1. Introduction
Lignocellulosic biomass has enormous potential as a replacement for fossil fuels due to its abundance and chemical composition. In general, all kinds of lignocellulosic biomass are predominately composed of three structural polymers: cellulose, hemicelluloses, and the aromatic polymer lignin [
1]. Since cellulose and hemicelluloses are sugar-based macromolecules, which amount to around 70% of the total biomass available, they can serve as raw materials for bioethanol production to replace the current source of starch-based biomass. Lignin, making up 20%–25% of plant cell walls, is a complex polymer synthesized mainly from three hydroxycinnamyl alcohols with different degree of methoxylation [
2,
3]. Each of these monolignols gives rise to a different type of lignin unit namely;
p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units, when incorporated into the polymer [
4]. The lignin polymer provides mechanical support to the plant and plays a role in protecting plants against pathogens [
5].
Until now, the characteristics of lignin have not yet been completely elucidated due to its compositional and structural complexity. The amount and composition of lignin vary among taxa, cell types, and individual cell wall layers. Softwood lignin consists almost exclusively of G-type lignin, hardwood lignin consists of G and S-type units (H units being minor components), while gramineae has all the three [
6]. In addition, different levels of regulations including (a) metabolic control, (b) regulation of individual enzymes in the biosynthetic pathway, and (c) regulation of gene expression also affect the lignin content, quality, and distribution [
7,
8]. Through these methods, the designed plants either deposit less lignin or produce lignins that are more amenable to chemical degradation [
9–
11]. The three major C
6–C
3 (phenylpropanoid) units of lignin are connected by ether and carbon-carbon linkages, such as β-
O-4′, 4-
O-5′, β-β′, β-1′, β-5′, and 5-5′ [
12]. In addition to the various types of bonds present within the lignin itself, there are associations between lignin and polysaccharides [
13], forming a lignin-carbohydrate complex with benzyl-ether, benzyl-ester, and phenyl-glycoside bonds [
14]. Currently, vast quantities of lignin are burnt in paper mills to produce process heat, power, steam and to recover pulping chemicals [
15]. If ethanol industries based on lignocellulosic biomass grow in the near future, the integration of transition of materials from lignin and overall heat balance need to be considered [
16]. Therefore, studying the isolation, structural characterization and potential utilization of lignin appears to be crucial, and has become very intense in industries such as, agriculture and the paper industry [
17].
The tight physical binding and chemical linkages between lignin and cell wall polysaccharides make it difficult to isolate lignin in an unaltered form. The isolation of lignin with high yields and minimal chemical modification is still a major challenge for structural characterization of lignin [
18]. Various standard methods have been traditionally used to isolate lignin from wood. A mild and widely used separation method was established by Björkman [
19], which is based on extensive grinding of plant material followed by extraction with dioxane/water. The milled wood lignin (MWL) is considered to be representative of the native lignin structure and has been extensively used in the elucidation of native lignin structure. Furthermore, enhancement on the lignin isolation based on hydrothermal pretreatment has been made as reported in previous studies [
20–
22]. The results showed that steam hydrolysis resulted in an extensive liberation of phenolic groups, and undoubtedly this was primarily due to cleavage of the aryl ether bonds in β-
O-4′ structures, whereas the contents of C–C condensed structures increased during steam treatment [
22]. Sugar degradation products (such as furfural) reacted with lignin, which increased the acid insoluble component of the plant [
23]. In addition, syringyl units situated mainly in S
2 layers in hardwood were more susceptible to hydrothermal degradation than guaiacyl units [
24,
25]. It was also shown that the MWL isolated from the hydrothermal treated solid residue was more condensed and had a lower molecular weight than the MWL isolated from the untreated material [
26]. Lignin reaction kinetics during the hydrothermal treatment has also been reported by Zhang
et al.[
27]. The results indicated that the lignin degradation mechanism consisted of a two phase reactions for the hydrothermal treatment: first, a very fast reaction phase where lignin was degraded into the soluble fragments and then a slower reaction phase where the soluble fragments reacted with one another via recondensation. Recently, ammonia has been considered to be a desirable pretreatment reagent [
28]. This is because it was effective in improving cellulose digestion and had high reaction selectivity towards lignin over carbohydrates. It has been demonstrated that the reactions of aqueous ammonia with lignin are the cleavage of C–O–C bonds in lignin and ether and ester bonds in the lignin-carbohydrate complex (LCC). In addition, its high volatility makes it easy to recover and reuse [
29].
Cotton stalks, the lignocellulosic by-products from cotton production, have a considerable economic and ecological importance. In China, it is estimated that more than 20 million tons (dry weight) of cotton stalk are generated annually [
30]. However, most of the cotton stalk is burned off, or even sometimes the stalk is left in the ground to decay, without being fully utilized. As a lignocellulosic feedstock with high potential, the utilization efficiency of this lignocellulosic biomass is highly dependent on its structural properties, such as the relative content, composition, accessibility, and reactivity of the three cell wall components. Particularly, the lignin content and its composition in terms of guaiacyl (G), syringyl (S), and
p-hydroxyphenyl (H) moieties as well as the nature of the different inter-unit linkages are important factors affecting delignification rate, hydrolysis rate, and chemical consumption [
31]. In this study, in order to investigate the potential for the utilization of cotton stalk, separation of lignin components from cotton stalk based on ammonia hydrothermal treatment was investigated. Under the alkalinity of ammonia, part of the linkages between lignin and carbohydrates are likely to be broken, so as to achieve the purpose of lignin’s separation. In addition, MWL from the same material was also isolated as a comparison. The structure of the lignin fractions obtained were elucidated by ion chromatography, gel permeation chromatography (GPC), Fourier transform infrared (FT-IR) spectroscopy,
13C nuclear magnetic resonance (
13C NMR) spectroscopy and
1H-
13C correlation two-dimensional nuclear magnetic resonance (
1H-
13C 2D NMR) spectroscopy.
3. Experimental Section
3.1. Materials
Cotton stalk was obtained from Hebei province, China. It was dried in an oven at 50 °C and cut into small pieces. The cotton stalk was then ground and screened to obtain particles ranging from 40 to 80 mesh. The chemical composition of the stalks was: cellulose 34.70%, hemicelluloses 38.62%, lignin 20.99%, and ash 1.90%. A simple extraction procedure is available that solubilizes essentially all the lipid, lignin, and hemicellulose, leaving the cellulose fibers intact. The milled powder was extracted with 90% (v/v) acetone/water in a Soxhlet apparatus for 24 h. The extracted stalk powder was dried under vacuum for several days. For determining the content of cellulose, the extractive-free cotton stalk was delignified using sodium chlorite under pH 3.8–4.2 adjusted by acetic acid for 2 h at 70 °C. Samples were then washed extensively with distilled water until the filtrate pH was neutral. The obtained holocellulose was then extracted with 10% potassium hydroxide at room temperature for 16 h with the vigorous stirring. In this procedure, hemicelluloses were dissolved in alkaline solution. The cellulose fibers could be obtained by filtration the suspension with 100% polyester cloth and washed thoroughly with distilled water until pH = 7. Finally, the cellulosic preparation was oven-dried at 50 °C overnight, and kept in a desiccator for the further experiment. Ash was determined according to TAPPI standard method T211 om-93. Klason lignin was determined according to the TAPPI standard method T-222 om-98. All chemicals used were analytical grade and standard sugars were purchased from Sigma-Aldrich Company (Beijing).
