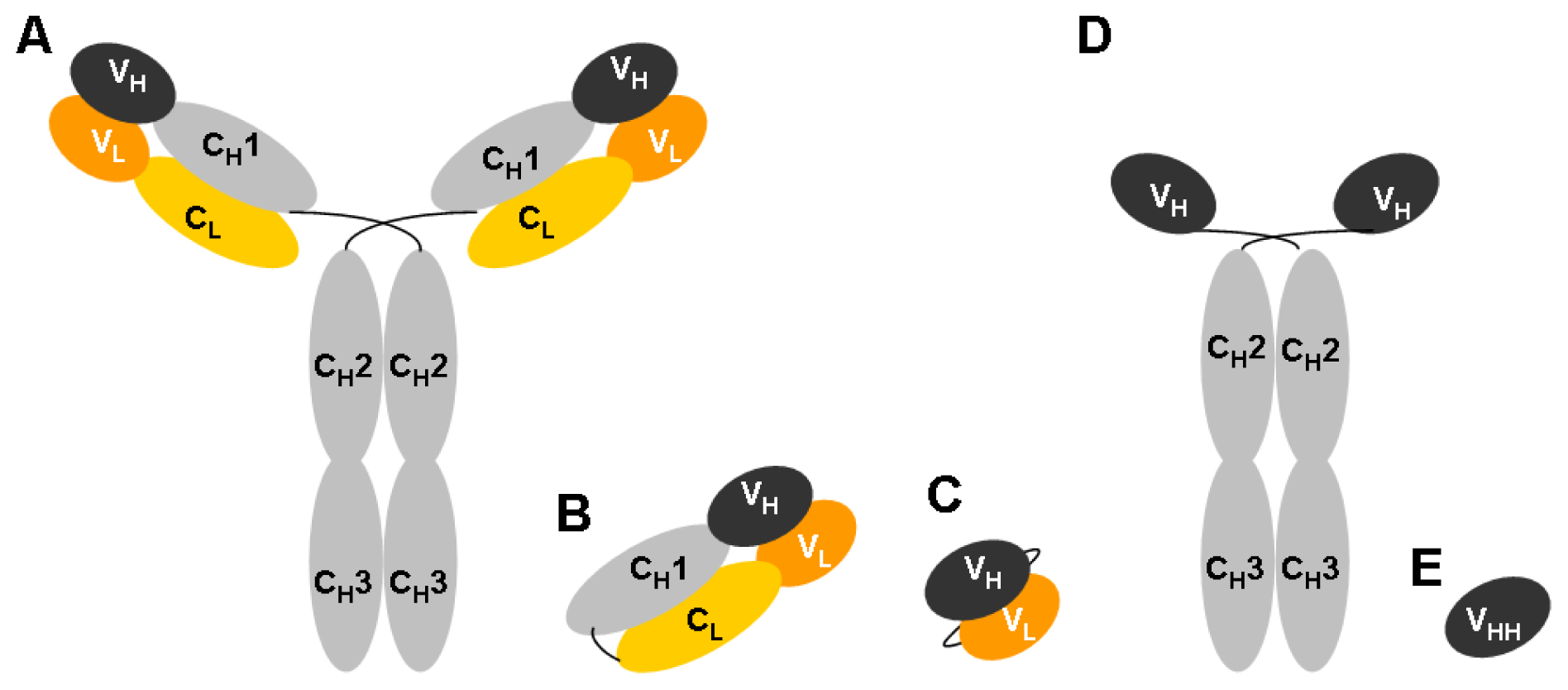
| VH | Variable Heavy chain domain |
| VHH | Variable Heavy-chain domains of Heavy-chain only Antibodies |
| Vif | Virion infectivity Factor |
| VL | Variable Light chain domain |
| vpr | Viral Protein of Regulation |
| VLP | Virus-Like Particle. |
References
- Gottlieb, M.S.; Schroff, R.; Schanker, H.M.; Weisman, J.D.; Fan, P.T.; Wolf, R.A.; Saxon, A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: Evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med 1981, 305, 1425–1431. [Google Scholar]
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a t-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar]
- Naeger, L.K.; Struble, K.A.; Murray, J.S.; Birnkrant, D.B. Running a tightrope: Regulatory challenges in the development of antiretrovirals. Antiviral Res 2010, 85, 232–240. [Google Scholar]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med 1998, 4, 1302–1307. [Google Scholar]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (uk-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother 2005, 49, 4721–4732. [Google Scholar]
- Summa, V.; Petrocchi, A.; Matassa, V.G.; Gardelli, C.; Muraglia, E.; Rowley, M.; Paz, O.G.; Laufer, R.; Monteagudo, E.; Pace, P. 4,5-dihydroxypyrimidine carboxamides and N-alkyl-5-hydroxypyrimidinone carboxamides are potent, selective HIV integrase inhibitors with good pharmacokinetic profiles in preclinical species. J. Med. Chem 2006, 49, 6646–6649. [Google Scholar]
- Tozzi, V.; Bellagamba, R.; Castiglione, F.; Amendola, A.; Ivanovic, J.; Nicastri, E.; Libertone, R.; D’Offizi, G.; Liuzzi, G.; Gori, C.; et al. Plasma HIV RNA decline and emergence of drug resistance mutations among patients with multiple virologic failures receiving resistance testing-guided haart. AIDS Res. Hum. Retrovir 2008, 24, 787–796. [Google Scholar]
- Dragic, T. An overview of the determinants of CCR5 and CXCR4 co-receptor function. J. Gen. Virol 2001, 82, 1807–1814. [Google Scholar]
- Pantophlet, R.; Wang, M.; Aguilar-Sino, R.O.; Burton, D.R. The human immunodeficiency virus type 1 envelope spike of primary viruses can suppress antibody access to variable regions. J. Virol 2009, 83, 1649–1659. [Google Scholar]
- Mahalanabis, M.; Jayaraman, P.; Miura, T.; Pereyra, F.; Chester, E.M.; Richardson, B.; Walker, B.; Haigwood, N.L. Continuous viral escape and selection by autologous neutralizing antibodies in drug-naive human immunodeficiency virus controllers. J. Virol 2009, 83, 662–672. [Google Scholar]
- Overbaugh, J.; Morris, L. The antibody response against HIV-1. Cold Spring Harb. Perspect. Med 2012, 2, a007039. [Google Scholar]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Beghetto, E.; Gargano, N. Lambda-display: A powerful tool for antigen discovery. Molecules 2011, 16, 3089–3105. [Google Scholar]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar]
- Poropatich, K.; Sullivan, D.J., Jr. Human immunodeficiency virus type 1 long-term non-progressors: The viral, genetic and immunological basis for disease non-progression. J. Gen. Virol 2011, 92, 247–268. [Google Scholar]
- Gorny, M.K.; Xu, J.Y.; Gianakakos, V.; Karwowska, S.; Williams, C.; Sheppard, H.W.; Hanson, C.V.; Zolla-Pazner, S. Production of site-selected neutralizing human monoclonal antibodies against the third variable domain of the human immunodeficiency virus type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 1991, 88, 3238–3242. [Google Scholar]
- Keller, P.M.; Arnold, B.A.; Shaw, A.R.; Tolman, R.L.; van Middlesworth, F.; Bondy, S.; Rusiecki, V.K.; Koenig, S.; Zolla-Pazner, S.; Conard, P.; et al. Identification of HIV vaccine candidate peptides by screening random phage epitope libraries. Virology 1993, 193, 709–716. [Google Scholar]
- Boots, L.J.; McKenna, P.M.; Arnold, B.A.; Keller, P.M.; Gorny, M.K.; Zolla-Pazner, S.; Robinson, J.E.; Conley, A.J. Anti-human immunodeficiency virus type 1 human monoclonal antibodies that bind discontinuous epitopes in the viral glycoproteins can identify mimotopes from recombinant phage peptide display libraries. AIDS Res. Hum. Retrovir 1997, 13, 1549–1559. [Google Scholar]
- Jellis, C.L.; Cradick, T.J.; Rennert, P.; Salinas, P.; Boyd, J.; Amirault, T.; Gray, G.S. Defining critical residues in the epitope for a HIV-neutralizing monoclonal antibody using phage display and peptide array technologies. Gene 1993, 137, 63–68. [Google Scholar]
- Moore, J.P.; Trkola, A.; Korber, B.; Boots, L.J.; Kessler, J.A., 2nd; McCutchan, F.E.; Mascola, J.; Ho, D.D.; Robinson, J.; Conley, A.J. A human monoclonal antibody to a complex epitope in the V3 region of gp120 of human immunodeficiency virus type 1 has broad reactivity within and outside clade b. J. Virol 1995, 69, 122–130. [Google Scholar]
- Grihalde, N.D.; Chen, Y.C.; Golden, A.; Gubbins, E.; Mandecki, W. Epitope mapping of anti-HIV and anti-HCV monoclonal antibodies and characterization of epitope mimics using a filamentous phage peptide library. Gene 1995, 166, 187–195. [Google Scholar]
- Laisney, I.L.; Benjamin, H.; Gefter, M.; Strosberg, A.D. Permissive residues within the minimal epitopes of neutralizing monoclonal antibodies to the V3 loop of HIV-1. Eur. J. Immunol 1996, 26, 1634–1640. [Google Scholar]
- Conley, A.J.; Kessler, J.A., 2nd; Boots, L.J.; Tung, J.S.; Arnold, B.A.; Keller, P.M.; Shaw, A.R.; Emini, E.A. Neutralization of divergent human immunodeficiency virus type 1 variants and primary isolates by iam-41-2f5, an anti-gp41 human monoclonal antibody. Proc. Natl. Acad. Sci. USA 1994, 91, 3348–3352. [Google Scholar]
- Menendez, A.; Chow, K.C.; Pan, O.C.; Scott, J.K. Human immunodeficiency virus type 1-neutralizing monoclonal antibody 2F5 is multispecific for sequences flanking the DKW core epitope. J. Mol. Biol 2004, 338, 311–327. [Google Scholar]
- Palacios-Rodriguez, Y.; Gazarian, T.; Huerta, L.; Gazarian, K. Constrained peptide models from phage display libraries highlighting the cognate epitope-specific potential of the anti-HIV-1 mab 2f5. Immunol. Lett 2011, 136, 80–89. [Google Scholar]
- Stern, B.; Denisova, G.; Buyaner, D.; Raviv, D.; Gershoni, J.M. Helical epitopes determined by low-stringency antibody screening of a combinatorial peptide library. FASEB J 1997, 11, 147–153. [Google Scholar]
- Gomez-Roman, V.R.; Cao, C.; Bai, Y.; Santamaria, H.; Acero, G.; Manoutcharian, K.; Weiner, D.B.; Ugen, K.E.; Gevorkian, G. Phage-displayed mimotopes recognizing a biologically active anti-HIV-1 gp120 murine monoclonal antibody. J. Acquir. Immune Defic. Syndr 2002, 31, 147–153. [Google Scholar]
- Zwick, M.B.; Bonnycastle, L.L.; Menendez, A.; Irving, M.B.; Barbas, C.F., 3rd; Parren, P.W.; Burton, D.R.; Scott, J.K. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J. Virol 2001, 75, 6692–6699. [Google Scholar]
- Bonnycastle, L.L.; Mehroke, J.S.; Rashed, M.; Gong, X.; Scott, J.K. Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J. Mol. Biol 1996, 258, 747–762. [Google Scholar]
- Dorgham, K.; Dogan, I.; Bitton, N.; Parizot, C.; Cardona, V.; Debre, P.; Hartley, O.; Gorochov, G. Immunogenicity of HIV type 1 gp120 CD4 binding site phage mimotopes. AIDS Res. Hum. Retrovir 2005, 21, 82–92. [Google Scholar]
- Enshell-Seijffers, D.; Denisov, D.; Groisman, B.; Smelyanski, L.; Meyuhas, R.; Gross, G.; Denisova, G.; Gershoni, J.M. The mapping and reconstitution of a conformational discontinuous b-cell epitope of HIV-1. J. Mol. Biol 2003, 334, 87–101. [Google Scholar]
- Wilkinson, R.A.; Evans, J.R.; Jacobs, J.M.; Slunaker, D.; Pincus, S.H.; Pinter, A.; Parkos, C.A.; Burritt, J.B.; Teintze, M. Peptides selected from a phage display library with an HIV-neutralizing antibody elicit antibodies to HIV gp120 in rabbits, but not to the same epitope. AIDS Res. Hum. Retrovir 2007, 23, 1416–1427. [Google Scholar]
- Ferrer, M.; Sullivan, B.J.; Godbout, K.L.; Burke, E.; Stump, H.S.; Godoy, J.; Golden, A.; Profy, A.T.; van Schravendijk, M.R. Structural and functional characterization of an epitope in the conserved C-terminal region of HIV-1 gp120. J. Pept. Res 1999, 54, 32–42. [Google Scholar]
- Menendez, A.; Calarese, D.A.; Stanfield, R.L.; Chow, K.C.; Scanlan, C.N.; Kunert, R.; Katinger, H.; Burton, D.R.; Wilson, I.A.; Scott, J.K. A peptide inhibitor of HIV-1 neutralizing antibody 2G12 is not a structural mimic of the natural carbohydrate epitope on gp120. FASEB J 2008, 22, 1380–1392. [Google Scholar]
- Rusche, J.R.; Javaherian, K.; McDanal, C.; Petro, J.; Lynn, D.L.; Grimaila, R.; Langlois, A.; Gallo, R.C.; Arthur, L.O.; Fischinger, P.J.; et al. Antibodies that inhibit fusion of human immunodeficiency virus-infected cells bind a 24-amino acid sequence of the viral envelope, gp120. Proc. Natl. Acad. Sci. USA 1988, 85, 3198–3202. [Google Scholar]
