Role of Key Residues at the Flavin Mononucleotide (FMN):Adenylyltransferase Catalytic Site of the Bifunctional Riboflavin Kinase/Flavin Adenine Dinucleotide (FAD) Synthetase from Corynebacterium ammoniagenes



Abstract
:
1. Introduction
2. Results and Discussion
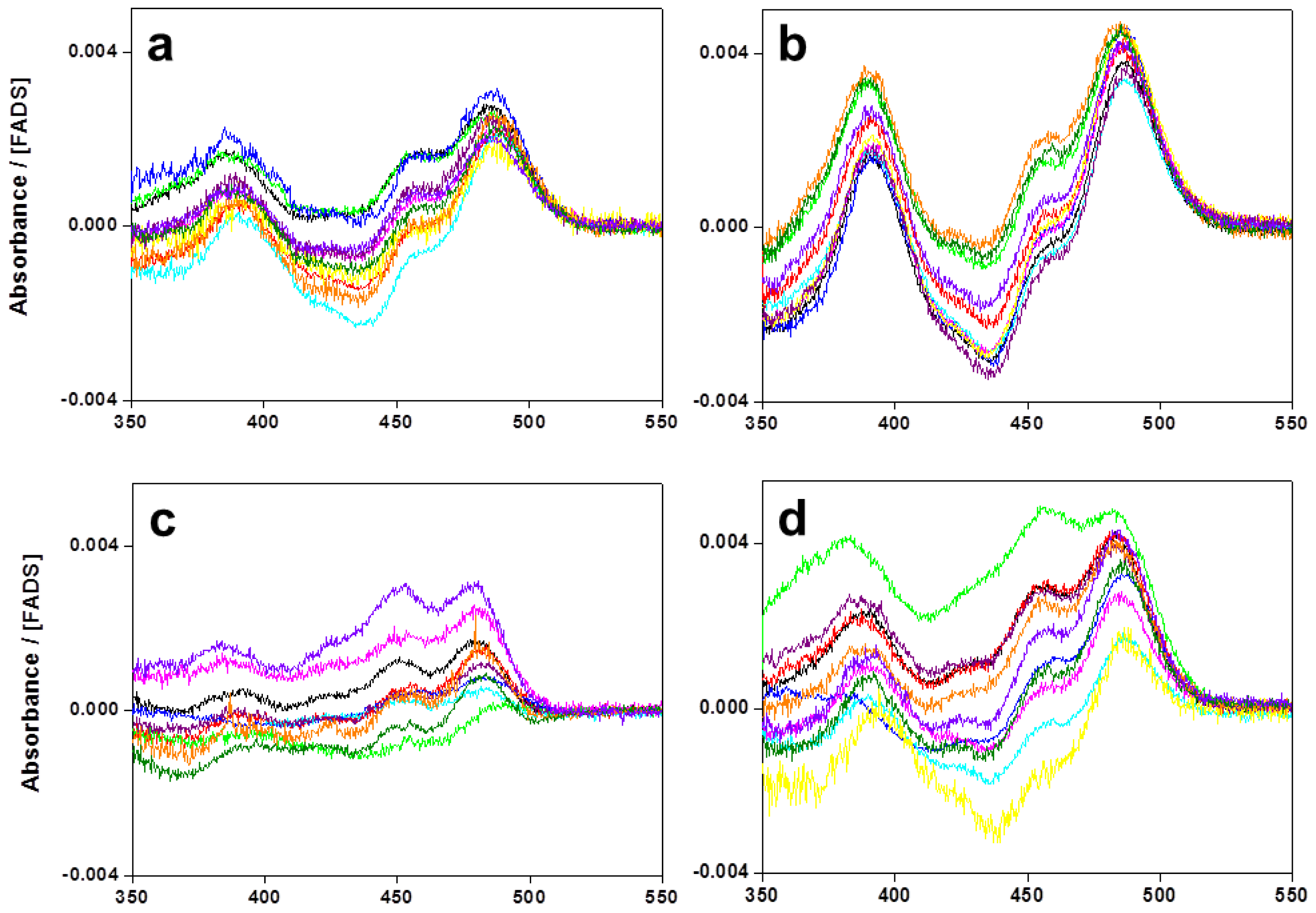
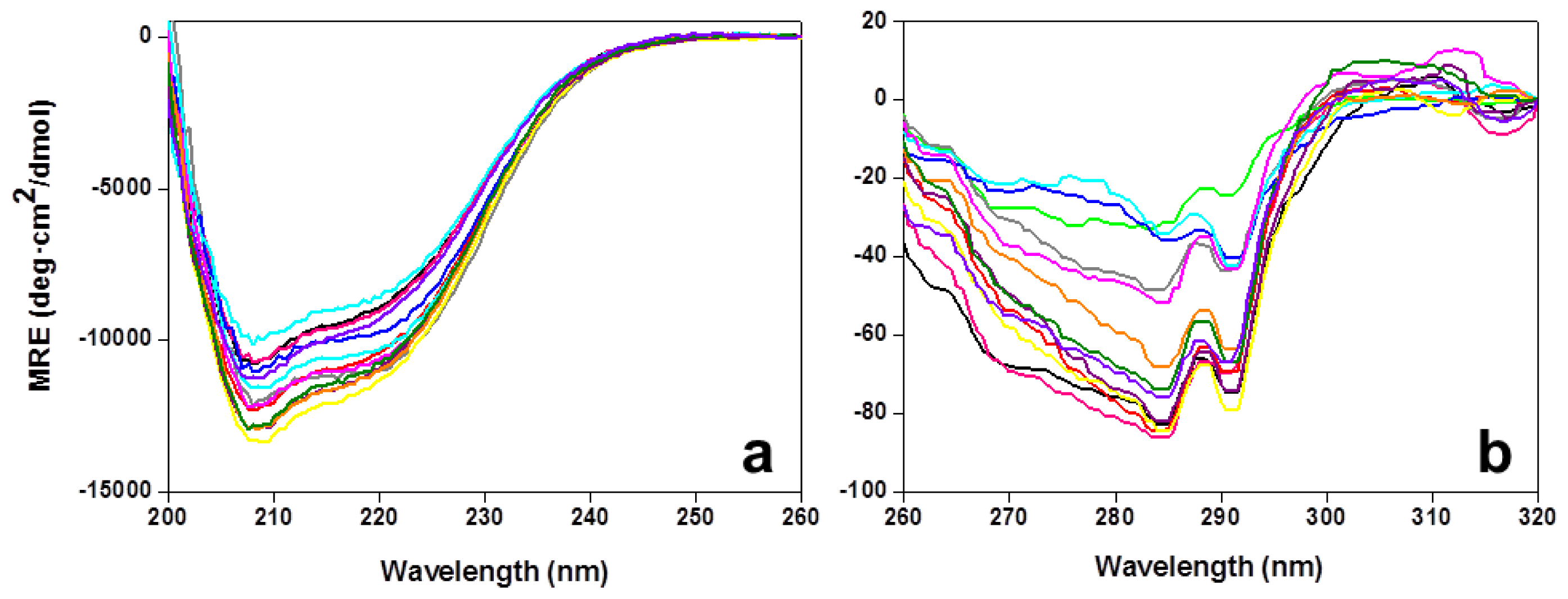