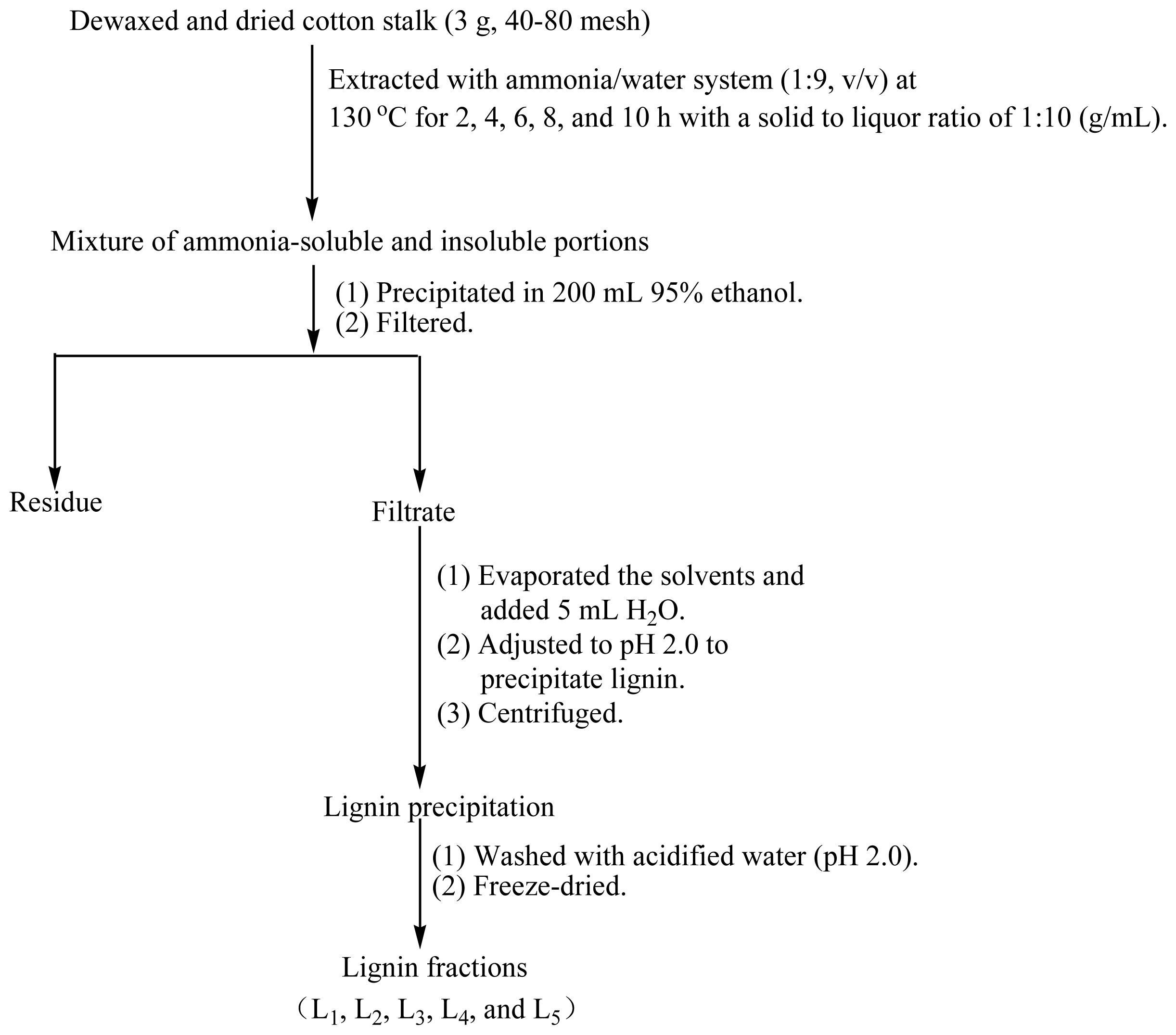
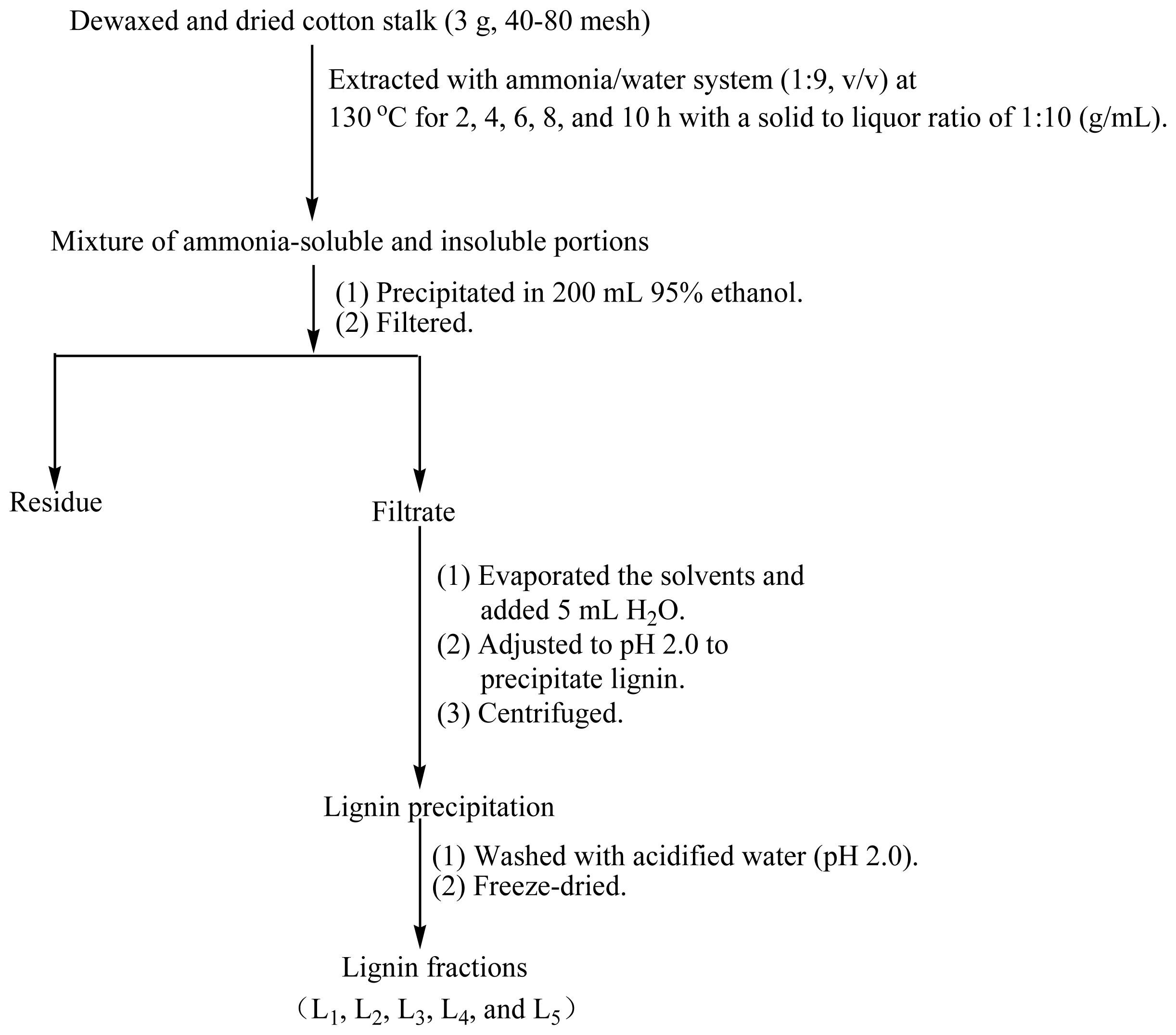
3.2. Isolation and Purification of Lignin Fractions
Figure 6 shows the fractional separation sequence for lignin preparations based on ammonia hydrothermal system treatment. It has been shown that furfural increased sharply beyond extraction temperature at 160 °C in hot water from woodchips [
42]. Therefore, the extraction conditions including temperature, solid to liquid ratio, ammonia concentration were set as follows based on pretreatment conditions in previously published papers [
42,
43]. Three grams of the prepared cotton stalk and 30 mL of 10% (
v/
v) ammonia was mixed and incubated in a Parr reactor at 130 °C for 2, 4, 6, 8, and 10 h, respectively. The brown solution was transferred into 200 mL of 95% ethanol under continuous agitation to precipitate the carbohydrates. The suspension was then filtered through filter