Cholesterol-Dependent Energy Transfer between Fluorescent Proteins—Insights into Protein Proximity of APP and BACE1 in Different Membranes in Niemann-Pick Type C Disease Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
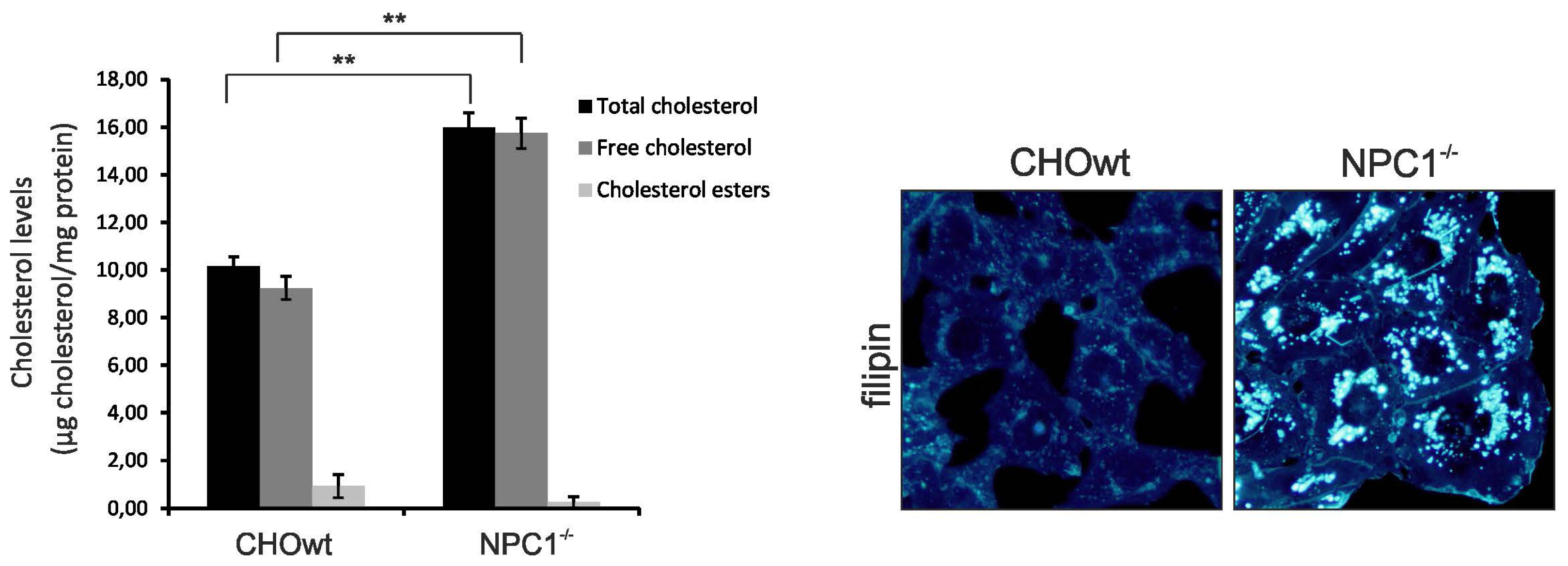
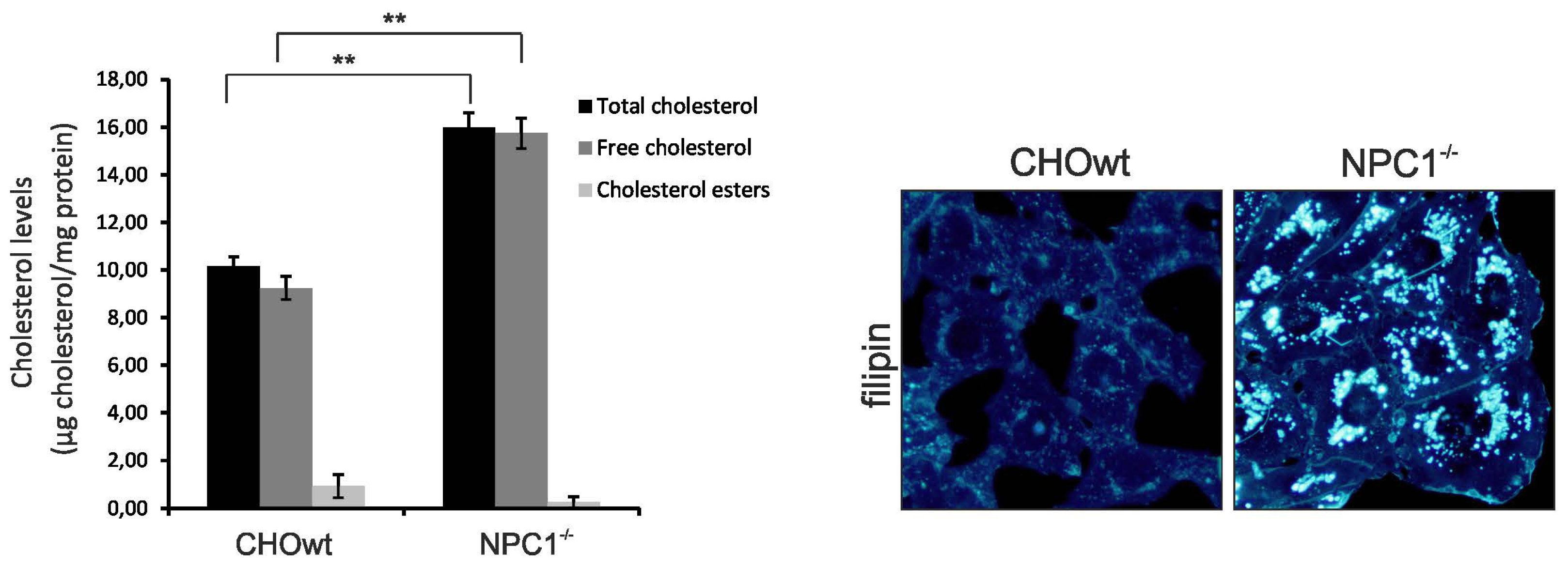
2.1. CHO-NPC1−/− Cells are Enriched in Total, as well as, Vesicular Cholesterol
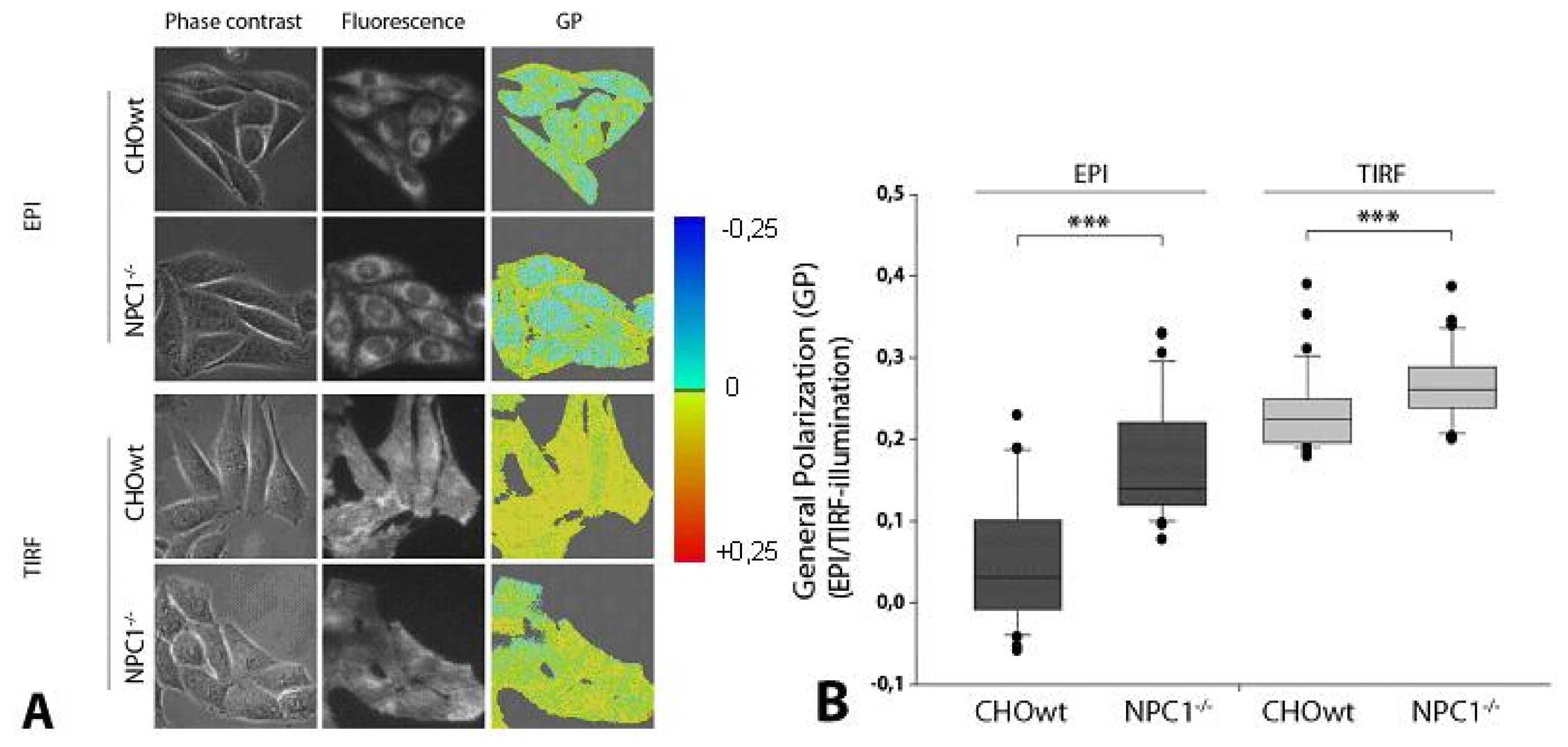
2.2. NPC1−/− Cells Display Higher Membrane Stiffness at Intracellular Membranes
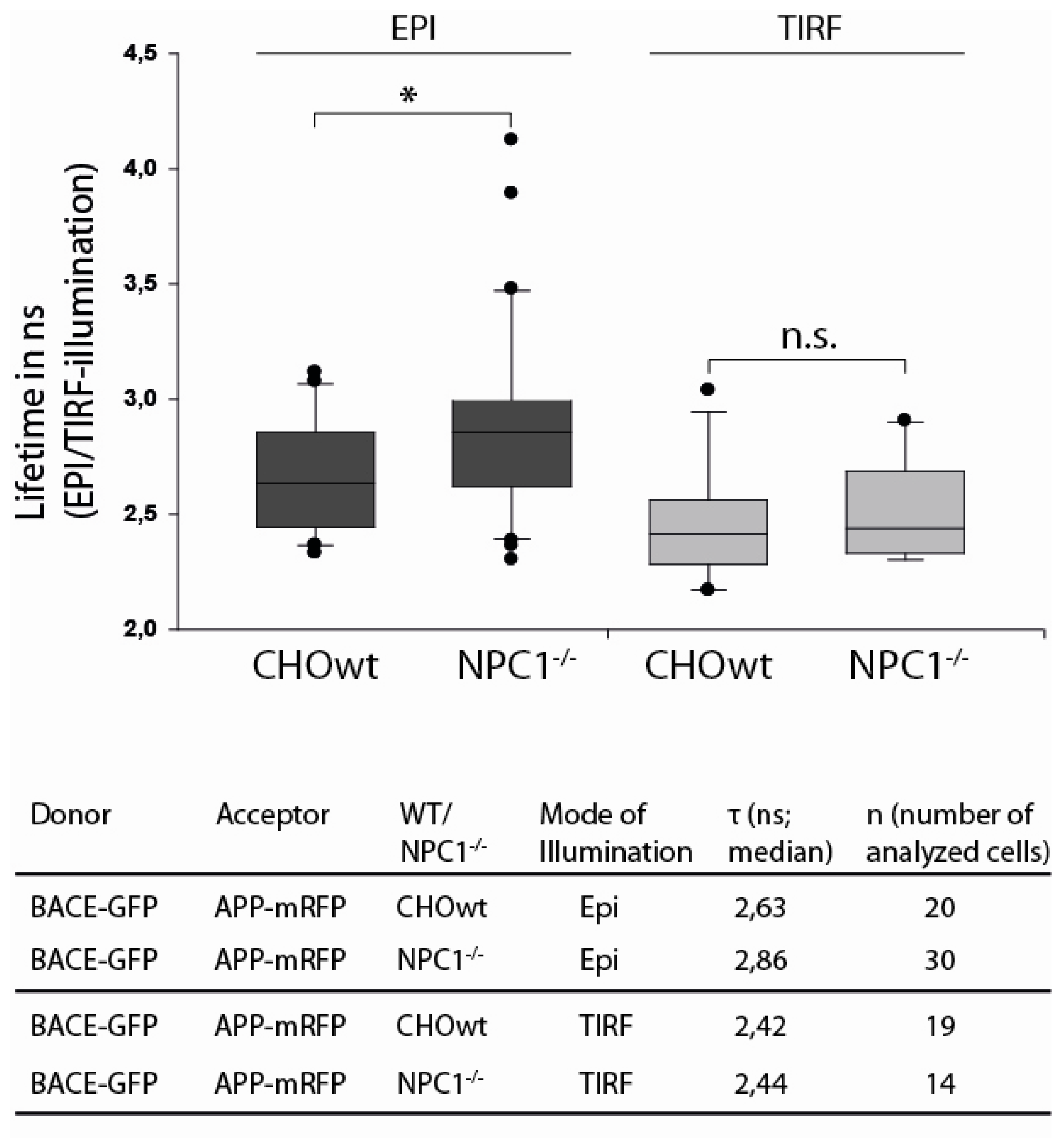
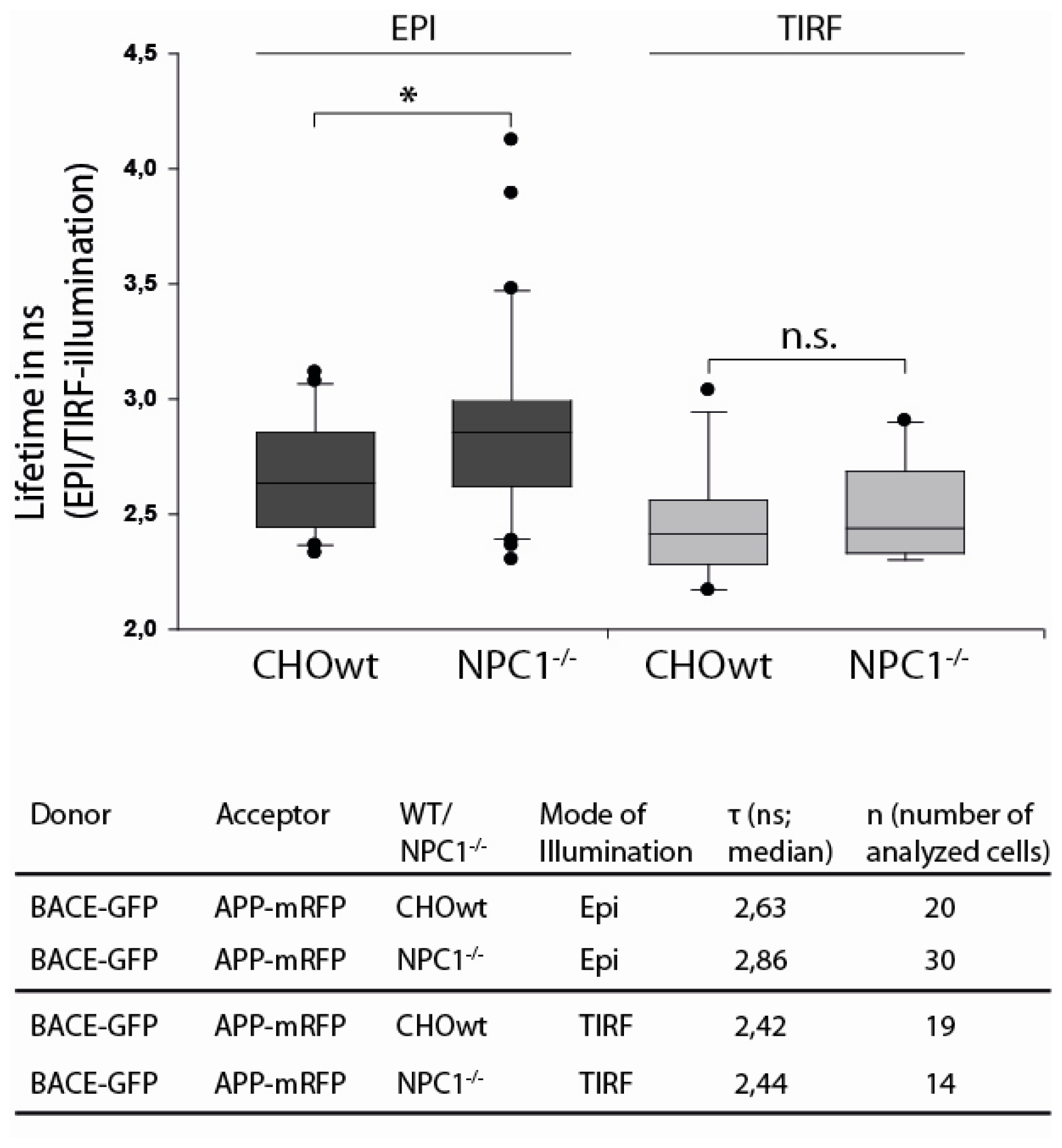
2.3. APP and BACE1 Proximity is Decreased in NPC1−/− Cells, but Not at the Cell Surface
3. Experimental Section
3.1. Expression Constructs
3.2. Cell Culture and Stable Transfection
3.3. Cholesterol Quantification
3.4. Filipin Staining
3.5. Laurdan Staining and GP Measurements
3.6. Microscopic Imaging and FRET Analysis
4. Conclusion
Supplementary Information
ijms-13-15801-s001.pdfAcknowledgment
References
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol 2001, 12, 105–112. [Google Scholar]
- Simons, K.; Ikonen, E. How cells handle cholesterol. Science 2000, 290, 1721–1726. [Google Scholar]
- Bjorkhem, I. Crossing the barrier: Oxysterols as cholesterol transporters and metabolic modulators in the brain. J. Int. Med 2006, 260, 493–508. [Google Scholar]
- Jurevics, H.; Morell, P. Cholesterol for synthesis of myelin is made locally, not imported into brain. J. Neurochem 1995, 64, 895–901. [Google Scholar]