- Javaherian, K.; Langlois, A.J.; McDanal, C.; Ross, K.L.; Eckler, L.I.; Jellis, C.L.; Profy, A.T.; Rusche, J.R.; Bolognesi, D.P.; Putney, S.D.; et al. Principal neutralizing domain of the human immunodeficiency virus type 1 envelope protein. Proc. Natl. Acad. Sci. USA 1989, 86, 6768–6772. [Google Scholar]
- Scott, C.F., Jr; Silver, S.; Profy, A.T.; Putney, S.D.; Langlois, A.; Weinhold, K.; Robinson, J.E. Human monoclonal antibody that recognizes the v3 region of human immunodeficiency virus gp120 and neutralizes the human T-lymphotropic virus type IIIMN strain. Proc. Natl. Acad. Sci. USA 1990, 87, 8597–8601. [Google Scholar]
- Muster, T.; Steindl, F.; Purtscher, M.; Trkola, A.; Klima, A.; Himmler, G.; Ruker, F.; Katinger, H. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol 1993, 67, 6642–6647. [Google Scholar]
- Denisova, G.; Stern, B.; Raviv, D.; Zwickel, J.; Smorodinsky, N.I.; Gershoni, J.M. Humoral immune response to immunocomplexed HIV envelope glycoprotein 120. AIDS Res. Hum. Retrovir 1996, 12, 901–909. [Google Scholar]
- Burton, D.R.; Barbas, C.F., 3rd; Persson, M.A.; Koenig, S.; Chanock, R.M.; Lerner, R.A. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. USA 1991, 88, 10134–10137. [Google Scholar]
- Barbas, C.F., 3rd; Bjorling, E.; Chiodi, F.; Dunlop, N.; Cababa, D.; Jones, T.M.; Zebedee, S.L.; Persson, M.A.; Nara, P.L.; Norrby, E.; et al. Recombinant human fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc. Natl. Acad. Sci. USA 1992, 89, 9339–9343. [Google Scholar]
- Bublil, E.M.; Yeger-Azuz, S.; Gershoni, J.M. Computational prediction of the cross-reactive neutralizing epitope corresponding to the [corrected] monclonal [corrected] antibody b12 specific for HIV-1 gp120. FASEB J 2006, 20, 1762–1774. [Google Scholar]
- Rizzuto, C.D.; Wyatt, R.; Hernandez-Ramos, N.; Sun, Y.; Kwong, P.D.; Hendrickson, W.A.; Sodroski, J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 1998, 280, 1949–1953. [Google Scholar]
- Moore, J.P.; McCutchan, F.E.; Poon, S.W.; Mascola, J.; Liu, J.; Cao, Y.; Ho, D.D. Exploration of antigenic variation in gp120 from clades a through f of human immunodeficiency virus type 1 by using monoclonal antibodies. J. Virol 1994, 68, 8350–8364. [Google Scholar]
- Thali, M.; Moore, J.P.; Furman, C.; Charles, M.; Ho, D.D.; Robinson, J.; Sodroski, J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol 1993, 67, 3978–3988. [Google Scholar]
- Scanlan, C.N.; Pantophlet, R.; Wormald, M.R.; Ollmann Saphire, E.; Stanfield, R.; Wilson, I.A.; Katinger, H.; Dwek, R.A.; Rudd, P.M.; Burton, D.R. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1→2 mannose residues on the outer face of gp120. J. Virol 2002, 76, 7306–7321. [Google Scholar]
- Trkola, A.; Purtscher, M.; Muster, T.; Ballaun, C.; Buchacher, A.; Sullivan, N.; Srinivasan, K.; Sodroski, J.; Moore, J.P.; Katinger, H. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol 1996, 70, 1100–1108. [Google Scholar]
- Sanders, R.W.; Venturi, M.; Schiffner, L.; Kalyanaraman, R.; Katinger, H.; Lloyd, K.O.; Kwong, P.D.; Moore, J.P. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol 2002, 76, 7293–7305. [Google Scholar]
- Scala, G.; Chen, X.; Liu, W.; Telles, J.N.; Cohen, O.J.; Vaccarezza, M.; Igarashi, T.; Fauci, A.S. Selection of HIV-specific immunogenic epitopes by screening random peptide libraries with HIV-1-positive sera. J. Immunol 1999, 162, 6155–6161. [Google Scholar]
- Chen, X.; Scala, G.; Quinto, I.; Liu, W.; Chun, T.W.; Justement, J.S.; Cohen, O.J.; van Cott, T.C.; Iwanicki, M.; Lewis, M.G.; et al. Protection of rhesus macaques against disease progression from pathogenic SHIV-89.6PD by vaccination with phage-displayed HIV-1 epitopes. Nat. Med 2001, 7, 1225–1231. [Google Scholar]
- Enshell-Seijffers, D.; Smelyanski, L.; Vardinon, N.; Yust, I.; Gershoni, J.M. Dissection of the humoral immune response toward an immunodominant epitope of HIV: A model for the analysis of antibody diversity in HIV+ individuals. FASEB J 2001, 15, 2112–2120. [Google Scholar]
- Palacios-Rodriguez, Y.; Gazarian, T.; Rowley, M.; Majluf-Cruz, A.; Gazarian, K. Collection of phage-peptide probes for HIV-1 immunodominant loop-epitope. J. Microbiol. Methods 2007, 68, 225–235. [Google Scholar]
- Humbert, M.; Antoni, S.; Brill, B.; Landersz, M.; Rodes, B.; Soriano, V.; Wintergerst, U.; Knechten, H.; Staszewski, S.; von Laer, D.; et al. Mimotopes selected with antibodies from HIV-1-neutralizing long-term non-progressor plasma. Eur. J. Immunol 2007, 37, 501–515. [Google Scholar]
- Humbert, M.; Rasmussen, R.A.; Ong, H.; Kaiser, F.M.; Hu, S.L.; Ruprecht, R.M. Inducing cross-clade neutralizing antibodies against HIV-1 by immunofocusing. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Dieltjens, T.; Heyndrickx, L.; Willems, B.; Gray, E.; Van Nieuwenhove, L.; Grupping, K.; Vanham, G.; Janssens, W. Evolution of antibody landscape and viral envelope escape in an HIV-1 CRF02_AG infected patient with 4E10-like antibodies. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Dieltjens, T.; Willems, B.; Coppens, S.; Van Nieuwenhove, L.; Humbert, M.; Dietrich, U.; Heyndrickx, L.; Vanham, G.; Janssens, W. Unravelling the antigenic landscape of the HIV-1 subtype a envelope of an individual with broad cross-neutralizing antibodies using phage display peptide libraries. J. Virol. Methods 2010, 169, 95–102. [Google Scholar]
- Gupta, S.; Arora, K.; Sampath, A.; Singh, S.S.; Gupta, A.; Chaudhary, V.K. Mapping of HIV-1 gag epitopes recognized by polyclonal antibodies using gene-fragment phage display system. Prep. Biochem. Biotechnol 2001, 31, 185–200. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic. Acids Res 2000, 28, 235–242. [Google Scholar]
- Schreiber, A.; Humbert, M.; Benz, A.; Dietrich, U. 3d-epitope-explorer (3dex): Localization of conformational epitopes within three-dimensional structures of proteins. J. Comput. Chem 2005, 26, 879–887. [Google Scholar]
- Song, R.J.; Chenine, A.L.; Rasmussen, R.A.; Ruprecht, C.R.; Mirshahidi, S.; Grisson, R.D.; Xu, W.; Whitney, J.B.; Goins, L.M.; Ong, H.; et al. Molecularly cloned SHIV-1157IPD3N4: A highly replication- competent, mucosally transmissible R5 simian-human immunodeficiency virus encoding HIV clade c env. J. Virol 2006, 80, 8729–8738. [Google Scholar]
- O’Connor, K.H.; Konigs, C.; Rowley, M.J.; Irving, J.A.; Wijeyewickrema, L.C.; Pustowka, A.; Dietrich, U.; Mackay, I.R. Requirement of multiple phage displayed peptide libraries for optimal mapping of a conformational antibody epitope on CCR5. J. Immunol. Methods 2005, 299, 21–35. [Google Scholar]
- Konigs, C.; Rowley, M.J.; Thompson, P.; Myers, M.A.; Scealy, M.; Davies, J.M.; Wu, L.; Dietrich, U.; Mackay, C.R.; Mackay, I.R. Monoclonal antibody screening of a phage-displayed random peptide library reveals mimotopes of chemokine receptor CCR5: Implications for the tertiary structure of the receptor and for an N-terminal binding site for HIV-1 gp120. Eur. J. Immunol 2000, 30, 1162–1171. [Google Scholar]
- Meta, A.; Torigoe, N.; Ito, Y.; Arakaki, R.; Nakashima, H.; Sugimura, K. Inhibition of M-tropic HIV-1 infection by the fd phage-gene 3 protein with MIP-1α-binding activity. Mol. Immunol 1999, 36, 1249–1254. [Google Scholar]
- Khurana, S.; Kennedy, M.; King, L.R.; Golding, H. Identification of a linear peptide recognized by monoclonal antibody 2D7 capable of generating CCR5-specific antibodies with human immunodeficiency virus-neutralizing activity. J. Virol 2005, 79, 6791–6800. [Google Scholar]
- Poloni, F.; Puddu, P.; Moretti, F.; Flego, M.; Romagnoli, G.; Tombesi, M.; Capone, I.; Chersi, A.; Felici, F.; Cianfriglia, M. Identification of a LFA-1 region involved in the HIV-1-induced syncytia formation through phage-display technology. Eur. J. Immunol 2001, 31, 57–63. [Google Scholar]
- Wu, L.; LaRosa, G.; Kassam, N.; Gordon, C.J.; Heath, H.; Ruffing, N.; Chen, H.; Humblias, J.; Samson, M.; Parmentier, M.; et al. Interaction of chemokine receptor CCR5 with its ligands: Multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J. Exp. Med 1997, 186, 1373–1381. [Google Scholar]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med 1997, 185, 1681–1691. [Google Scholar]
- Pantaleo, G.; Butini, L.; Graziosi, C.; Poli, G.; Schnittman, S.M.; Greenhouse, J.J.; Gallin, J.I.; Fauci, A.S. Human immunodeficiency virus (HIV) infection in CD4+ t lymphocytes genetically deficient in LFA-1: LFA-1 is required for HIV-mediated cell fusion but not for viral transmission. J. Exp. Med 1991, 173, 511–514. [Google Scholar]
- Barbas, C.F., 3rd; Collet, T.A.; Amberg, W.; Roben, P.; Binley, J.M.; Hoekstra, D.; Cababa, D.; Jones, T.M.; Williamson, R.A.; Pilkington, G.R.; et al. Molecular profile of an antibody response to HIV-1 as probed by combinatorial libraries. J. Mol. Biol 1993, 230, 812–823. [Google Scholar]
- Bera, T.K.; Kennedy, P.E.; Berger, E.A.; Barbas, C.F., 3rd; Pastan, I. Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol. Med 1998, 4, 384–391. [Google Scholar]