paper using a Buchner funnel under reduced pressure. The lignin-containing solution was combined with the washing solution and concentrated under reduced pressure, followed by an addition of 5 mL deionized water. The lignin was precipitated at pH 1.5–2.0 by dropwise addition of 1 M HCl and then centrifuged at 4000 rpm for 20 min. The lignin precipitation was washed twice with acidified water (pH 2.0), centrifuged under the conditions described above and freeze-dried. Milled wood lignin (MWL) was also prepared from the same material referring to the literature for comparison [
20]. The procedures are as follows: the extractive-free cotton stalk powder was subjected to milling in a planetary ball mill using ZrO
2 balls for 10 h, followed by dioxane (96%
v/
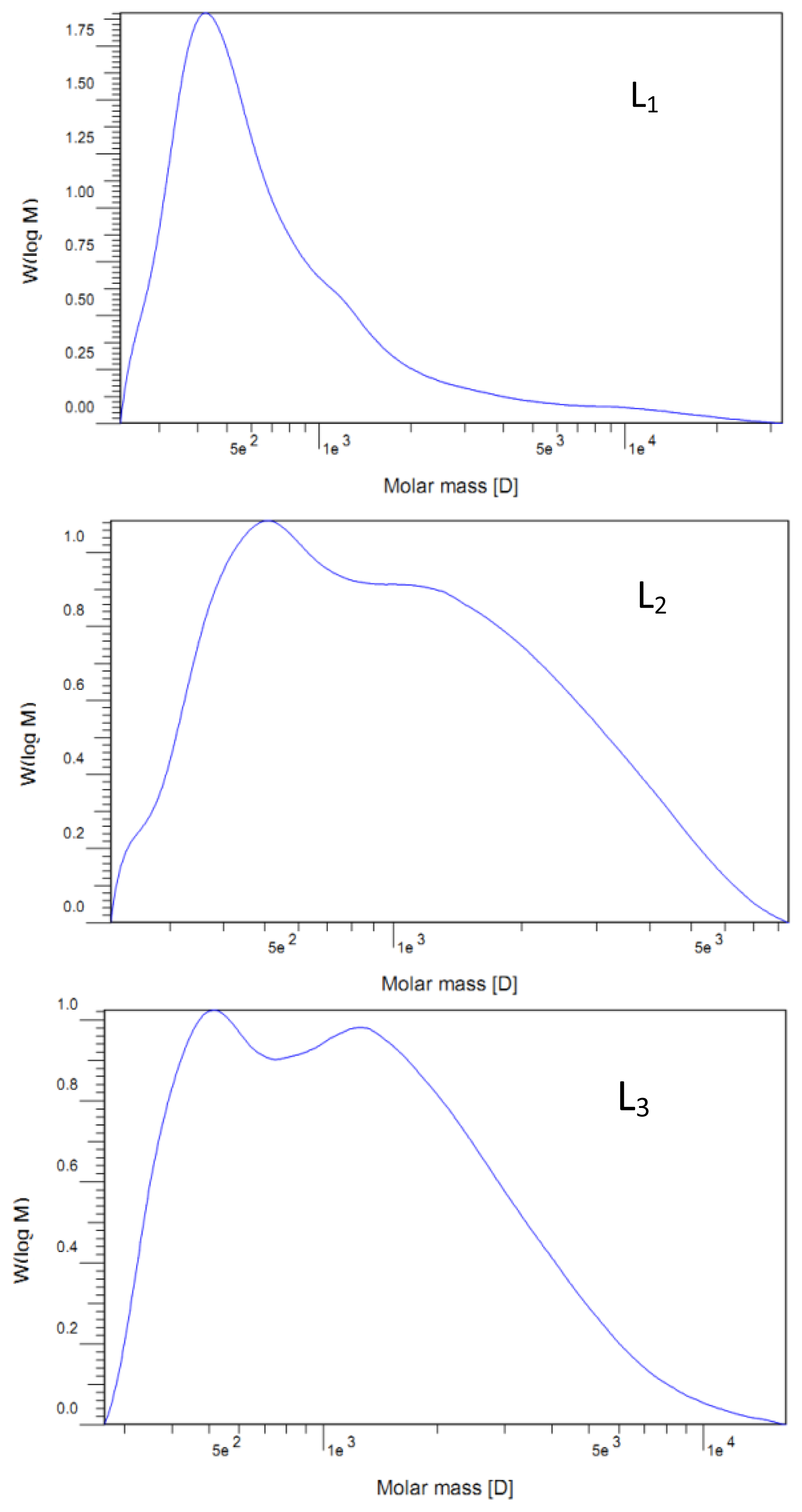
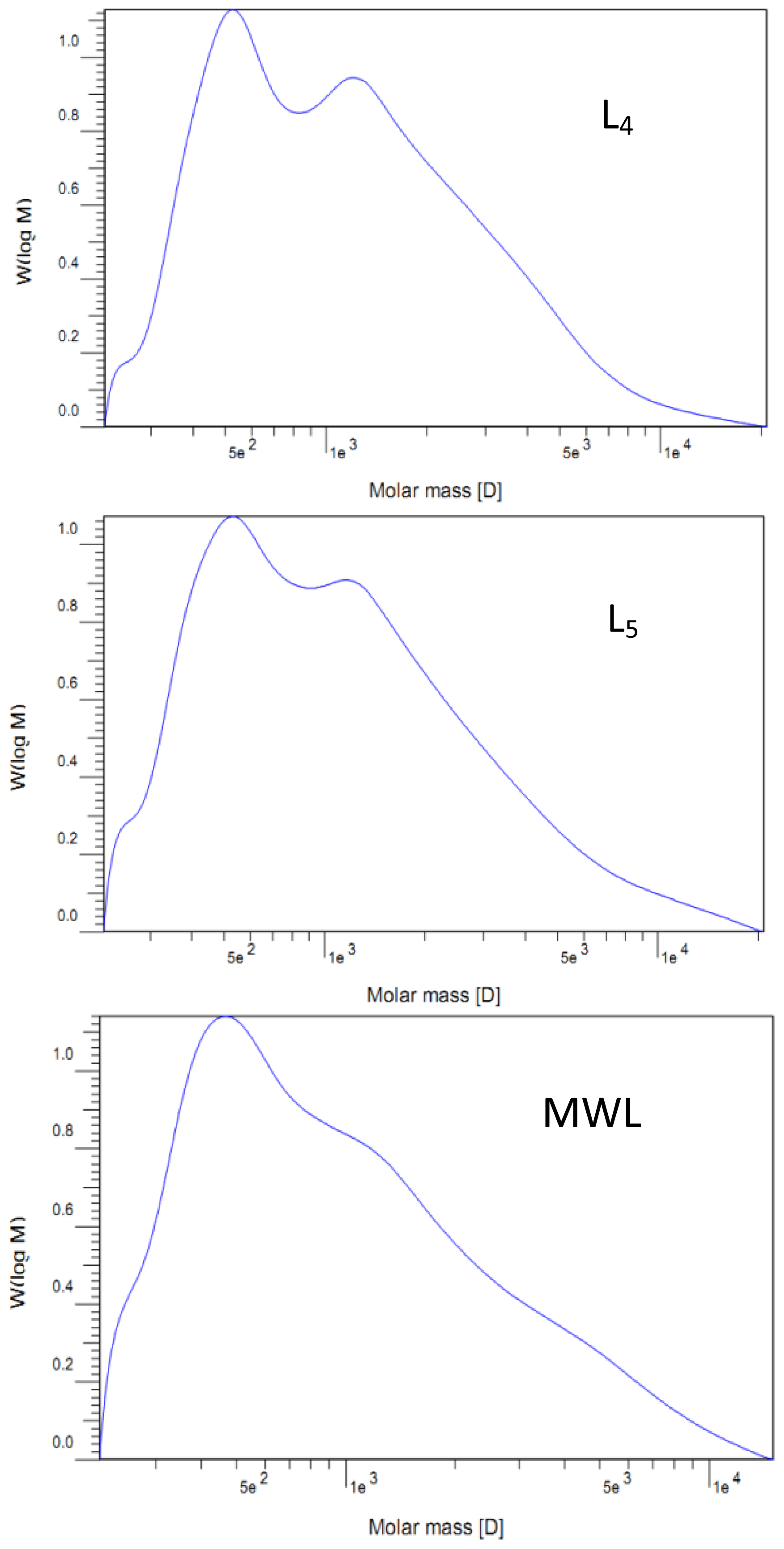