- Pfrieger, F.W. Role of cholesterol in synapse formation and function. Biochim. Biophys. Acta 2003, 1610, 271–280. [Google Scholar]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. Cns synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the cholesterol biosynthetic pathway in huntington’s disease. J. Neurosci 2005, 25, 9932–9939. [Google Scholar]
- Leoni, V.; Mariotti, C.; Nanetti, L.; Salvatore, E.; Squitieri, F.; Bentivoglio, A.R.; Bandettini del Poggio, M.; Piacentini, S.; Monza, D.; Valenza, M.; et al. Whole body cholesterol metabolism is impaired in huntington’s disease. Neurosci. Lett 2011, 494, 245–249. [Google Scholar]
- Hu, G. Total cholesterol and the risk of parkinson’s disease: A review for some new findings. Parkinson’s Dis 2010, 2010, 836962. [Google Scholar]
- Bar-On, P.; Crews, L.; Koob, A.O.; Mizuno, H.; Adame, A.; Spencer, B.; Masliah, E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of parkinson’s disease. J. Neurochem 2008, 105, 1656–1667. [Google Scholar]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P., Jr; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s disease: The cholesterol connection. Nat. Neurosci 2003, 6, 345–351. [Google Scholar]
- Reitz, C.; Tang, M.X.; Schupf, N.; Manly, J.J.; Mayeux, R.; Luchsinger, J.A. Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch. Neurol 2010, 67, 1491–1497. [Google Scholar]
- Notkola, I.L.; Sulkava, R.; Pekkanen, J.; Erkinjuntti, T.; Ehnholm, C.; Kivinen, P.; Tuomilehto, J.; Nissinen, A. Serum total cholesterol, apolipoprotein e epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 1998, 17, 14–20. [Google Scholar]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hanninen, T.; Hallikainen, M.; Alhainen, K.; Iivonen, S.; Mannermaa, A.; Tuomilehto, J.; Nissinen, A.; et al. Apolipoprotein e epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life alzheimer disease. Ann. Intern. Med 2002, 137, 149–155. [Google Scholar]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-pick disease type c1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med 2008, 14, 1247–1255. [Google Scholar]
- Sturley, S.L.; Patterson, M.C.; Pentchev, P. Unraveling the sterol-trafficking defect in niemann-pick c disease. Proc. Natl. Acad. Sci. USA 2009, 106, 2093–2094. [Google Scholar]
- Vance, J.E. Lipid imbalance in the neurological disorder, niemann-pick c disease. FEBS Lett 2006, 580, 5518–5524. [Google Scholar]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci 1991, 12, 383–388. [Google Scholar]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol 2003, 160, 113–123. [Google Scholar]
- Rouvinski, A.; Gahali-Sass, I.; Stav, I.; Metzer, E.; Atlan, H.; Taraboulos, A. Both raft- and non-raft proteins associate with chaps-insoluble complexes: Some app in large complexes. Biochem. Biophys. Res. Commun 2003, 308, 750–758. [Google Scholar]
- Bouillot, C.; Prochiantz, A.; Rougon, G.; Allinquant, B. Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J. Biol. Chem 1996, 271, 7640–7644. [Google Scholar]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol 2001, 11, 1288–1293. [Google Scholar]
- Simons, M.; Keller, P.; de Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar]
- Tolppanen, A.M.; Solomon, A.; Soininen, H.; Kivipelto, M. Midlife vascular risk factors and Alzheimer’s disease: Evidence from epidemiological studies. J. Alzheimer’s Dis 2012, 32, 531–540. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein e type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar]
