UVA Irradiation of Dysplastic Keratinocytes: Oxidative Damage versus Antioxidant Defense

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
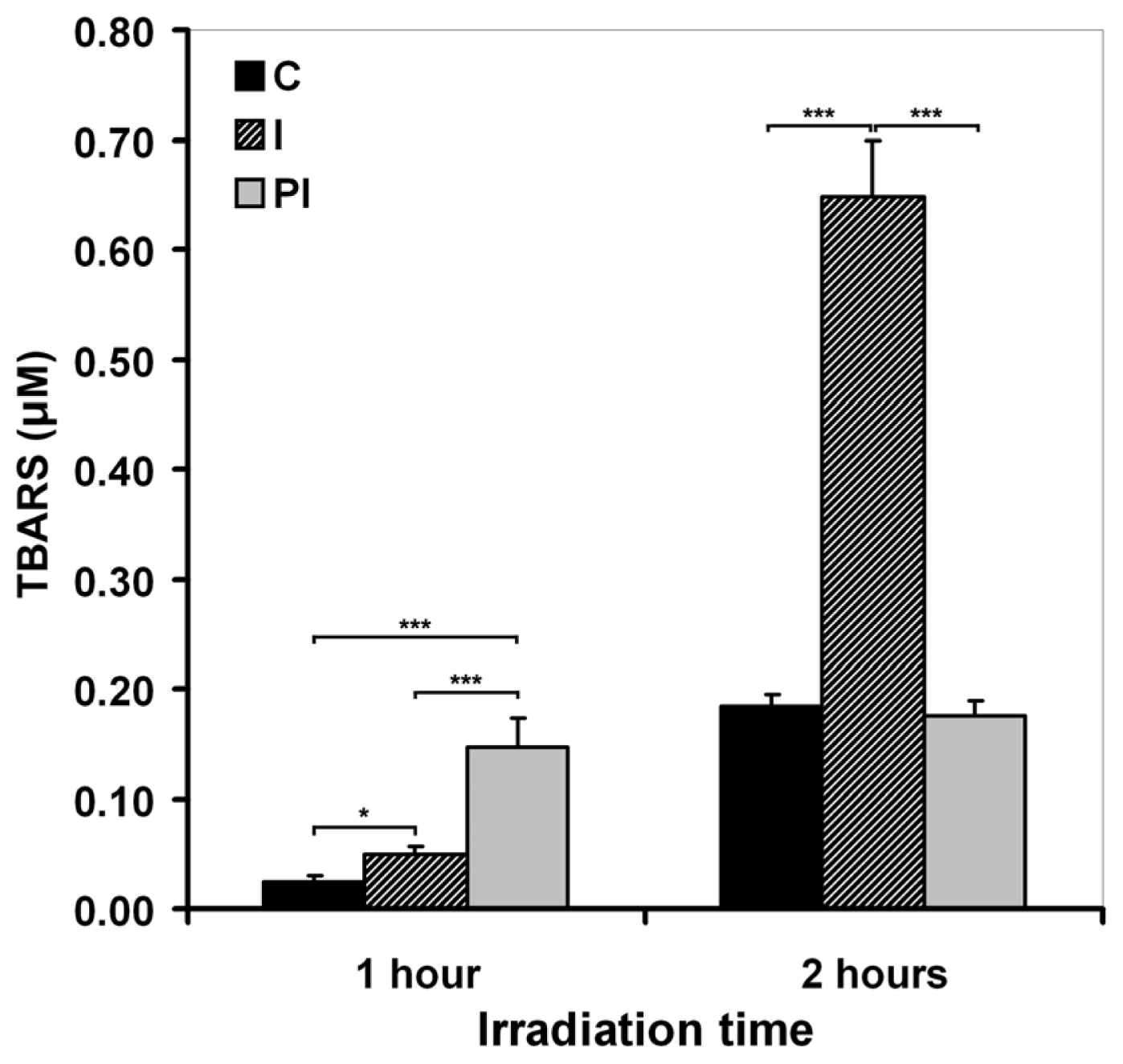
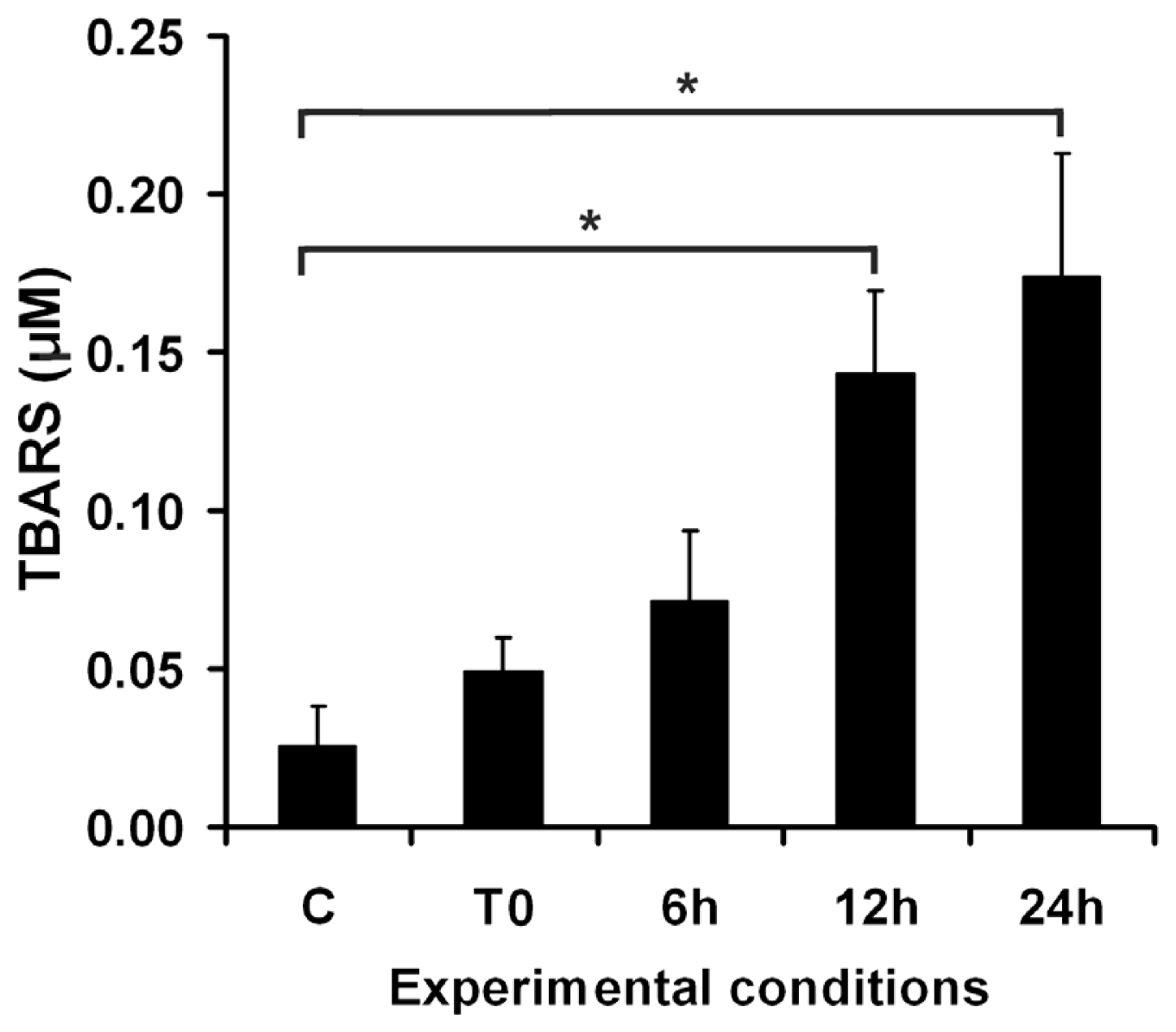
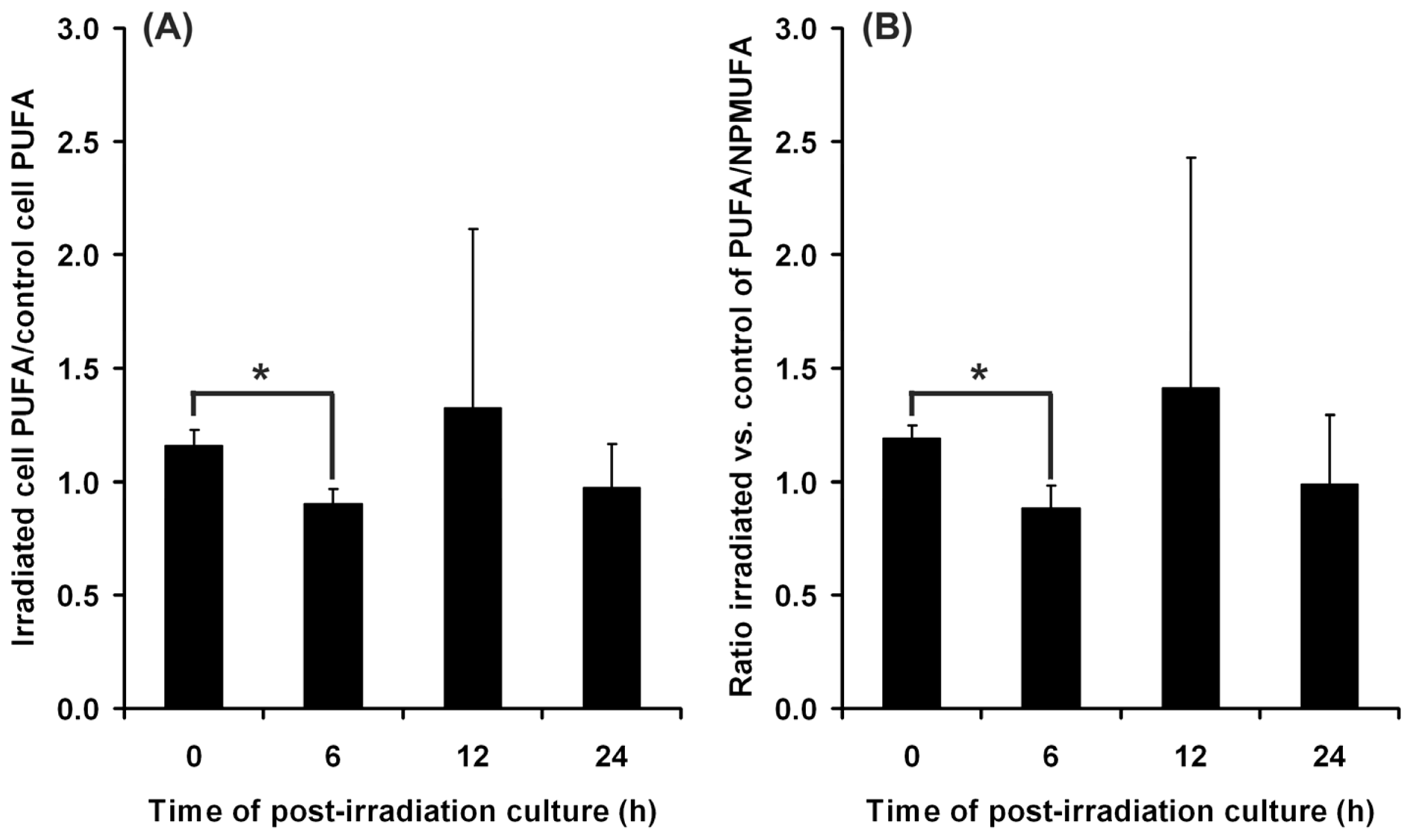
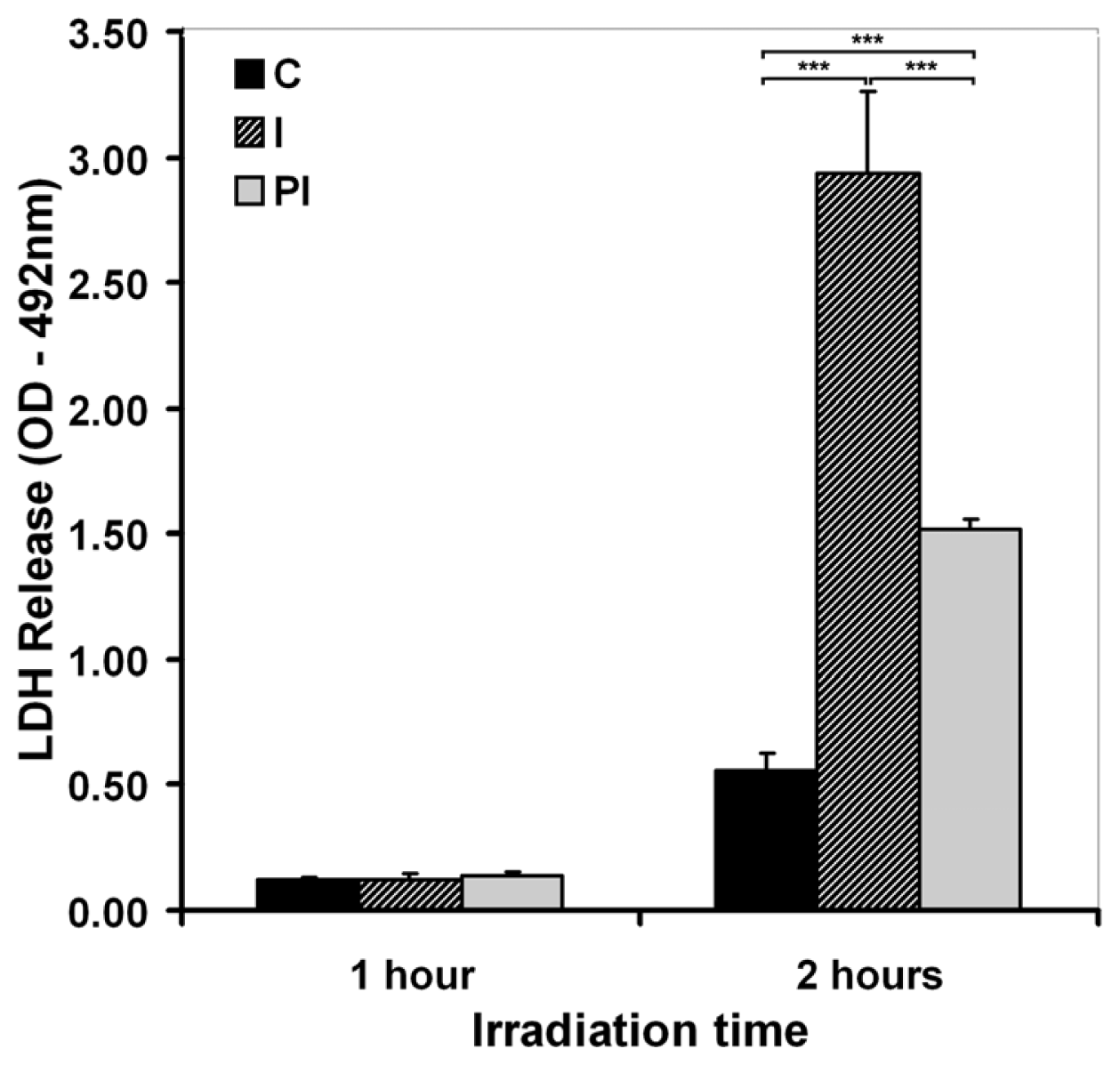
2.1. Effects of UVA on Keratinocyte Cell Membrane
2.1.1. Cell Membrane Biochemistry
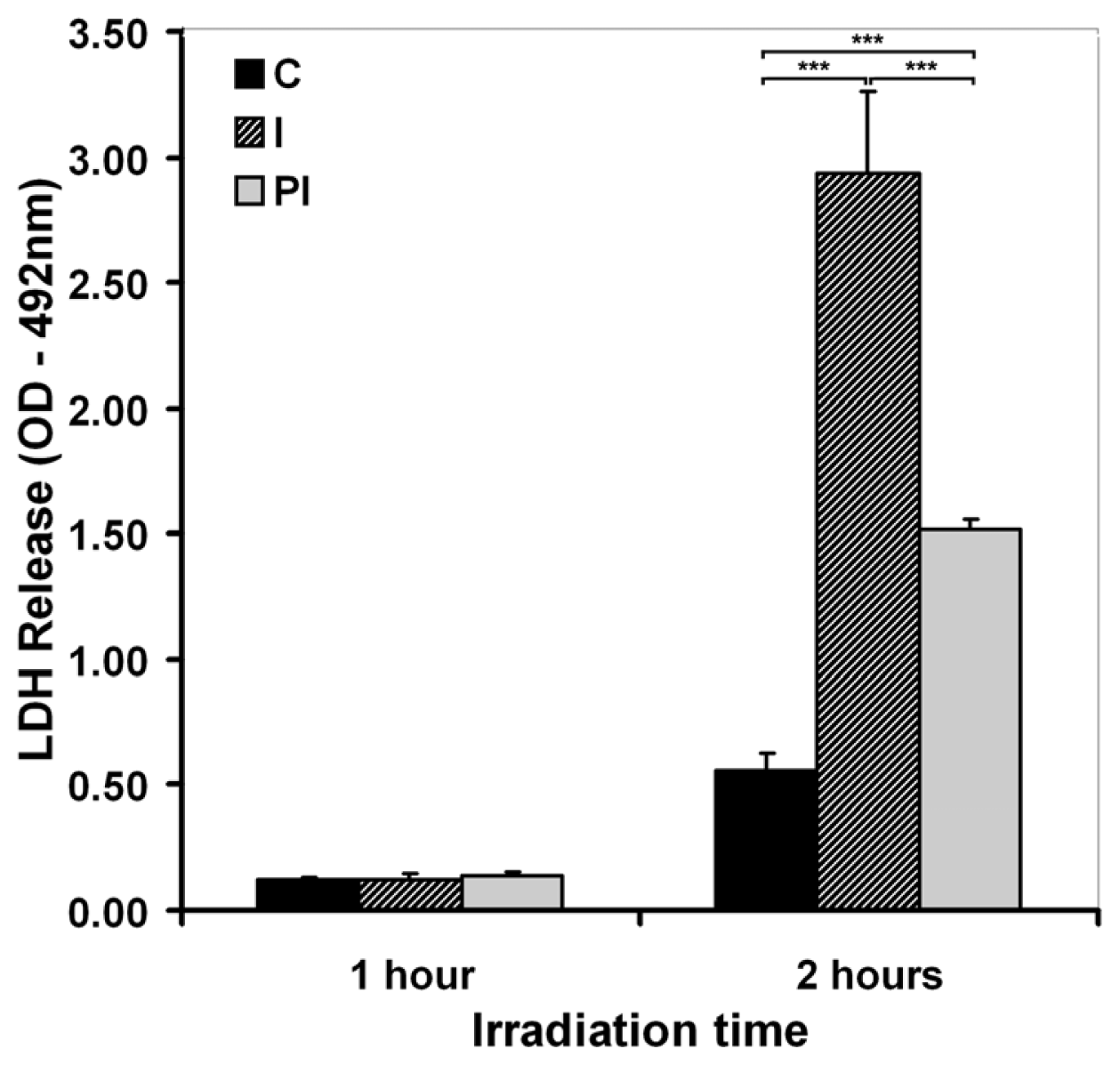
2.1.2. Cell Membrane Integrity and Permeability
2.2. Effects of UVA on Catalase Activity and Expression
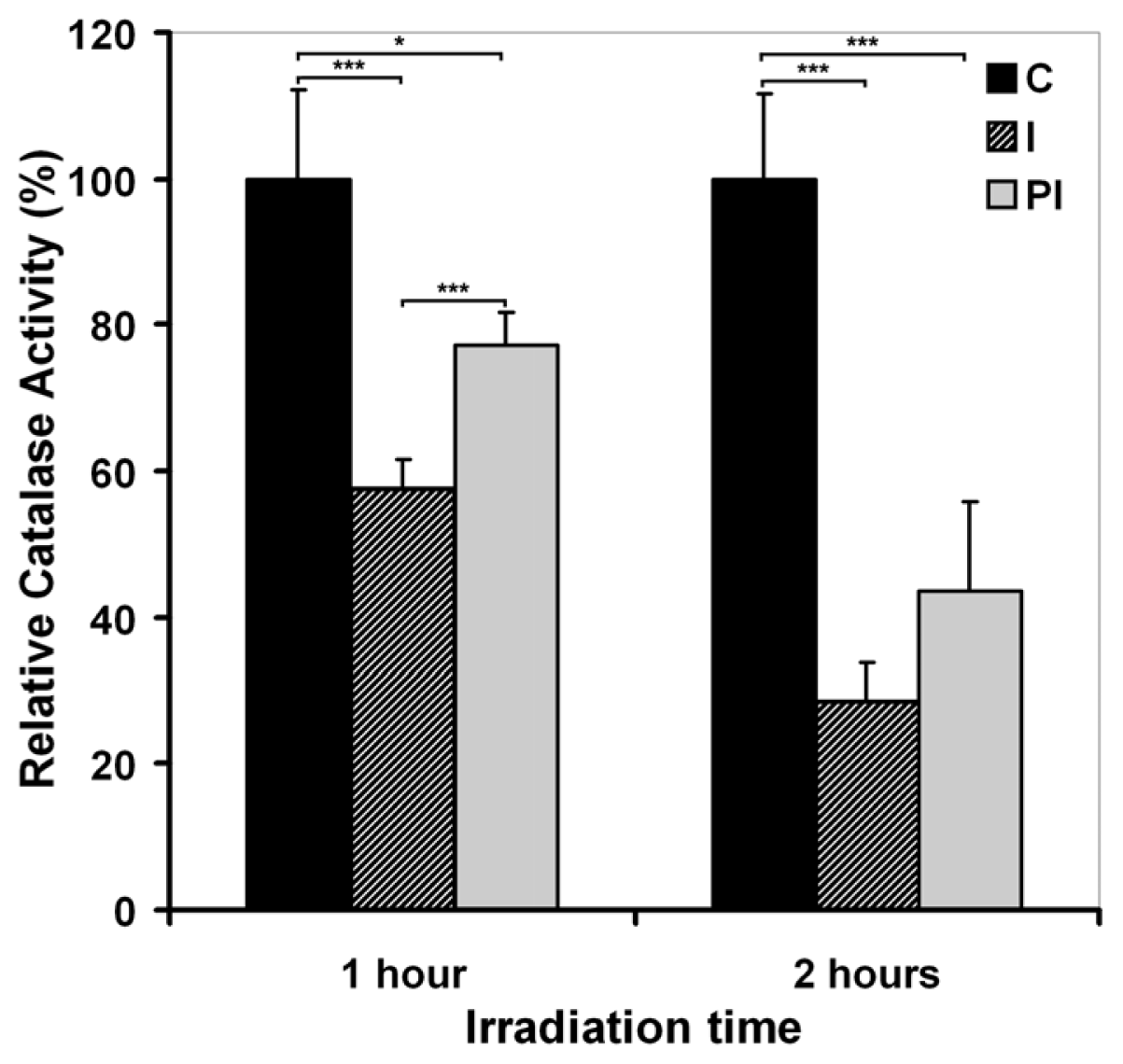
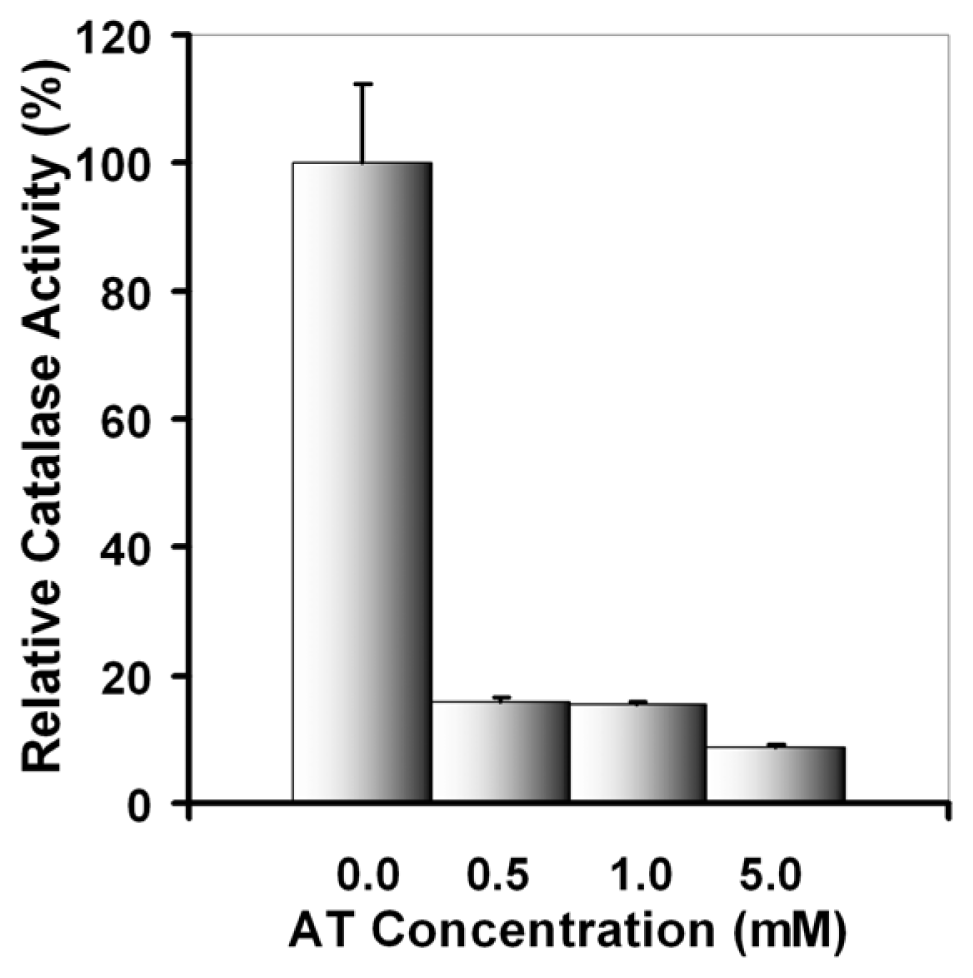
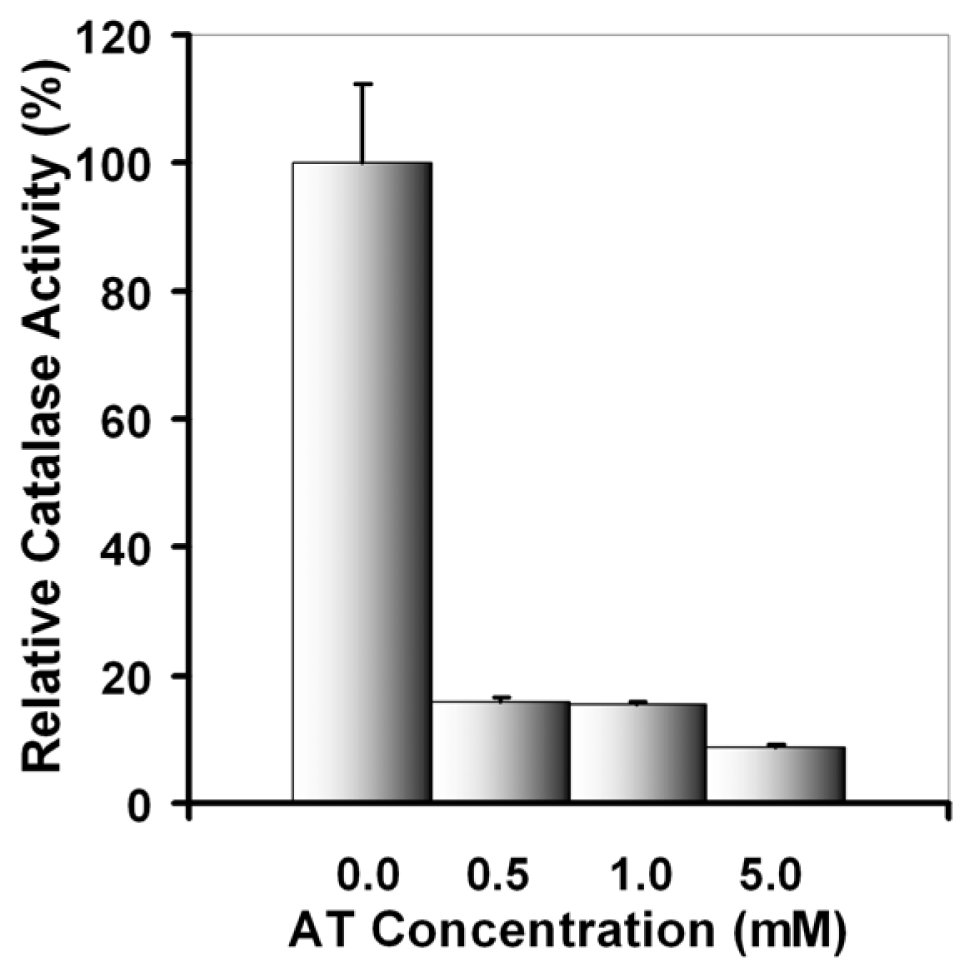
2.2.1. UVA Effect on Catalase Activity
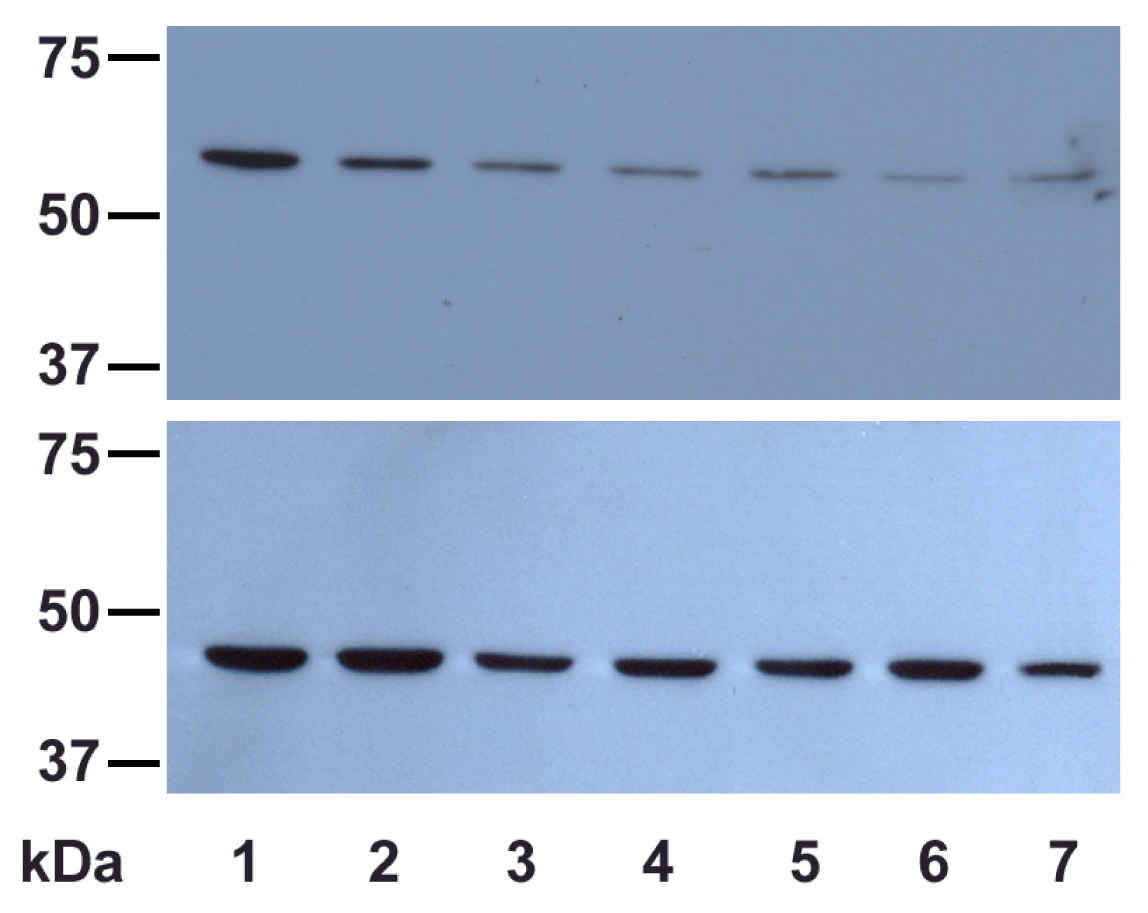
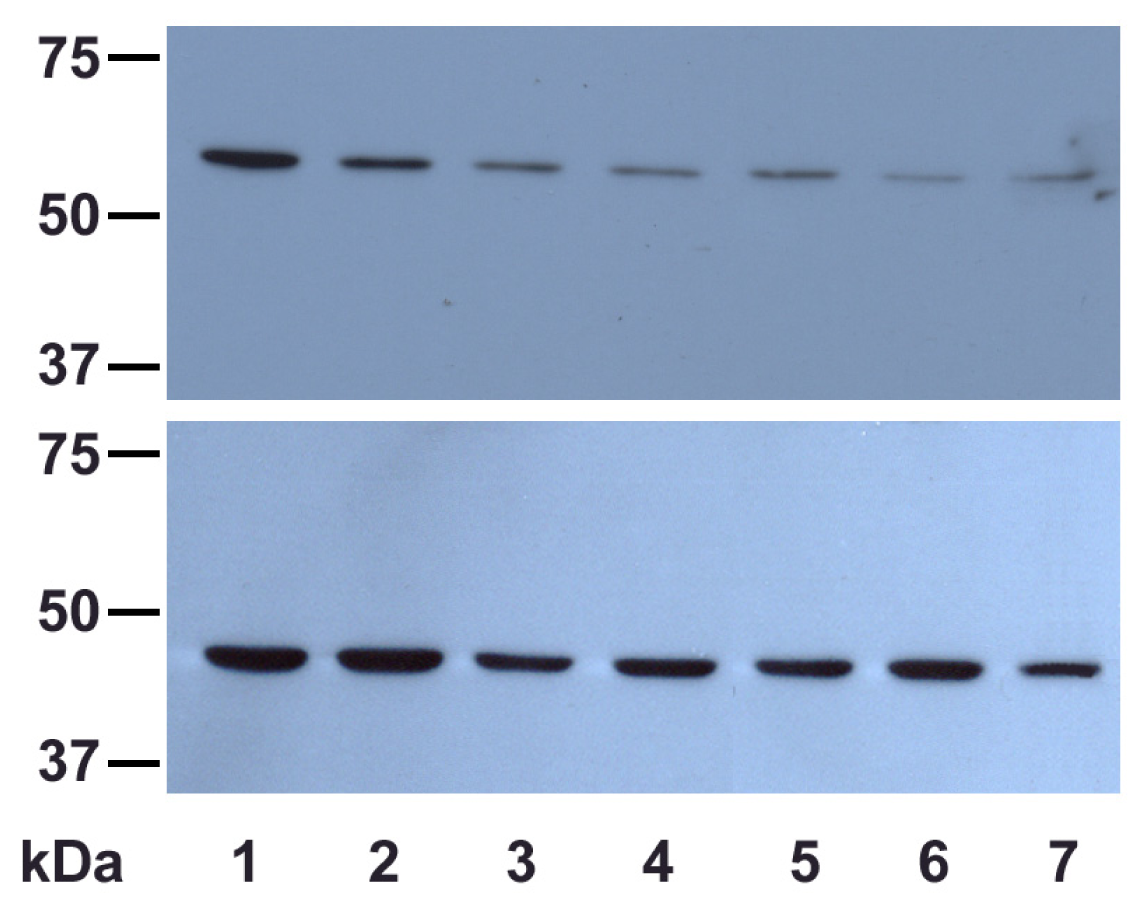
2.2.2. UVA Effect on Catalase Expression
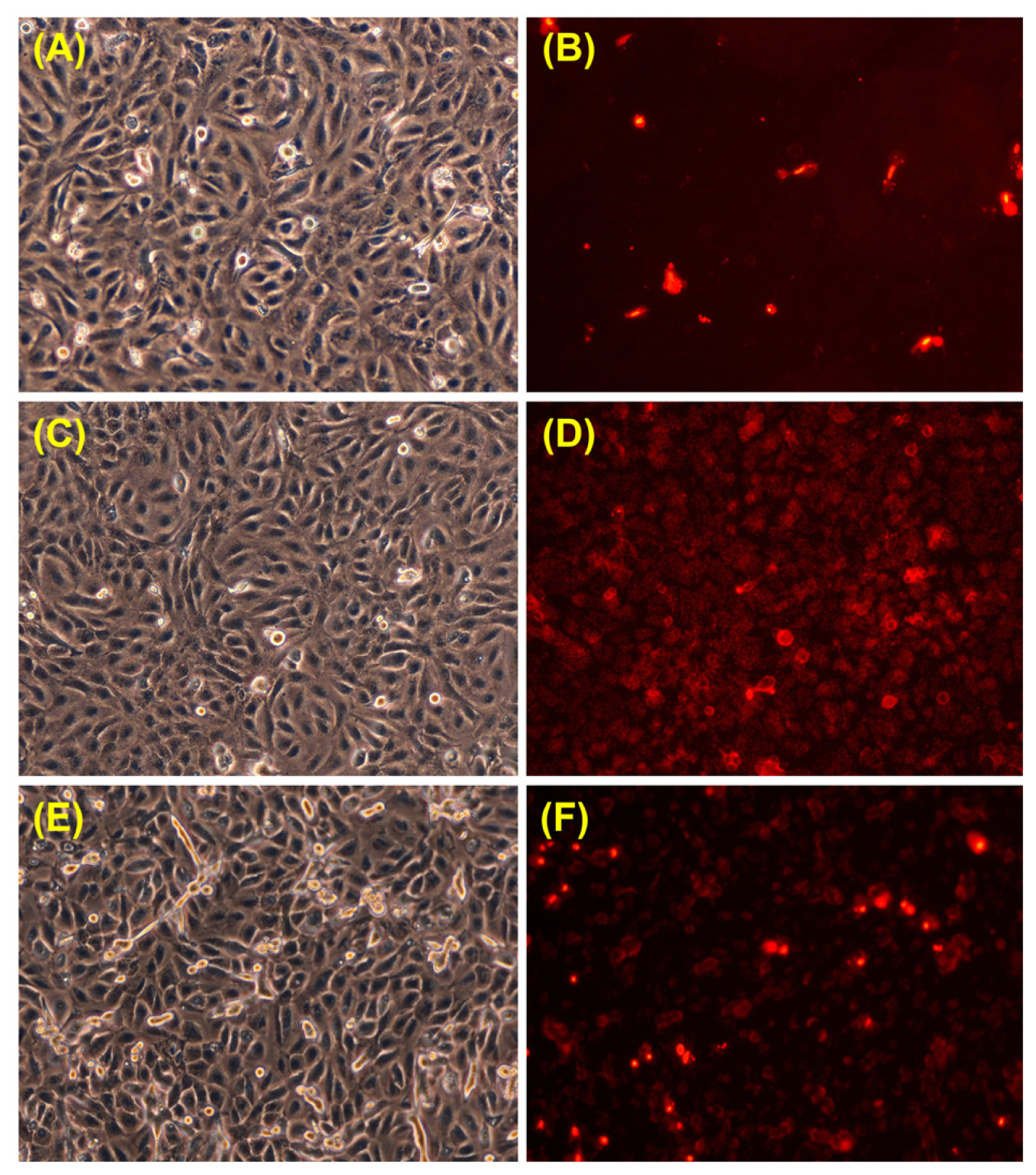
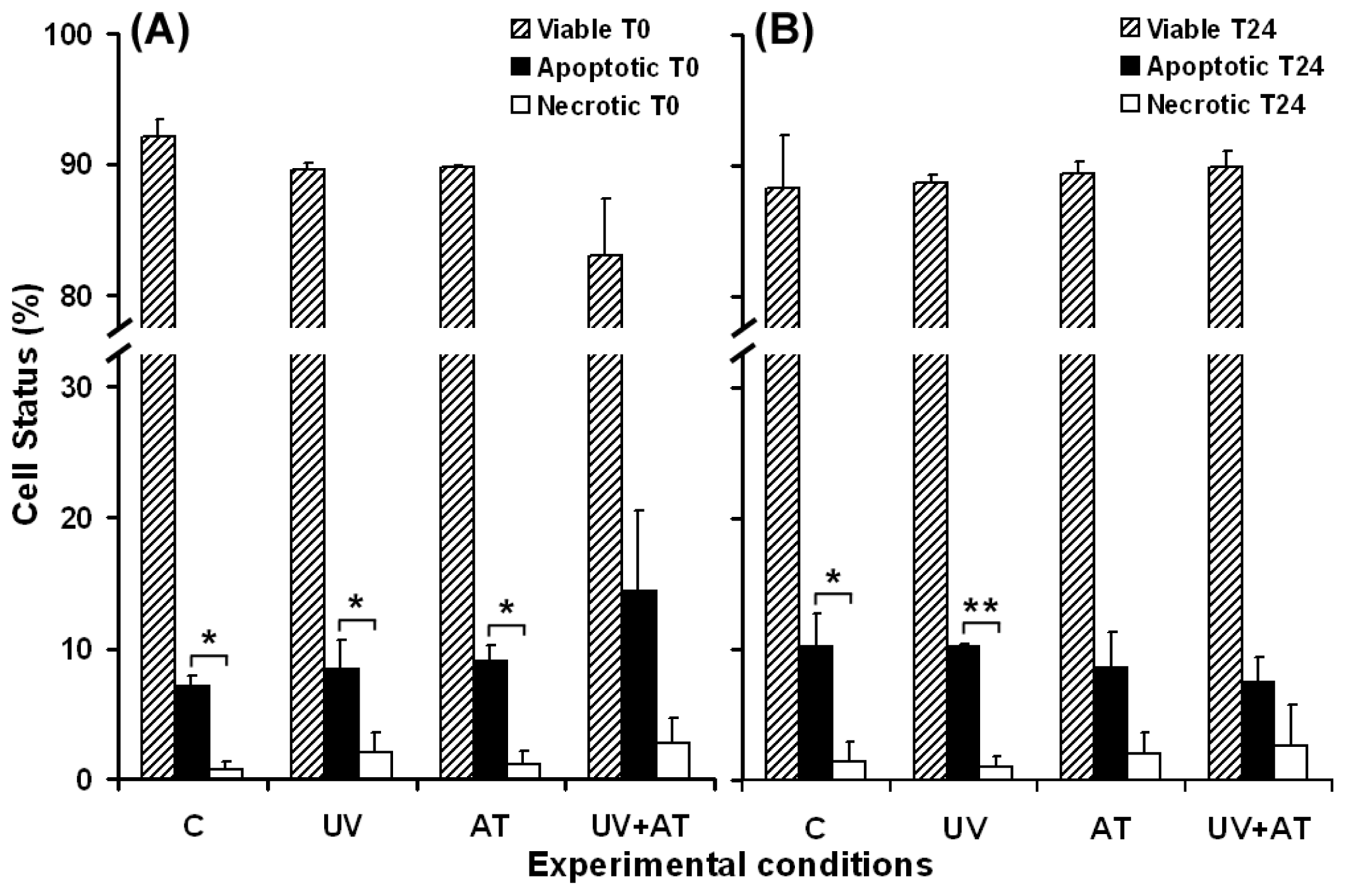
2.3. Effects of UVA Exposure and Catalase Inhibition on Keratinocyte Fate
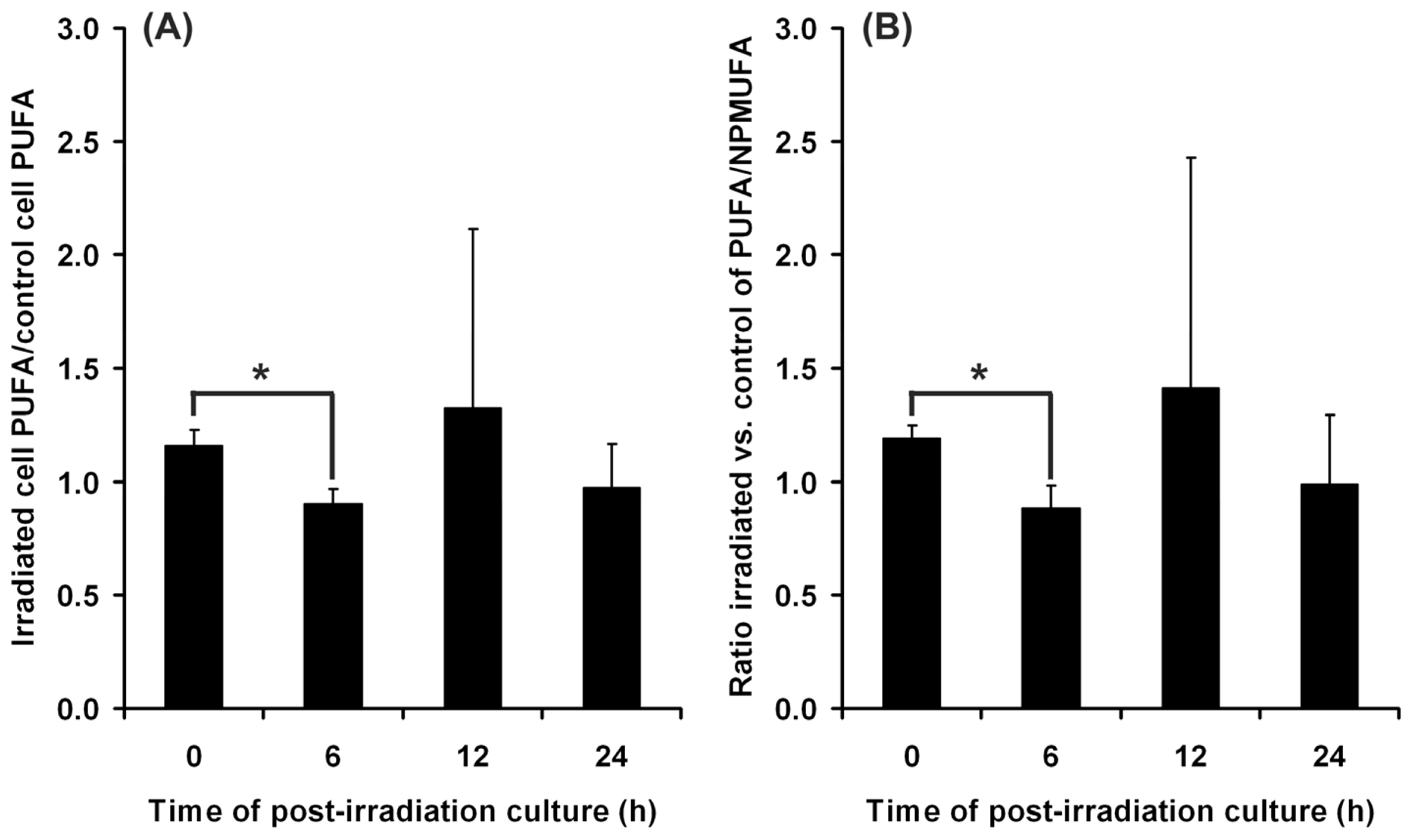
2.4. Adaptive Response of DOK Cells to UVA Radiation
3. Experimental Section
3.1. Cell Culture
3.2. UVA Source and Irradiation Conditions
3.3. TBARS Assay
3.4. Fatty Acid Gas Chromatography
3.5. LDH Release Assay
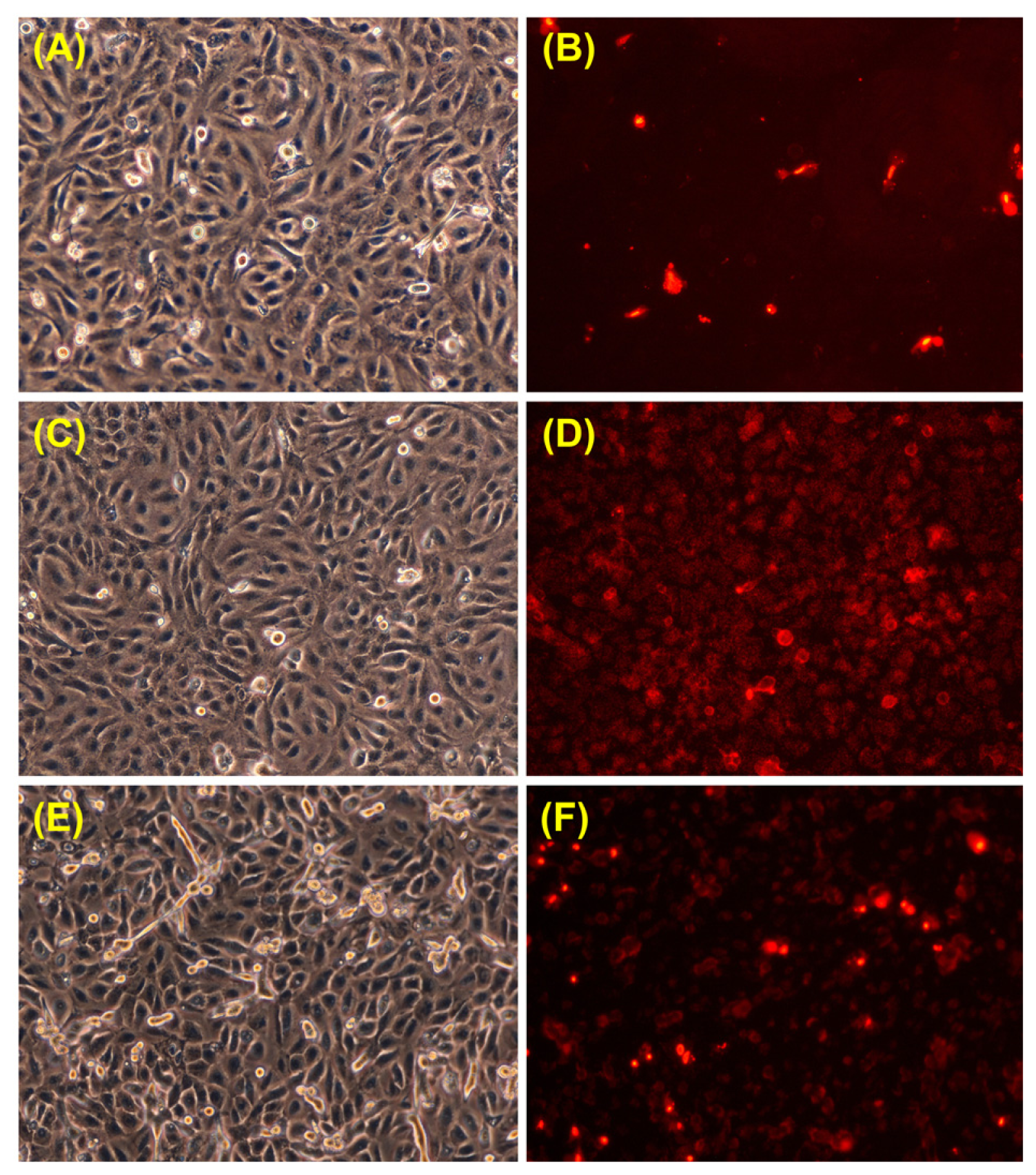
3.6. Propidium Iodide Staining
3.7. Catalase Activity Assay
3.8. Electrophoresis and Western Blotting
3.9. Flow Cytometry Assay
3.10. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Krutmann, J. Ultraviolet A radiation-induced biological effects in human skin: Relevance for photoaging and photodermatosis. J. Dermatol. Sci 2000, 23, S22–S26. [Google Scholar]
- Honda, A.; Abe, R.; Makino, T.; Norisugi, O.; Fujita, Y.; Watanabe, H.; Nishihira, J.; Jwakura, Y.; Yamagishi, S.; Shimizu, H.; Shimizu, T. Interleukin-1β and macrophage migration inhibitory factor (MIF) in dermal fibroblasts mediate UVA-induced matrix metalloproteinase-1 expression. J. Dermatol. Sci 2008, 49, 63–72. [Google Scholar]
- Onoue, S.; Nonaka, R.; Sato, F.; Koide, C.; Hayashi, A.; Wachi, H. Involvement of reactive oxygen species in abnormal tropoelastin deposition induced by UVA-photosensitizers. J. Health Sci 2009, 55, 946–951. [Google Scholar]