- Yang, W.P.; Green, K.; Pinz-Sweeney, S.; Briones, A.T.; Burton, D.R.; Barbas, C.F., 3rd. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol 1995, 254, 392–403. [Google Scholar]
- Barbas, C.F., 3rd; Hu, D.; Dunlop, N.; Sawyer, L.; Cababa, D.; Hendry, R.M.; Nara, P.L.; Burton, D.R. In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc. Natl. Acad. Sci. USA 1994, 91, 3809–3813. [Google Scholar]
- Ditzel, H.J.; Binley, J.M.; Moore, J.P.; Sodroski, J.; Sullivan, N.; Sawyer, L.S.; Hendry, R.M.; Yang, W.P.; Barbas, C.F., 3rd; Burton, D.R. Neutralizing recombinant human antibodies to a conformational V2- and CD4-binding site-sensitive epitope of HIV-1 gp120 isolated by using an epitope-masking procedure. J. Immunol 1995, 154, 893–906. [Google Scholar]
- Nishiyama, Y.; Karle, S.; Planque, S.; Taguchi, H.; Paul, S. Antibodies to the superantigenic site of HIV-1 gp120: Hydrolytic and binding activities of the light chain subunit. Mol. Immunol 2007, 44, 2707–2718. [Google Scholar]
- Karle, S.; Planque, S.; Nishiyama, Y.; Taguchi, H.; Zhou, Y.X.; Salas, M.; Lake, D.; Thiagarajan, P.; Arnett, F.; Hanson, C.V.; et al. Cross-clade HIV-1 neutralization by an antibody fragment from a lupus phage display library. AIDS 2004, 18, 329–331. [Google Scholar]
- Koh, W.W.; Steffensen, S.; Gonzalez-Pajuelo, M.; Hoorelbeke, B.; Gorlani, A.; Szynol, A.; Forsman, A.; Aasa-Chapman, M.M.; de Haard, H.; Verrips, T.; et al. Generation of a family-specific phage library of llama single chain antibody fragments that neutralize HIV-1. J. Biol. Chem 2010, 285, 19116–19124. [Google Scholar]
- Forsman, A.; Beirnaert, E.; Aasa-Chapman, M.M.; Hoorelbeke, B.; Hijazi, K.; Koh, W.; Tack, V.; Szynol, A.; Kelly, C.; McKnight, A.; et al. Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J. Virol 2008, 82, 12069–12081. [Google Scholar]
- Krykbaev, R.; McKeating, J.; Jones, I. Mutant CD4 molecules with improved binding to HIV envelope protein gp120 selected by phage display. Virology 1997, 234, 196–202. [Google Scholar]
- Thompson, J.; Pope, T.; Tung, J.S.; Chan, C.; Hollis, G.; Mark, G.; Johnson, K.S. Affinity maturation of a high-affinity human monoclonal antibody against the third hypervariable loop of human immunodeficiency virus: Use of phage display to improve affinity and broaden strain reactivity. J. Mol. Biol 1996, 256, 77–88. [Google Scholar]
- Ditzel, H.J.; Parren, P.W.; Binley, J.M.; Sodroski, J.; Moore, J.P.; Barbas, C.F., 3rd; Burton, D.R. Mapping the protein surface of human immunodeficiency virus type 1 gp120 using human monoclonal antibodies from phage display libraries. J. Mol. Biol 1997, 267, 684–695. [Google Scholar]
- Ferrer, M.; Harrison, S.C. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J. Virol 1999, 73, 5795–5802. [Google Scholar]
- Moulard, M.; Phogat, S.K.; Shu, Y.; Labrijn, A.F.; Xiao, X.; Binley, J.M.; Zhang, M.Y.; Sidorov, I.A.; Broder, C.C.; Robinson, J.; et al. Broadly cross-reactive HIV-1-neutralizing human monoclonal fab selected for binding to gp120-CD4-CCR5 complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 6913–6918. [Google Scholar]
- Dervillez, X.; Klaukien, V.; Durr, R.; Koch, J.; Kreutz, A.; Haarmann, T.; Stoll, M.; Lee, D.; Carlomagno, T.; Schnierle, B.; et al. Peptide ligands selected with cd4-induced epitopes on native dualtropic HIV-1 envelope proteins mimic extracellular coreceptor domains and bind to HIV-1 gp120 independently of coreceptor usage. J. Virol 2010, 84, 10131–10138. [Google Scholar]
- Zwick, M.B.; Labrijn, A.F.; Wang, M.; Spenlehauer, C.; Saphire, E.O.; Binley, J.M.; Moore, J.P.; Stiegler, G.; Katinger, H.; Burton, D.R.; et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol 2001, 75, 10892–10905. [Google Scholar]
- Nelson, J.D.; Brunel, F.M.; Jensen, R.; Crooks, E.T.; Cardoso, R.M.; Wang, M.; Hessell, A.; Wilson, I.A.; Binley, J.M.; Dawson, P.E.; et al. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol 2007, 81, 4033–4043. [Google Scholar]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent d-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA 2007, 104, 16828–16833. [Google Scholar]
- Welch, B.D.; Francis, J.N.; Redman, J.S.; Paul, S.; Weinstock, M.T.; Reeves, J.D.; Lie, Y.S.; Whitby, F.G.; Eckert, D.M.; Hill, C.P.; et al. Design of a potent d-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol 2010, 84, 11235–11244. [Google Scholar]
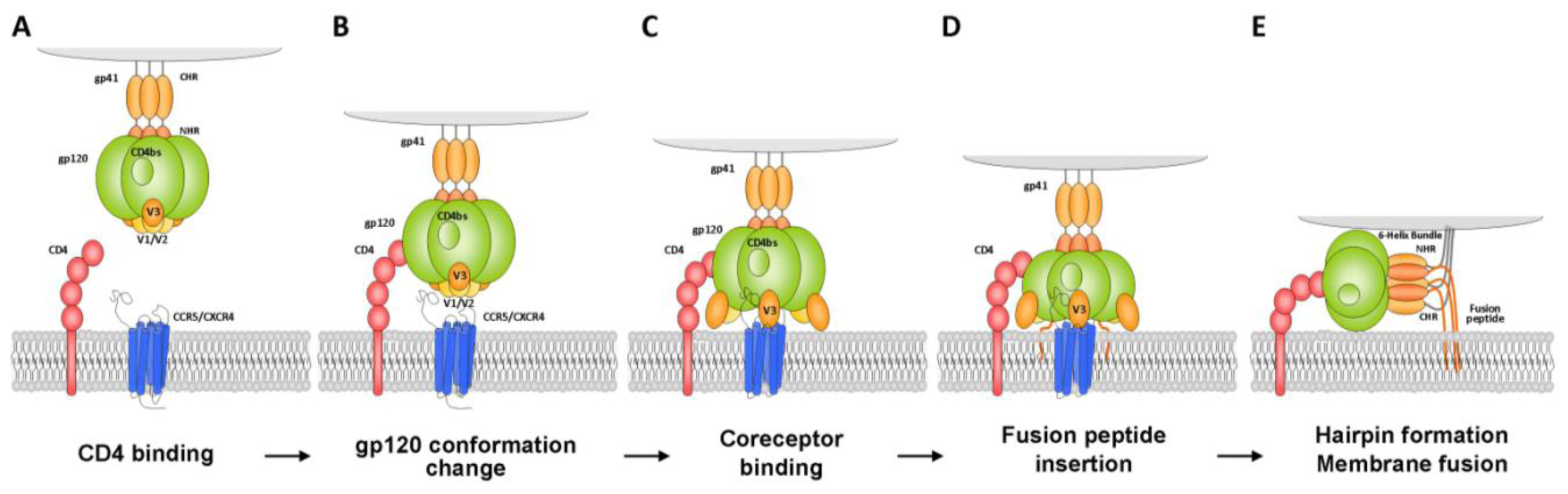
- Eckert, D.M.; Malashkevich, V.N.; Hong, L.H.; Carr, P.A.; Kim, P.S. Inhibiting HIV-1 entry: Discovery of d-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 1999, 99, 103–115. [Google Scholar]
- Miller, M.D.; Geleziunas, R.; Bianchi, E.; Lennard, S.; Hrin, R.; Zhang, H.; Lu, M.; An, Z.; Ingallinella, P.; Finotto, M.; et al. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc. Natl. Acad. Sci. USA 2005, 102, 14759–14764. [Google Scholar]
- Eckert, D.M.; Kim, P.S. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc. Natl. Acad. Sci. USA 2001, 98, 11187–11192. [Google Scholar]
- Liu, Y.; Regula, L.K.; Stewart, A.; Lai, J.R. Synthetic fab fragments that bind the HIV-1 gp41 heptad repeat regions. Biochem. Biophys. Res. Commun 2011, 413, 611–615. [Google Scholar]
- Huang, J.H.; Liu, Z.Q.; Liu, S.; Jiang, S.; Chen, Y.H. Identification of the HIV-1 gp41 core-binding motif—HXXNPF. FEBS Lett 2006, 580, 4807–4814. [Google Scholar]
- Huang, J.H.; Yang, H.W.; Liu, S.; Li, J.; Jiang, S.; Chen, Y.H. The mechanism by which molecules containing the HIV gp41 core-binding motif hxxnpf inhibit HIV-1 envelope glycoprotein-mediated syncytium formation. Biochem. J 2007, 403, 565–571. [Google Scholar]
- Louis, J.M.; Bewley, C.A.; Gustchina, E.; Aniana, A.; Clore, G.M. Characterization and HIV-1 fusion inhibitory properties of monoclonal fabs obtained from a human non-immune phage library selected against diverse epitopes of the ectodomain of HIV-1 gp41. J. Mol. Biol 2005, 353, 945–951. [Google Scholar]
- Gustchina, E.; Louis, J.M.; Lam, S.N.; Bewley, C.A.; Clore, G.M. A monoclonal fab derived from a human nonimmune phage library reveals a new epitope on gp41 and neutralizes diverse human immunodeficiency virus type 1 strains. J. Virol 2007, 81, 12946–12953. [Google Scholar]
- Gustchina, E.; Louis, J.M.; Frisch, C.; Ylera, F.; Lechner, A.; Bewley, C.A.; Clore, G.M. Affinity maturation by targeted diversification of the CDR-H2 loop of a monoclonal fab derived from a synthetic naive human antibody library and directed against the internal trimeric coiled-coil of gp41 yields a set of fabs with improved HIV-1 neutralization potency and breadth. Virology 2009, 393, 112–119. [Google Scholar]
- Gustchina, E.; Bewley, C.A.; Clore, G.M. Sequestering of the prehairpin intermediate of gp41 by peptide N36Mut(e,g) potentiates the human immunodeficiency virus type 1 neutralizing activity of monoclonal antibodies directed against the N-terminal helical repeat of gp41. J. Virol 2008, 82, 10032–10041. [Google Scholar]
- Nelson, J.D.; Kinkead, H.; Brunel, F.M.; Leaman, D.; Jensen, R.; Louis, J.M.; Maruyama, T.; Bewley, C.A.; Bowdish, K.; Clore, G.M.; et al. Antibody elicited against the gp41 N-heptad repeat (NHR) coiled-coil can neutralize HIV-1 with modest potency but non-neutralizing antibodies also bind to NHR mimetics. Virology 2008, 377, 170–183. [Google Scholar]
- Zhang, M.Y.; Vu, B.K.; Choudhary, A.; Lu, H.; Humbert, M.; Ong, H.; Alam, M.; Ruprecht, R.M.; Quinnan, G.; Jiang, S.; et al. Cross-reactive human immunodeficiency virus type 1-neutralizing human monoclonal antibody that recognizes a novel conformational epitope on gp41 and lacks reactivity against self-antigens. J. Virol 2008, 82, 6869–6879. [Google Scholar]
- Choudhry, V.; Zhang, M.Y.; Sidorov, I.A.; Louis, J.M.; Harris, I.; Dimitrov, A.S.; Bouma, P.; Cham, F.; Choudhary, A.; Rybak, S.M.; et al. Cross-reactive HIV-1 neutralizing monoclonal antibodies selected by screening of an immune human phage library against an envelope glycoprotein (gp140) isolated from a patient (R2) with broadly HIV-1 neutralizing antibodies. Virology 2007, 363, 79–90. [Google Scholar]
- Zhang, M.Y.; Choudhry, V.; Sidorov, I.A.; Tenev, V.; Vu, B.K.; Choudhary, A.; Lu, H.; Stiegler, G.M.; Katinger, H.W.; Jiang, S.; et al. Selection of a novel gp41-specific HIV-1 neutralizing human antibody by competitive antigen panning. J. Immunol. Methods 2006, 317, 21–30. [Google Scholar]
- BouHamdan, M.; Xue, Y.; Baudat, Y.; Hu, B.; Sire, J.; Pomerantz, R.J.; Duan, L.X. Diversity of HIV-1 Vpr interactions involves usage of the WXXF motif of host cell proteins. J. Biol. Chem 1998, 273, 8009–8016. [Google Scholar]