- Poirier, J.; Davignon, J.; Bouthillier, D.; Kogan, S.; Bertrand, P.; Gauthier, S. Apolipoprotein e polymorphism and alzheimer’s disease. Lancet 1993, 342, 697–699. [Google Scholar]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein e in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein e: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar]
- Von Arnim, C.A.; von Einem, B.; Weber, P.; Wagner, M.; Schwanzar, D.; Spoelgen, R.; Strauss, W.L.; Schneckenburger, H. Impact of cholesterol level upon app and bace proximity and app cleavage. Biochem. Biophys. Res. Commun 2008, 370, 207–212. [Google Scholar]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides abeta 42 and abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar]
- Malnar, M.; Kosicek, M.; Mitterreiter, S.; Omerbasic, D.; Lichtenthaler, S.F.; Goate, A.; Hecimovic, S. Niemann-pick type c cells show cholesterol dependent decrease of app expression at the cell surface and its increased processing through the beta-secretase pathway. Biochim. Biophys. Acta 2010, 1802, 682–691. [Google Scholar]
- Vanier, M.T.; Millat, G. Niemann-pick disease type c. Clin. Genet 2003, 64, 269–281. [Google Scholar]
- Walkley, S.U.; Suzuki, K. Consequences of npc1 and npc2 loss of function in mammalian neurons. Biochim. Biophys. Acta 2004, 1685, 48–62. [Google Scholar]
- Jin, L.W.; Shie, F.S.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with niemann-pick type c defects is associated with endosomal abnormalities. Am. J. Pathol 2004, 164, 975–985. [Google Scholar]