- He, Y.Y.; Pi, J.; Huang, J.L.; Diwan, B.A.; Waalkes, M.P.; Chignell, C.F. Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance. Oncogene 2006, 25, 3680–3688. [Google Scholar]
- Jans, J.; Garinis, G.A.; Schul, W.; van Oudenaren, A.; Moorhouse, M.; Smid, M.; Sert, Y.G.; van der Velde, A.; Rijksen, Y.; de Gruijl, F.R.; et al. Differential role of basal keratinocytes in UV-induced immunosuppression and skin cancer. Mol. Cell Biol 2006, 26, 8515–8526. [Google Scholar]
- Brash, D.E.; Heffernan, T.P.; Nghiem, P. Carcinogenesis: UV Radiation. In Textbook of Aging Skin; Farage, M.A., Miller, K.W., et al., Eds.; Springer-Verlag: Berlin, Germany, 2010; pp. 567–578. [Google Scholar]
- Sage, E.; Girard, P.M.; Francesconi, S. Unravelling UVA-induced mutagenesis. Photochem. Photobiol. Sci 2012, 11, 74–80. [Google Scholar]
- Wickelgren, I. Skin biology: A healthy tan? Science 2007, 315, 1214–1216. [Google Scholar]
- Nishimura, H.; Yasui, H.; Sakurai, H. Generation and distribution of reactive oxygen species in the skin of hairless mice under UVA: Studies on in vivo chemiluminescent detection and tape stripping methods. Exp. Dermatol 2006, 15, 891–899. [Google Scholar]
- Valencia, A.; Kochevar, I.E. Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes. J. Invest. Dermatol 2008, 128, 214–222. [Google Scholar]
- Dalle Carbonare, M.; Pathak, M.A. Skin photosensitizing agents and the role of reactive oxygen species in photoaging. J. Photochem. Photobiol. B 1992, 14, 105–124. [Google Scholar]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem. Photobiol. Sci 2006, 5, 215–237. [Google Scholar]
- Yu, H.; Xia, O.; Yan, J.; Herreno-Saenz, D.; Wu, Y.S.; Tanq, I.W.; Fu, P.P. Photoirradiation of polycyclic aromatic hydrocarbons with UVA light—A pathway leading to the generation of reactive oxygen species, lipid peroxidation and DNA damage. Int. J. Environ. Res. Public Health 2006, 3, 348–354. [Google Scholar]
- Herrling, T.; Fuchs, J.; Rehberg, J.; Groth, N. UV-induced free radicals in the skin detected by ESR spectroscopy and imaging using nitroxides. Free Radic. Biol. Med 2003, 35, 59–67. [Google Scholar]