- Krichevsky, A.; Graessmann, A.; Nissim, A.; Piller, S.C.; Zakai, N.; Loyter, A. Antibody fragments selected by phage display against the nuclear localization signal of the HIV-1 Vpr protein inhibit nuclear import in permeabilized and intact cultured cells. Virology 2003, 305, 77–92. [Google Scholar]
- Desjobert, C.; de Soultrait, V.R.; Faure, A.; Parissi, V.; Litvak, S.; Tarrago-Litvak, L.; Fournier, M. Identification by phage display selection of a short peptide able to inhibit only the strand transfer reaction catalyzed by human immunodeficiency virus type 1 integrase. Biochemistry 2004, 43, 13097–13105. [Google Scholar]
- Pilkington, G.R.; Duan, L.; Zhu, M.; Keil, W.; Pomerantz, R.J. Recombinant human Fab antibody fragments to HIV-1 Rev and Tat regulatory proteins: Direct selection from a combinatorial phage display library. Mol. Immunol 1996, 33, 439–450. [Google Scholar]
- Bai, J.; Sui, J.; Zhu, R.Y.; Tallarico, A.S.; Gennari, F.; Zhang, D.; Marasco, W.A. Inhibition of Tat-mediated transactivation and HIV-1 replication by human anti-hCyclinT1 intrabodies. J. Biol. Chem 2003, 278, 1433–1442. [Google Scholar]
- Kolb, G.; Boiziau, C. Selection by phage display of peptides targeting the HIV-1 Tar element. RNA Biol 2005, 2, 28–33. [Google Scholar]
- Lener, D.; Benarous, R.; Calogero, R.A. Use of a constrain phage displayed-peptide library for the isolation of peptides binding to HIV-1 nucleocapsid protein (NCp7). FEBS Lett 1995, 361, 85–88. [Google Scholar]
- Pustowka, A.; Dietz, J.; Ferner, J.; Baumann, M.; Landersz, M.; Konigs, C.; Schwalbe, H.; Dietrich, U. Identification of peptide ligands for target RNA structures derived from the HIV-1 packaging signal ψ by screening phage-displayed peptide libraries. ChemBioChem 2003, 4, 1093–1097. [Google Scholar]
- Dietz, J.; Koch, J.; Kaur, A.; Raja, C.; Stein, S.; Grez, M.; Pustowka, A.; Mensch, S.; Ferner, J.; Moller, L.; et al. Inhibition of HIV-1 by a peptide ligand of the genomic RNA packaging signal ψ. ChemMedChem 2008, 3, 749–755. [Google Scholar]
- Park, M.Y.; Kwon, J.; Lee, S.; You, J.; Myung, H. Selection and characterization of peptides specifically binding to HIV-1 Ψ (Ψ) RNA. Virus Res 2004, 106, 77–81. [Google Scholar]
- Yoshikawa, M.; Mukai, Y.; Tsunoda, S.; Tsutsumi, Y.; Yoshioka, Y.; Okada, N.; Nakagawa, S. Modifying the antigen-immunization schedule improves the variety of monoclonal antibodies obtained from immune-phage antibody libraries against HIV-1 Nef and Vif. J. Biosci. Bioeng 2011, 111, 597–599. [Google Scholar]
- Bouchet, J.; Basmaciogullari, S.E.; Chrobak, P.; Stolp, B.; Bouchard, N.; Fackler, O.T.; Chames, P.; Jolicoeur, P.; Benichou, S.; Baty, D. Inhibition of the nef regulatory protein of HIV-1 by a single-domain antibody. Blood 2011, 117, 3559–3568. [Google Scholar]
- Nunoya, J.; Nakashima, T.; Kawana-Tachikawa, A.; Kiyotani, K.; Ito, Y.; Sugimura, K.; Iwamoto, A. Short communication: Generation of recombinant monoclonal antibodies against an immunodominant HLA-A*2402-restricted HIV type 1 CTL epitope. AIDS Res. Hum. Retrovir 2009, 25, 897–904. [Google Scholar]
- Yang, S.; Sun, Y.; Zhang, H. The multimerization of human immunodeficiency virus type I Vif protein: A requirement for Vif function in the viral life cycle. J. Biol. Chem 2001, 276, 4889–4893. [Google Scholar]
- Yang, B.; Gao, L.; Li, L.; Lu, Z.; Fan, X.; Patel, C.A.; Pomerantz, R.J.; DuBois, G.C.; Zhang, H. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J. Biol. Chem 2003, 278, 6596–6602. [Google Scholar]
- Ohba, H.; Soga, T.; Tomozawa, T.; Nishikawa, Y.; Yasuda, A.; Kojima, A.; Kurata, T.; Chiba, J. An immunodominant neutralization epitope on the “thumb” subdomain of human immunodeficiency virus type 1 reverse transcriptase revealed by phage display antibodies. J. Gen. Virol 2001, 82, 813–820. [Google Scholar]
- Chiba, J.; Yamaguchi, A.; Suzuki, Y.; Nakano, M.; Zhu, W.; Ohba, H.; Saito, A.; Shinagawa, H.; Yamakawa, Y.; Kobayashi, T.; et al. A novel neutralization epitope on the “thumb” subdomain of human immunodeficiency virus type 1 reverse transcriptase revealed by a monoclonal antibody. J. Gen. Virol 1996, 77 Pt 12, 2921–2929. [Google Scholar]
- Herschhorn, A.; Admon, A.; Hizi, A. Recombinant human antibodies against the reverse transcriptase of human immunodeficiency virus type-1. Biochim. Biophys. Acta 2003, 1648, 154–163. [Google Scholar]
- Jensen, A.; Jensen, T.H.; Kjems, J. HIV-1 Rev nuclear export signal binding peptides isolated by phage display. J. Mol. Biol 1998, 283, 245–254. [Google Scholar]
- Vercruysse, T.; Pawar, S.; de Borggraeve, W.; Pardon, E.; Pavlakis, G.N.; Pannecouque, C.; Steyaert, J.; Balzarini, J.; Daelemans, D. Measuring cooperative Rev protein-protein interactions on rev responsive RNA by fluorescence resonance energy transfer. RNA Biol 2011, 8, 316–324. [Google Scholar]
- Vercruysse, T.; Pardon, E.; Vanstreels, E.; Steyaert, J.; Daelemans, D. An intrabody based on a llama single-domain antibody targeting the N-terminal α-helical multimerization domain of HIV-1 Rev prevents viral production. J. Biol. Chem 2010, 285, 21768–21780. [Google Scholar]
- Stahl, S.J.; Watts, N.R.; Rader, C.; DiMattia, M.A.; Mage, R.G.; Palmer, I.; Kaufman, J.D.; Grimes, J.M.; Stuart, D.I.; Steven, A.C.; et al. Generation and characterization of a chimeric rabbit/human fab for co-crystallization of HIV-1 Rev. J. Mol. Biol 2010, 397, 697–708. [Google Scholar]
- Sticht, J.; Humbert, M.; Findlow, S.; Bodem, J.; Muller, B.; Dietrich, U.; Werner, J.; Krausslich, H.G. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol 2005, 12, 671–677. [Google Scholar]
- Huang, L.; Zhang, L.; Chen, C.H. Potential drug targets on the HIV-1 envelope glycoproteins, gp120 and gp41. Curr. Pharm. Des 2003, 9, 1453–1462. [Google Scholar]
- Kadow, J.; Wang, H.G.; Lin, P.F. Small-molecule HIV-1 gp120 inhibitors to prevent HIV-1 entry: An emerging opportunity for drug development. Curr. Opin. Investig. Drugs 2006, 7, 721–726. [Google Scholar]
- Koefoed, K.; Farnaes, L.; Wang, M.; Svejgaard, A.; Burton, D.R.; Ditzel, H.J. Molecular characterization of the circulating anti-HIV-1 gp120-specific b cell repertoire using antibody phage display libraries generated from pre-selected HIV-1 gp120 binding PBLs. J. Immunol. Methods 2005, 297, 187–201. [Google Scholar]
- Parren, P.W.; Gauduin, M.C.; Koup, R.A.; Poignard, P.; Fisicaro, P.; Burton, D.R.; Sattentau, Q.J. Relevance of the antibody response against human immunodeficiency virus type 1 envelope to vaccine design. Immunol. Lett 1997, 57, 105–112. [Google Scholar]
- Bermas, B.L.; Petri, M.; Berzofsky, J.A.; Waisman, A.; Shearer, G.M.; Mozes, E. Binding of glycoprotein 120 and peptides from the HIV-1 envelope by autoantibodies in mice with experimentally induced systemic lupus erythematosus and in patients with the disease. AIDS Res. Hum. Retrovir 1994, 10, 1071–1077. [Google Scholar]
- Zhou, Y.X.; Karle, S.; Taguchi, H.; Planque, S.; Nishiyama, Y.; Paul, S. Prospects for immunotherapeutic proteolytic antibodies. J. Immunol. Methods 2002, 269, 257–268. [Google Scholar]
- Paul, S.; Tramontano, A.; Gololobov, G.; Zhou, Y.X.; Taguchi, H.; Karle, S.; Nishiyama, Y.; Planque, S.; George, S. Phosphonate ester probes for proteolytic antibodies. J. Biol. Chem 2001, 276, 28314–28320. [Google Scholar]
- Paul, S.; Karle, S.; Planque, S.; Taguchi, H.; Salas, M.; Nishiyama, Y.; Handy, B.; Hunter, R.; Edmundson, A.; Hanson, C. Naturally occurring proteolytic antibodies: Selective immunoglobulin m-catalyzed hydrolysis of HIV gp120. J. Biol. Chem 2004, 279, 39611–39619. [Google Scholar]
- Paul, S.; Nishiyama, Y.; Planque, S.; Taguchi, H. Theory of proteolytic antibody occurrence. Immunol. Lett 2006, 103, 8–16. [Google Scholar]
- Gao, Q.S.; Sun, M.; Rees, A.R.; Paul, S. Site-directed mutagenesis of proteolytic antibody light chain. J. Mol. Biol 1995, 253, 658–664. [Google Scholar]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar]
- Vanlandschoot, P.; Stortelers, C.; Beirnaert, E.; Ibanez, L.I.; Schepens, B.; Depla, E.; Saelens, X. Nanobodies(r): New ammunition to battle viruses. Antiviral Res 2011, 92, 389–407. [Google Scholar]
- Biorn, A.C.; Cocklin, S.; Madani, N.; Si, Z.; Ivanovic, T.; Samanen, J.; van Ryk, D.I.; Pantophlet, R.; Burton, D.R.; Freire, E.; et al. Mode of action for linear peptide inhibitors of HIV-1 gp120 interactions. Biochemistry 2004, 43, 1928–1938. [Google Scholar]
- Garg, R.; Juncadella, I.J.; Ramamoorthi, N.; Ashish; Ananthanarayanan, S.K.; Thomas, V.; Rincon, M.; Krueger, J.K.; Fikrig, E.; Yengo, C.M.; et al. Cutting edge: CD4 is the receptor for the tick saliva immunosuppressor, Salp15. J. Immunol 2006, 177, 6579–6583. [Google Scholar]
- Juncadella, I.J.; Garg, R.; Bates, T.C.; Olivera, E.R.; Anguita, J. The ixodes scapularis salivary protein, Salp15, prevents the association of HIV-1 gp120 and CD4. Biochem. Biophys. Res. Commun 2008, 367, 41–46. [Google Scholar]