v) extraction. After that, the dioxane was removed by evaporation. The obtained crude MWL was then dissolved in 90% acetic acid and precipitated drop-wise into water. The precipitate was separated by centrifugation and was dried in a vacuum oven at 35 °C. The lignin extraction efficiency from cotton stalk dissolved in the ammonia hydrothermal system at the reaction time of 2, 4, 6, 8, and 10 h showed that the yield of five lignin fractions and MWL were 14.6, 15.2, 15.7, 16.2, 16.6, and 8.2% (Klason lignin), respectively. These lignin fractions treated with ammonia hydrothermal system at the reaction time of 2, 4, 6, 8, and 10 h were denoted as L
1, L
2, L
3, L
4, and L
5, respectively.
3.3. Sugar Analysis
The monosaccharide associated with lignin fractions was determined by hydrolysis with dilute sulfuric acid. Lignin samples weighing 4–6 mg were hydrolyzed with 1.475 mL of 6.1% H2SO4 for 2.5 h at 105 °C. Subsequently, the mixture was filtered through a 0.22 μm PTFE filter to remove unhydrolyzed residues, and the filtrate containing the liberated neutral sugars was diluted 50-fold and analyzed by high-performance anion exchange chromatography (HPAEC) system (Dionex ICS3000, U.S.) with a pulsed amperometric detector and an ion exchange Carbopac PA-1 column (4 × 250 mm).
3.4. Determination of Molecular Weight
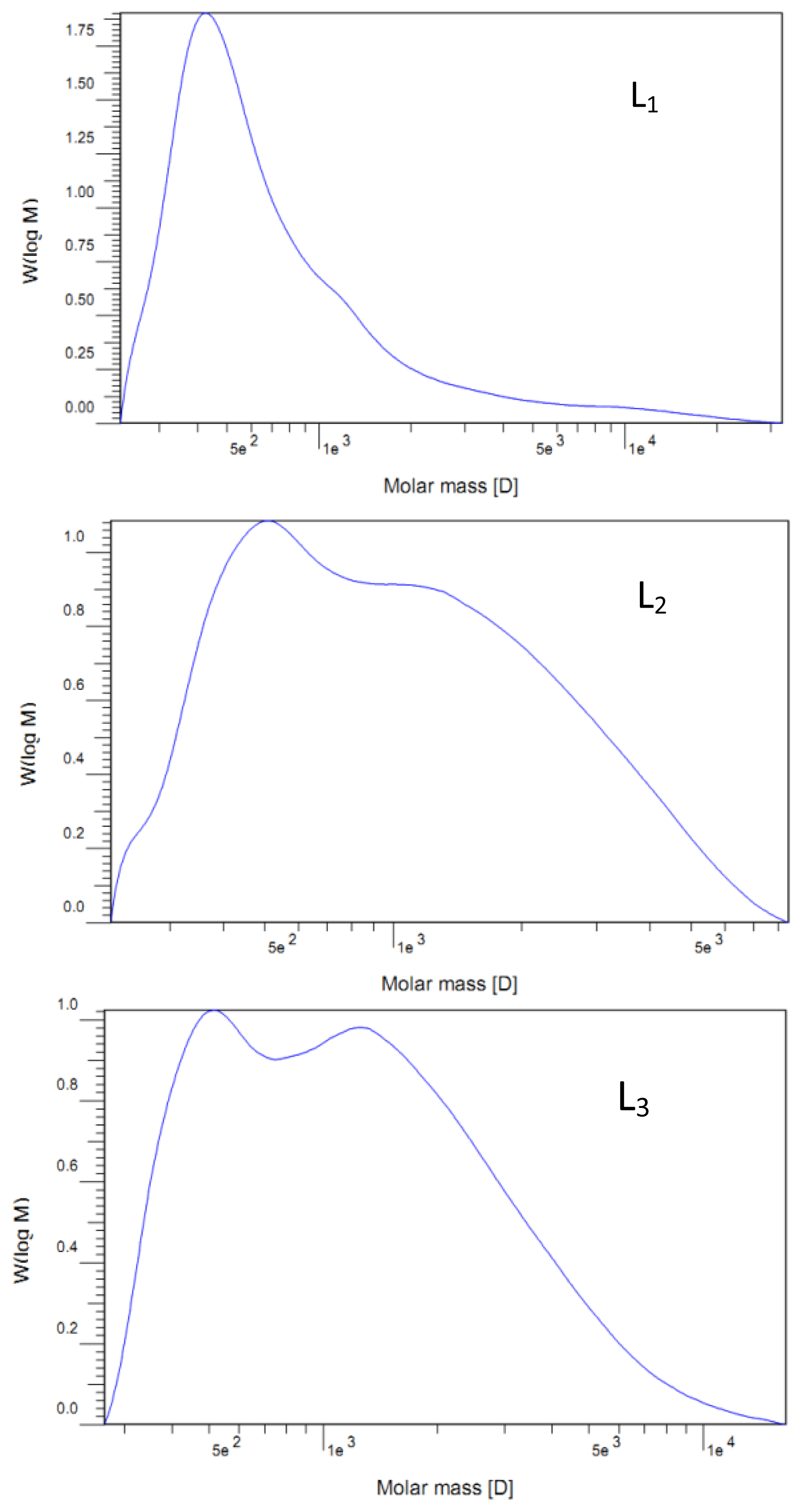
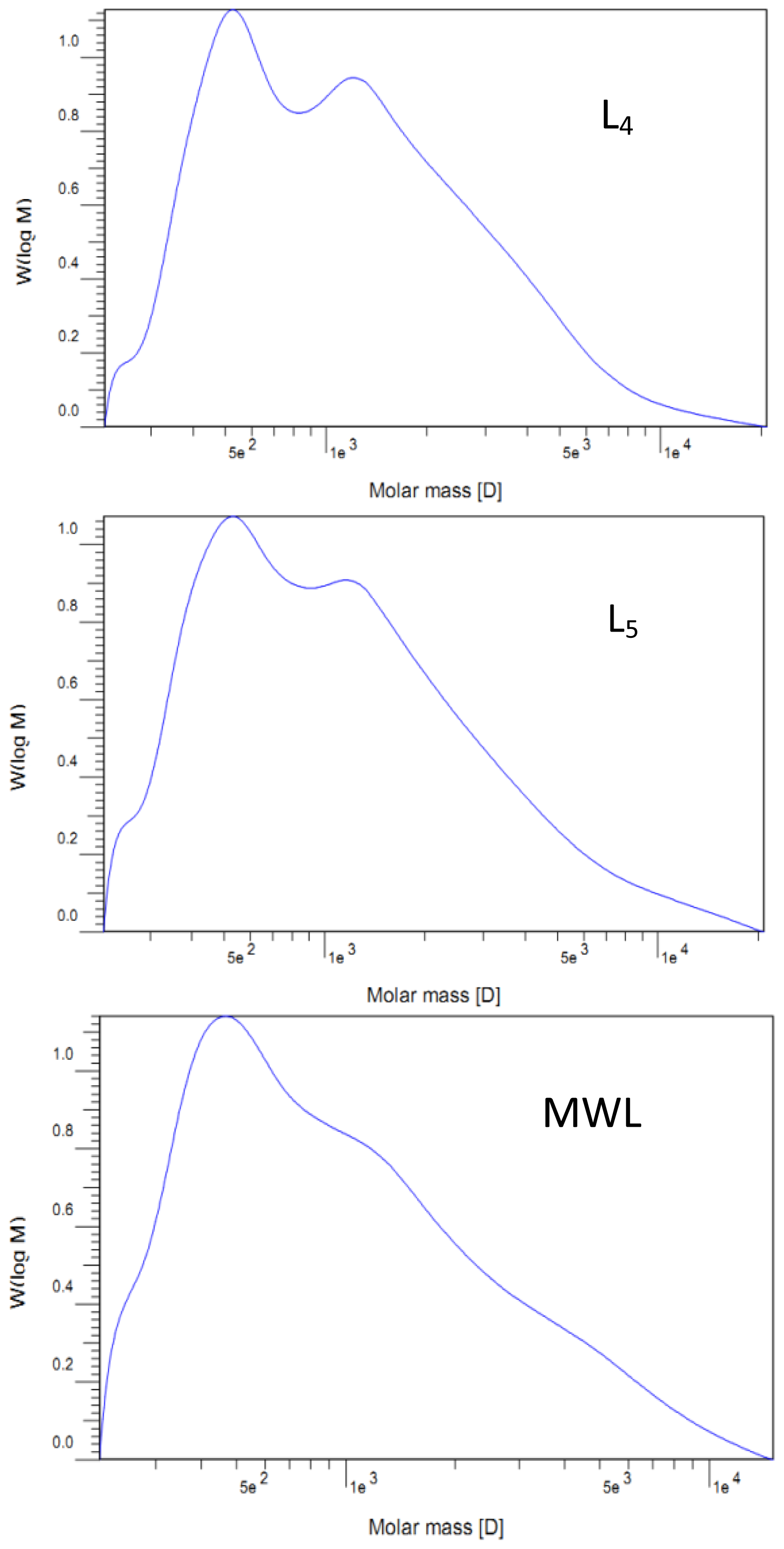
The molecular weights (Mw and Mn) of the lignin fractions were determined by gel permeation chromatography (GPC, Agilent1200, USA) with a refraction index detector (RID) on a PL-gel 10 μm Mixed-B 7.5 mm ID column, calibrated with PL polystyrene standards. The samples were dissolved with tetrahydrofuran (THF) with a concentration of 0.2%, and 20 μL aliquots of the lignin solutions were injected. The column was operated at ambient temperature and eluted with THF at a flow rate of 1.0 mL/min.