- Sakurai, H.; Yasui, H.; Yamada, Y.; Nishimura, H.; Shigemoto, M. Detection of reactive oxygen species in the skin of live mice and rats exposed to UVA light: A research review on chemiluminescence and trials for UVA protection. Photochem. Photobiol. Sci 2005, 4, 715–720. [Google Scholar]
- Baier, J.; Maisch, T.; Maier, M.; Engel, E.; Landthaler, M.; Baumler, W. Singlet oxygen generation by UVA light exposure of endogenous photosensitizers. Biophys. J 2006, 91, 1452–1459. [Google Scholar]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med 2000, 28, 463–499. [Google Scholar]
- Assefa, Z.; van Laethem, A.; Garmyn, M.; Agostinis, P. Ultraviolet radiation-induced apoptosis in keratinocytes: On the role of cytosolic factors. Biochim. Biophys. Acta 2005, 1755, 90–106. [Google Scholar]
- Kurita, M.; Shimauchi, T.; Kobayashi, M.; Atarashi, K.; Mori, K.; Tokura, Y. Induction of keratinocyte apoptosis by photosensitizing chemicals plus UVA. J. Dermatol. Sci 2007, 45, 105–112. [Google Scholar]
- Schneider, L.A.; Dissemond, J.; Brenneisen, P.; Hainzl, A.; Briviba, K.; Wlaschek, M.; Scharffetter-Kochanek, K. Adaptive cellular protection against UVA-1-induced lipid peroxidation in human dermal fibroblasts shows donor-to-donor variability and is glutathione dependent. Arch. Dermatol. Res 2006, 297, 324–328. [Google Scholar]
- Leccia, M.T.; Yaar, M.; Allen, N.; Gleason, M.; Gilchrest, B.A. Solar simulated irradiation modulates gene expression and activity of antioxidant enzymes in cultured human dermal fibroblasts. Exp. Dermatol 2001, 10, 272–279. [Google Scholar]
- Applegate, L.A.; Frenk, E. Cellular defense mechanisms of the skin against oxidant stress and in particular UVA radiation. Eur. J. Dermatol 1995, 5, 97–103. [Google Scholar]
- Rezvani, H.R.; Cario-André, M.; Pain, C.; Ged, C.; deVerneuil, H.; Taïeb, A. Protection of normal human reconstructed epidermis from UV by catalase overexpression. Cancer Gene Ther 2007, 14, 174–186. [Google Scholar]
- Morel, Y.; Barouki, R. Repression of gene expression by oxidative stress. Biochem. J 1999, 342, 481–496. [Google Scholar]
