Aptamer-Based Molecular Recognition of Lysergamine, Metergoline and Small Ergot Alkaloids

 ,
,
Abstract
:
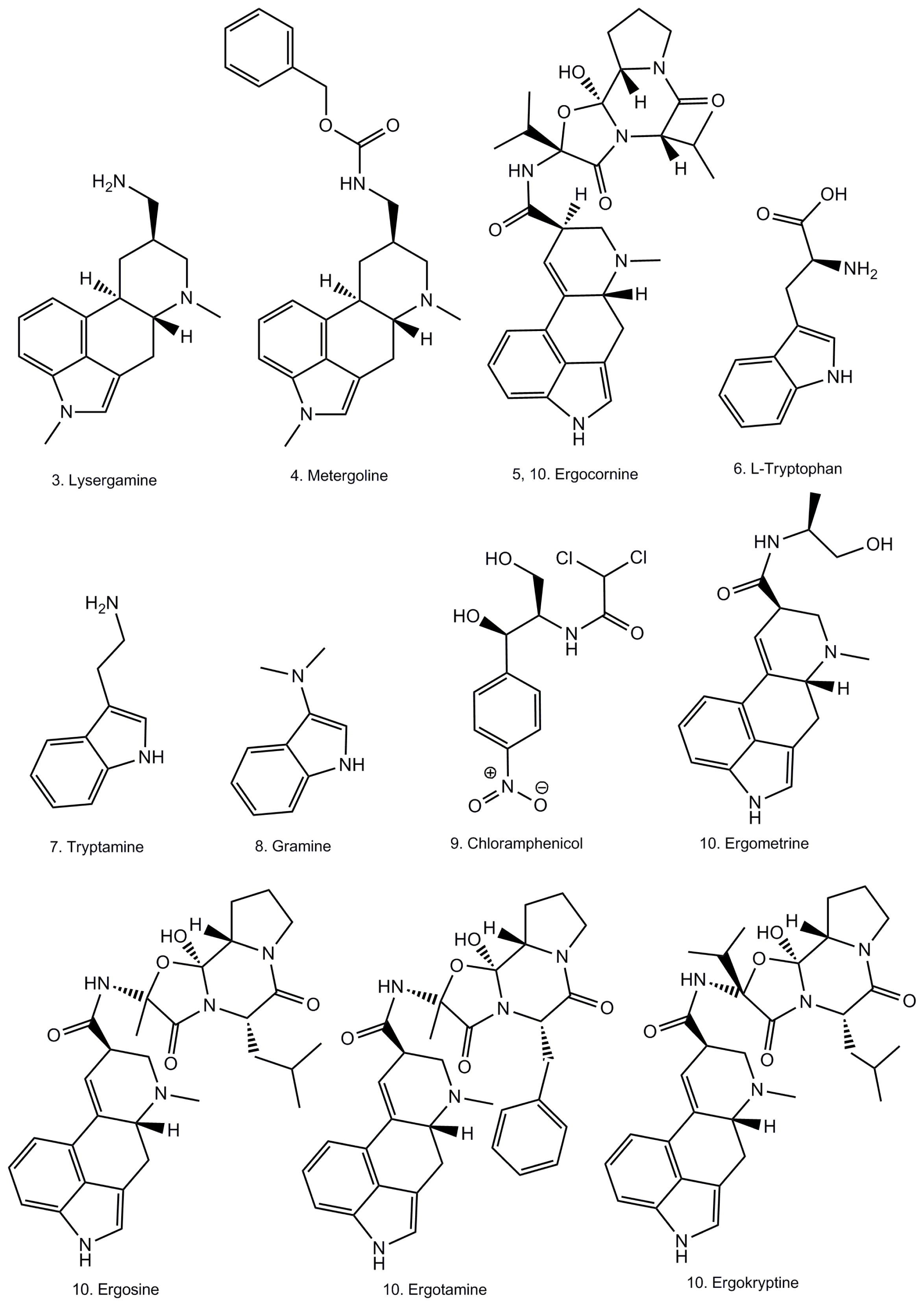
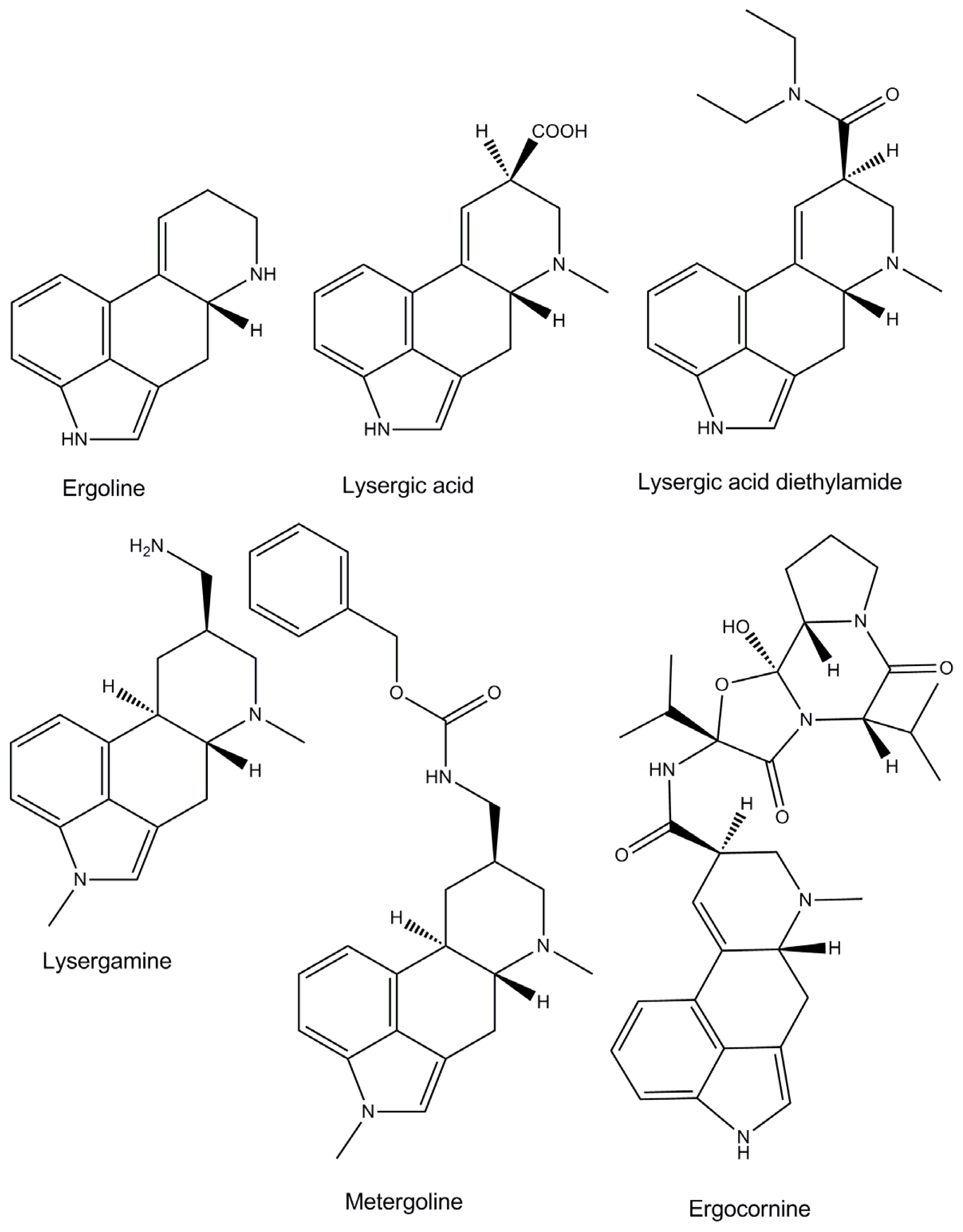
1. Introduction
2. Results and Discussion
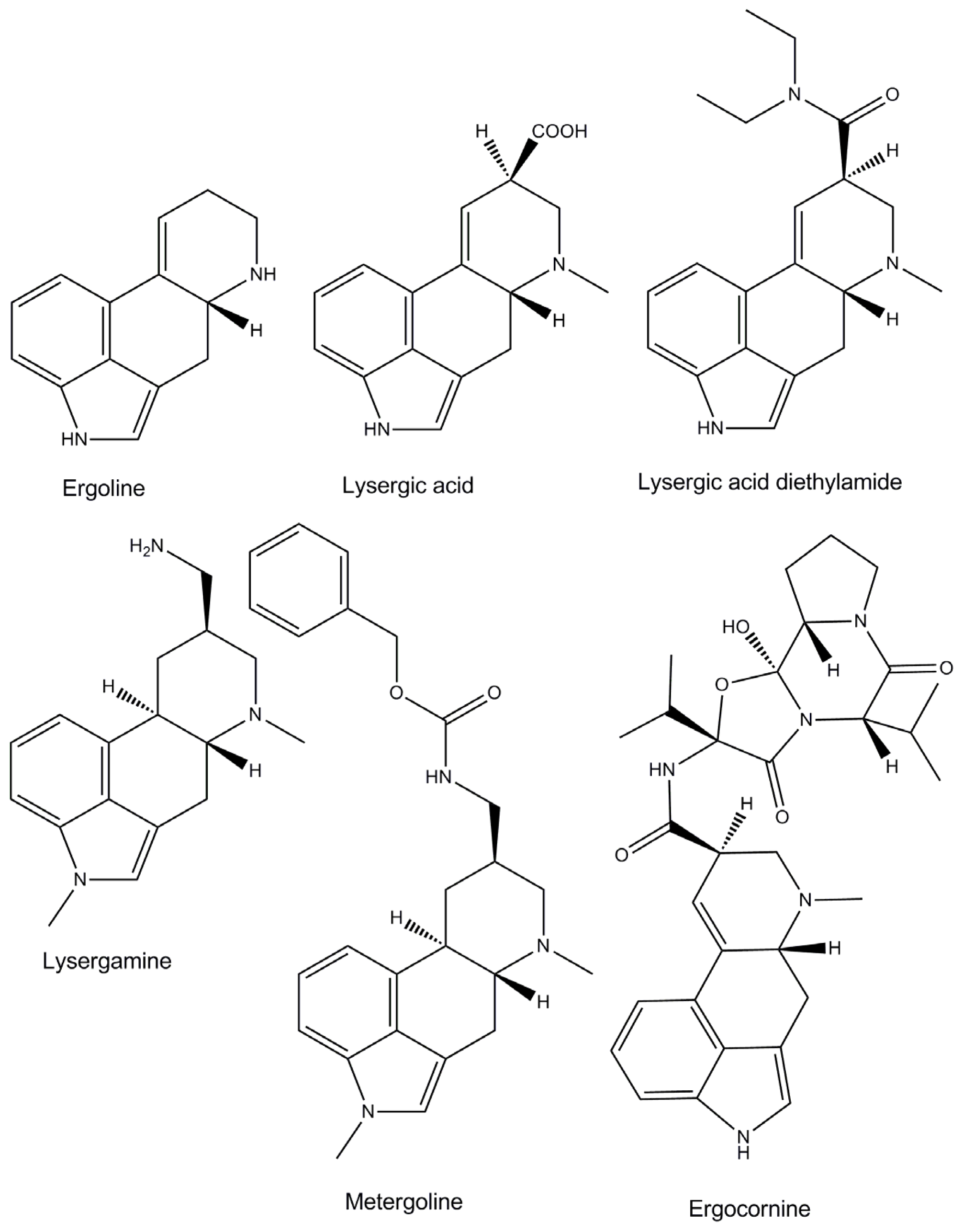
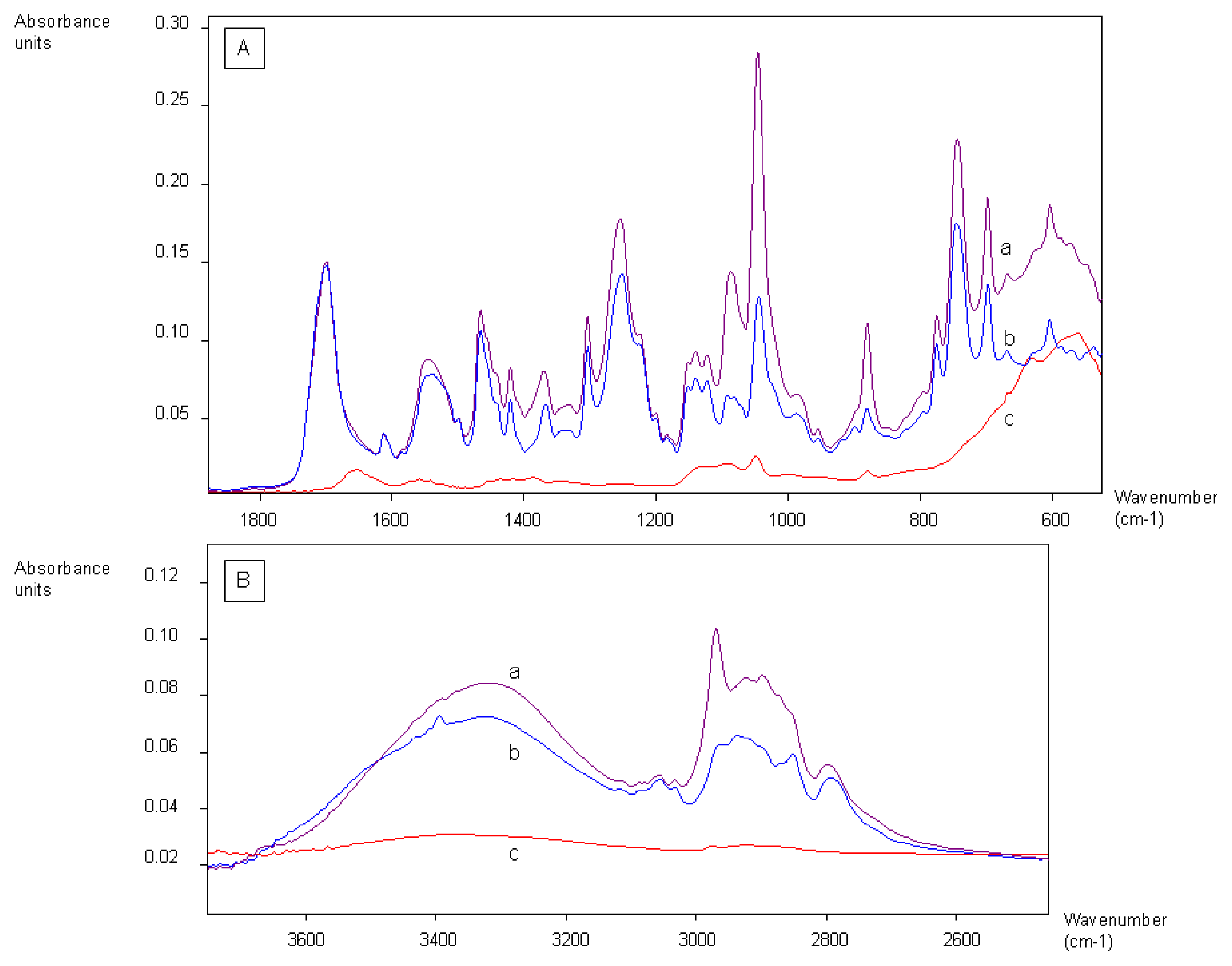
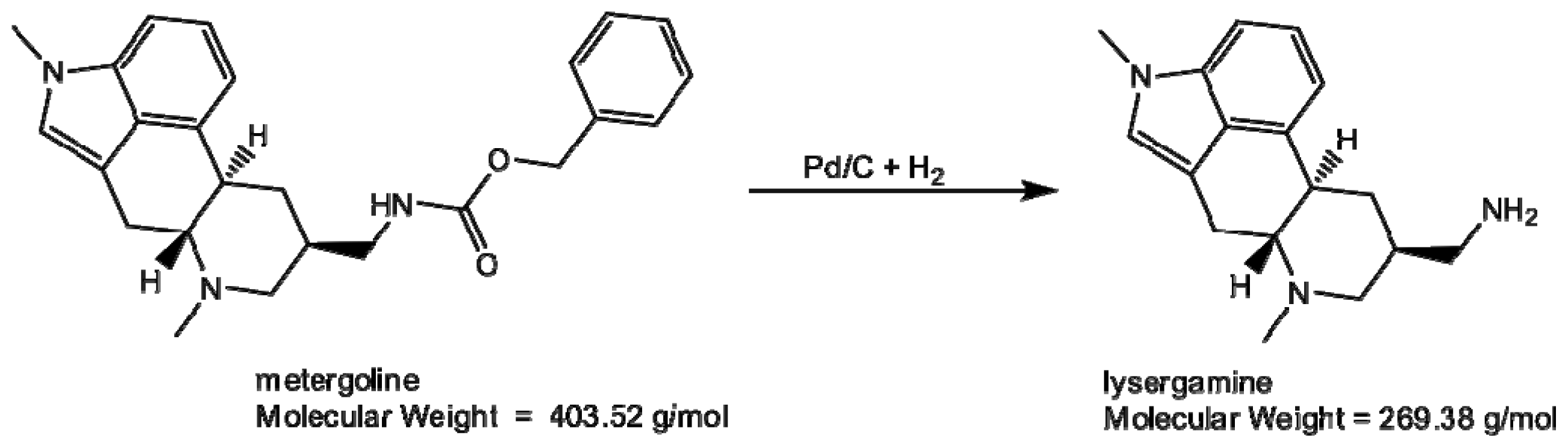
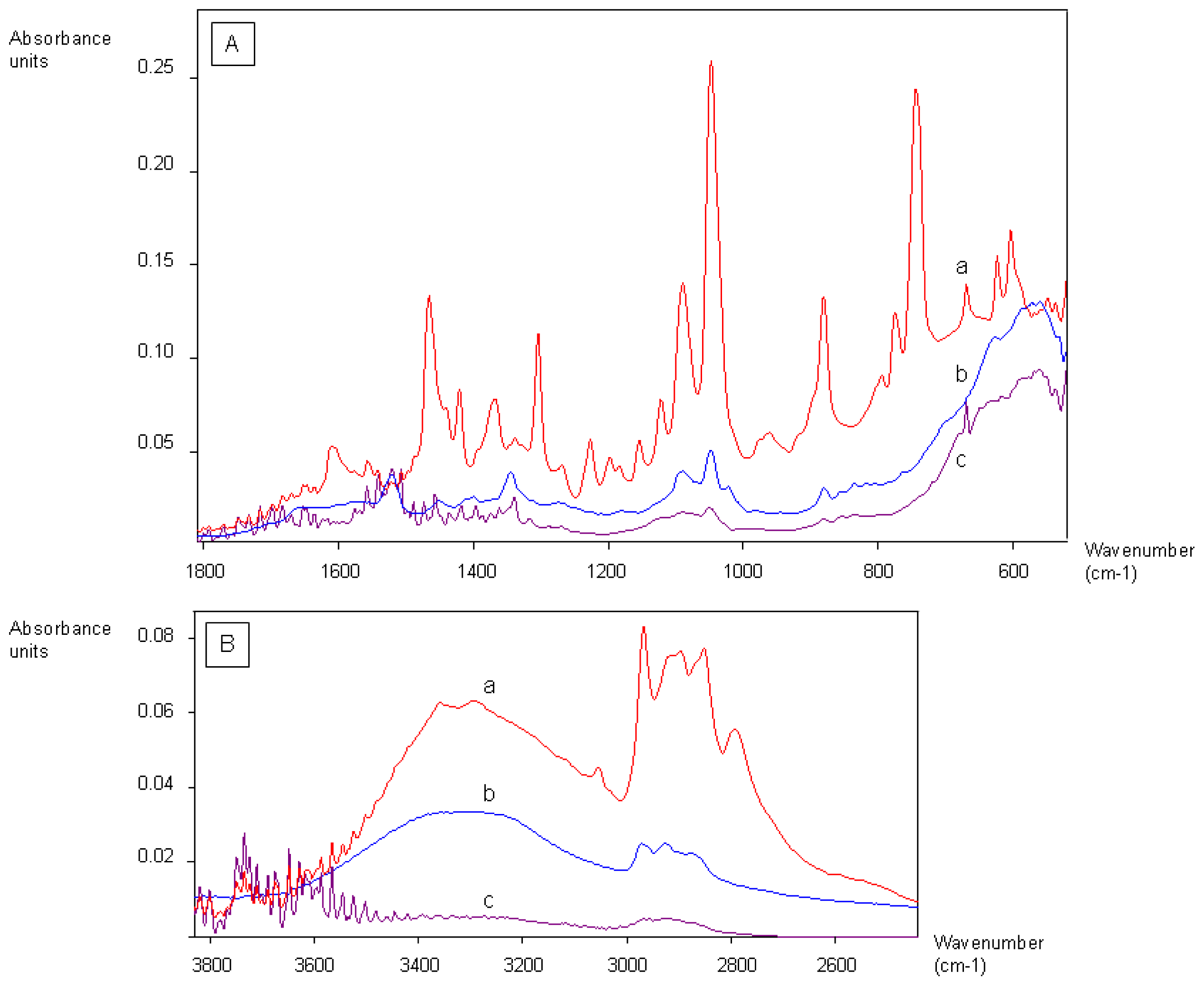
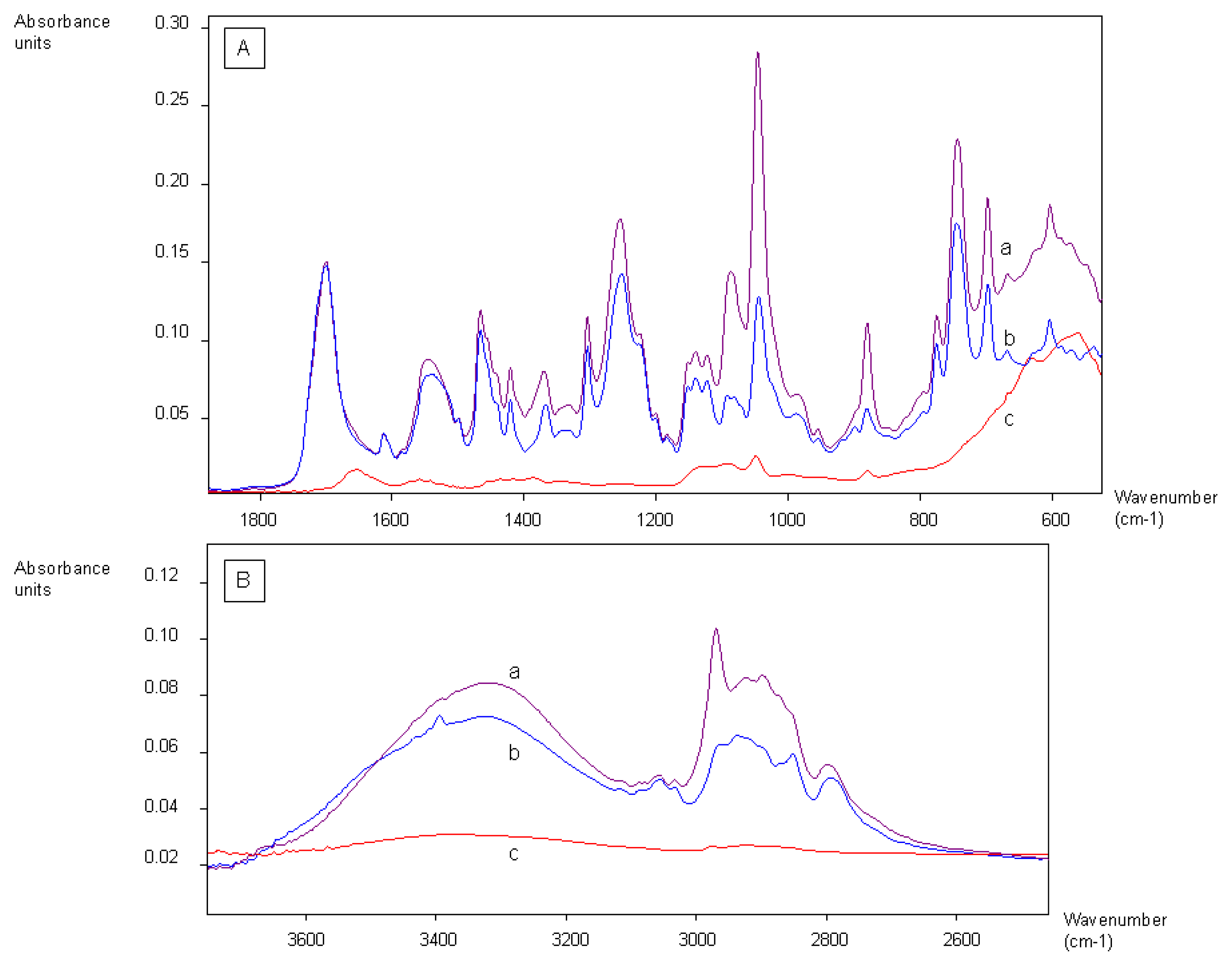
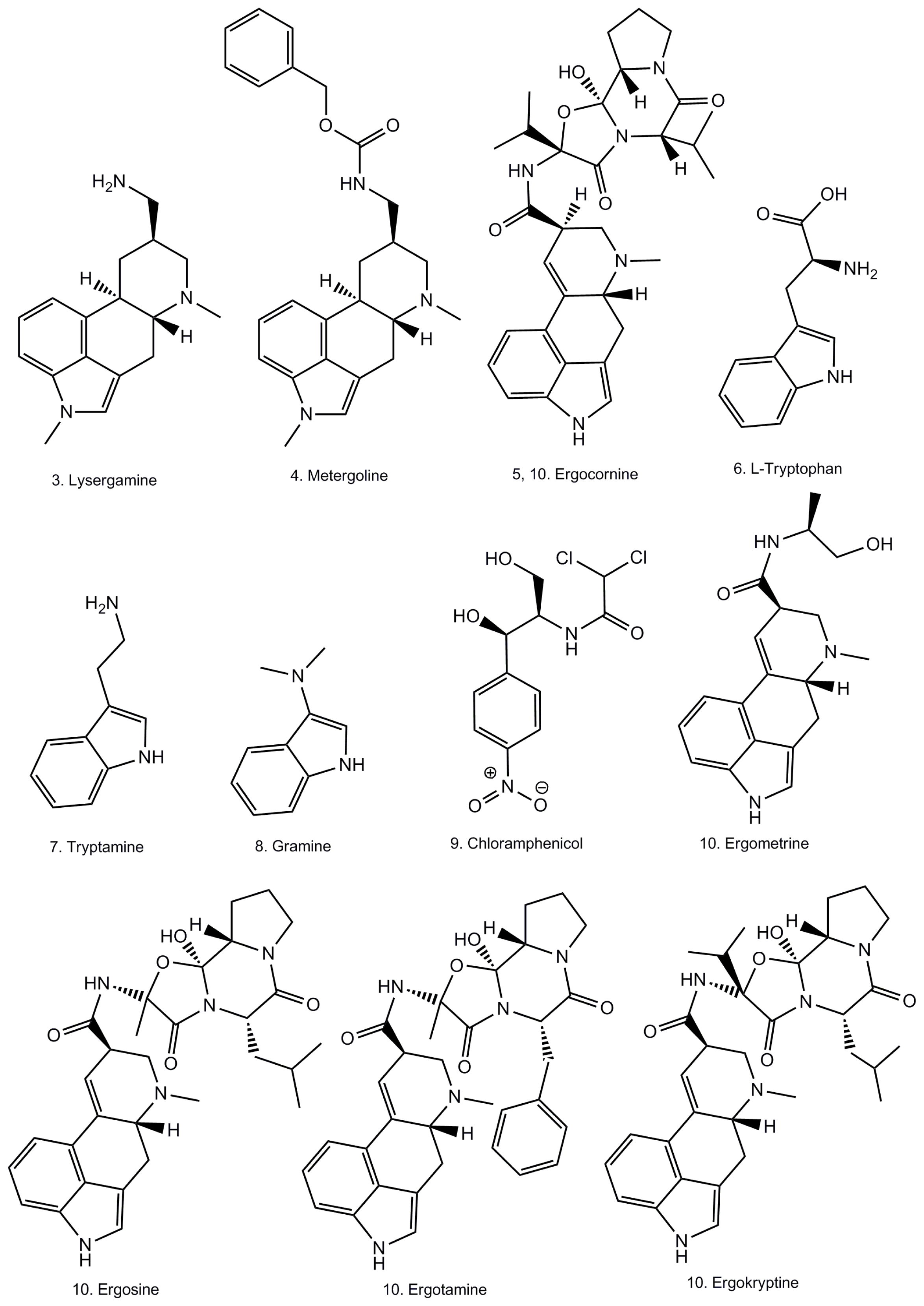
2.1. Coating of Magnetic Beads with Lysergamine and Metergoline
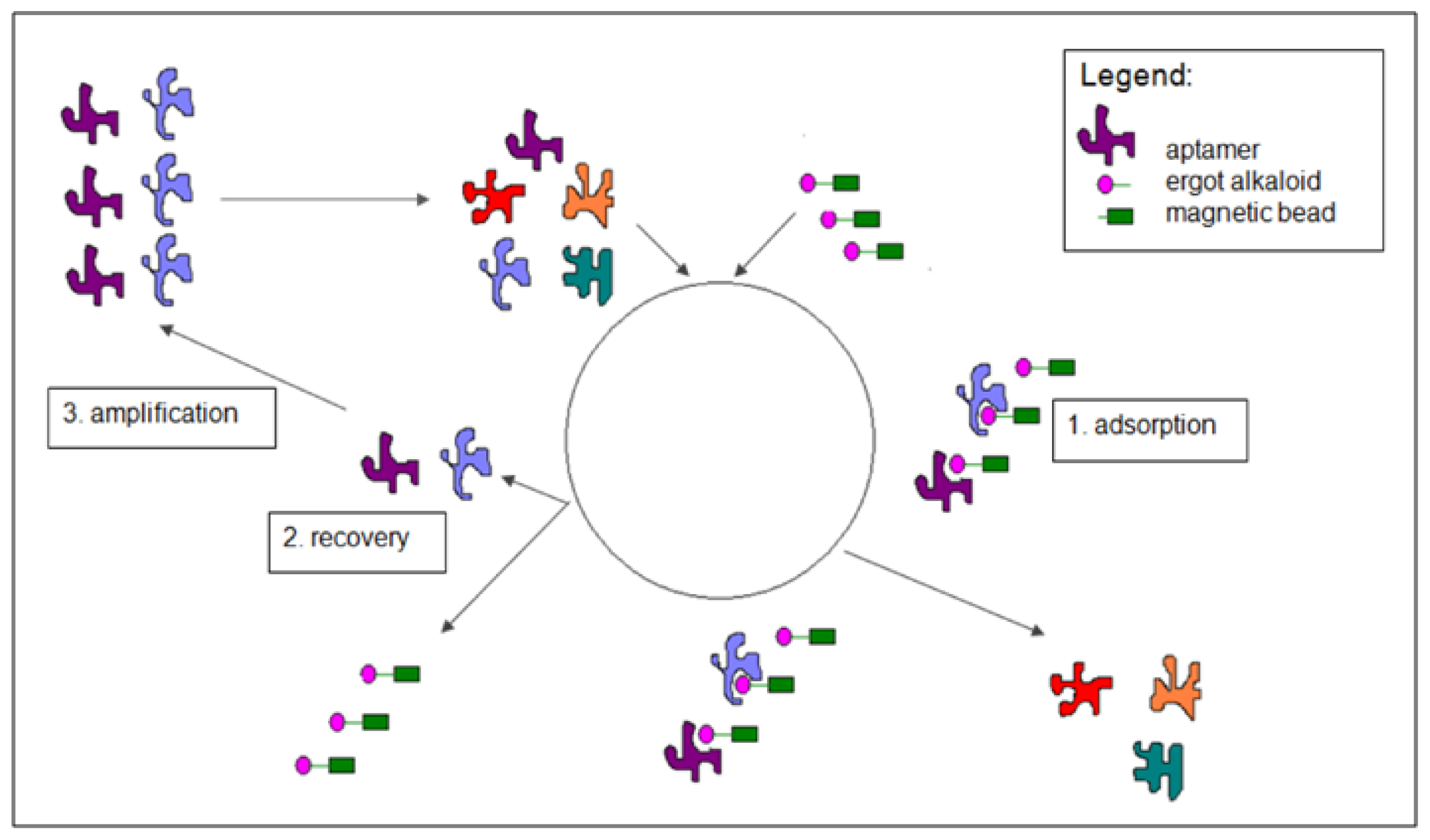
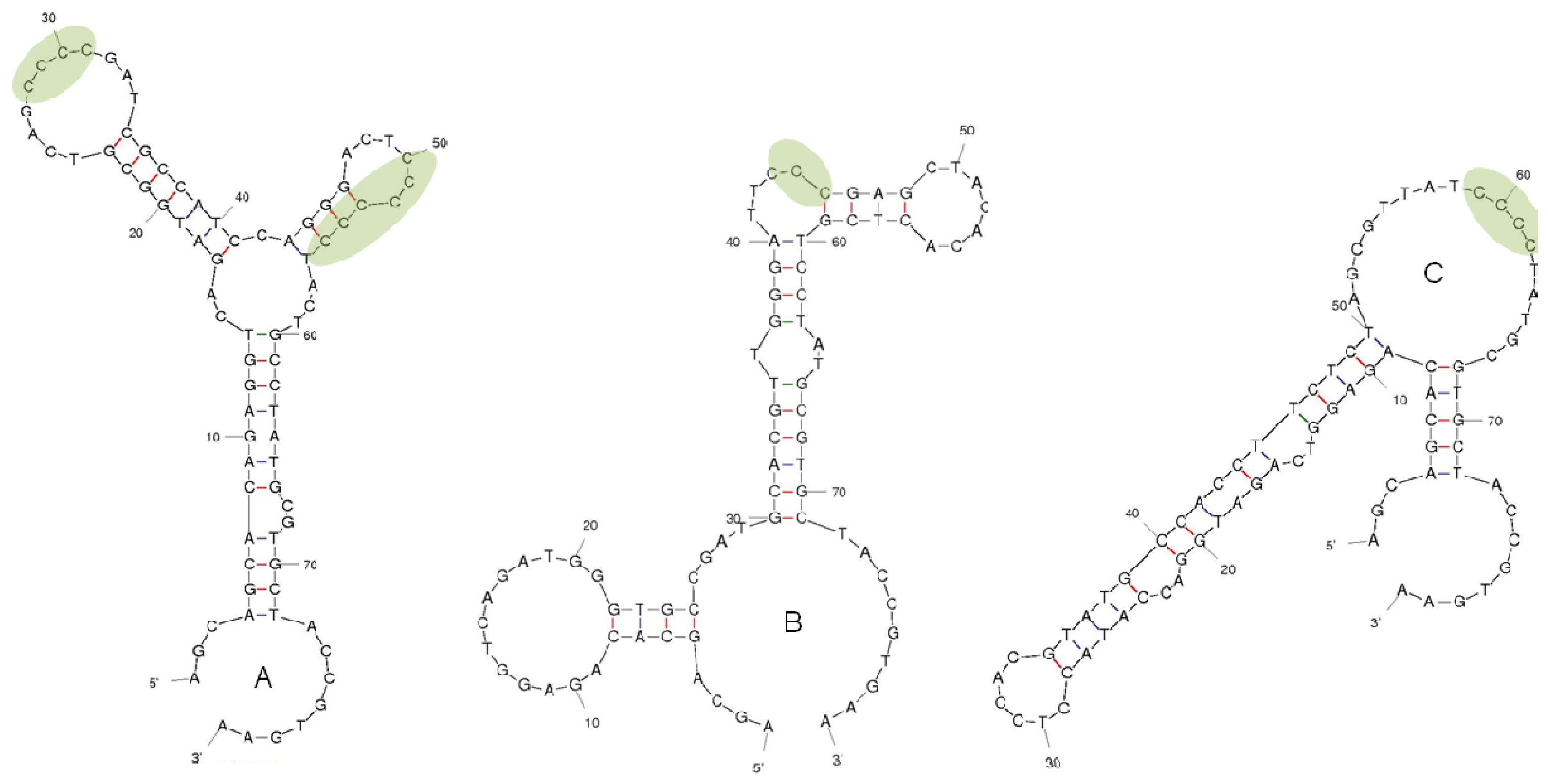
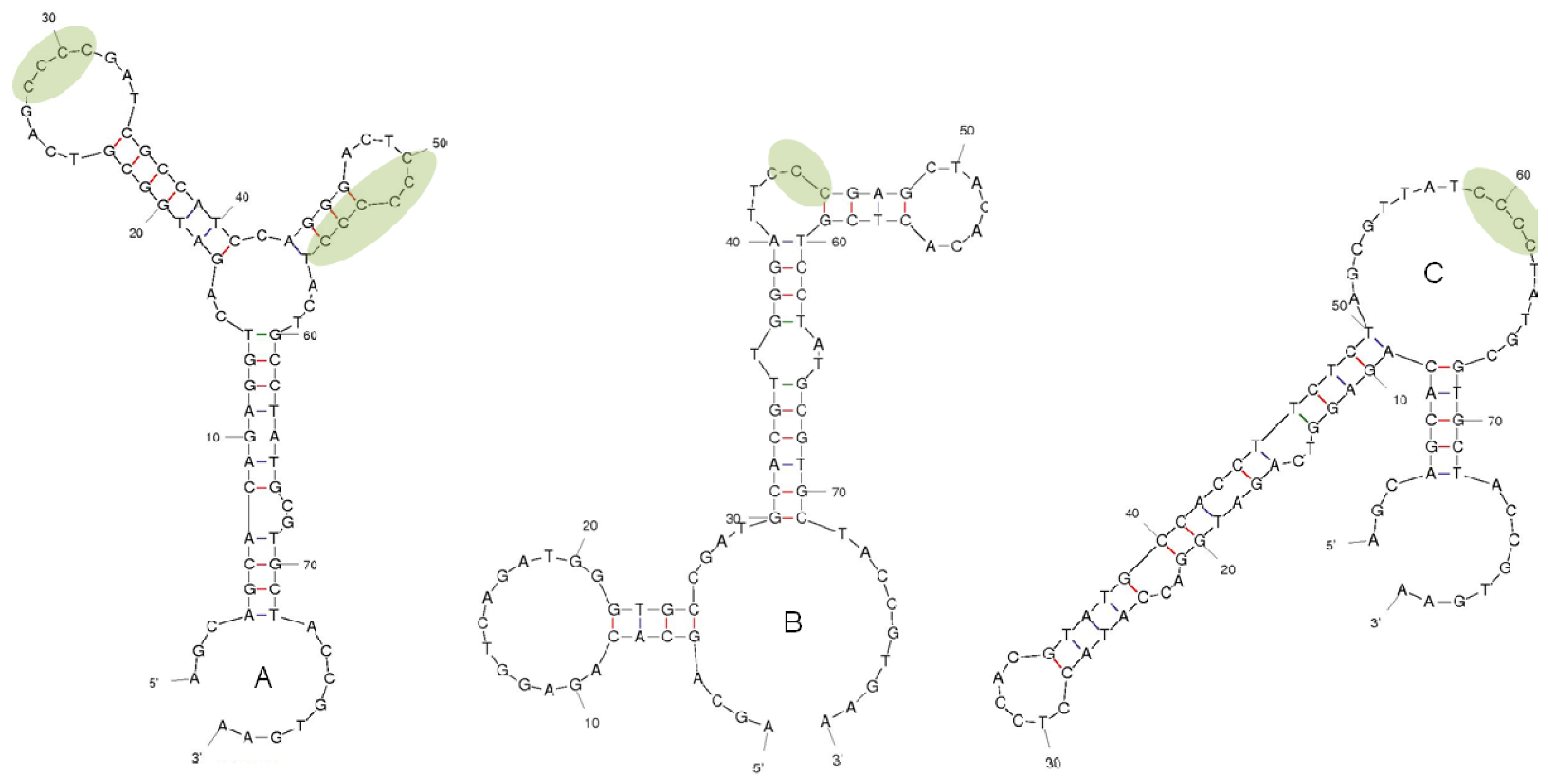
2.2. Selection of DNA Aptamers for Lysergamine and Metergoline
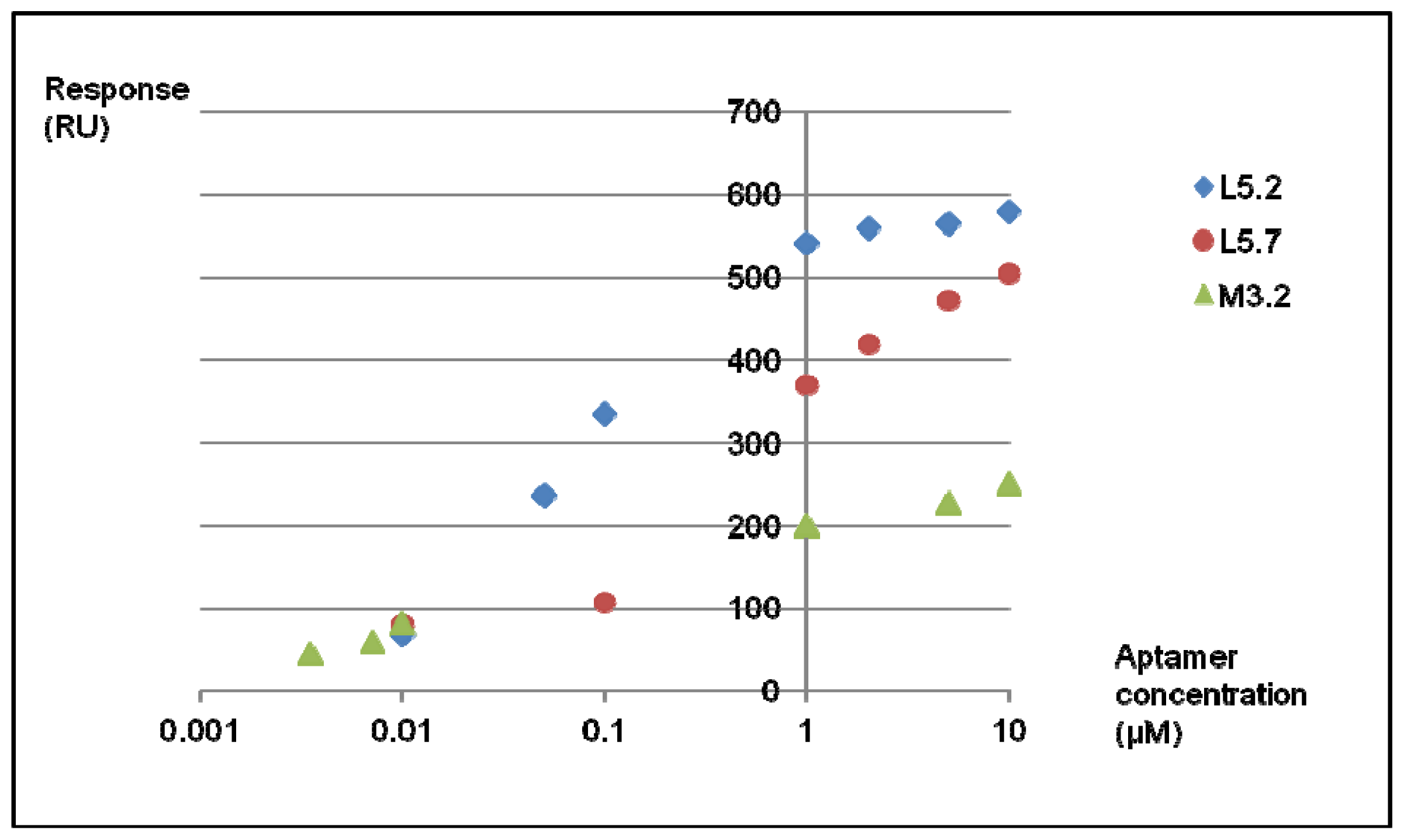
2.3. SPR Analysis of the Complexes of Aptamers and Lysergamine
2.3.1. Coating of SPR Streptavidin Chip with Biotinylated Derivatives of Lysergamine and Ethanolamine
2.3.2. SPR Measurements
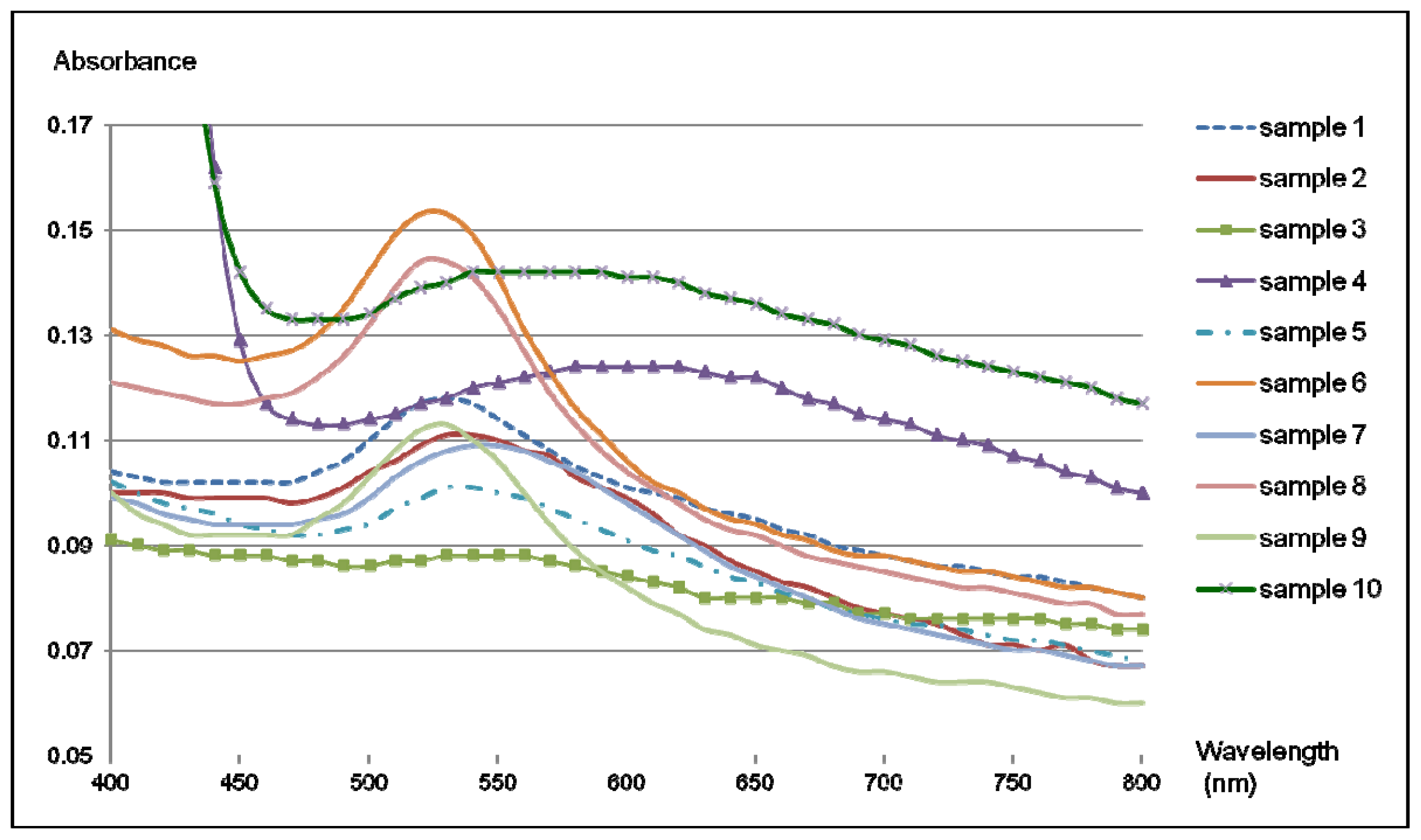
2.4. Colorimetric Reaction with Gold Nanoparticles
3. Experimental Section
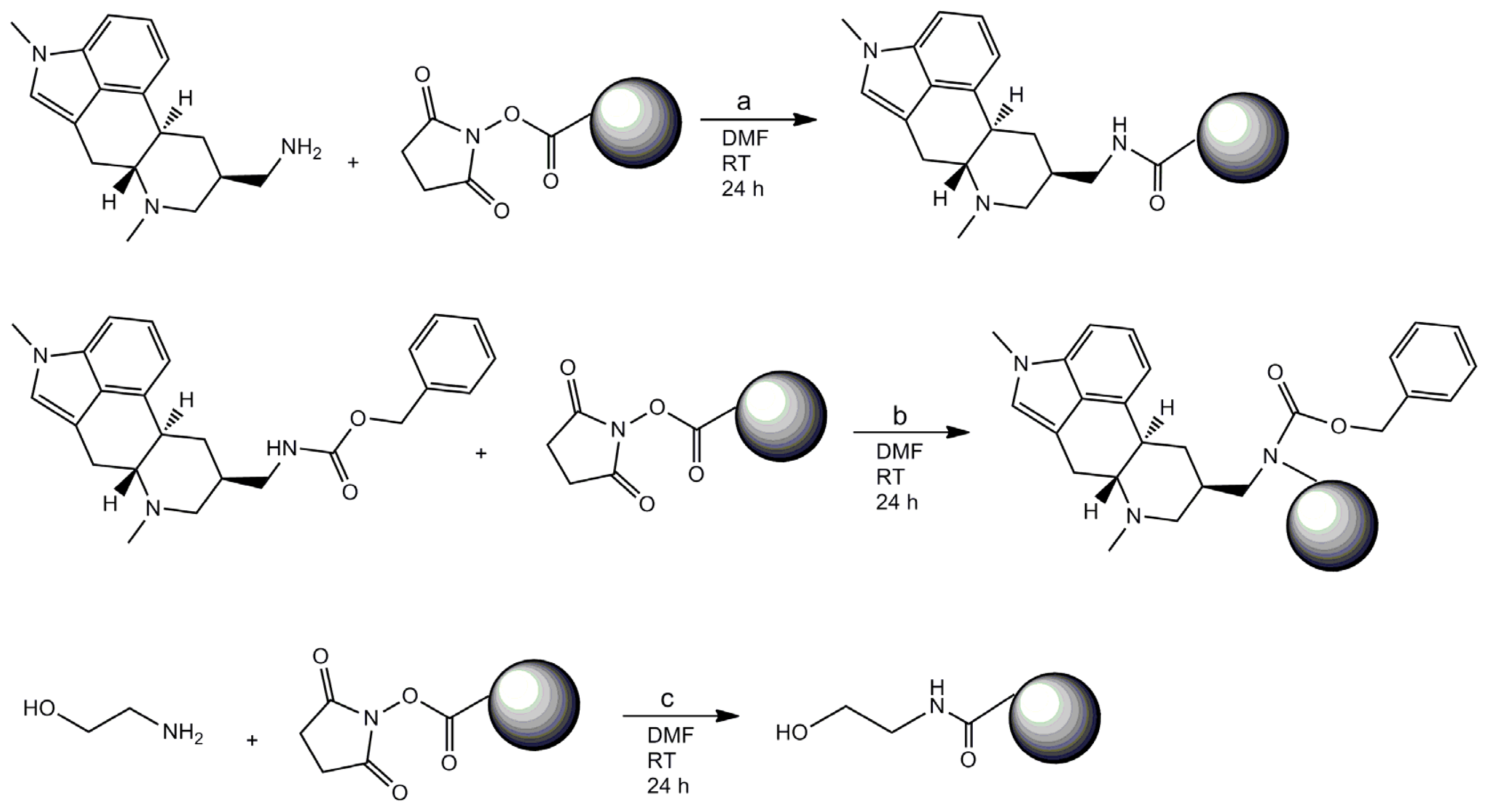
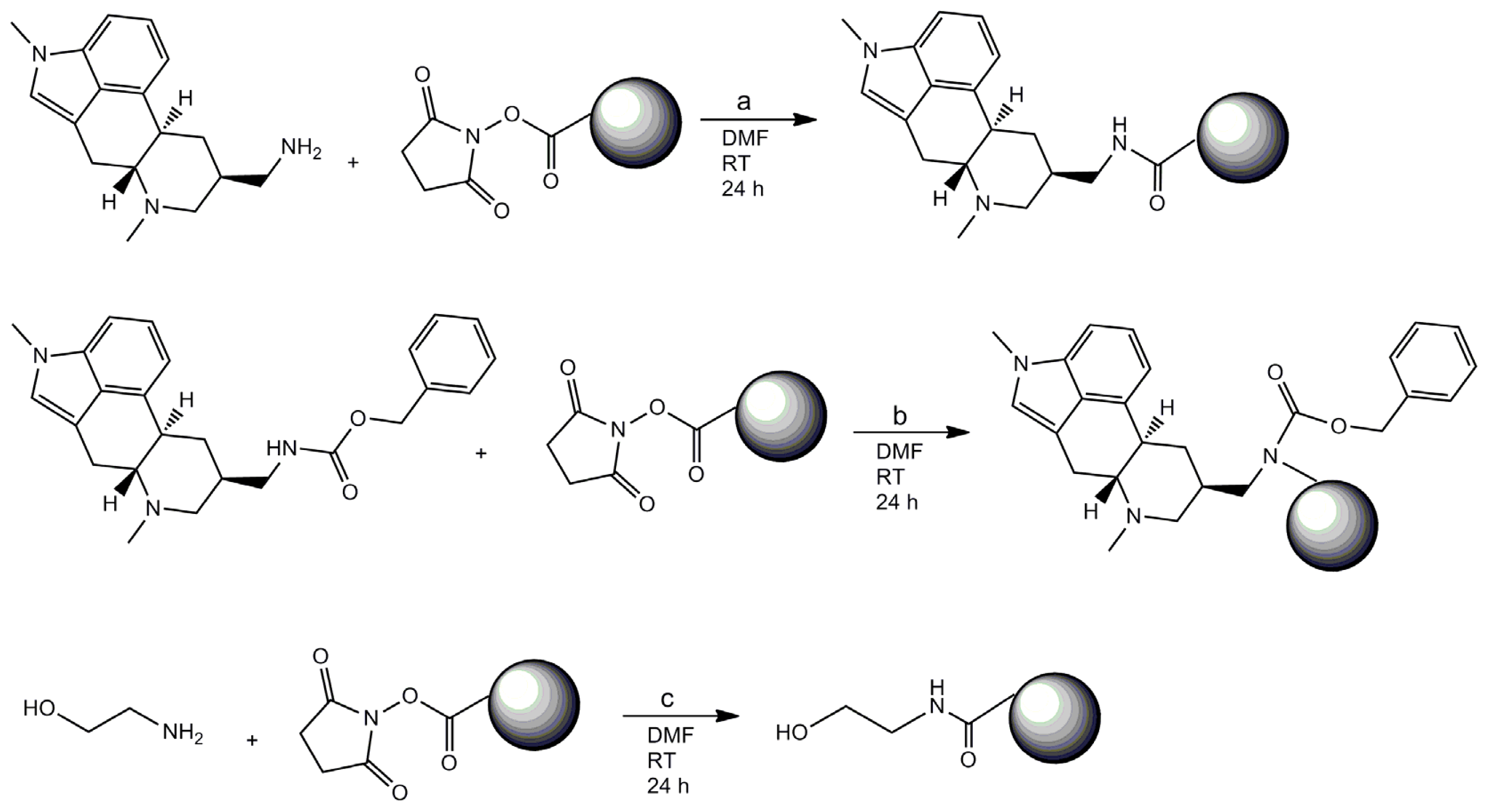
3.1. Coating of Magnetic Beads with Lysergamine and Metergoline
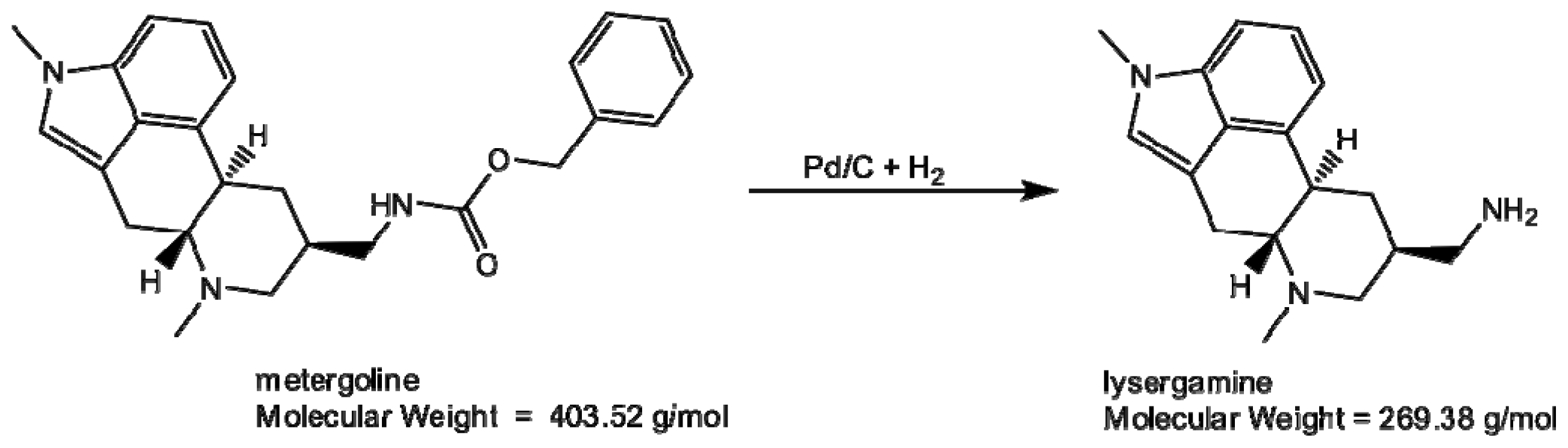