3.5. FT-IR Spectral Characterization
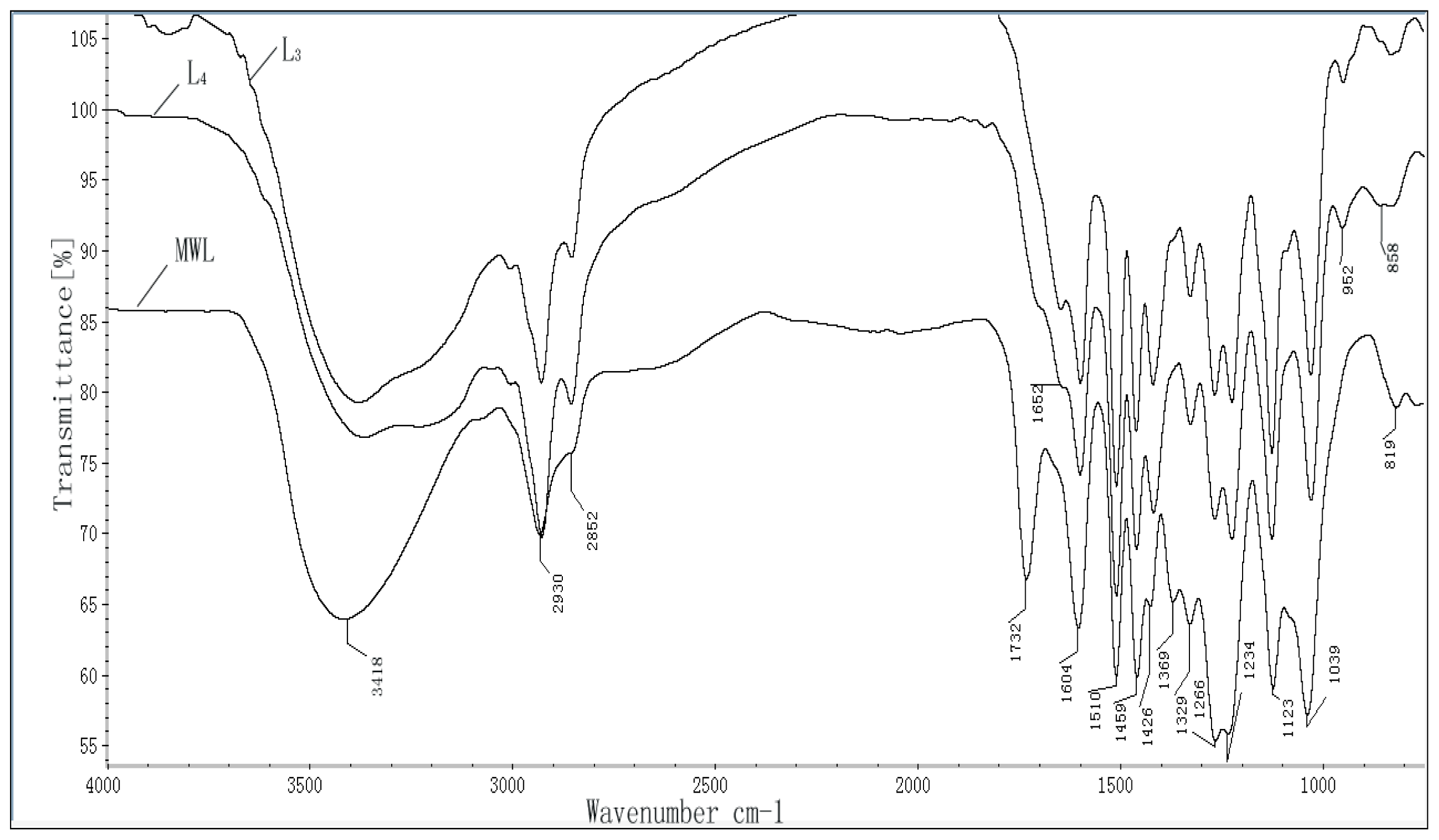
Fourier transform infrared (FT-IR) spectra of the lignin fractions were obtained from an FT-IR spectrophotometer (Tensor 27, Bruker, Germany) using a KBr disk containing 1% finely ground samples. Thirty-two scans were taken of each sample, recorded from 4000 to 400 cm−1 at a resolution of 2 cm−1 in the transmission mode.
3.6. Nuclear Magnetic Resonance Spectra
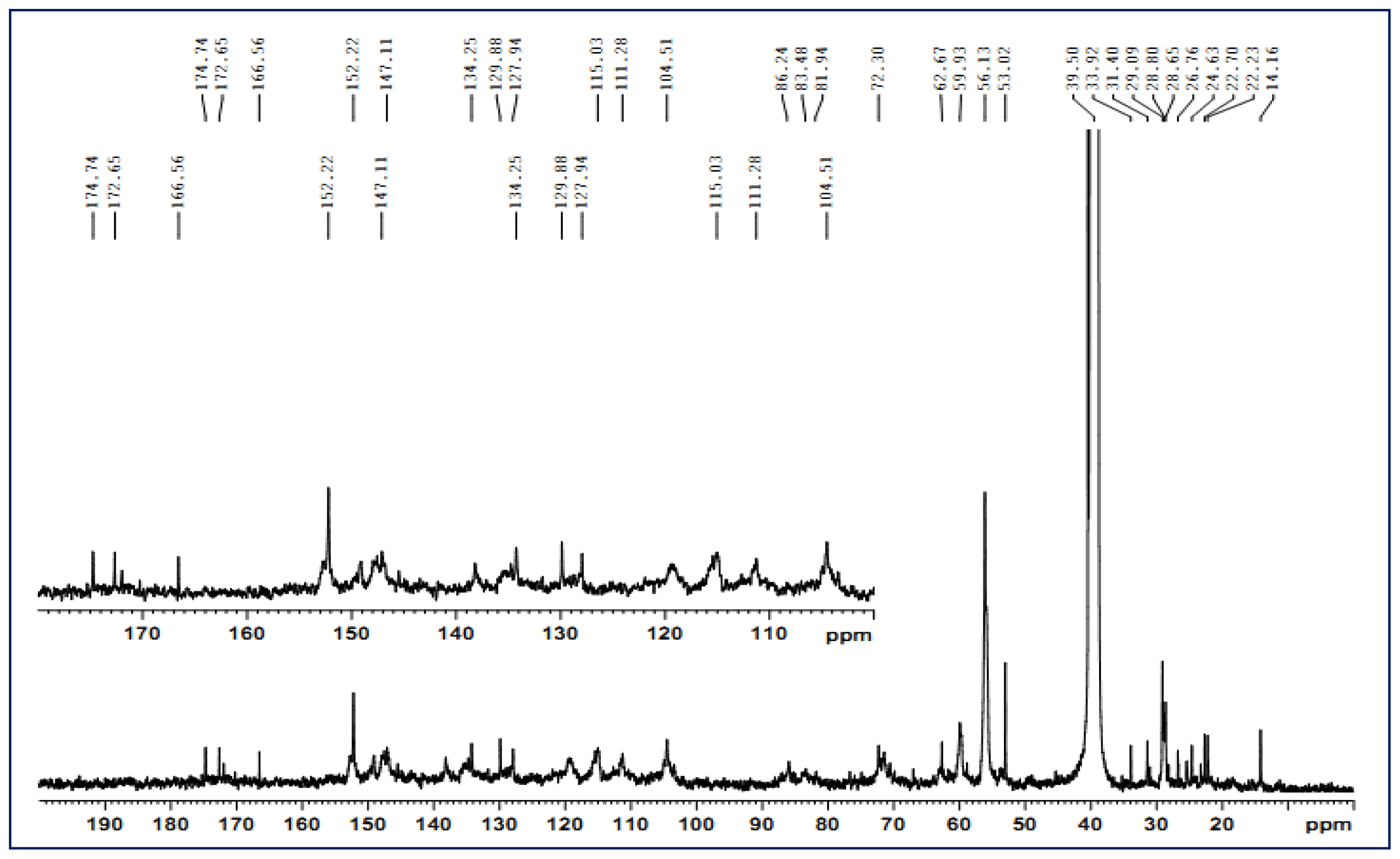