- Llorente, M.; Sanchez-Palomino, S.; Manes, S.; Lucas, P.; Kremer, L.; de Alboran, I.M.; Toran, J.L.; Alcami, J.; Del Real, G.; Martinez, A.C. Natural human antibodies retrieved by phage display libraries from healthy donors: Polyreactivity and recognition of human immunodeficiency virus type 1gp120 epitopes. Scand. J. Immunol 1999, 50, 270–279. [Google Scholar]
- Berberian, L.; Valles-Ayoub, Y.; Sun, N.; Martinez-Maza, O.; Braun, J. A VH clonal deficit in human immunodeficiency virus-positive individuals reflects a B-cell maturational arrest. Blood 1991, 78, 175–179. [Google Scholar]
- David, D.; Demaison, C.; Bani, L.; Zouali, M.; Theze, J. Selective variations in vivo of VH3 and VH1 gene family expression in peripheral B cell igM, igD and igG during HIV infection. Eur. J. Immunol 1995, 25, 1524–1528. [Google Scholar]
- Toran, J.L.; Kremer, L.; Sanchez-Pulido, L.; de Alboran, I.M.; del Real, G.; Llorente, M.; Valencia, A.; de Mon, M.A.; Martinez, A.C. Molecular analysis of HIV-1 gp120 antibody response using isotype igM and igG phage display libraries from a long-term non-progressor HIV-1-infected individual. Eur. J. Immunol 1999, 29, 2666–2675. [Google Scholar]
- Toran, J.L.; Sanchez-Pulido, L.; Kremer, L.; del Real, G.; Valencia, A.; Martinez, A.C. Improvement in affinity and HIV-1 neutralization by somatic mutation in the heavy chain first complementarity-determining region of antibodies triggered by HIV-1 infection. Eur. J. Immunol 2001, 31, 128–137. [Google Scholar]
- Chen, W.; Zhu, Z.; Liao, H.; Quinnan, G.V., Jr; Broder, C.C.; Haynes, B.F.; Dimitrov, D.S. Cross-reactive human igm-derived monoclonal antibodies that bind to HIV-1 envelope glycoproteins. Viruses 2010, 2, 547–565. [Google Scholar]
- Chen, W.; Streaker, E.D.; Russ, D.E.; Feng, Y.; Prabakaran, P.; Dimitrov, D.S. Characterization of germline antibody libraries from human umbilical cord blood and selection of monoclonal antibodies to viral envelope glycoproteins: Implications for mechanisms of immune evasion and design of vaccine immunogens. Biochem. Biophys. Res. Commun 2012, 417, 1164–1169. [Google Scholar]
- Zhu, Z.; Qin, H.R.; Chen, W.; Zhao, Q.; Shen, X.; Schutte, R.; Wang, Y.; Ofek, G.; Streaker, E.; Prabakaran, P.; et al. Cross-reactive HIV-1-neutralizing human monoclonal antibodies identified from a patient with 2F5-like antibodies. J. Virol 2011, 85, 11401–11408. [Google Scholar]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov 2004, 3, 215–225. [Google Scholar]
- Schumacher, T.N.; Mayr, L.M.; Minor, D.L., Jr; Milhollen, M.A.; Burgess, M.W.; Kim, P.S. Identification of D-peptide ligands through mirror-image phage display. Science 1996, 271, 1854–1857. [Google Scholar]
- Root, M.J.; Kay, M.S.; Kim, P.S. Protein design of an HIV-1 entry inhibitor. Science 2001, 291, 884–888. [Google Scholar]
- Fellouse, F.A.; Li, B.; Compaan, D.M.; Peden, A.A.; Hymowitz, S.G.; Sidhu, S.S. Molecular recognition by a binary code. J. Mol. Biol 2005, 348, 1153–1162. [Google Scholar]
- Luftig, M.A.; Mattu, M.; di Giovine, P.; Geleziunas, R.; Hrin, R.; Barbato, G.; Bianchi, E.; Miller, M.D.; Pessi, A.; Carfi, A. Structural basis for HIV-1 neutralization by a gp41 fusion intermediate-directed antibody. Nat. Struct. Mol. Biol 2006, 13, 740–747. [Google Scholar]
- Lu, M.; Ji, H.; Shen, S. Subdomain folding and biological activity of the core structure from human immunodeficiency virus type 1 gp41: Implications for viral membrane fusion. J. Virol 1999, 73, 4433–4438. [Google Scholar]
- Knappik, A.; Ge, L.; Honegger, A.; Pack, P.; Fischer, M.; Wellnhofer, G.; Hoess, A.; Wolle, J.; Pluckthun, A.; Virnekas, B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and cdrs randomized with trinucleotides. J. Mol. Biol 2000, 296, 57–86. [Google Scholar]
- Louis, J.M.; Nesheiwat, I.; Chang, L.; Clore, G.M.; Bewley, C.A. Covalent trimers of the internal N-terminal trimeric coiled-coil of gp41 and antibodies directed against them are potent inhibitors of HIV envelope-mediated cell fusion. J. Biol. Chem 2003, 278, 20278–20285. [Google Scholar]
- Louis, J.M.; Bewley, C.A.; Clore, G.M. Design and properties of N(CCG)-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J. Biol. Chem 2001, 276, 29485–29489. [Google Scholar]
- Bewley, C.A.; Louis, J.M.; Ghirlando, R.; Clore, G.M. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J. Biol. Chem 2002, 277, 14238–14245. [Google Scholar]
- Montefiori, D.C.; Pantaleo, G.; Fink, L.M.; Zhou, J.T.; Zhou, J.Y.; Bilska, M.; Miralles, G.D.; Fauci, A.S. Neutralizing and infection-enhancing antibody responses to human immunodeficiency virus type 1 in long-term nonprogressors. J. Infect. Dis 1996, 173, 60–67. [Google Scholar]
- Agostini, I.; Navarro, J.M.; Rey, F.; Bouhamdan, M.; Spire, B.; Vigne, R.; Sire, J. The human immunodeficiency virus type 1 Vpr transactivator: Cooperation with promoter-bound activator domains and binding to TFIIB. J. Mol. Biol 1996, 261, 599–606. [Google Scholar]
- Stewart, S.A.; Poon, B.; Jowett, J.B.; Chen, I.S. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol 1997, 71, 5579–5592. [Google Scholar]
- Cohen, E.A.; Dehni, G.; Sodroski, J.G.; Haseltine, W.A. Human immunodeficiency virus Vpr product is a virion-associated regulatory protein. J. Virol 1990, 64, 3097–3099. [Google Scholar]
- Cohen, E.A.; Terwilliger, E.F.; Jalinoos, Y.; Proulx, J.; Sodroski, J.G.; Haseltine, W.A. Identification of HIV-1 Vpr product and function. J. Acquir. Immune Defic. Syndr 1990, 3, 11–18. [Google Scholar]
- Bouhamdan, M.; Benichou, S.; Rey, F.; Navarro, J.M.; Agostini, I.; Spire, B.; Camonis, J.; Slupphaug, G.; Vigne, R.; Benarous, R.; et al. Human immunodeficiency virus type 1 Vpr protein binds to the Uracil DNA glycosylase DNA repair enzyme. J. Virol 1996, 70, 697–704. [Google Scholar]
- Refaeli, Y.; Levy, D.N.; Weiner, D.B. The glucocorticoid receptor type II complex is a target of the HIV-1 Vpr gene product. Proc. Natl. Acad. Sci. USA 1995, 92, 3621–3625. [Google Scholar]
- Wang, L.; Mukherjee, S.; Jia, F.; Narayan, O.; Zhao, L.J. Interaction of virion protein Vpr of human immunodeficiency virus type 1 with cellular transcription factor sp1 and trans-activation of viral long terminal repeat. J. Biol. Chem 1995, 270, 25564–25569. [Google Scholar]
- Nissim, A.; Hoogenboom, H.R.; Tomlinson, I.M.; Flynn, G.; Midgley, C.; Lane, D.; Winter, G. Antibody fragments from a “single pot” phage display library as immunochemical reagents. EMBO J 1994, 13, 692–698. [Google Scholar]
- Garber, M.E.; Wei, P.; Jones, K.A. HIV-1 tat interacts with cyclin t1 to direct the p-TEFb CTD kinase complex to TAR RNA. Cold Spring Harb. Symp. Quant. Biol 1998, 63, 371–380. [Google Scholar]
- Druillennec, S.; Dong, C.Z.; Escaich, S.; Gresh, N.; Bousseau, A.; Roques, B.P.; Fournie-Zaluski, M.C. A mimic of HIV-1 nucleocapsid protein impairs reverse transcription and displays antiviral activity. Proc. Natl. Acad. Sci. USA 1999, 96, 4886–4891. [Google Scholar]
- Druillennec, S.; Caneparo, A.; de Rocquigny, H.; Roques, B.P. Evidence of interactions between the nucleocapsid protein ncp7 and the reverse transcriptase of HIV-1. J. Biol. Chem 1999, 274, 11283–11288. [Google Scholar]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem 1998, 67, 1–25. [Google Scholar]
- Cimarelli, A.; Sandin, S.; Hoglund, S.; Luban, J. Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol 2000, 74, 3046–3057. [Google Scholar]
- Hashiguchi, S.; Nakashima, T.; Nitani, A.; Yoshihara, T.; Yoshinaga, K.; Ito, Y.; Maeda, Y.; Sugimura, K. Human Fc epsilon rialpha-specific human single-chain Fv (ScFv) antibody with antagonistic activity toward igE/Fc epsilon Rialpha-binding. J. Biochem 2003, 133, 43–49. [Google Scholar]
- Gargano, N.; Biocca, S.; Bradbury, A.; Cattaneo, A. Human recombinant antibody fragments neutralizing human immunodeficiency virus type 1 reverse transcriptase provide an experimental basis for the structural classification of the DNA polymerase family. J. Virol 1996, 70, 7706–7712. [Google Scholar]
- Heaphy, S.; Finch, J.T.; Gait, M.J.; Karn, J.; Singh, M. Human immunodeficiency virus type 1 regulator of virion expression, Rev, forms nucleoprotein filaments after binding to a purine-rich “Bubble” Located within the Rev-responsive region of viral mRNAs. Proc. Natl. Acad. Sci. USA 1991, 88, 7366–7370. [Google Scholar]
- De Haard, J.J.; Kazemier, B.; Oudshoorn, P.; Boender, P.; van Gemen, B.; Koolen, M.J.; van der Groen, G.; Hoogenboom, H.R.; Arends, J.W. Selection of human anti-human immunodeficiency virus type 1 envelope single-chain antibodies from a peripheral blood cell-based phage repertoire. J. Gen. Virol 1998, 79 Pt 12, 2883–2894. [Google Scholar]