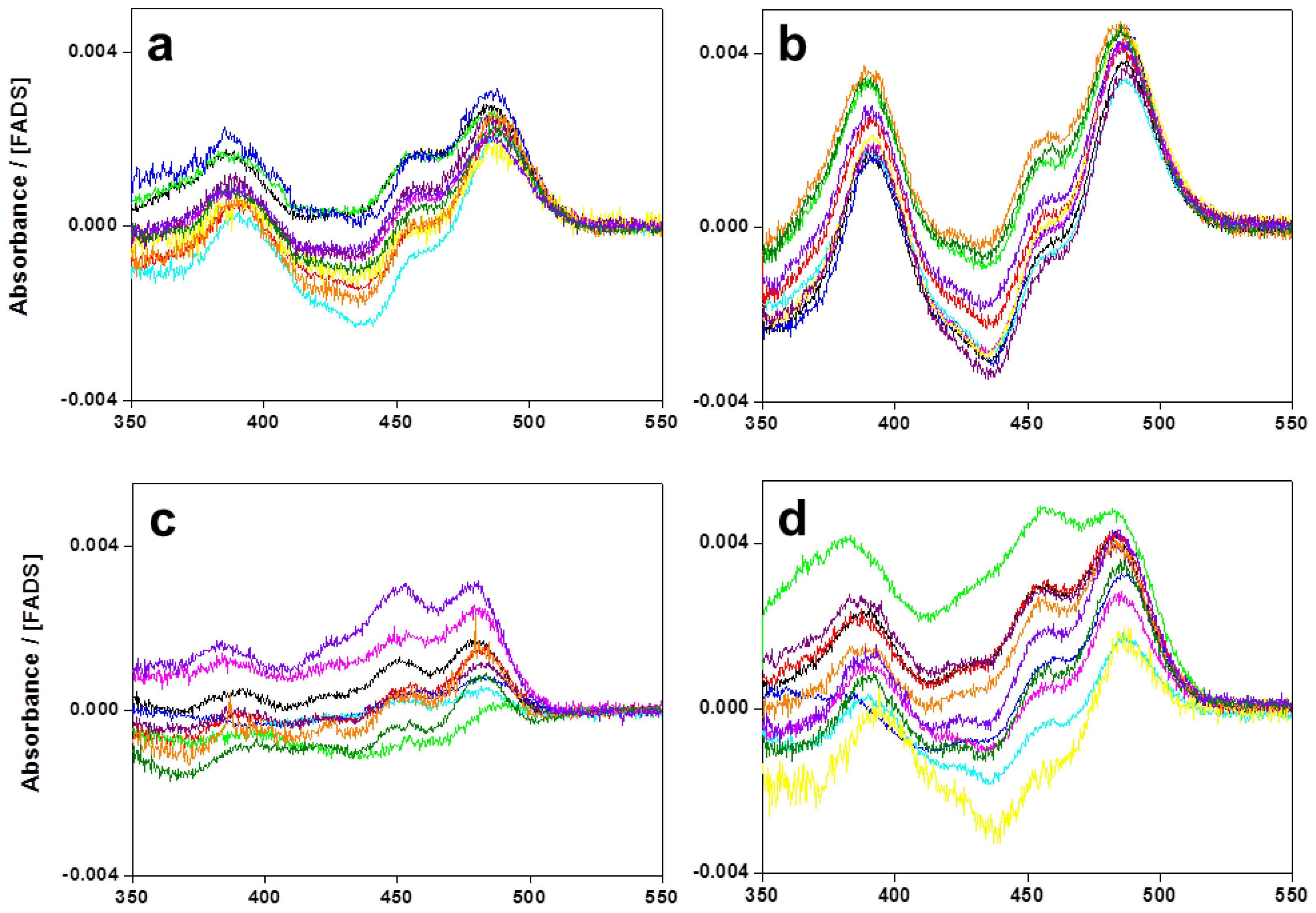
2.1. Expression, Purification, and Spectral Properties of the CaFADS Mutants
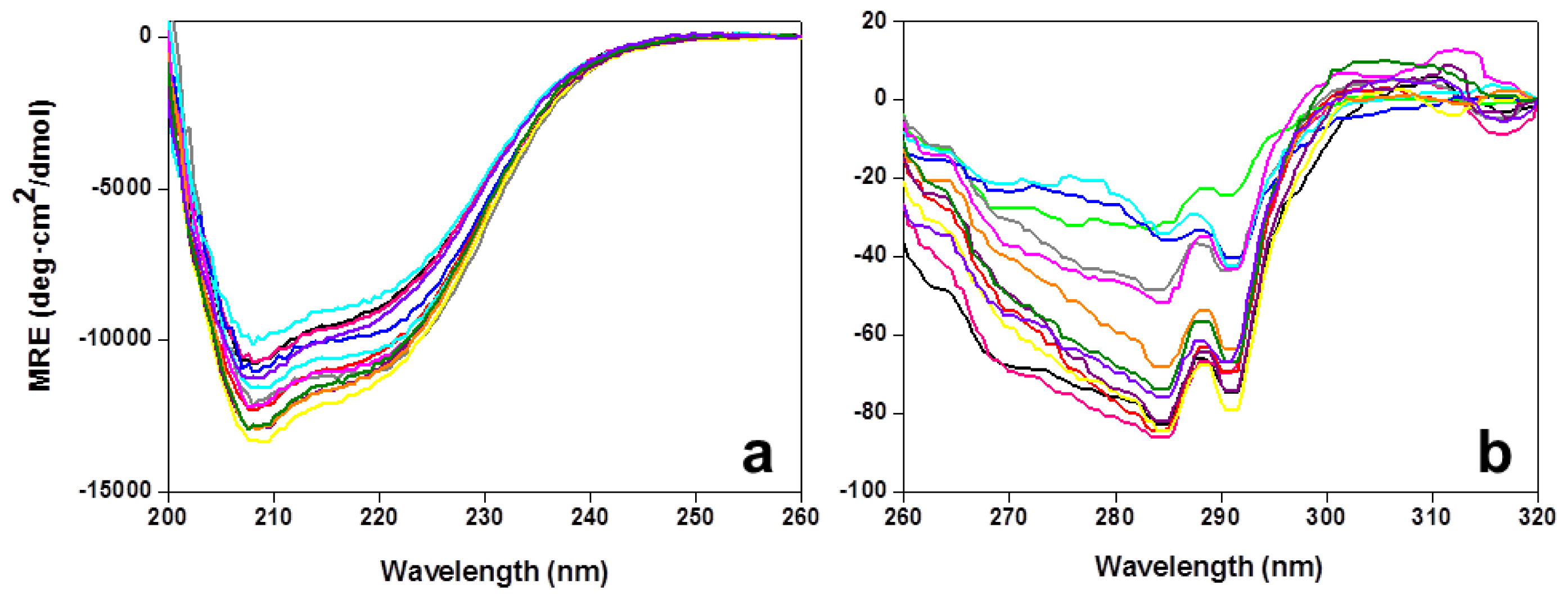
2.2. Stability of the CaFADS Mutants
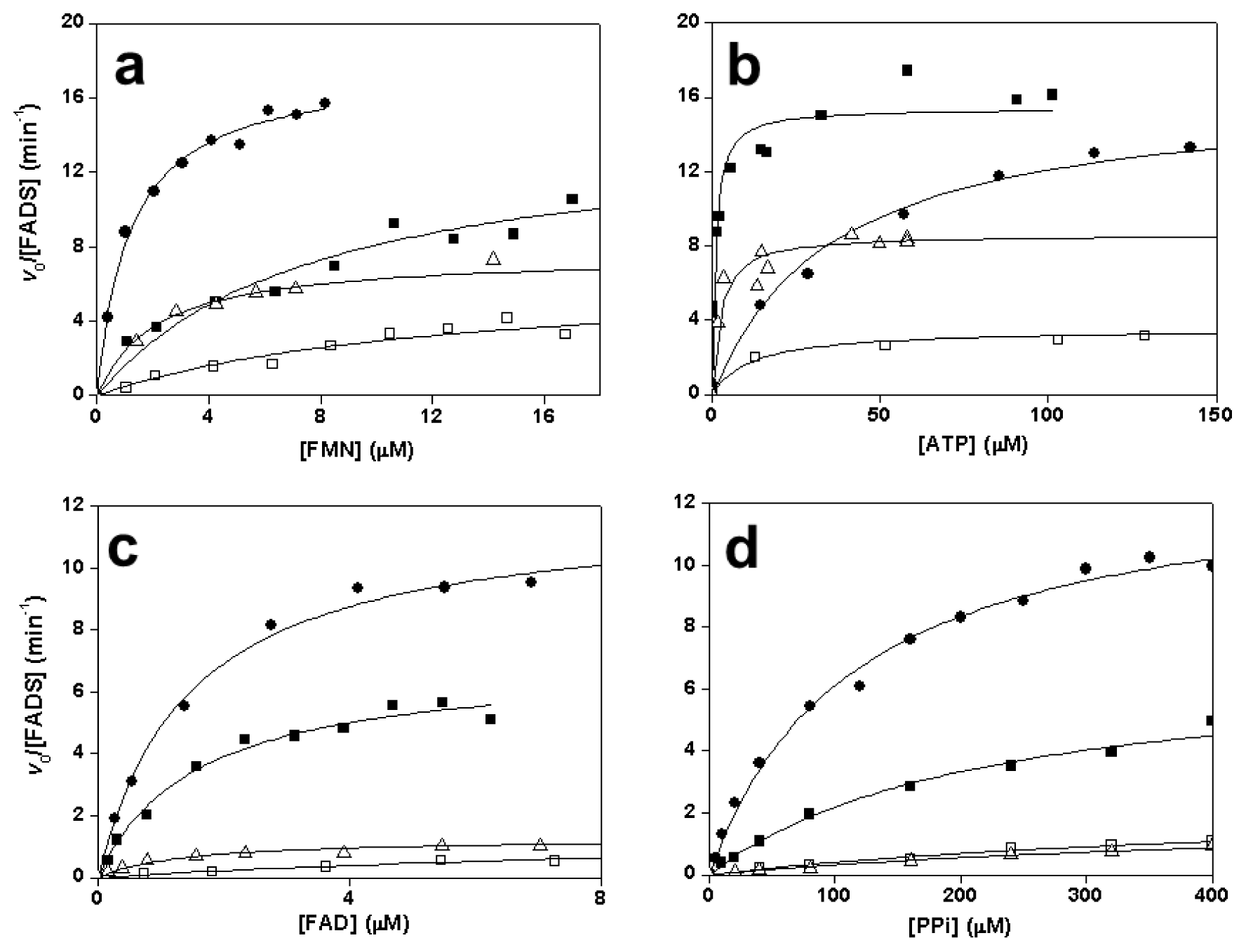
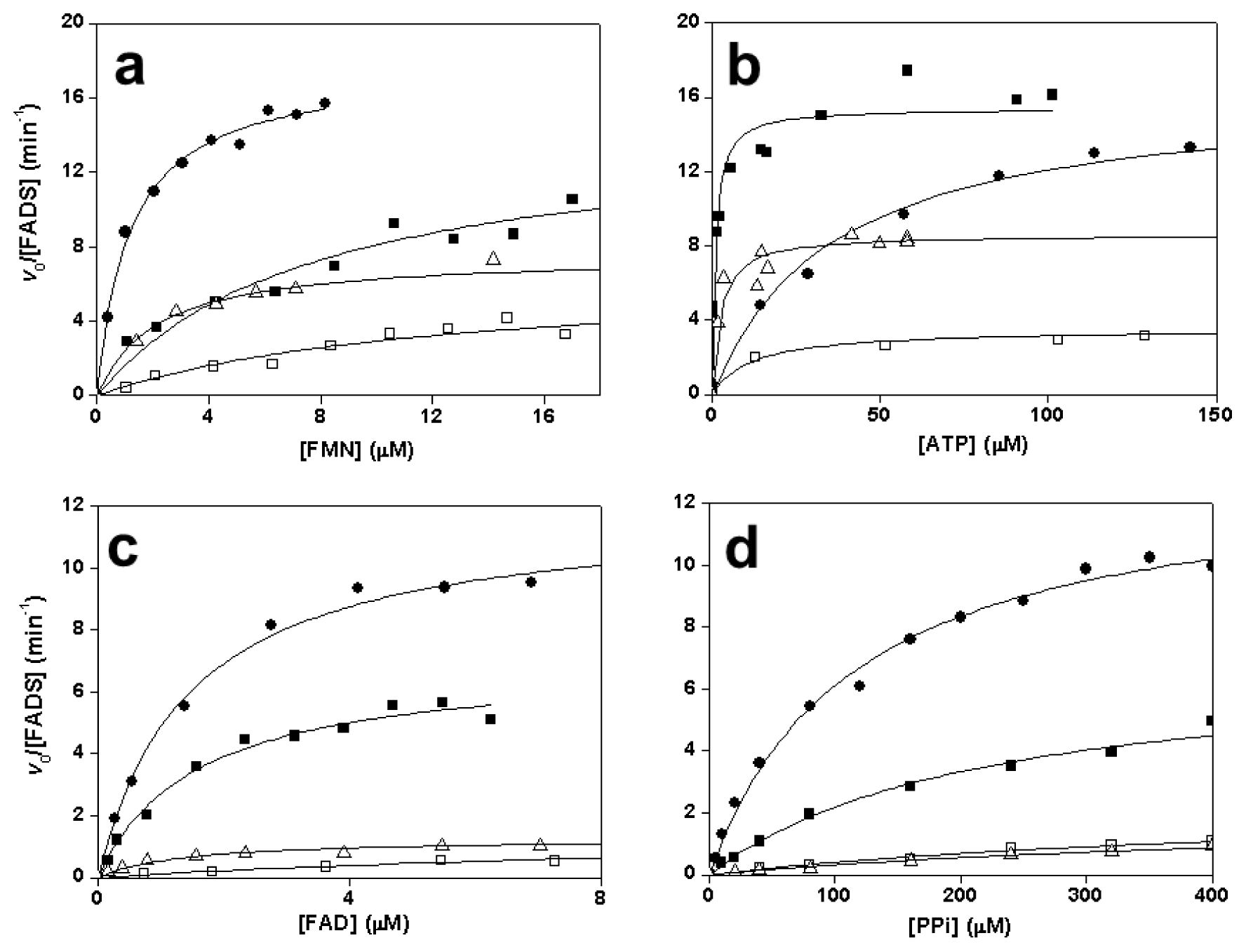
2.3. Effects of Mutations on the FMNAT Activity of CaFADS