3.1.1. Production of Lysergamine and Preparation of Lysergamine Coated Beads
3.1.2. Preparation of Lysergamine Coated Magnetic Beads
3.1.3. Preparation of Metergoline Coated Magnetic Beads
3.1.4. Preparation of Ethanolamine Coated Beads
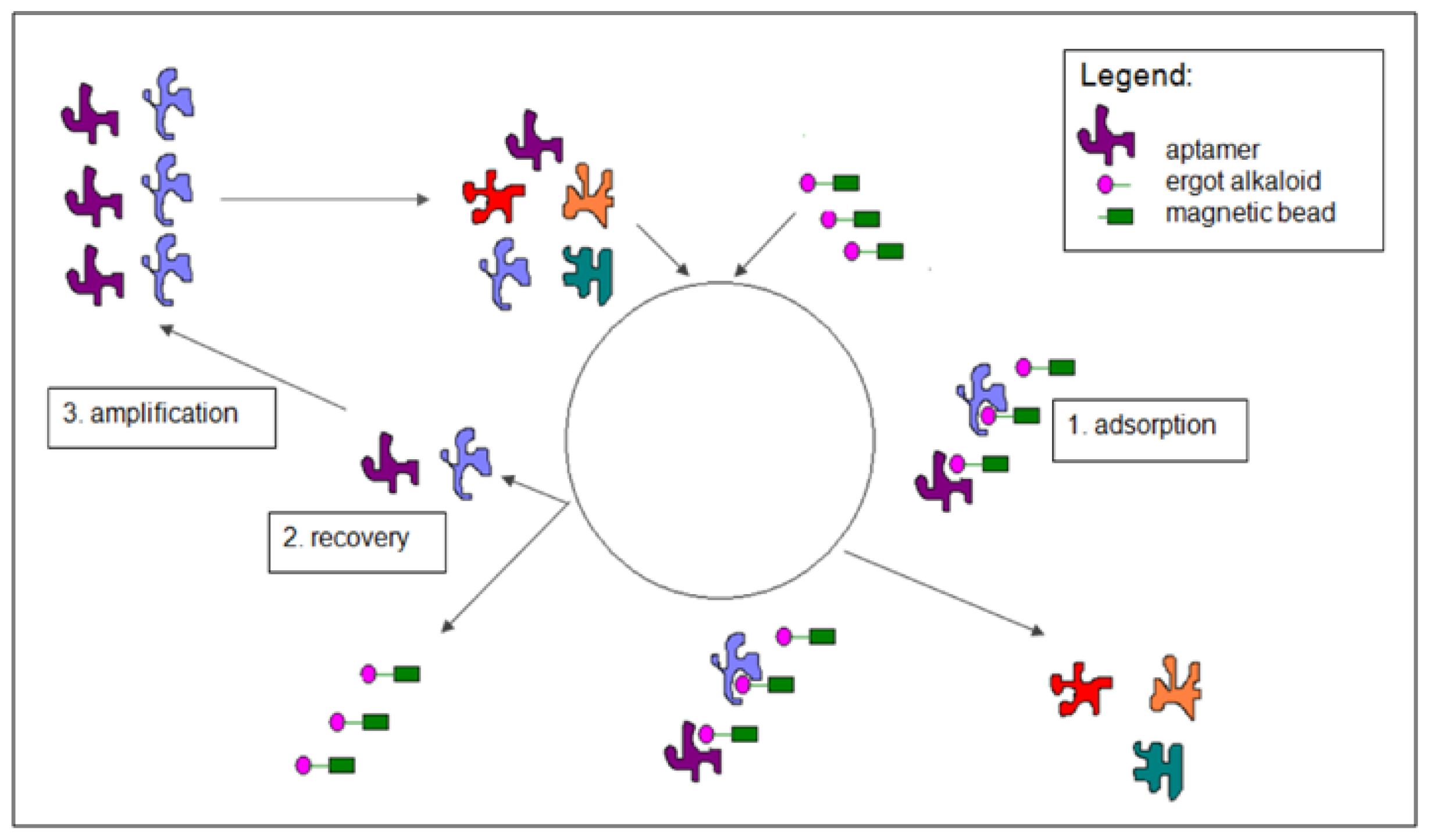
3.2. Selection of DNA Aptamers for Lysergamine and Metergoline
3.2.1. Selection Procedure
3.2.2. Elution of ssDNA
3.2.3. Precipitation of ssDNA
3.2.4. Amplification of the selected ssDNA aptamers by PCR
3.2.5. Preparation of ssDNA from Biotinylated PCR Product
3.2.6. Cloning and Sequencing of Aptamers
3.3. SPR Analysis of the Complexes of Aptamers and Lysergamine
3.3.1. Preparation of Biotinylated Lysergamine Linker
3.3.2. Preparation of Biotinylated Ethanolamine Linker
3.3.3. Coating SPR Chip with Biotinylated Linkers
3.4. Colorimetric Reaction with Aptamer Linker to Gold Nanoparticles
4. Conclusions
Supplementary Information
ijms-13-17138-s001.pdfAcknowledgments
References
- Scott, P. Analysis of ergot alkaloids—A review. Mycotoxin Res 2007, 23, 113–121. [Google Scholar]
- Van Dongen, P.W.J.; de Groot, A.N.J.A. History of ergot alkaloids from ergotism to ergometrine. Eur. J. Obstet. Gynecol. Reprod. Biol 1995, 60, 109–116. [Google Scholar]
- Mulac, D.; Humpf, H.-U. Cytotoxicity and accumulation of ergot alkaloids in human primary cells. Toxicology 2011, 282, 112–121. [Google Scholar]