- Kodam, A.; Maulik, M.; Peake, K.; Amritraj, A.; Vetrivel, K.S.; Thinakaran, G.; Vance, J.E.; Kar, S. Altered levels and distribution of amyloid precursor protein and its processing enzymes in niemann-pick type c1-deficient mouse brains. Glia 2010, 58, 1267–1281. [Google Scholar]
- Burns, M.; Gaynor, K.; Olm, V.; Mercken, M.; LaFrancois, J.; Wang, L.; Mathews, P.M.; Noble, W.; Matsuoka, Y.; Duff, K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci 2003, 23, 5645–5649. [Google Scholar]
- Yamazaki, T.; Chang, T.Y.; Haass, C.; Ihara, Y. Accumulation and aggregation of amyloid beta-protein in late endosomes of niemann-pick type c cells. J. Biol. Chem 2001, 276, 4454–4460. [Google Scholar]
- Mattsson, N.; Olsson, M.; Gustavsson, M.K.; Kosicek, M.; Malnar, M.; Mansson, J.E.; Blomqvist, M.; Gobom, J.; Andreasson, U.; Brinkmalm, G.; et al. Amyloid-beta metabolism in niemann-pick c disease models and patients. Metab. Brain Dis 2012, 27, 573–585. [Google Scholar]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of bace: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem 2005, 280, 36815–36823. [Google Scholar]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase adam 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar]
- Weber, P.; Wagner, M.; Schneckenburger, H. Fluorescence imaging of membrane dynamics in living cells. J. Biomed. Optic 2010, 15, 046017. [Google Scholar]
- Parasassi, T.; De Stasio, G.; Ravagnan, G.; Rusch, R.M.; Gratton, E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of laurdan fluorescence. Biophys. J 1991, 60, 179–189. [Google Scholar]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Physik 1948, 6, 55–75. [Google Scholar]
- Bodovitz, S.; Klein, W.L. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem 1996, 271, 4436–4440. [Google Scholar]