- Bickers, D.R.; Athar, M. Oxidative stress in pathogenesis of skin disease. J. Invest. Dermatol 2006, 126, 2565–2575. [Google Scholar]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar]
- Hanselmann, C.; Mauch, C.; Werner, S. Haem oxygenase-1: A novel player in cutaneous wound repair and psoriasis? Biochem. J 2001, 353, 459–466. [Google Scholar]
- Sedlak, T.W.; Snyder, S.H. Bilirubin benefits: Cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 2004, 113, 1776–1782. [Google Scholar]
- Kawanishi, S.; Hiraku, Y. Sequence specific DNA damage induced by UVA radiation in the presence of endogenous and exogenous photosensitizers. Curr. Probl. Dermatol 2001, 29, 74–82. [Google Scholar]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar]
- Campalans, A.; Amouroux, R.; Bravard, A.; Epe, B.; Radicella, J.P. UVA irradiation induces relocalisation of the DNA repair protein hOGG1 to nuclear speckles. J. Cell Sci 2007, 120, 23–32. [Google Scholar]
- Marrot, L.; Meunier, J.R. Skin DNA photodamage and its biological consequences. J. Am. Acad. Dermatol 2008, 58, S139–S148. [Google Scholar]
- Besaratinia, A.; Synold, T.W.; Xi, B.; Pfeifer, G.P. G-to-T tranversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet A radiations in mammalian cells. Biochemistry 2004, 43, 8169–8177. [Google Scholar]
- Huang, X.X.; Bernerd, F.; Halliday, G.M. Ultraviolet A within sunlight induces mutations in the epidermal basal layer of engineered human skin. Am. J. Pathol 2009, 174, 1534–1543. [Google Scholar]
- Mitchell, D. Revisiting the photochemistry of solar UVA in human skin. Proc. Natl. Acad. Sci. USA 2006, 103, 13567–13568. [Google Scholar]
- Molho-Pessach, V.; Lotem, M. Ultraviolet radiation and cutaneous carcinogenesis. Curr. Probl. Dermatol 2007, 35, 14–27. [Google Scholar]
- Ibbotson, S.H.; Lambert, C.R.; Moran, M.N.; Lynch, M.C.; Kochevar, I.E. Benzoyl peroxide increases UVA-induced plasma membrane damage and lipid oxidation in murine leukemia L1210 cells. J. Invest. Dermatol 1998, 110, 79–83. [Google Scholar]
- Gniadecki, R.; Christoffersen, N.; Wulf, H.C. Cholesterol-rich plasma membrane domains (lipid rafts) in keratinocytes: Importance in the baseline and UVA-induced generation of reactive oxygen species. J. Invest. Dermatol 2002, 118, 582–588. [Google Scholar]