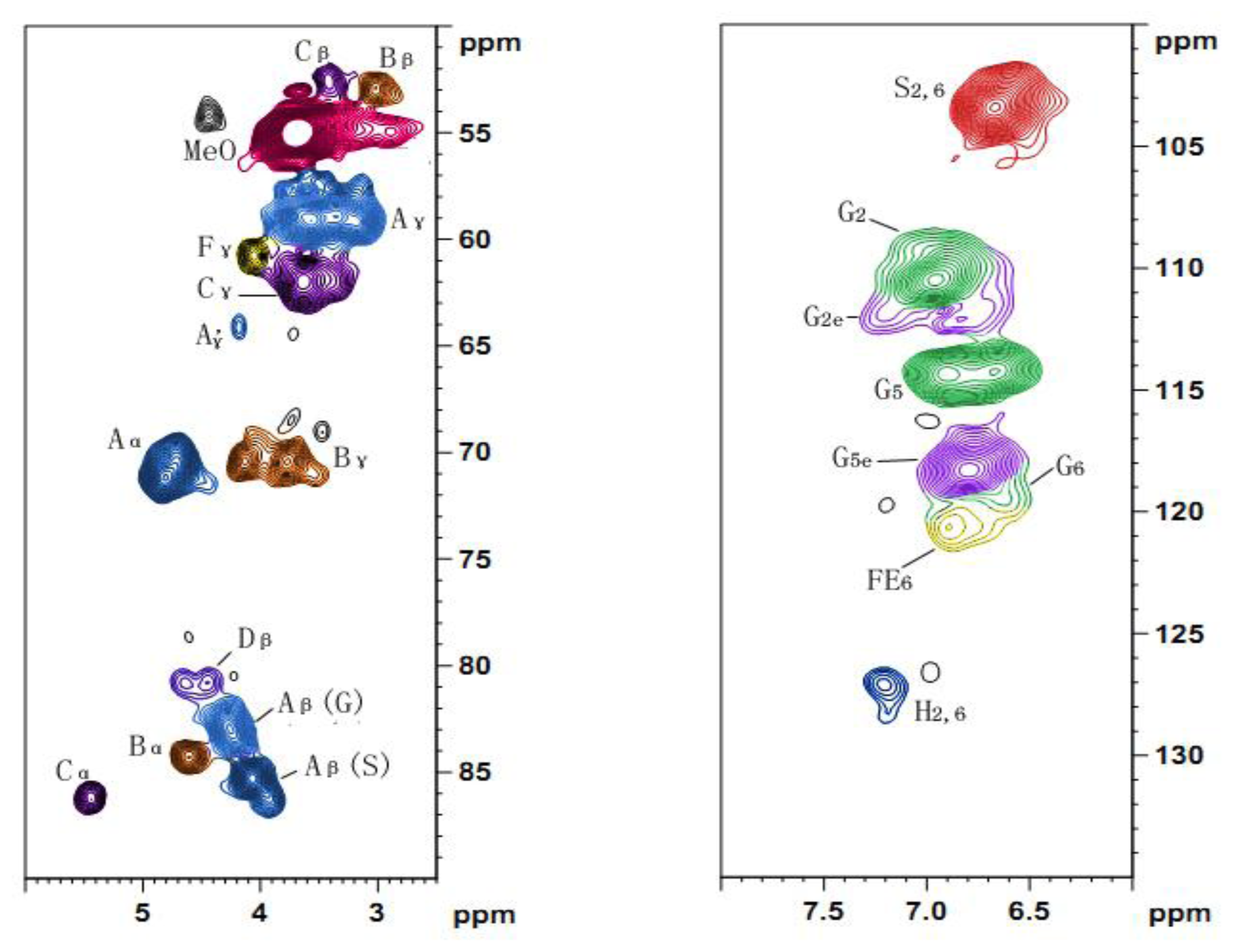
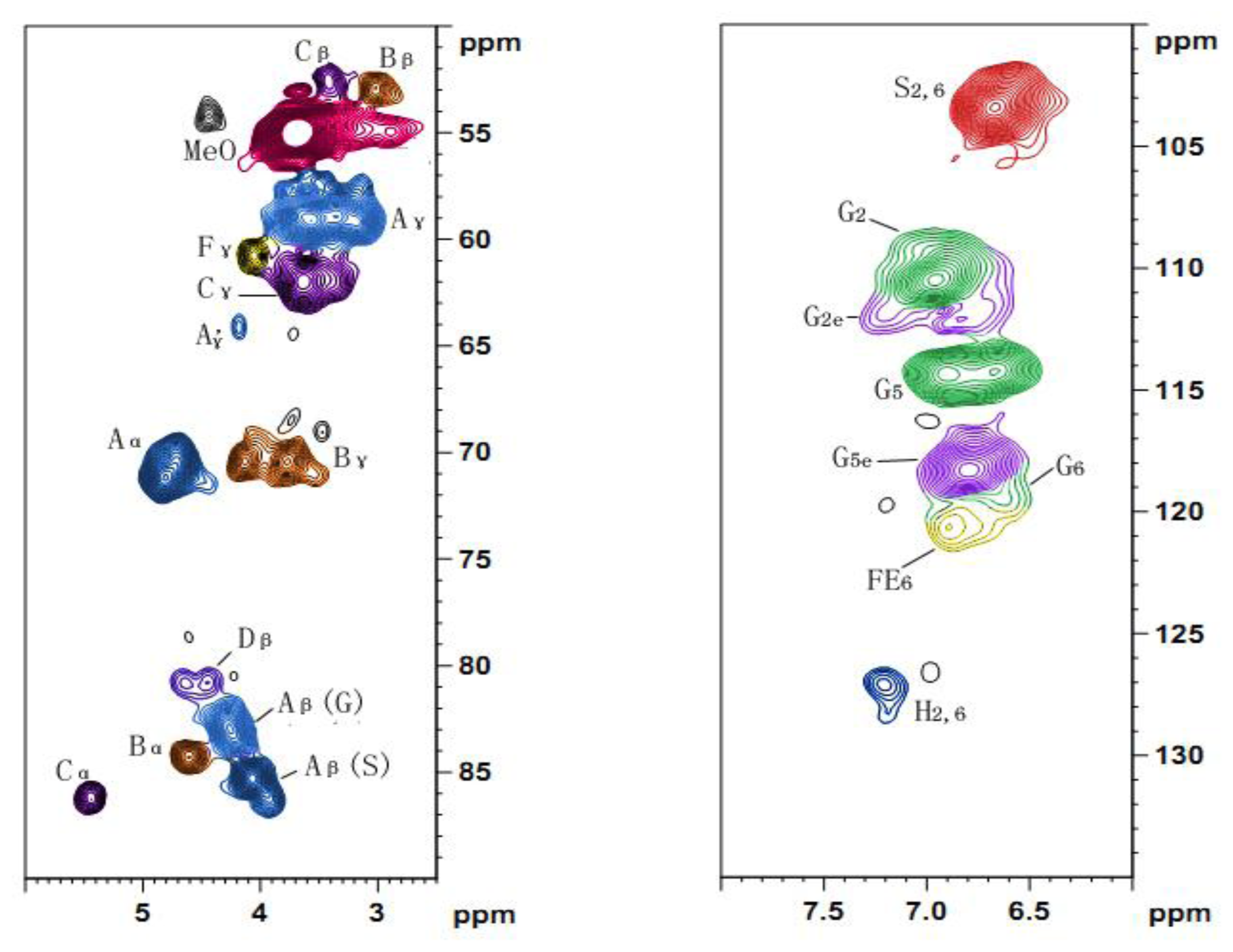
The solution-state 13C-NMR spectroscopy was carried out on a Bruker AV III 400 MHz NMR spectrometer. The sample (80 mg) was dissolved in 0.5 mL of dimethyl sulfoxide-d6 (DMSO, 99.8%), and the spectrum was recorded at 25 °C after 30,000 scans. A 30° pulse flipping angle, a 9.2 μs pulse width, a 1.89 s delay time between scans, a 1.63 s acquisition time and a 2 s relaxation time were used.