- Mainka, S.; Dänicke, S.; Böhme, H.; Ueberschär, K.H.; Liebert, F. On the composition of ergot and the effects of feeding two different ergot sources on piglets. Anim. Feed Sci. Technol 2007, 139, 52–68. [Google Scholar]
- Mantegani, S.; Brambilla, E.; Varasi, M. Ergoline derivatives: Receptor affinity and selectivity. II Farmaco 1999, 54, 288–296. [Google Scholar]
- Mohamed, R.; Gremaud, E.; Richoz-Payot, J.; Tabet, J.-C.; Guy, P.A. Quantitative determination of five ergot alkaloids in rye flour by liquid chromatography-electrospray ionisation tandem mass spectrometry. J. Chromatogr 2006, 1114, 62–72. [Google Scholar]
- Luzi, E.; Minunni, M.; Tombelli, S.; Mascini, M. New trends in affinity sensing: Aptamers for ligand binding. Trends Anal. Chem 2003, 22, 810–818. [Google Scholar]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng 2007, 24, 381–403. [Google Scholar]
- Torres-Chavolla, E.; Alocilja, E.C. Aptasensors for detection of microbial and viral pathogens. Biosens. Bioelectron 2009, 24, 3175–3182. [Google Scholar]
- Michael, F. Oligonucleotide aptamers that recognize small molecules. Curr. Opin. Struct. Biol 1999, 9, 324–329. [Google Scholar]