- Khurana, S.; Needham, J.; Mathieson, B.; Rodriguez-Chavez, I.R.; Catanzaro, A.T.; Bailer, R.T.; Kim, J.; Polonis, V.; Cooper, D.A.; Guerin, J.; et al. Human immunodeficiency virus (HIV) vaccine trials: A novel assay for differential diagnosis of HIV infections in the face of vaccine-generated antibodies. J. Virol 2006, 80, 2092–2099. [Google Scholar]
- Vyroubalova, E.C.; Hartley, O.; Mermod, N.; Fisch, I. Identification of peptide ligands to the chemokine receptor CCR5 and their maturation by gene shuffling. Mol. Immunol 2006, 43, 1573–1578. [Google Scholar]
- Wang, F.Y.; Zhang, T.Y.; Luo, J.X.; He, G.A.; Gu, Q.L.; Xiao, F. Selection of CC chemokine receptor 5-binding peptide from a phage display peptide library. Biosci. Biotechnol. Biochem 2006, 70, 2035–2041. [Google Scholar]
- Mirzabekov, T.; Kontos, H.; Farzan, M.; Marasco, W.; Sodroski, J. Paramagnetic proteoliposomes containing a pure, native, and oriented seven-transmembrane segment protein, CCR5. Nat. Biotechnol 2000, 18, 649–654. [Google Scholar]
- Zhang, Y.; Pool, C.; Sadler, K.; Yan, H.P.; Edl, J.; Wang, X.; Boyd, J.G.; Tam, J.P. Selection of active ScFv to g-protein-coupled receptor CCR5 using surface antigen-mimicking peptides. Biochemistry 2004, 43, 12575–12584. [Google Scholar]
- Chevigne, A.; Fischer, A.; Mathu, J.; Counson, M.; Beaupain, N.; Plesseria, J.M.; Schmit, J.C.; Deroo, S. Selection of a CXCR4 antagonist from a human heavy chain CDR3-derived phage library. FEBS J 2011, 278, 2867–2878. [Google Scholar]
- Huang, Y.; Paxton, W.A.; Wolinsky, S.M.; Neumann, A.U.; Zhang, L.; He, T.; Kang, S.; Ceradini, D.; Jin, Z.; Yazdanbakhsh, K.; et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med 1996, 2, 1240–1243. [Google Scholar]
- Steinberger, P.; Sutton, J.K.; Rader, C.; Elia, M.; Barbas, C.F., 3rd. Generation and characterization of a recombinant human CCR5-specific antibody. A phage display approach for rabbit antibody humanization. J. Biol. Chem 2000, 275, 36073–36078. [Google Scholar]
- Hartley, O.; Dorgham, K.; Perez-Bercoff, D.; Cerini, F.; Heimann, A.; Gaertner, H.; Offord, R.E.; Pancino, G.; Debre, P.; Gorochov, G. Human immunodeficiency virus type 1 entry inhibitors selected on living cells from a library of phage chemokines. J. Virol 2003, 77, 6637–6644. [Google Scholar]
- Gaertner, H.; Cerini, F.; Escola, J.M.; Kuenzi, G.; Melotti, A.; Offord, R.; Rossitto-Borlat, I.; Nedellec, R.; Salkowitz, J.; Gorochov, G.; et al. Highly potent, fully recombinant anti-HIV chemokines: Reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. USA 2008, 105, 17706–17711. [Google Scholar]
- Jahnichen, S.; Blanchetot, C.; Maussang, D.; Gonzalez-Pajuelo, M.; Chow, K.Y.; Bosch, L.; de Vrieze, S.; Serruys, B.; Ulrichts, H.; Vandevelde, W.; et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 20565–20570. [Google Scholar]
- Ellmark, P.; Andersson, H.; Abayneh, S.; Fenyo, E.M.; Borrebaeck, C.A. Identification of a strongly activating human anti-CD40 antibody that suppresses HIV type 1 infection. AIDS Res. Hum. Retrovir 2008, 24, 367–373. [Google Scholar]
- Ellmark, P.; Ottosson, C.; Borrebaeck, C.A.; Malmborg Hager, A.C.; Furebring, C. Modulation of the CD40-CD40 ligand interaction using human anti-CD40 single-chain antibody fragments obtained from the n-coder phage display library. Immunology 2002, 106, 456–463. [Google Scholar]
- Garbelli, A.; Beermann, S.; di Cicco, G.; Dietrich, U.; Maga, G. A motif unique to the human dead-box protein DDX3 is important for nucleic acid binding, ATP hydrolysis, RNA/DNA unwinding and HIV-1 replication. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Chevigne, A.; Fievez, V.; Schmit, J.C.; Deroo, S. Engineering and screening the N-terminus of chemokines for drug discovery. Biochem. Pharmacol 2011, 82, 1438–1456. [Google Scholar]
- Tamamura, H.; Fujii, N. The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Expert Opin. Ther. Targets 2005, 9, 1267–1282. [Google Scholar]
- Steen, A.; Schwartz, T.W.; Rosenkilde, M.M. Targeting CXCR4 in HIV cell-entry inhibition. Mini Rev. Med. Chem 2009, 9, 1605–1621. [Google Scholar]
- Deroo, S.; Fischer, A.; Beaupain, N.; Counson, M.; Boutonnet, N.; Pletinckx, J.; Loverix, S.; Beirnaert, E.; de Haard, H.; Schmit, J.C.; et al. Non-immunized natural human heavy chain CDR3 repertoires allow the isolation of high affinity peptides mimicking a human influenza hemagglutinin epitope. Mol. Immunol 2008, 45, 1366–1373. [Google Scholar]
- Rubinstein, D.B.; Leblanc, P.; Wright, D.G.; Guillaume, T.; Strotchevoi, A.; Boosalis, M. Anti-CD34+ fabs generated against hematopoietic stem cells in HIV-derived combinatorial immunoglobulin library suggest antigen-selected autoantibodies. Mol. Immunol 1998, 35, 955–964. [Google Scholar]
- Abayneh, S.A.; Ellmark, P.; Karlsson, U.; Andersson, H.; Borrebaeck, C.A.; Karlsson, I.; Fenyo, E.M. Sensitivity of HIV type 1 primary isolates to human anti-CD40 antibody-mediated suppression is related to coreceptor use. AIDS Res. Hum. Retrovir 2008, 24, 447–452. [Google Scholar]
- Matthews, D.J.; Wells, J.A. Substrate phage: Selection of protease substrates by monovalent phage display. Science 1993, 260, 1113–1117. [Google Scholar]
- Beck, Z.Q.; Lin, Y.C.; Elder, J.H. Molecular basis for the relative substrate specificity of human immunodeficiency virus type 1 and feline immunodeficiency virus proteases. J. Virol 2001, 75, 9458–9469. [Google Scholar]
- Beck, Z.Q.; Hervio, L.; Dawson, P.E.; Elder, J.H.; Madison, E.L. Identification of efficiently cleaved substrates for HIV-1 protease using a phage display library and use in inhibitor development. Virology 2000, 274, 391–401. [Google Scholar]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun 1967, 27, 157–162. [Google Scholar]
- Tozser, J.; Weber, I.T.; Gustchina, A.; Blaha, I.; Copeland, T.D.; Louis, J.M.; Oroszlan, S. Kinetic and modeling studies of s3-s3′ subsites of HIV proteinases. Biochemistry 1992, 31, 4793–4800. [Google Scholar]
- Kleinschmidt, W.J.; Douthart, R.J.; Murphy, E.B. Interferon production by t4 coliphage. Nature 1970, 228, 27–30. [Google Scholar]
- Clark, J.R.; March, J.B. Bacterial viruses as human vaccines? Expert Rev. Vaccines 2004, 3, 463–476. [Google Scholar]
- Jepson, C.D.; March, J.B. Bacteriophage lambda is a highly stable DNA vaccine delivery vehicle. Vaccine 2004, 22, 2413–2419. [Google Scholar]
- Minenkova, O.O.; Ilyichev, A.A.; Kishchenko, G.P.; Petrenko, V.A. Design of specific immunogens using filamentous phage as the carrier. Gene 1993, 128, 85–88. [Google Scholar]
- De Berardinis, P.; Sartorius, R.; Fanutti, C.; Perham, R.N.; Del Pozzo, G.; Guardiola, J. Phage display of peptide epitopes from HIV-1 elicits strong cytolytic responses. Nat. Biotechnol 2000, 18, 873–876. [Google Scholar]
- Di Marzo Veronese, F.; Willis, A.E.; Boyer-Thompson, C.; Appella, E.; Perham, R.N. Structural mimicry and enhanced immunogenicity of peptide epitopes displayed on filamentous bacteriophage. The V3 loop of HIV-1 gp120. J. Mol. Biol 1994, 243, 167–172. [Google Scholar]
- Cano, A.; Viveros, M.; Acero, G.; Govezensky, T.; Munguia, M.E.; Gonzalez, E.; Soto, L.; Gevorkian, G.; Manoutcharian, K. Antigenic properties of phage displayed peptides comprising disulfide-bonded loop of the immunodominant region of HIV-1 gp41. Immunol. Lett 2004, 95, 207–212. [Google Scholar]
- Majumdar, S.; Hajduczki, A.; Mendez, A.S.; Weiss, G.A. Phage display of functional, full-length human and viral membrane proteins. Bioorg. Med. Chem. Lett 2008, 18, 5937–5940. [Google Scholar]
- Perham, R.N.; Terry, T.D.; Willis, A.E.; Greenwood, J.; di Marzo Veronese, F.; Appella, E. Engineering a peptide epitope display system on filamentous bacteriophage. FEMS Microbiol. Rev 1995, 17, 25–31. [Google Scholar]
- Ren, Z.; Black, L.W. Phage T4 SOC and HOC display of biologically active, full-length proteins on the viral capsid. Gene 1998, 215, 439–444. [Google Scholar]
- Peabody, D.S.; Manifold-Wheeler, B.; Medford, A.; Jordan, S.K.; do Carmo Caldeira, J.; Chackerian, B. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J. Mol. Biol 2008, 380, 252–263. [Google Scholar]
- Mattiacio, J.; Walter, S.; Brewer, M.; Domm, W.; Friedman, A.E.; Dewhurst, S. Dense display of HIV-1 envelope spikes on the lambda phage scaffold does not result in the generation of improved antibody responses to HIV-1 Env. Vaccine 2011, 29, 2637–2647. [Google Scholar]
- Ren, Z.J.; Lewis, G.K.; Wingfield, P.T.; Locke, E.G.; Steven, A.C.; Black, L.W. Phage display of intact domains at high copy number: A system based on soc, the small outer capsid protein of bacteriophage T4. Protein Sci 1996, 5, 1833–1843. [Google Scholar]
- Pedroza-Roldan, C.; Charles-Nino, C.; Saavedra, R.; Govezensky, T.; Vaca, L.; Avaniss-Aghajani, E.; Gevorkian, G.; Manoutcharian, K. Variable epitope library-based vaccines: Shooting moving targets. Mol. Immunol 2009, 47, 270–282. [Google Scholar]