- Frears, E.R.; Stephens, D.J.; Walters, C.E.; Davies, H.; Austen, B.M. The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport 1999, 10, 1699–1705. [Google Scholar]
- Abad-Rodriguez, J.; Ledesma, M.D.; Craessaerts, K.; Perga, S.; Medina, M.; Delacourte, A.; Dingwall, C.; De Strooper, B.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol 2004, 167, 953–960. [Google Scholar]
- Guardia-Laguarta, C.; Coma, M.; Pera, M.; Clarimon, J.; Sereno, L.; Agullo, J.M.; Molina-Porcel, L.; Gallardo, E.; Deng, A.; Berezovska, O.; et al. Mild cholesterol depletion reduces amyloid-beta production by impairing app trafficking to the cell surface. J. Neurochem 2009, 110, 220–230. [Google Scholar]
- Malnar, M.; Kosicek, M.; Lisica, A.; Posavec, M.; Krolo, A.; Njavro, J.; Omerbasic, D.; Tahirovic, S.; Hecimovic, S. Cholesterol-depletion corrects app and bace1 misstrafficking in npc1-deficient cells. Biochim. Biophys. Acta 2012, 1822, 1270–1283. [Google Scholar]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lecart, S.; Saudou, F.; Duyckaerts, C.; Leveque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursor protein-bace1 clustering in lipid rafts and rapid endocytosis. FASEB J 2011, 25, 1295–1305. [Google Scholar]
- Kosicek, M.; Malnar, M.; Goate, A.; Hecimovic, S. Cholesterol accumulation in niemann pick type c (npc) model cells causes a shift in app localization to lipid rafts. Biochem. Biophys. Res. Commun 2010, 393, 404–409. [Google Scholar]