- Van Dorst, B.; Mehta, J.; Bekaert, K.; Rouah-Martin, E.; de Coen, W.; Dubruel, P.; Blust, R.; Robbens, J. Recent advances in recognition elements of food and environmental biosensors: A review. Biosens. Bioelectron 2010, 26, 1178–1194. [Google Scholar]
- Musheev, M.U.; Krylov, S.N. Selection of aptamers by systematic evolution of ligands by exponential enrichment: Addressing the polymerase chain reaction issue. Anal. Chim. Acta 2006, 564, 91–96. [Google Scholar]
- Ellington, A.D.; Szostak, J.W. In Vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar]
- Hamula, C.L.A.; Zhang, H.; Li, F.; Wang, Z.; Chris Le, X.; Li, X.-F. Selection and analytical applications of aptamers binding microbial pathogens. Trends Anal. Chem 2011, 30, 1587–1597. [Google Scholar]
- Djordjevic, M. SELEX experiments: New prospects, applications and data analysis in inferring regulatory pathways. Biomol. Eng 2007, 24, 179–189. [Google Scholar]
- Wilson, W.D. Analyzing biomolecular interactions. Science 2002, 295, 2103–2105. [Google Scholar]
- Hušák, M.; Jegorov, A.; Brus, J.; van Beek, W.; Pattison, P.; Christensen, M.; Favre-Nicolin, V.; Maixner, J. Metergoline II: Structure solution from powder diffraction data with preferred orientation and from microcrystal. Struct. Chem 2008, 19, 517–525. [Google Scholar]