- Sathaliyawala, T.; Rao, M.; Maclean, D.M.; Birx, D.L.; Alving, C.R.; Rao, V.B. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: A novel in vitro approach to construct multicomponent HIV vaccines. J. Virol 2006, 80, 7688–7698. [Google Scholar]
- Caldeira Jdo, C.; Medford, A.; Kines, R.C.; Lino, C.A.; Schiller, J.T.; Chackerian, B.; Peabody, D.S. Immunogenic display of diverse peptides, including a broadly cross-type neutralizing human papillomavirus l2 epitope, on virus-like particles of the RNA bacteriophage PP7. Vaccine 2010, 28, 4384–4393. [Google Scholar]
- Charles-Nino, C.; Pedroza-Roldan, C.; Viveros, M.; Gevorkian, G.; Manoutcharian, K. Variable epitope libraries: New vaccine immunogens capable of inducing broad human immunodeficiency virus type 1-neutralizing antibody response. Vaccine 2011, 29, 5313–5321. [Google Scholar]
- Ishii, T.; Yanagida, M. The two dispensable structural proteins (soc and hoc) of the T4 phage capsid; their purification and properties, isolation and characterization of the defective mutants, and their binding with the defective heads in vitro. J. Mol. Biol 1977, 109, 487–514. [Google Scholar]
- Zhu, P.; Liu, J.; Bess, J., Jr; Chertova, E.; Lifson, J.D.; Grise, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar]
- Deroo, S.; Muller, C.P. Antigenic and immunogenic phage displayed mimotopes as substitute antigens: Applications and limitations. Comb. Chem. High Throughput Screen 2001, 4, 75–110. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Library | Target presentation | Mimotopes | Epitopes | Additional | Vaccination attempts | Results | Reference |
|---|---|---|---|---|---|---|---|---|
| MAb447-52D |
|
| GPxR | gp120 V3 loop | NA | Rabbit immunization with 1 peptide | NAbs induced | [17,18] |
| MAb 58.2 | 20-mer RPL |
| (Y/L)(V/L/I)GPGRxF | gp120 V3 loop | Characterization of non-essential aminoacids in the epitope using a peptide array | NA | NA | [19] |
| MAb 19B | 15-mer RPL | Ab captured on polystyrene beads | xIx4GxxFYxT | gp120 V3 loop | Type II β turn structure of the epitope | NA | NA | [18,20] |
| MAb 1001 | 30-mer RPL | Ab coated onto microwells | (R/K/H)xGR | gp120 V3 loop | Peptides fused to E. coli AP. Affinities for the Ab determined by SPR | NA | NA | [21] |
| 6-mer RPL | NA |
| gp120 V3 loop | QR insertion characteristic of the LAI isolate | NA | NA | [22] |
| MAb 268 | 6-mer RPL | Biotinylated Ab captured on streptavidin-coated beads | 268.1 (HLGPGR), 268.2 (KAIHRI) | gp120 V3 loop | NA | Rabbit immunization | gp120-specific Abs | [23] |
| MAb 2F5 | 15mer | Ab captured on polystyrene beads | ELDKW, D(K/R)W | ELDKWA (gp41 MPER) | 2F5 affinity for gp41 and gp41 peptide determined by SPR | NA | NA | [24] |
| MAb 2F5 | 17 constrained and linear RPL, x12-AADKW and AADKW-x12 sublibraries | Biotinylated Ab captured on streptavidin-coated beads | 3 peptides (AADKW-x12) | ELDKWA (gp41 MPER) | Ala substitutions and deletions studies | NA | NA | [25] |
| MAb 2F5 | 12-mer and 7-mer-c RPL | Ab coated onto microwells | DKWA, LDKWA | ELDKWA (gp41 MPER) | Epitope specificity depends on the structural context of the library | Mice and rabbit immunization | Inhibition of cell fusion | [26] |
| 20-mer RPL | Rabbit anti-mouse IgG, Fc-specific Ab coated on Petri dishes |
| C1 domain of gp120 | Pepscan analysis, quantitative binding analysis | NA | NA | [27] |
| MAb ID6 | 12-mer, 7-mer, 7-mer-c RPL | Mab captured on rat anti-mouse Ab coated microwells | TxxFxxWxxD, FxDWxF | C1 domain of gp120 | ADCC-inducing Ab | NA | NA | [28] |
| MAb b12 |
| Biotinylated IgG1b12 captured on avidin-coated microwells |
| gp120 CD4 binding site | Competition with gp120 for the binding to IgG1b12, determination of the in-solution binding affinity. | (2) Phage B2.1 immunization of mice and rabbits | Anti-peptide Abs elicited | [29,30] |
| MAb b12 | x10ALLRYx10, x3(M/V)WSDx3 and xLXVWxDExx RPL | Ab coated onto tubes | GLLVWSDEL | gp120 CD4 binding site | NA | Mice immunization | Env-specific Abs | [31] |
| MAb b12 | 12-mer-c RPL | Ab captured on Protein G-coated Petri dishes | WSDL | gp120 CD4 binding site | Prediction of epitope clusters with Mapitope | NA | NA | [32] |
| MAb 5145A | 9-mer and 10-mer-c RPL | Ab coated onto Petri dishes | AECGPAEPRGAWVC, AECGPYEPRGDWTCC | gp120 CD4 binding site | NA | Rabbit immunization with peptides-HSP fusion constructs | gp120-specific Abs elicited, recognizing a different epitope | [33] |
| MAb 803-15.6 | 7-mer RPL |
| AxxKxRH | gp120 residues 502–508 | Quantitative binding analysis | NA | NA | [34] |
| 12-mer-c RPL | Ab captured on Protein G-coated Petri dishes | NA |
| Prediction of epitope clusters with Mapitope | NA | NA | [32] |
| 15-mer RPL | Ab captured on polystyrene beads |
|
| NA | NA | NA | [18] |
| MAb 2G12 | Set of RPL | Ab captured on protein A-coated beads | 2G12.1 (ACPPSHVLDMRSGTCL) | Glycosylated gp120 | NA | Rabbit immunization | No HIV-specific Abs | [35] |
| Library | Biopanning support | Samples | Target | Biopanning procedure | Mimotopes | Epitopes | Additional | Vaccination attempts | Results | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 9-mer, 9-merc RPLs | Tosylactivated beads coupled to Anti human IgG, Fc-specific Ab | LTNP (serum) | IgG | Panning on one LTNP, reactivities assessed on a second LTNP | 3 linear, 7 putative conformational | Linear: gp41 ID GKLIC region, gp120 V1 and C2 regions | Reactivities of positive clones assessed on 22 LTNP, 25 HIV, 50 HD. Affinity purification of Nabs with phages | Phage immunization of C57BL and BalB/c mice | NAbs induced | [50] |
| 12-mer-c RPL | Protein G coated on Petri dishes | HIV + (serum) | IgG | Panning on HIV+ serum IgG | 4 linear, 2 unassigned | Linear: gp41 ID GKLIC region | Reactivities of the clones assessed on the sera of 30 HIV+ | NA | NA | [52] |
| 12-mer, 7- mer, 7-mer-c RPLs | Microwells coated with purified IgG | HIV+ (HAART) | IgG | Panning on purified HIV+ IgG, before and after HAART | Linear, CxxKxxC | Linear: gp41 ID GKLIC region | Reactivities of 1 insert with IgG of 22 HIV+ | Phage immunization of C57BL/6J mice | Abs against epitope | [53] |
| 12-mer, 7- mer, 7-mer-c RPLs | Tosylactivated beads coupled to Anti human IgG, Fc-specific Ab | LTNP (plasma) | IgG | Panning on pool of 8 LTNP plasma, negative selection on HIV - plasma pool | 160 linear, 124 putative conformational, 160 unassigned | gp120 V3 loop, gp41 GKLIC, gp120 MPER | Reactivities with plasma of 7 HIV+ plasma | Phage immunization of mice | NAbs induced | [54] |
| 12-mer, 7- mer, 7-mer-c RPLs | Tosylactivated beads coupled to Anti monkey IgG Ab | Clade C SHIV-infected rhesus macaque | IgG | Panning on one SHIV + plasma, negative selection on SHIV - monkey plasma pool | 72 linear, 6 putative conformational | gp120 V2, V3, C-term, gp41 GKLIC, MPER | Reactivities of inserts tested as fusion proteins | DNA prime-phage boost of BALB/c mice | NAbs induced | [55] |
| 12-mer RPL | Tosylactivated beads coupled to Anti human IgG, Fc-specific Ab | HIV subtype CRF02AG-infected plasma with 4E10-like BNtAbs | IgG | Panning on pooled longitudinal samples of one HIV + plasma, negative selection on HIV - plasma pool | Linear (SLxxLRL, KxWWxA, Kx3IGPHxxY) | gp41 MPER and LLP2 regions; gp120 C1 and V3 regions | NA | NA | NA | [56] |
| 12-mer, 7- mer, 7-mer-c RPLs | Tosylactivated beads coupled to Anti human IgG, Fc-specific Ab | HIV subtype A-infected plasma with 4E10-like BNtAbs | IgG | Panning on pooled longitudinal samples of one HIV + plasma, negative selection on HIV - plasma pool | 38 linear, 22 putative conformational (Kx3Hx3Y, KxxHxGPx3F, CxGxLxCTxNxP) | gp41 ID epitope; gp120 V2 and V3 regions | Competition with rgp120/gp140 for antibodies binding; peptide neutralization inhibition assays | NA | NA | [57] |
| DNAse-fragmented Gag DNA | Purified IgG captured on microwells | Serum of rabbits immunized with p24 | IgG | Panning on rabbit anti p24 IgG | p24 fragments | N- and C-terminus of p24 | Reactivities of inserts tested as proteins in fusion with GST | NA | NA | [58] |
| Target | Library | Target presentation | Mimotopes | Epitopes | Additional | Vaccination attempts | Results | Reference |
|---|---|---|---|---|---|---|---|---|
| MAbs 3A9, 5C7 | 7-mer, 7-mer-c, 9-mer-c, 12-mer RPL | Phage-antibody complexes captured on beads coated with anti-mouse IgG Ab | CHASIYDFGSC, CPHWLRDLRVC | CCR5 N-terminus (SIYD) and ECL1 (FG) CCR5 N-terminus (P), ECL1 (HW) and ECL3 (DLR) | Reactivities assessed against inserts synthesized as peptides, binding and competition assays with peptides. Entry inhibitor | NA | NA | [62,63] |
| MAb 2D7 | 15-mer RPL | Ab coated onto Petri dishes | M23 (FCALDGDFGWLAPAC) | CCR5 ECL2 | Neutralization of HIV-1 infection | NA | NA | [64] |
| MAb 2D7 | 12-mer RPL | Ab coated onto microwells | EWQKEGLVTLWL | CCR5 ECL2 | NA | Rabbit immunization | NAbs with 2D7-like functions | [65] |