2.4. Effects of Mutations on the FADpp Activity of CaFADS
2.5. Effects of Mutations on the RFK Activity of CaFADS
2.6. Effects of Mutations on Binding Parameters of the Flavins to CaFADS
2.7. Effects of Mutations on the ATP Binding Parameters to CaFADS
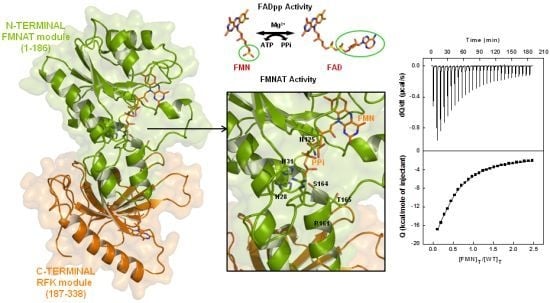
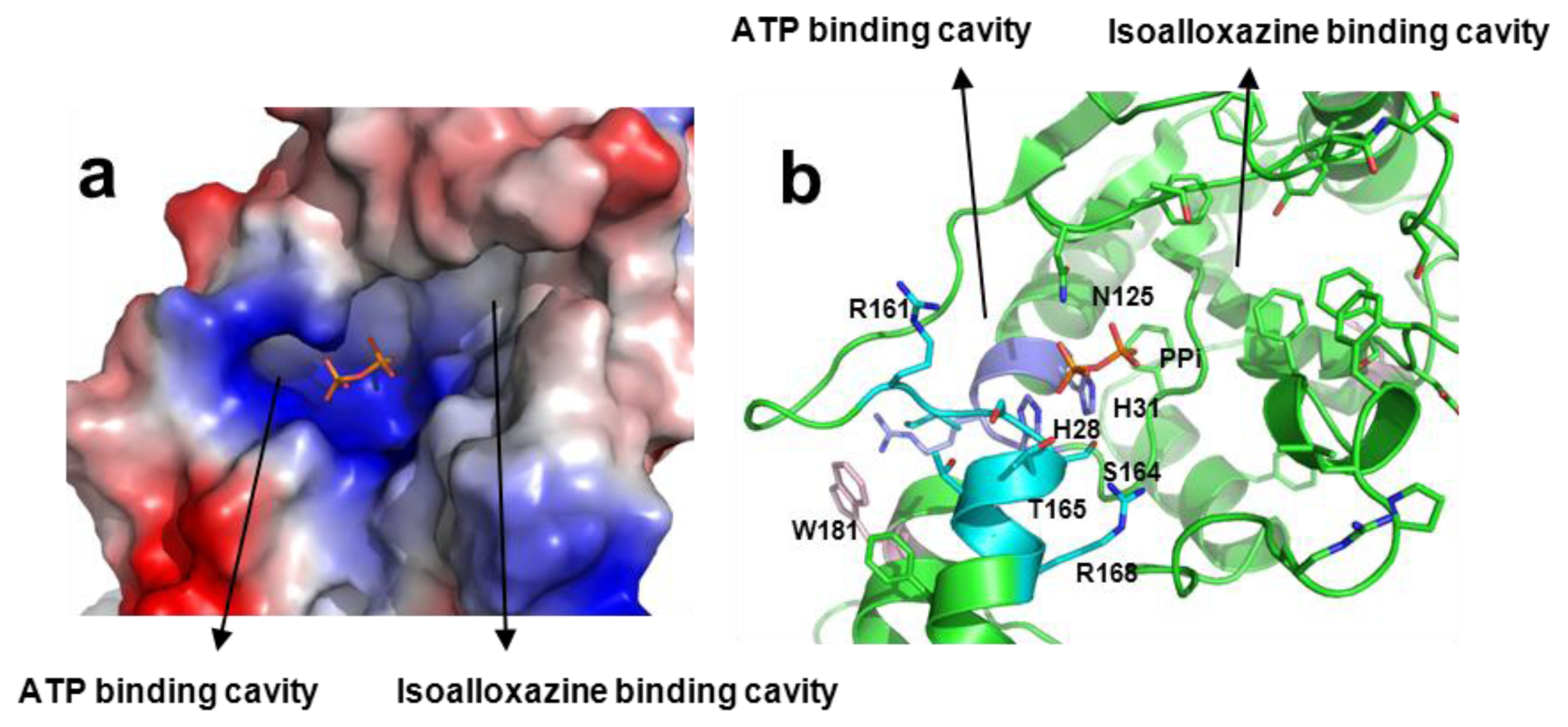