- Xiao, P.; Lv, X.; Deng, Y. Immobilization of chymotrypsin on silica beads based on high affinity and specificity aptamer and its applications. Anal. Lett 2012, 45, 1264–1273. [Google Scholar]
- Oster, J.; Parker, J.; à Brassard, L. Polyvinyl-alcohol-based magnetic beads for rapid and efficient separation of specific or unspecific nucleic acid sequences. J. Magn. Magn. Mater 2001, 225, 145–150. [Google Scholar]
- Hooker, J.M.; Reibel, A.T.; Hill, S.M.; Schueller, M.J.; Fowler, J.S. One-pot, direct incorporation of [11C]CO2 into carbamates. Angew. Chem. Int. Ed 2009, 48, 3482–3485. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl. Acids Res 2003, 31, 3406–3415. [Google Scholar]
- Cruz-Aguado, J.A.; Penner, G. Determination of ochratoxin A with a DNA aptamer. J. Agric. Food Chem 2008, 56, 10456–10461. [Google Scholar]
- Ogawa, A.; Tomita, N.; Kikuchi, N.; Sando, S.; Aoyama, Y. Aptamer selection for the inhibition of cell adhesion with fibronectin as target. Bioorg. Med. Chem. Lett 2004, 14, 4001–4004. [Google Scholar]
- Huizenga, D.E.; Szostak, J.W. A DNA aptamer that binds adenosine and ATP. Biochemistry 1995, 34, 656–665. [Google Scholar]
- Fialová, M.; Kypr, J.; Vorlíčková, M. The thrombin binding aptamer GGTTGGTGTGGTTGG forms a bimolecular guanine tetraplex. Biochem. Biophys. Res. Commun 2006, 344, 50–54. [Google Scholar]