| MAb MHM23 | 9-mer, 9-mer-c RPL | Phages-Ab complexes captured on streptavidin-coated dishes | PPFxYRK | CD18 (AA 200–206) | Inhibition of HIV-1-induced syncytium formation | NA | NA | [66] |
| Target | Library | Target presentation | Phagotopes | Affinity (KD) | Inhibition (IC50) | Virus isolates/clades | Additional | Reference |
|---|---|---|---|---|---|---|---|---|
| gp120 (CD4 BS) | LTNP Fab | Recombinant gp120 | b12 | <10 nM | ~20 nM | MN, IIIb | NA | [41,42,70] |
| gp120 (CD4 BS) | Fab (CDR walking of b12 Fab) | Recombinant gp120 | 3D3 | 15 pM | ~0.1–0.9 nM | MN, IIIb | 3B3 ScFv engineered as fusion immunotoxin | [71–73] |
| gp120 (CD4 BS) | LTNP Fab | Recombinant gp120 | L78 | 4–300 nM | ~2 ug/mL | MN, IIIb | epitope-masking strategy | [74] |
| gp120 (CD4 BS) |
| Recombinant gp120 | JL413 SKL6 | NA |
|
| gp120-hydrolyzing Abs | [75,76] |
| gp120 (CD4 BS) | VHH (immunized llamas) | Recombinant gp120 | A12 D7 C8 | 0.1–1 nM | 0.003–38 μg/mL | A, B, C, D, CFR02_AG and CRF07_BC | sublibrary engineered to increase potency | [77,78] |
| gp120 (CD4 BS) | CD4 V1 and V1-V2 variants | Recombinant gp120 | E6, B6, 22, F8, D11 | NA | 0.2–1 μg/mL | BH10 | NA | [79] |
| gp120 (V3 loop) | MAb 447-52D-derived ScFv | V3 loop peptide | 402P5H7 | 0.28–3.1 nM | NA | MN | VL shuffling and HCDR3 “spiking” of MAb 447-52D | [80] |
| gp120 (V3 loop) | LTNP Fab | Recombinant gp120 | DO142-10 Fab loop 2 | 11 nM 1.9 nM | 0.2–8 μg/mL 1–5 μg/mL | MN, IIIb | epitope-masking strategy | [70,81] |
| gp120 (CD4-i epitope) | 7-mer, 9-mer-c and 12-mer RPL | Recombinant gp120 | 12p1 | NA | 1.1–1.6 μM | YU2 | NA | [82] |
| gp120 (CD4-i epitope) | HIV-1 infected FDA-2 patient FAb | gp120-CD4-CCR5 complexes | X5 | nanomolar | 0.29–125 μg/mL | A, B, C, D, F and G | NA | [83] |
| gp120 (CD4-i epitope) | 7-mer, 7-mer-c and 12-mer RPL | sCD4-89.6 Env on retroviral particles | XD3 | NA | 50 μM | NL4-3, D117III | NA | [84] |
| gp41 MPER |
| MPER peptide |
| NA |
| B, C and E | NA | [85] [86] |
| gp41 Heptad Repeats |
|
|
| NA |
|
| (1) D-peptide | [87–89] [90,91] [92] [93,94] |
| gp41 Heptad Repeats | (1), (2) Naïve human Fabs (3) Fab 3674-derived Fabs |
|
|
|
|
| NA | [95] [96–98] |
| gp41 Heptad Repeats |
|
|
| NA |
|
| (2) competitive antigen panning | [99] [100–102] |
| Vpr |
|
|
| NA | NA | NA |
| [103] [104] |
| Int | 7-mer RPL | Recombinant Int | FHNHGKQ | NA | NA | NA | Inhibition of strand transfer activity | [105] |
|
|
|
|
| NA | NA | (2) inhibition of Tat-mediated transactivation and HIV-1 replication when transfected | [106] [107] [108] |
|
|
|
| NA | NA | NA | (2) reduction of virus release by infected cells | [109] [110–112] |
|
|
|
| (1) 2nM | NA | NA | (2) inhibition of HIV- 1 replication | [113–115] [116,117] |
| RT |
| Recombinant RT |
| NA | NA | NA | Inhibition of RDDP and/or DDDP activity | [118,119] [120] |
| Rev |
| Rev |
| NA | NA | NA | NA | [106,121–124] |
| Gag | 12-mer RPL | CA protein (AA 162–190) | CAI | NA | NA | NA | In vitro inhibition of capsid assembly | [125] |
| Target | Library | Target presentation | Biopanning procedure | Phagotope | Affinity | Inhibition | Virus isolates/clades | Additional | Reference |
|---|---|---|---|---|---|---|---|---|---|
| CCR5 | Human ScFv | Liposome | Five rounds | NA | NA | NA | NA | NA | [180] |
| CCR5 | Human VH-Rabbit ST6 HCDRs | CCR5 Nterm-GST fusion | Four rounds, acidic elution | Rabbit ST6 Human ST6/34 | 2.7 nM 8.5 nM | NA | NA | Inhibit CCR5 export when expressed as intrabodies | [184] |
| CCR5 | Randomized and extended CCL5 chemokine | HEK-CCR5, CHO-CCR5 | Recovery of internalized phages, acidic elution | CCL5 P1-CCL5 P2-CCL5 | IC50 = 4.1 nM IC50 = 2 nM IC50 = 0.2 nM | NA IC50 = 7.0 nM IC50 = 0.6 nM | BaL | P1: triggers reduced signaling P2: superagonist inducing CCR5 sequestration | [185] |
| CCR5 | Randomized and extended CCL5 chemokine | HEK-CCR5, CHO-CCR5 cells | Recovery of internalized phages, acidic elution | 6P4-CCL5 5P12-CCL5 5P14-CCL5 | NA NA NA | IC50 = 21 pM IC50 = 28 pM IC50 = 26 pM | BaL | Superagonist + sequestration No signaling + no sequestration Internalization + no signaling | [186] |
| CCR5 | Mice and rats ScFV | ECL1, ECL2, ECL3 biotinylated peptides | Alkaline elution | A1 (ECL1) B7 (ECL2) L9 (ECL2) | NA NA NA | 25% a 35% a 25% a | BaL | All active ScFv were selected on cyclic peptides | [181] |
| CCR5 | 12-mer RPL | CHO-CCR5 cells | Four rounds, acidic elution | AFDWTFVPSLIL | IC50 = 2.5 μM | NA | NA | Antagonist of CCR5 | [179] |
| CCR5 | Partially randomized
| CHO-CCR5 cells |
|
| NA | No inh IC50 = 2.5 μM | ADA | Does not act as CCR5 agonist or antagonist | [178] |
| CXCR4 | Llamas immunized VHH | HEK-CXCR4 cells | Two rounds and counterselection | 238D2 238D4 238D2-15GS-238D4 | KI = 10 nM KI = 6 nM KI = 0.3 nM | IC50 = 30 nM IC50 = 40 nM IC50 = 0.2 nM | NL4.3 | Inhibit chemotaxis and mobilize stem cells | [187] |
| CXCR4 | Non immunized HCDR3 | Linear ECL2 peptide | Four rounds, acidic elution | YYCARDRGGTYPGR YWCQG | KD = 5 μM | NA | NA | Antagonist of CXCR4 | [182] |
| CD40 | Human ScFv | Biotinylated CD40-Fc fusion protein | Four rounds, trypsin elution | B44 | KD = 60 nM | 3–4 fold reduction of infection | R5- tropic | Activates normal B cells, Does not block CD40- CD40L binding | [188,189] |
| DDX3 | Mix of 12-mer and 7- mer RPL | His tagged ALRAMKENGR peptide | Acidic elution | INS1: SDVPTQVGGRRRRRRRRR | NA | IC50 = 20 μM | LAI | Homologies to XPO1/CRM1 | [190] |
| Target | Library | Biopanning | Natural substrates P4-P3-P2-P1--P1′-P2′-P3′-P4′ | Selected substrates | Kcat/Km (μM−1s−1) | Derived Inhibitors | KI (nM) | Reference |
|---|---|---|---|---|---|---|---|---|
| HIV-1 protease Q7K | Random hexapeptides fused to the MAb 3-E7 epitope and displayed on Fd phage | 5 rounds with progressive decrease of the protease concentration and contact time | (RT/IN) RKIL↓FLDG (MA/CA) SQNY↓PIVQN (CA/P2) ARVL↓AEAM (P2/NC) ATIM↓MQRG (P1/P6) PQNF↓LQSR (in P6) KELY↓PLTS (TF/PR) SFNF↓PQIT (PR/RT) TLNF↓YVDG (in RT) AETF↓YVDG | GSGIF↓LETSL | 1.3 | [199] | ||
| GSGVF↓VEMPL | 3.9 | GSGIFΨ (CH2NH)LETSL | 5 | |||||
| GSGVF↓VVNGL | 18 | GSGVFΨ (CH2NH)VEMPL | 18 | |||||
| GSGLF↓TEYGL | 0.002 | GSGVFΨ (CH2NH)VVNGL | 100 | |||||
| IRKIL↓FLDG | 0.002 | GSGLFΨ (CH2NH)TEYGL | 200 | |||||
| HIV-1/FIV proteases | 4 rounds with progressive decrease of the protease concentration | Protease | IC50 (nM) | [198] | ||||
| SGNF↓VVNGLVK | HIV-1 only | |||||||
| KSGVF↓VENGLVK | HIV-1 only | |||||||
| KSGVF↓VQNGLVK | HIV-1 only | |||||||
| KSGNFVVN↓GLVK | FIV only | |||||||
| KSGV↓SVNGK | FIV only | |||||||
| KSGVFHVN↓GLVK | FIV only | GSGVF Ψ (CH2OH)VVNGL | HIV/FIV | |||||
| KSGVFQVN↓GLVK | FIV only | GSGVF Ψ (CH2NH)VVNGL | 160/5000 | |||||
| KSGVF↓VVNGLVK | HIV-1/FIV | 700/400 | ||||||
| KSFVF↓VVNGLVK | HIV-1/FIV | |||||||
| KSGIF↓VVNGLVK | HIV-1/FIV | |||||||
| KSGVF↓HVNGK | HIV-1/FIV | |||||||
| KSGVF↓QVNGLVK | HIV-1/FIV | |||||||
| KSGVF↓VVQGLVK | HIV-1/FIV | |||||||
| KSGVF↓VVTGLVK | HIV-1/FIV |
| P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ |
|---|---|---|---|---|---|---|---|
| S11 | G11 | V5 | Y5 | F4 | V5 | T4 | S3 |
| I3 | F4 | V3 | E4 | S3 | T2 | ||
| N3 | M2 | L3 | A1 | M1 | G2 | ||
| H1 | Q1 | E1 | P1 | ||||
| N1 | N1 | ||||||
| Q1 | D1 | ||||||
| A1 |
| Phage | Type | Carrier protein | HIV-1 antigen | Display level | Animal Model | Reference |
|---|---|---|---|---|---|---|
| M13 | Filamentous | pIII | p17 (GEDRW) | 3 to 5 copies/phage | Rabbit | [205] |
| fd | Filamentous | pIII | V3 loop of gp120 | 3 to 5 copies/phage | Mice | [207] |
| fd | Filamentous | pIII | V3 loop of gp120 | 3 to 5 copies/phage | Mice | [210] |
| fd | Filamentous | pVIII | RT (ILKEPVHGV) | 2700 copies/phage | Human cells | [206,214] |
| M13 | Filamentous | pVIII | V3 loop variants | 2700 copies/phage | Mice | [215] |
| T4 | Lytic/Icosahedral | SOC | V3 loop of gp120 | 960 copies/phage | Mice | [214] |
| T4 | Lytic/Icosahedral | HOC | p24 | 150 copies/phage | Mice | [216] |
| MS2 | VLP/Icosahedral | A-B loop | V3 loop of gp120 | 90 copies/VLP | Mice | [212] |
| PP7 | VLP/Icosahedral | A-B loop | V3 loop of gp120 | - | Mice | [217] |
| Lambda | Lytic/Icosahedral | gpD | gp140 trimer | 30 copies/phage | Rabbit | [213] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delhalle, S.; Schmit, J.-C.; Chevigné, A. Phages and HIV-1: From Display to Interplay. Int. J. Mol. Sci. 2012, 13, 4727-4794. https://doi.org/10.3390/ijms13044727
Delhalle S, Schmit J-C, Chevigné A. Phages and HIV-1: From Display to Interplay. International Journal of Molecular Sciences. 2012; 13(4):4727-4794. https://doi.org/10.3390/ijms13044727
Chicago/Turabian StyleDelhalle, Sylvie, Jean-Claude Schmit, and Andy Chevigné. 2012. "Phages and HIV-1: From Display to Interplay" International Journal of Molecular Sciences 13, no. 4: 4727-4794. https://doi.org/10.3390/ijms13044727