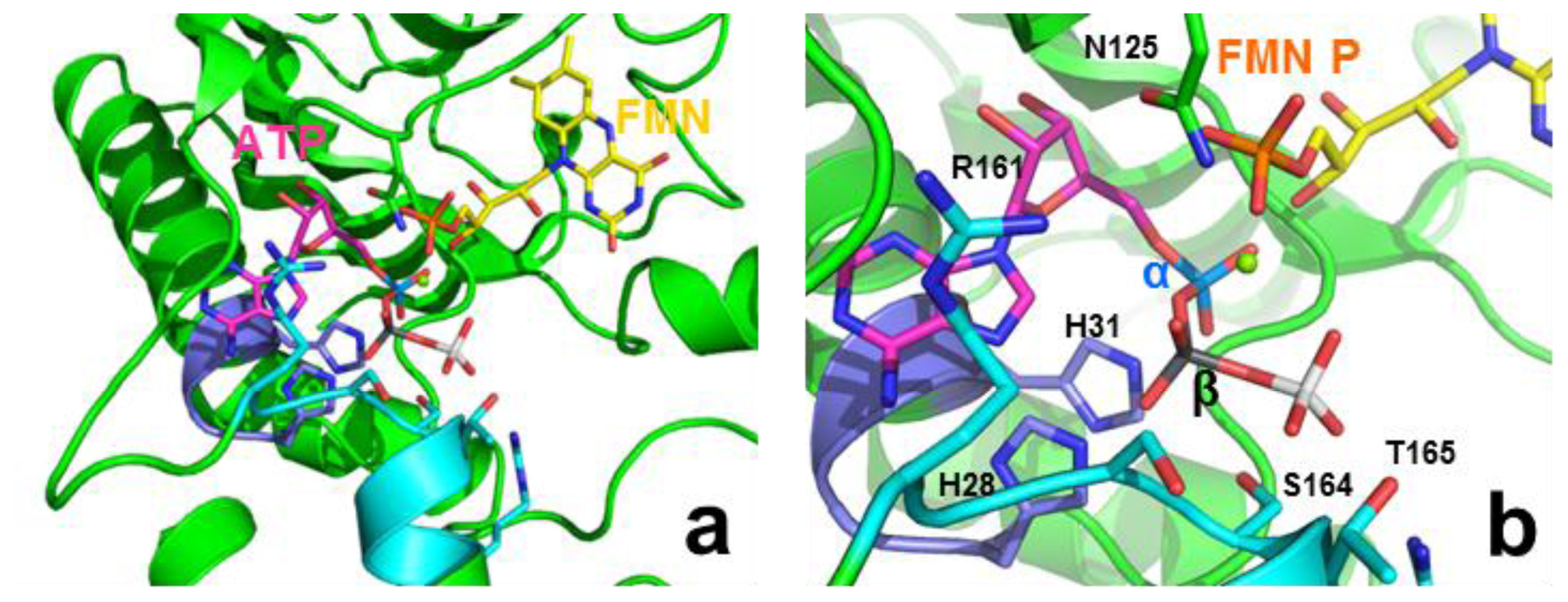
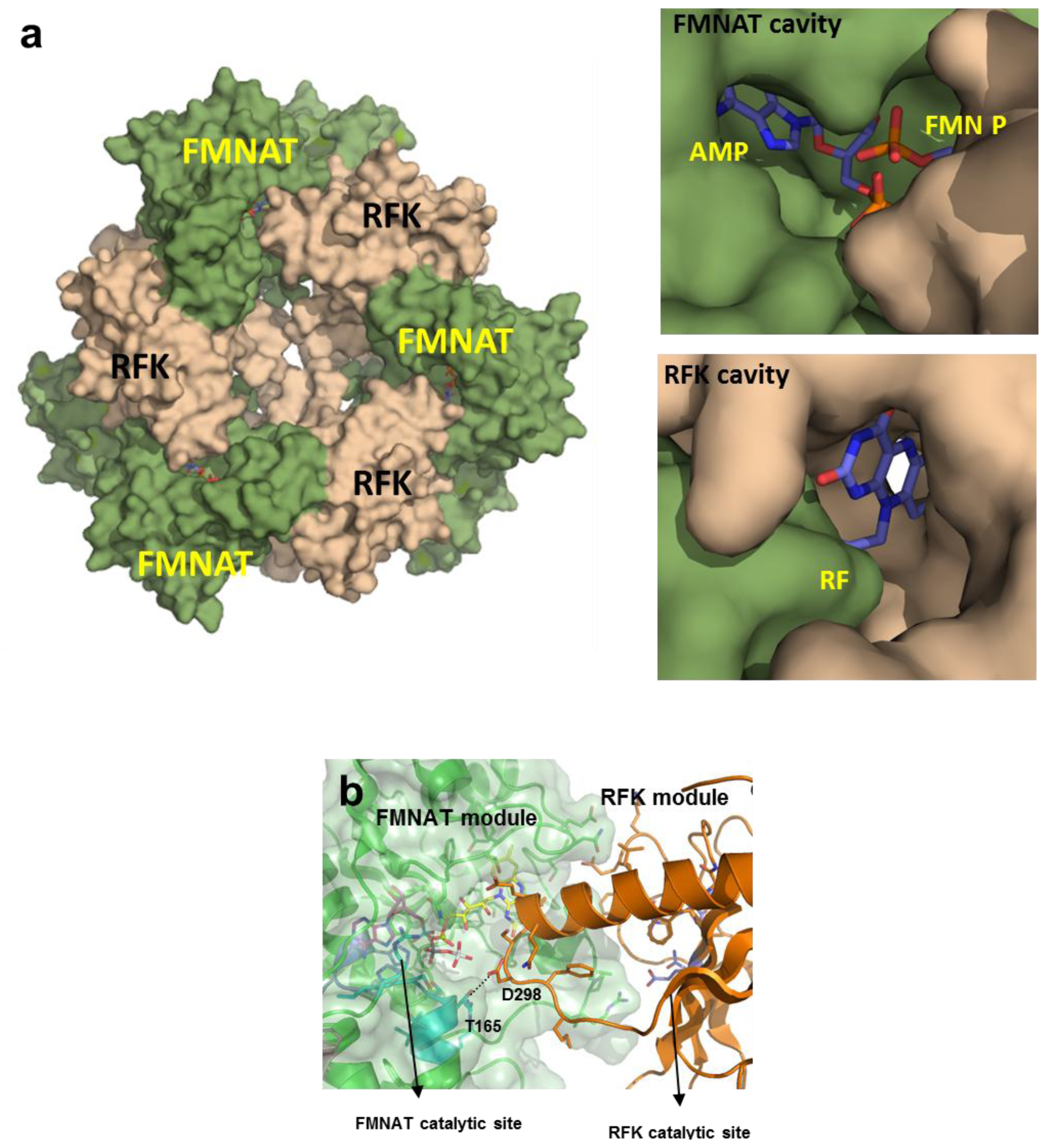
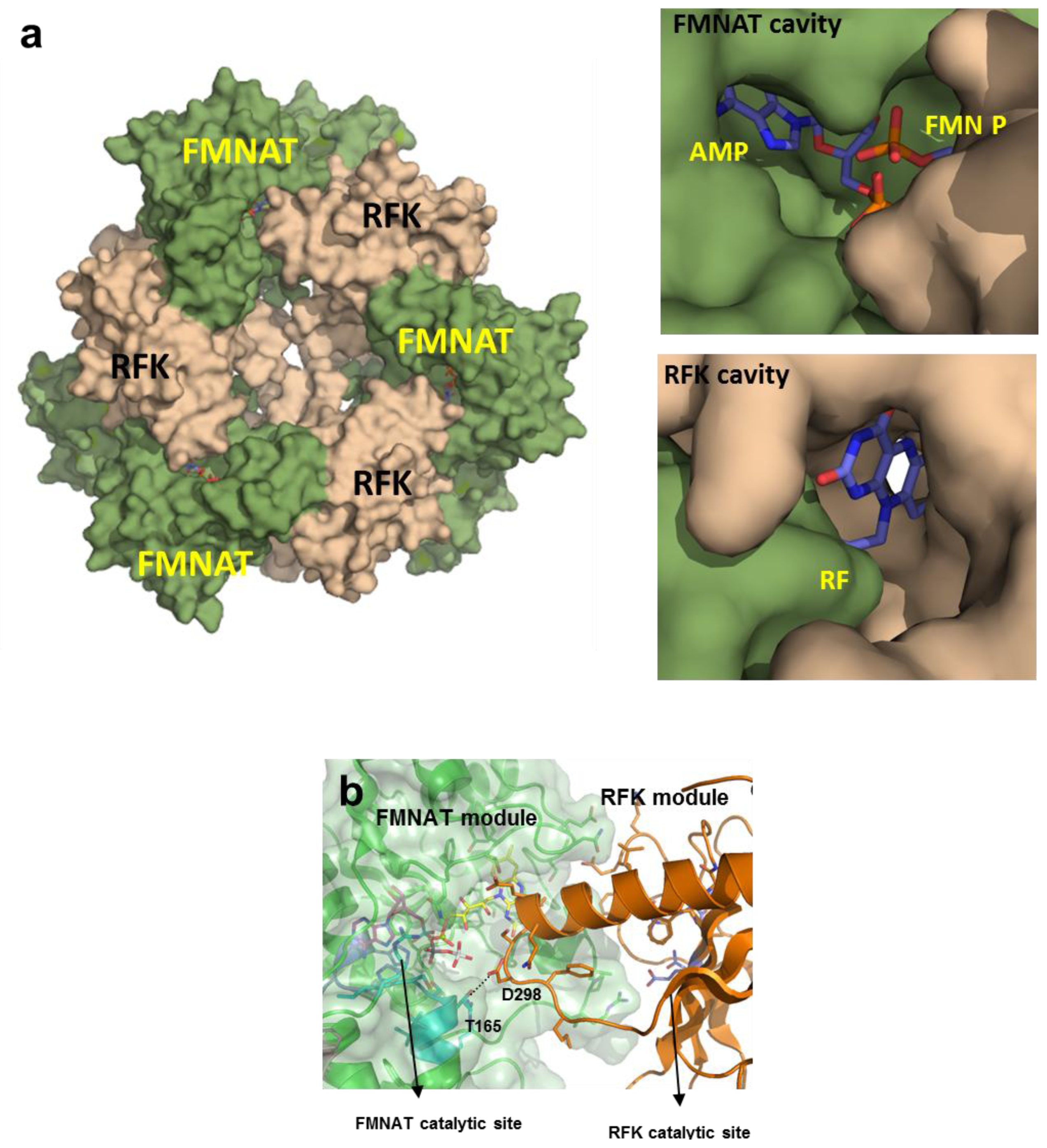
2.8. Structural Bases for the Role of Key Residues for the FMNAT and FADpp Activities of CaFADS
3. Experimental Section
3.1. Biological Material
3.2. Spectral Analysis