- Von Arnim, C.A.; Tangredi, M.M.; Peltan, I.D.; Lee, B.M.; Irizarry, M.C.; Kinoshita, A.; Hyman, B.T. Demonstration of bace (beta-secretase) phosphorylation and its interaction with gga1 in cells by fluorescence-lifetime imaging microscopy. J. Cell. Sci 2004, 117, 5437–5445. [Google Scholar]
- Von Arnim, C.A.; Spoelgen, R.; Peltan, I.D.; Deng, M.; Courchesne, S.; Koker, M.; Matsui, T.; Kowa, H.; Lichtenthaler, S.F.; Irizarry, M.C.; et al. Gga1 acts as a spatial switch altering amyloid precursor protein trafficking and processing. J. Neurosci 2006, 26, 9913–9922. [Google Scholar]
- Kinoshita, A.; Whelan, C.M.; Smith, C.J.; Mikhailenko, I.; Rebeck, G.W.; Strickland, D.K.; Hyman, B.T. Demonstration by fluorescence resonance energy transfer of two sites of interaction between the low-density lipoprotein receptor-related protein and the amyloid precursor protein: Role of the intracellular adapter protein fe65. J. Neurosci 2001, 21, 8354–8361. [Google Scholar]
- Neufeld, E.B.; Wastney, M.; Patel, S.; Suresh, S.; Cooney, A.M.; Dwyer, N.K.; Roff, C.F.; Ohno, K.; Morris, J.A.; Carstea, E.D.; et al. The niemann-pick c1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem 1999, 274, 9627–9635. [Google Scholar]
- Stock, K.; Sailer, R.; Strauss, W.S.; Lyttek, M.; Steiner, R.; Schneckenburger, H. Variable-angle total internal reflection fluorescence microscopy (va-tirfm): Realization and application of a compact illumination device. J. Microsc 2003, 211, 19–29. [Google Scholar]
- Von Einem, B.; Schwanzar, D.; Rehn, F.; Beyer, A.S.; Weber, P.; Wagner, M.; Schneckenburger, H.; von Arnim, C.A. The role of low-density receptor-related protein 1 (lrp1) as a competitive substrate of the amyloid precursor protein (app) for bace1. Exp. Neurol 2010, 225, 85–93. [Google Scholar]