- Pandey, B.N.; Mishra, K.P. In vitro studies on radiation induced membrane oxidative damage in apoptotic death of mouse thymocytes. Int. J. Low Radiat 2003, 1, 113–119. [Google Scholar]
- Larsson, P.; Anderson, E.; Johansson, U.; Öllinger, K.; Rosdahl, I. Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis. Exp. Dermatol 2005, 14, 117–123. [Google Scholar]
- Budai, M.; Reynaud-Angelin, A.; Szabó, Z.; Tóth, S.; Rontó, G.; Sage, E.; Gróf, P. Effect of UVA radiation on membrane fluidity and radical decay in human fibroblasts as detected by spin labeled stearic acids. J. Photochem. Photobiol. B 2004, 77, 27–38. [Google Scholar]
- Vile, G.F.; Tyrrell, R.M. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic. Biol. Med 1995, 18, 721–730. [Google Scholar]
- Armeni, T.; Damiani, E.; Battino, M.; Greci, L.; Principato, G. Lack of in vitro protection by a common sunscreen ingredient on UVA-induced cytotoxicity in keratinocytes. Toxicology 2004, 203, 165–178. [Google Scholar]
- Polte, T.; Tyrrell, R.M. Involvement of lipid peroxidation and organic peroxides in UVA-induced matrix metalloproteinase-1 expression. Free Radic. Biol. Med 2004, 36, 1566–1574. [Google Scholar]
- Girotti, A.W. Photosensitized oxidation of membrane lipids: Reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J. Photochem. Photobiol. B 2001, 63, 103–113. [Google Scholar]
- Hoerter, J.D.; Ward, C.S.; Bale, K.D.; Gizachew, A.N.; Graham, R.; Reynolds, J.; Ward, M.E.; Choi, C.; Kagabo, J.L.; Sauer, M.; et al. Effect of UVA fluence rate on indicators of oxidative stress in human dermal fibroblasts. Int. J. Biol. Sci 2008, 4, 63–70. [Google Scholar]
- Doherty, G.J.; McMahon, H.T. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu. Rev. Biophys 2008, 37, 65–95. [Google Scholar]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protein Cell 2010, 1, 888–897. [Google Scholar]
- Aronov, S. Catalase kinetics of photooxidation. Science 1965, 150, 72–73. [Google Scholar]
- Gantchev, T.G.; van Lier, J.E. Catalase inactivation following photosensitization with tetrasulfonated metallophtalocyan. Photochem. Photobiol 1995, 62, 123–134. [Google Scholar]