3.3. Determination of Steady-State Kinetics Parameters for the RFK, FMNAT and FADpp Activities of FADS
3.4. Differential Scanning Calorimetry (DSC)
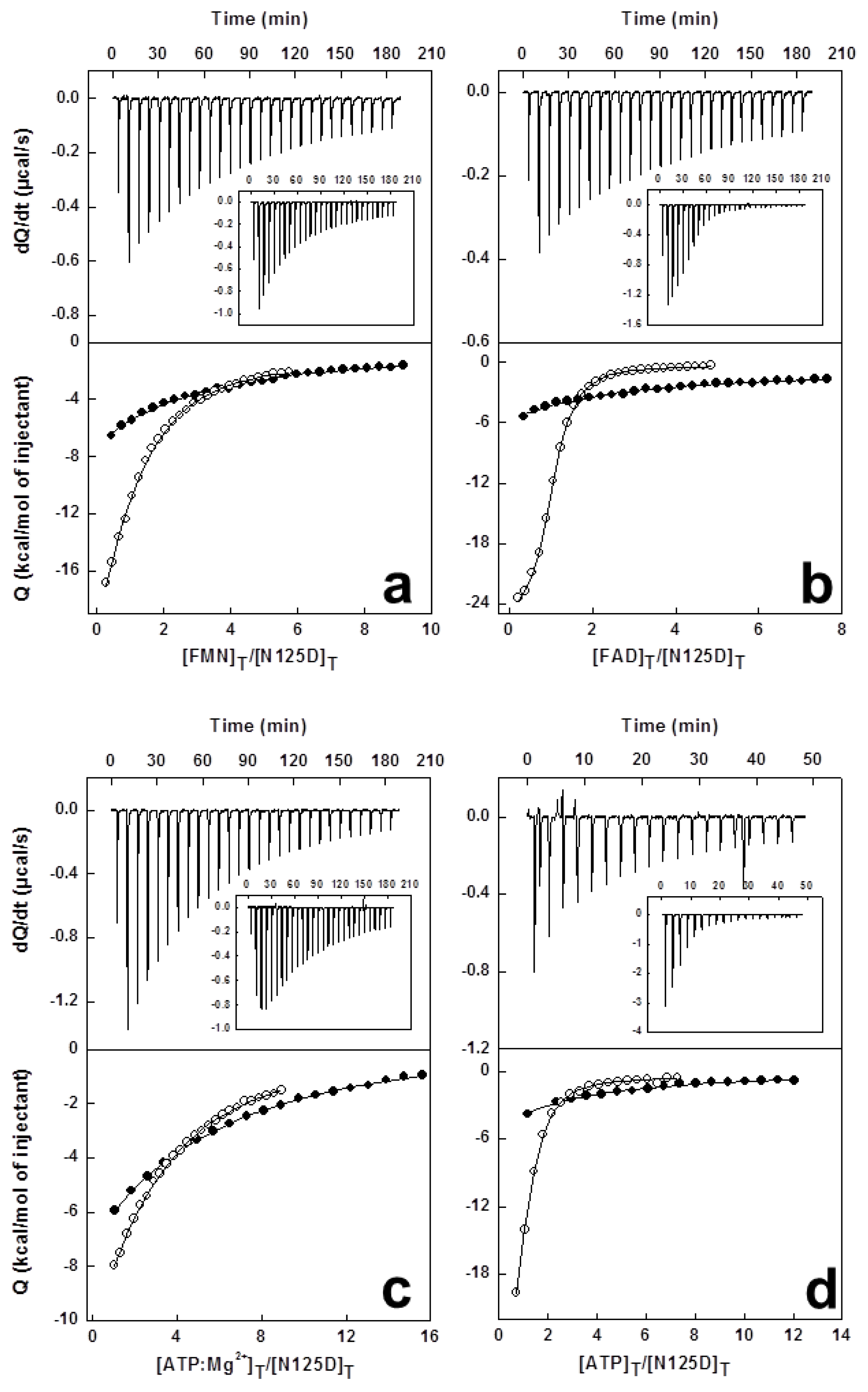
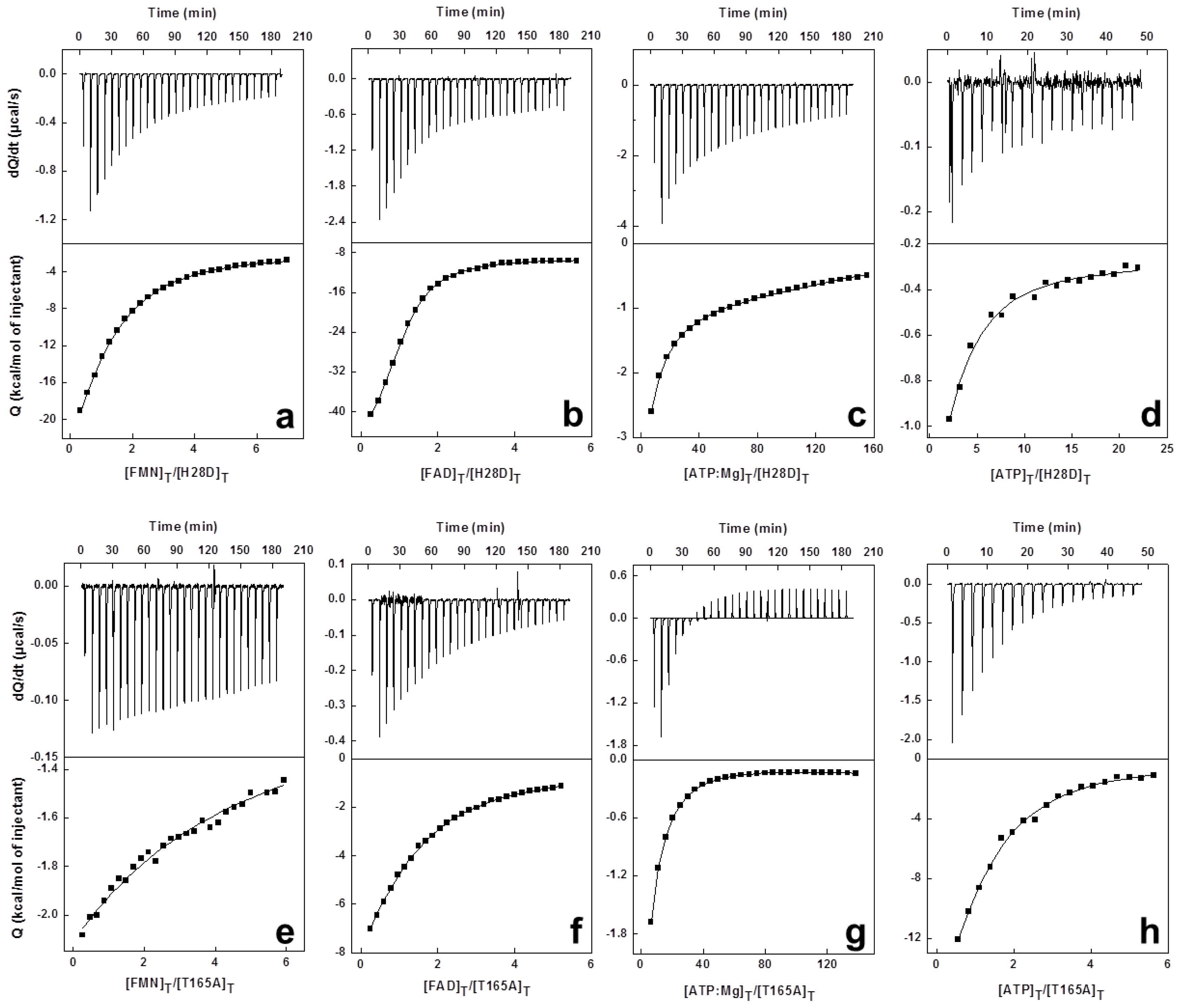
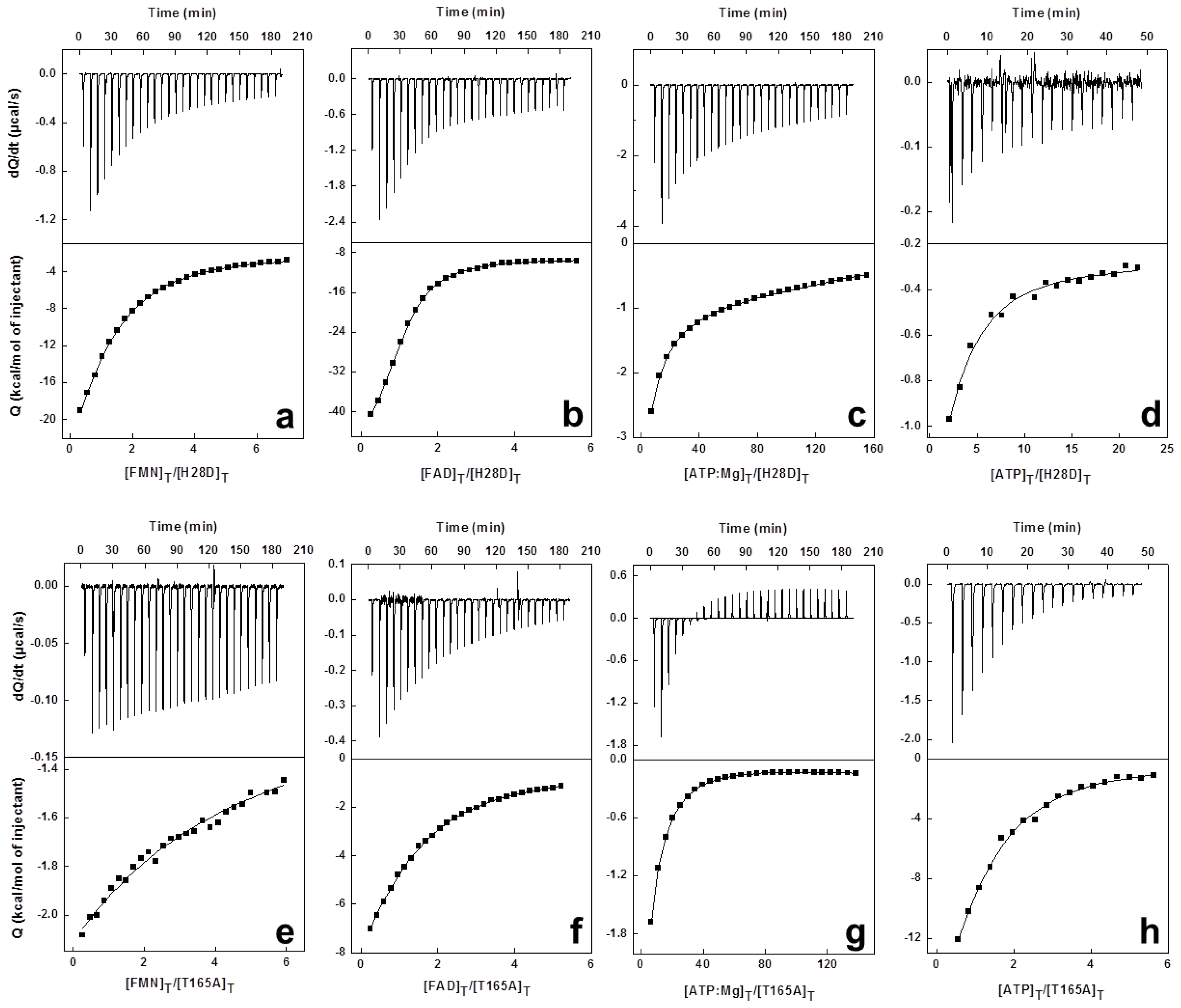
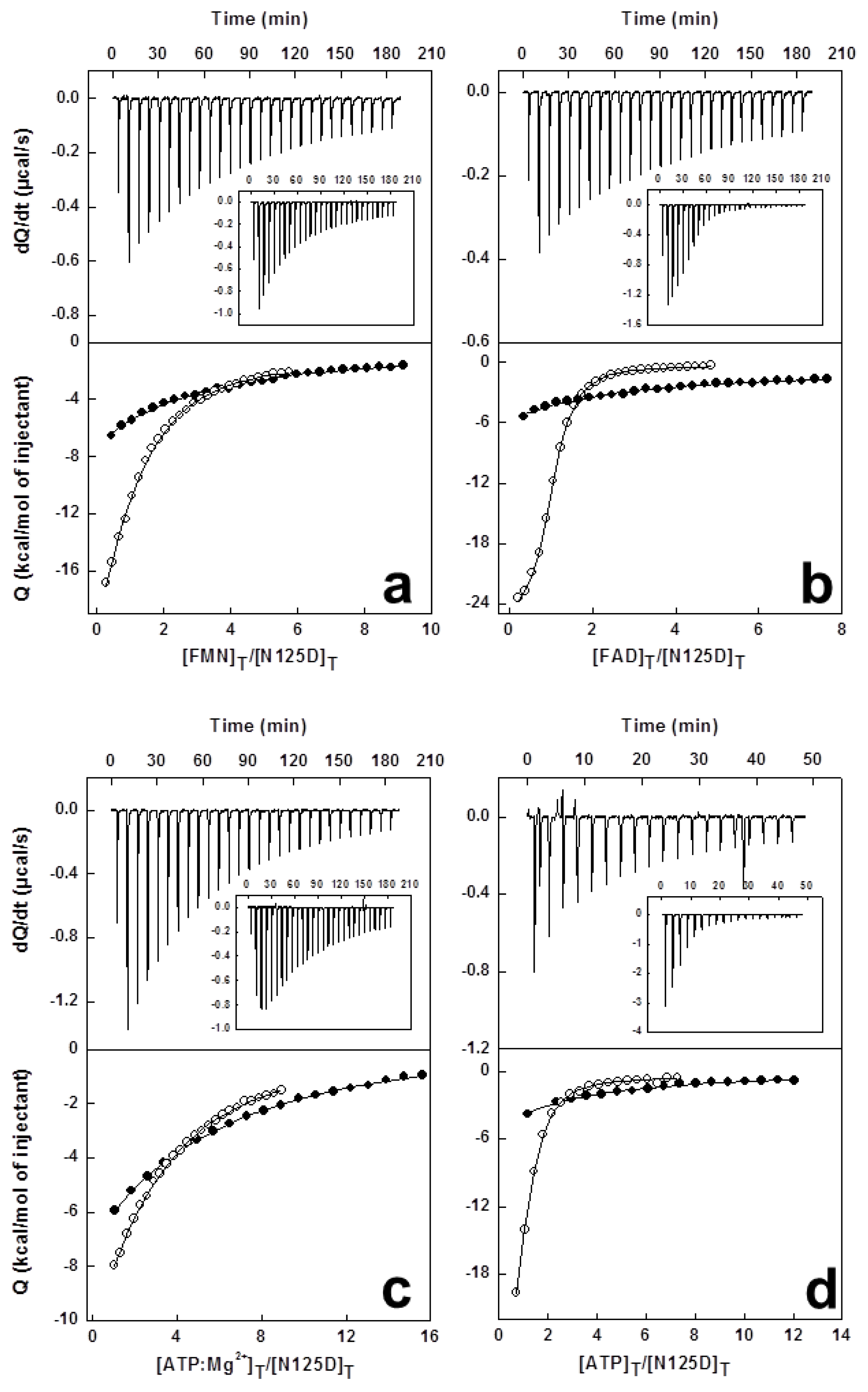
3.5. High Sensitivity Isothermal Titration Calorimetry (ITC)
4. Conclusions
Supplementary Information
1. Experimental Section
1.1. Biological Material
- 5′-GTGTCTTCGACGGCGTGGCGCGCGGGCATCAGAAATTG-3′ for H28A;
- 5′-GTGTCTTCGACGGCGTGGACCGCGGGCATCAGAAATTG-3′ for H28D;
- 5′-GGCGTGCATCGCGGGGCGCAGAAATTGATTAATGCC-3′ for H31A;
- 5′-GGCGTGCATCGCGGGGACCAGAAATTGATTAATGC-3′ for H31D;
- 5′-GACGATGAAGGCGTGGCGATCTCTTCCACGACC-3′ for R161A;
- 5′-CTTGACGATGAAGGCGTGGACATCTCTTCCACGACCG-3′ for R161D;
- 5′-GTGAGGATCTCTGCGACGACCGTGCGCGAGTTTC-3′ for S164A;
- 5′-CGTGAGGATCTCTGATACGACCGTGCGCGAGTTTC-3′ for S164D;
- 5′-GTGAGGATCTCTTCCGCCACCGTGCGCGAGTTTCTATC-3′ for T165A and;
- 5′-GTGAGGATCTCTTCCGATACCGTGCGCGAGTTTCTATC-3′ for T165D.