The 2D HSQC spectrum was acquired on the same NMR spectrometer in the HSQC GE experiment mode. The spectral widths for the heteronuclear single quantum correlation (HSQC) (semi-quantitative mode) were 5000 and 20,000 Hz for the 1H- and 13C-dimensions, respectively. The number of collected complex points was 1024 for the 1H- dimension with a recycle delay (d1) of 5 s, the number of transients for the HSQC spectrum was 128, and 256 time increments were recorded in the 13C-dimension. The 1JC–H used was 146 Hz. Prior to Fourier transformation, the data matrixes were zero filled up to 1024 points in the 13C-dimension. Data processing was performed using standard Bruker Topspin-NMR software.
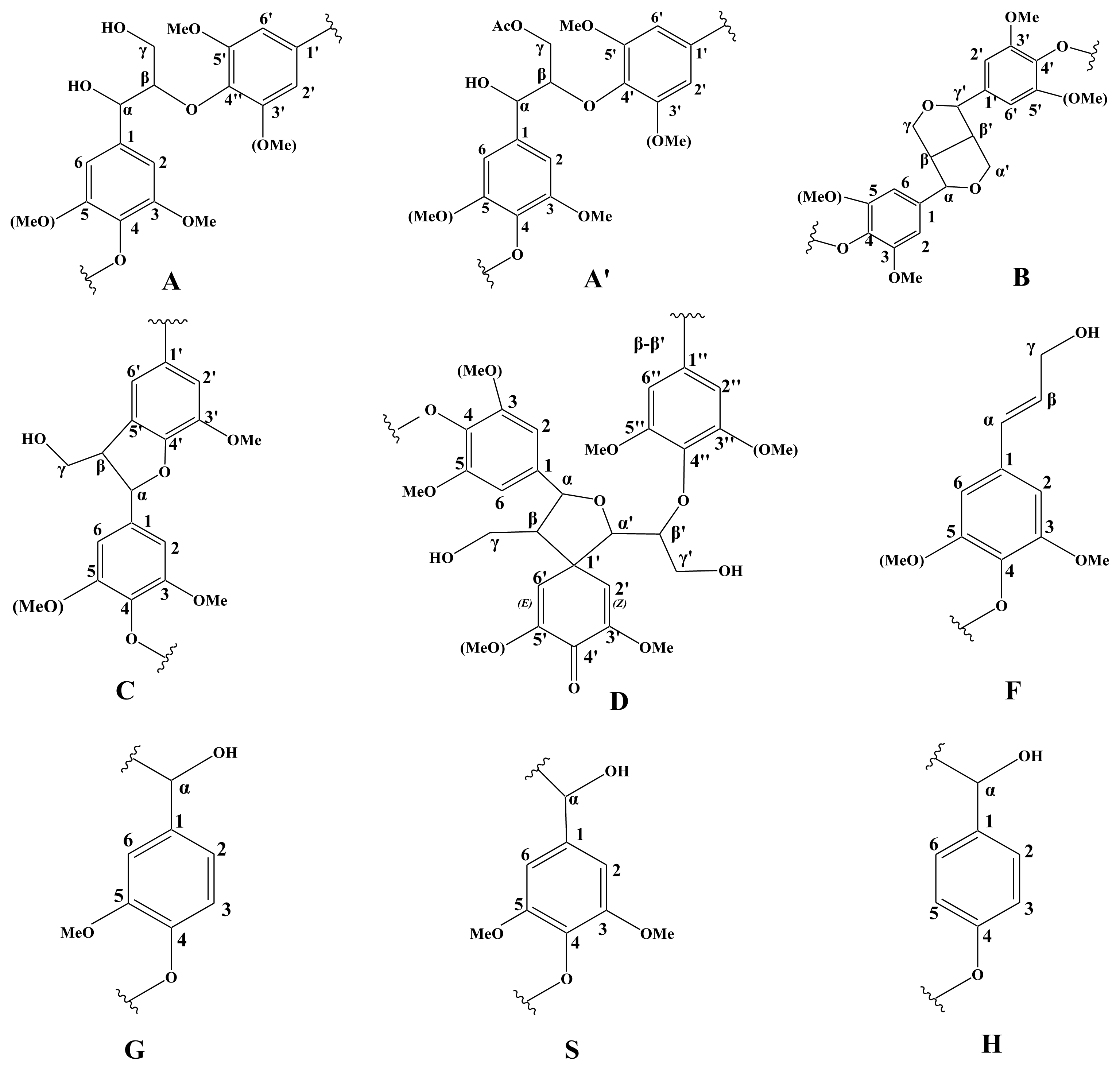
A semi-quantitative analysis of the HSQC cross-signal intensities was also performed. Since the cross-signal intensity depended on the particular 1JC–H value, as well as on the T2 relaxation time, a direct analysis of the intensities was impossible. Therefore, the integration of the cross-signals was performed separately for the different regions of the HSQC spectra, which contained signals that corresponded to chemically analogous carbon-proton pairs (in similar samples). For these signals, the 1JC–H coupling value was relatively similar and was used semi-quantitatively to estimate the relative abundance of the different lignin fractions. The inter-unit linkages of the aliphatic oxygenated region were estimated from Cα–Hα correlations to avoid possible interference from homo-nuclear 1H–1H couplings, and the relative abundance of side chains involved in inter-unit linkages and terminal structures was calculated. The C–H correlations from S and G type units in the aromatic region were used to estimate the S/G ratio of lignin and the percentage of oxidized units. Moreover, the volume integrals were corrected for proton numbers to obtain an accurate S/G ratio.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}