- Stojanovic, M.N.; de Prada, P.; Landry, D.W. Aptamer-based folding fluorescent sensor for cocaine. J. Am. Chem. Soc 2001, 123, 4928–4931. [Google Scholar]
- McKeague, M.; Bradley, C.R.; de Girolamo, A.; Visconti, A.; Miller, J.D.; DeRosa, M.C. Screening and initial binding assessment of fumonisin B1 aptamers. Int. J. Mol. Sci 2010, 11, 4864–4881. [Google Scholar]
- Mehta, J.; Rouah-Martin, E.; van Dorst, B.; Maes, B.; Herrebout, W.; Scippo, M.-L.; Dardenne, F.; Blust, R.; Robbens, J. Selection and characterization of PCB-binding DNA aptamers. Anal. Chem 2012, 84, 1669–1676. [Google Scholar]
- Wilchek, M.; Bayer, E.A. The avidin-biotin complex in bioanalytical applications. Anal. Biochem 1988, 171, 1–32. [Google Scholar]
- Hamula, C.L.A.; Guthrie, J.W.; Zhang, H.; Li, X.-F.; Le, X.C. Selection and analytical applications of aptamers. Trends Anal. Chem 2006, 25, 681–691. [Google Scholar]
- Liu, J.; Lu, Y. Preparation of aptamer-linked gold nanoparticle purple aggregates for colorimetric sensing of analytes. Nat. Protoc 2006, 1, 246–252. [Google Scholar]
- Liu, J.; Lu, Y. Fast colorimetric sensing of adenosine and cocaine based on a general sensor design involving aptamers and nanoparticles. Angew. Chem 2006, 118, 96–100. [Google Scholar]