- Schneckenburger, H.; Wagner, M.; Kretzschmar, M.; Strauss, W.S.; Sailer, R. Laser-assisted fluorescence microscopy for measuring cell membrane dynamics. Photochem. Photobiol. Sci 2004, 3, 817–822. [Google Scholar]
- Schneckenburger, H.; Gschwend, M.H.; Sailer, R.; Mock, H.P.; Strauss, W.S. Time-gated fluorescence microscopy in cellular and molecular biology. Cell. Mol. Biol. (Noisy-le-grand) 1998, 44, 795–805. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Von Einem, B.; Weber, P.; Wagner, M.; Malnar, M.; Kosicek, M.; Hecimovic, S.; VonArnim, C.A.F.; Schneckenburger, H. Cholesterol-Dependent Energy Transfer between Fluorescent Proteins—Insights into Protein Proximity of APP and BACE1 in Different Membranes in Niemann-Pick Type C Disease Cells. Int. J. Mol. Sci. 2012, 13, 15801-15812. https://doi.org/10.3390/ijms131215801
Von Einem B, Weber P, Wagner M, Malnar M, Kosicek M, Hecimovic S, VonArnim CAF, Schneckenburger H. Cholesterol-Dependent Energy Transfer between Fluorescent Proteins—Insights into Protein Proximity of APP and BACE1 in Different Membranes in Niemann-Pick Type C Disease Cells. International Journal of Molecular Sciences. 2012; 13(12):15801-15812. https://doi.org/10.3390/ijms131215801
Chicago/Turabian StyleVon Einem, Bjoern, Petra Weber, Michael Wagner, Martina Malnar, Marko Kosicek, Silva Hecimovic, Christine A. F. VonArnim, and Herbert Schneckenburger. 2012. "Cholesterol-Dependent Energy Transfer between Fluorescent Proteins—Insights into Protein Proximity of APP and BACE1 in Different Membranes in Niemann-Pick Type C Disease Cells" International Journal of Molecular Sciences 13, no. 12: 15801-15812. https://doi.org/10.3390/ijms131215801