- Aubailly, M.; Haigle, J.; Giordani, A.; Morlière, P.; Santus, R. UV photolysis of catalase revised: A spectral study of photolytic intermediates. J. Photochem. Photobiol. B 2000, 56, 61–67. [Google Scholar]
- Zigman, S.; Reddan, J.; Schultz, J.B.; McDaniel, T. Structural and functional changes in catalase induced by near-UV radiation. Photochem. Photobiol 1996, 63, 818–824. [Google Scholar]
- Maresca, V.; Flori, E.; Briganti, S.; Camera, E.; Cario-André, M.; Taïeb, A.; Picardo, M. UVA-induced modification of catalase charge properties in the epidermis is correlated with the skin phototype. J. Invest. Dermatol 2006, 126, 182–190. [Google Scholar]
- Margoliash, E.; Novogrodsky, A.; Schejter, A. Irreversible reaction of 3-amino-1,2,4-triazole and related inhibitors with the protein of catalase. Biochem. J 1960, 74, 339–350. [Google Scholar]
- Lubinsky, S.; Bewley, G. Genetics of catalase in Drosophila melanogaster: Rates of synthesis and degradation of the enzyme in flies aneuploid and euploid for the structural gene. Genetics 1979, 91, 723–742. [Google Scholar]
- Courgeon, A.M.; Rollet, E.; Becker, J.; Maisonhaute, C.; Best-Belpomme, M. Hydrogen peroxide (H2O2) induces actin and some heat-shock proteins in Drosophila cells. Eur. J. Biochem 1988, 171, 163–170. [Google Scholar]
- Batandier, C.; Fontaine, E.; Keriel, C.; Leverve, X.M. Determination of mitochondrial reactive oxygen species: Methodological aspects. J. Cell. Mol. Med 2002, 6, 175–187. [Google Scholar]
- Shorrocks, J.; Paul, N.D.; McMillan, T.J. The dose rate of UVA treatment influences the cellular response of HaCaT keratinocytes. J. Invest. Dermatol 2008, 128, 685–693. [Google Scholar]
- Chang, S.E.; Foster, S.; Betts, D.; Marnock, W.E. DOK, a cell line established from human dysplastic oral mucosa, shows a partially transformed non-malignant phenotype. Int. J. Cancer 1992, 52, 896–902. [Google Scholar]
- Burns, J.E.; Clark, L.J.; Yeudall, W.A.; Mitchell, R.; Mackenzie, K.; Chang, S.E.; Parkinson, E.K. The p53 status of cultured human premalignant oral keratinocytes. Br. J. Cancer 1994, 70, 591–595. [Google Scholar]