- Rankin, C.J.; Fuller, E.N.; Hamor, K.H.; Gabarra, S.A.; Shields, T.P. A simple fluorescent biosensor for theophylline based on its RNA aptamer. Nucleos. Nucleot. Nucl 2006, 25, 1407–1424. [Google Scholar]
- Diana di Mavingu, J.; Malysheva, S.V.; Sanders, M.; Larionova, D.; Robbens, J.; Dubruel, P.; van Peteghem, C.; de Saeger, S. Development and validation of a new LC-MS/MS method for the simultaneous determination of six major ergot alkaloids and their corresponding epimers. Application to some food and feed commodities. Food Chem. 2012, 135, 292–303. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fitting Model | Two-site specific binding | One-site specific binding | |
|---|---|---|---|
| Best fit values for | Aptamer M3.2 | Aptamer L5.2 | Aptamer L5.7 |
| Bmax (RU) | 205.2 | 585.8 | 531.0 |
| Kd | 44 nmol2/L2 | 73 nmol/L | 499 nmol/L |
| R2 | 0.997 | 0.993 | 0.991 |
| Number of points analyzed | 6 | 7 | 6 |
| Degree of freedom | 2 | 5 | 4 |
| Ergot alkaloid | Concentration (μg/kg) | Total epimers | % of ergot alkaloid content |
|---|---|---|---|
| Ergometrine | 106 | 132 | 4.3 |
| Ergometrinine | 26 | ||
| Ergosine | 1567 | 1567 | 51 |
| Ergosinine | <LOQ | ||
| Ergotamine | 22 | 24 | 0.8 |
| Ergotaminine | 2 | ||
| Ergocornine | 65 | 84 | 2.7 |