- De Laat, A.; Kroon, E.D.; de Gruijl, F.R. Cell cycle effects and concomitant p53 expression in hairless murine skin after longwave UVA (365 nm) irradiation: A comparison with UVB irradiation. Photochem. Photobiol 1997, 65, 730–735. [Google Scholar]
- Zhang, H. p53 plays a central role in UVA and UVB induced cell damage and apoptosis in melanoma cells. Cancer Lett 2006, 244, 229–238. [Google Scholar]
- Sun, W.; Yang, J. Functional mechanisms for human tumor suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar]
- Kim, K.W.; Ha, K.Y.; Lee, J.S.; Rhyu, K.W.; An, H.S.; Woo, Y.K. The apoptotic effects of oxidative stress and antiapoptotic effects of caspase inhibitors on rat notochordal cells. Spine 2007, 32, 2443–2448. [Google Scholar]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar]
- Kagan, V.E.; Fabisiak, J.P.; Shvedova, A.A.; Tyurina, Y.Y.; Tyurin, V.A.; Schor, N.F.; Kawai, K. Oxidative signaling pathway for externalization of plasma membrane phosphatidylserine during apoptosis. FEBS Lett 2000, 477, 1–7. [Google Scholar]
- Verhoven, B.; Schlegel, R.A.; Williamson, P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J. Exp. Med 1995, 182, 1597–1601. [Google Scholar]
- Bender, K.; Blattner, C.; Knebel, A.; Iordanov, M.; Herrlich, P.; Rahmsdorf, H.J. UV-induced signal transduction. J. Photochem. Photobiol. B 1997, 37, 1–17. [Google Scholar]
- Le Panse, R.; Dubertret, L.; Coulomb, B. p38 mitogen-activated protein kinase activation by ultraviolet A in human dermal fibroblasts. Photochem. Photobiol 2003, 78, 168–174. [Google Scholar]
- Grether-Beck, S.; Timmer, A.; Felsner, I.; Brenden, H.; Brammertz, D.; Krutmann, J. Ultraviolet A-induced signaling involves a ceramide-mediated autocrine loop leading to ceramide de novo synthesis. J. Invest. Dermatol 2005, 125, 545–553. [Google Scholar]
- Krutmann, J. The interaction of UVA and UVB wavebands with particular emphasis on signalling. Prog. Biophys. Mol. Biol 2006, 92, 105–107. [Google Scholar]
- Silvers, A.L.; Bowden, G.T. UVA irradiation-induced activation of activator protein-1 is correlated with induced expression of AP-1 family members in the human keratinocyte cell line HaCaT. Photochem. Photobiol 2002, 75, 302–310. [Google Scholar]
- Yu, L.; Venkataraman, S.; Coleman, M.C.; Spitz, D.R.; Wertz, P.W.; Domann, F.E. Glutathione peroxidase-1 inhibits UVA-induced AP-alpha expression in human keratinocytes. Biochem. Biophys. Res. Commun 2006, 351, 1066–1071. [Google Scholar]
- ScienCell Research Laboratories. Human Oral Keratinocytes. Available online: http://www.sciencellonline.com/site/productDetails.php?keyword=2610 accessed on 3 December 2012.
- Gavrila, C.; Gruia, I.; Lungu, C.P. Optical emission phenomena modeling in TVA carbon plasma. Adv. Appl. Plasma Sci. 2011, 8, 91–92. [Google Scholar]
- Gavrila, C.; Gruia, I.; Lungu, C.P. Determining the radial distribution of the emission coefficient from a plasma source. Optoelecron. Adv. Mater 2009, 8, 835–838. [Google Scholar]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem 1979, 95, 351–358. [Google Scholar]
- Aebi, H. Catalase In Vitro. In Methods in Enzymology; Packer, L., Ed.; Academic Press: New York, NY, USA, 1984; Volume 105, Chapter 13; pp. 121–126. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem 1951, 193, 265–275. [Google Scholar]




© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nechifor, M.T.; Niculiţe, C.M.; Urs, A.O.; Regalia, T.; Mocanu, M.; Popescu, A.; Manda, G.; Dinu, D.; Leabu, M. UVA Irradiation of Dysplastic Keratinocytes: Oxidative Damage versus Antioxidant Defense. Int. J. Mol. Sci. 2012, 13, 16718-16736. https://doi.org/10.3390/ijms131216718
Nechifor MT, Niculiţe CM, Urs AO, Regalia T, Mocanu M, Popescu A, Manda G, Dinu D, Leabu M. UVA Irradiation of Dysplastic Keratinocytes: Oxidative Damage versus Antioxidant Defense. International Journal of Molecular Sciences. 2012; 13(12):16718-16736. https://doi.org/10.3390/ijms131216718
Chicago/Turabian StyleNechifor, Marina T., Cristina M. Niculiţe, Andreea O. Urs, Teodor Regalia, Mihaela Mocanu, Alexandra Popescu, Gina Manda, Diana Dinu, and Mircea Leabu. 2012. "UVA Irradiation of Dysplastic Keratinocytes: Oxidative Damage versus Antioxidant Defense" International Journal of Molecular Sciences 13, no. 12: 16718-16736. https://doi.org/10.3390/ijms131216718