| Ergocorninine | 19 | ||
| Ergokryptine | 1251 | 1259 | 41 |
| Ergokryptinine | 8 | ||
| Ergocristine | <LOQ | 3 | 0.1 |
| Ergocristinine | 3 | ||
| Total | 3068 | 3068 | 100 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rouah-Martin, E.; Mehta, J.; Van Dorst, B.; De Saeger, S.; Dubruel, P.; Maes, B.U.W.; Lemiere, F.; Goormaghtigh, E.; Daems, D.; Herrebout, W.; et al. Aptamer-Based Molecular Recognition of Lysergamine, Metergoline and Small Ergot Alkaloids. Int. J. Mol. Sci. 2012, 13, 17138-17159. https://doi.org/10.3390/ijms131217138
Rouah-Martin E, Mehta J, Van Dorst B, De Saeger S, Dubruel P, Maes BUW, Lemiere F, Goormaghtigh E, Daems D, Herrebout W, et al. Aptamer-Based Molecular Recognition of Lysergamine, Metergoline and Small Ergot Alkaloids. International Journal of Molecular Sciences. 2012; 13(12):17138-17159. https://doi.org/10.3390/ijms131217138
Chicago/Turabian StyleRouah-Martin, Elsa, Jaytry Mehta, Bieke Van Dorst, Sarah De Saeger, Peter Dubruel, Bert U. W. Maes, Filip Lemiere, Erik Goormaghtigh, Devin Daems, Wouter Herrebout, and et al. 2012. "Aptamer-Based Molecular Recognition of Lysergamine, Metergoline and Small Ergot Alkaloids" International Journal of Molecular Sciences 13, no. 12: 17138-17159. https://doi.org/10.3390/ijms131217138