1.2. Kinetic Analysis
1.3. Isothermal Titration Calorimetry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tm,app (°C) | |
|---|---|
| WT | 42.5 |
| H28A | 48.9 |
| H28D | 41.5 |
| H31A | 45.9 |
| N125A | 40.2 |
| N125D | 43.5 |
| R161A | 49.2 |
| R161D | 43.9 |
| S164A | 38.9 |
| S164D | 49.7 |
| T165A | 40.1 |
| T165D | 44.9 |
| kcatb,c (min−1) | KmRF b,c (μM) | Kib,c (μM) | kcat/KmRF b (min−1 μM−1) | kcatc,d (min−1) | KmATP c,d (μM) | kcat/KmATP d (min−1 μM−1) | |
|---|---|---|---|---|---|---|---|
| WT a | 301 | 13 | 4.0 | 23 | 68 | 14 | 4.9 |
| H28A | 427 | 23 | 1.8 | 18 | 45 | 14 | 3.1 |
| H28D | 287 | 25 | 1.9 | 12 | 55 | 12 | 4.7 |
| H31A | 169 | 4.5 | 6.3 | 37 | 58 | 14 | 4.3 |
| N125A | 111 | 1.7 | 4.6 | 65 | 46 | 24 | 1.9 |
| N125D | 415 | 16 | 3.3 | 26 | 80 | 35 | 2.3 |
| R161A | 300 | 12 | 5.1 | 25 | 65 | 12 | 5.6 |
| R161D | 300 | 13 | 5.1 | 23 | 68 | 11 | 6.1 |
| S164A | 259 | 8.8 | 4.4 | 29 | 59 | 12 | 5.0 |
| S164D | 185 | 5.6 | 5.8 | 33 | 55 | 10 | 5.3 |
| T165A | 171 | 10 | 3.2 | 17 | 66 | 12 | 5.4 |
| T165D | 180 | 7.7 | 4.1 | 23 | 45 | 11 | 4.1 |
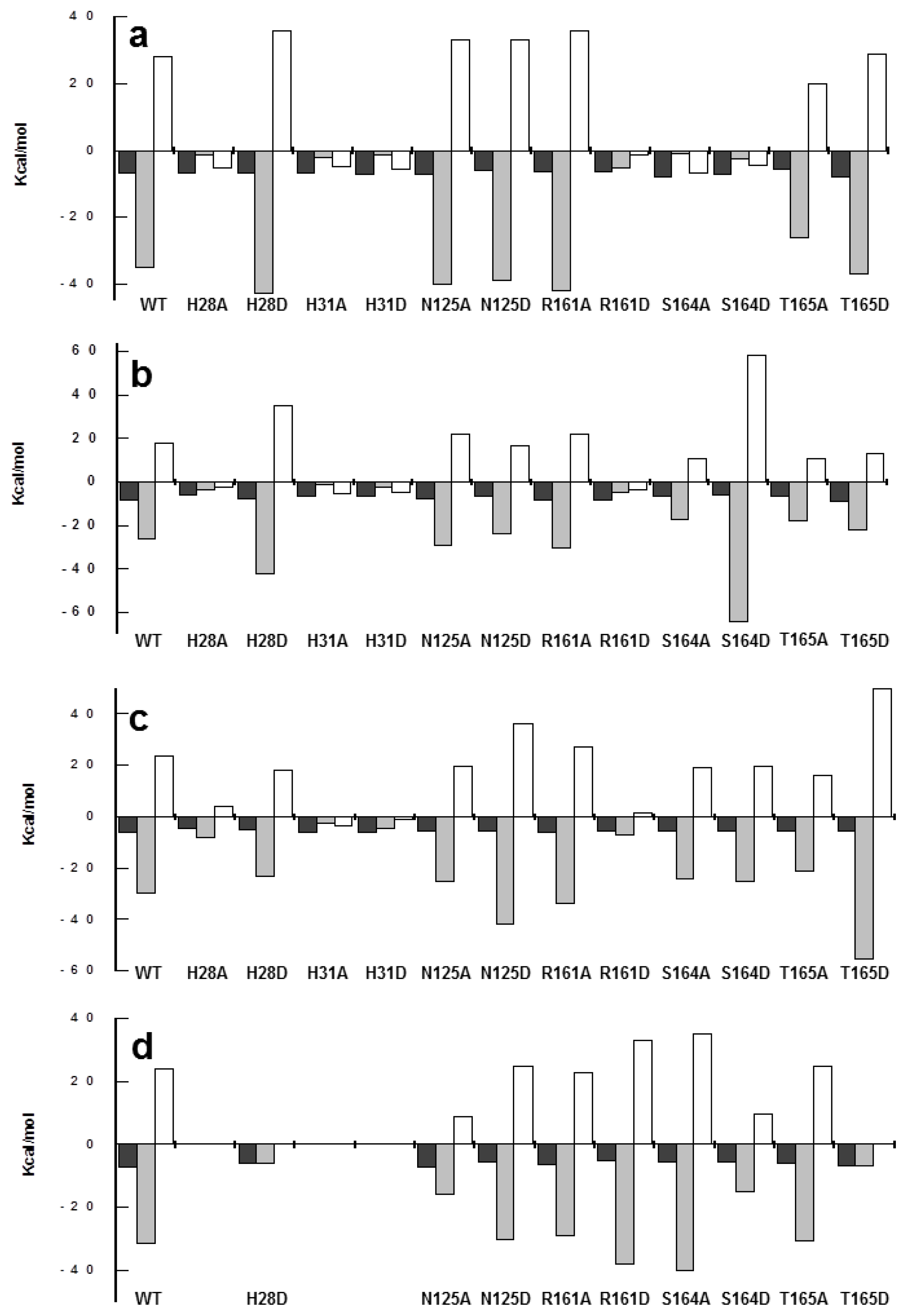
| FADS:FMN 10 mM Mg2+ | FADS:FAD 10 mM Mg2+ | FADS:ATP 10 mM Mg2+ | FADS:ATP 0 mM Mg2+ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔG (kcal/mol) | ΔH (kcal/mol) | −TΔS (kcal/mol) | ΔG (kcal/mol) | ΔH (kcal/mol) | −TΔS (kcal/mol) | ΔG (kcal/mol) | ΔH (kcal/mol) | −TΔS (kcal/mol) | ΔG (kcal/mol) | ΔH (kcal/mol) | −TΔS (kcal/mol) | |
| WT | −7.0 | −35 | 28 | −8.4 | −26 | 18 | −6.2 | −30 | 23 | −7.2 | −31 | 24 |
| H28A | −6.7 | −1.4 | −5.3 | −5.9 | −3.5 | −2.4 | −4.3 | −8.3 | 4.0 | --- | --- | --- |
| H28D | −7.0 | −43 | 36 | −7.8 | −42 | 35 | −5.3 | −23 | 18 | −6.0 | −6.1 | 0.1 |
| H31A | −6.7 | −2.0 | −4.7 | −6.5 | −1.2 | −5.3 | −5.9 | −2.6 | −3.3 | --- | --- | --- |
| H31D | −7.1 | −1.4 | −5.7 | −6.7 | −2.1 | −4.6 | −5.9 | −4.6 | −1.3 | --- | --- | --- |
| N125A | −7.2 | −40 | 33 | −7.6 | −29 | 22 | −5.3 | −25 | 20 | −7.1 | −16 | 8.7 |
| N125D | −6.0 | −39 | 33 | −6.7 | −24 | 17 | −5.7 | −42 | 36 | −5.5 | −30 | 25 |
| R161A | −6.5 | −42 | 36 | −8.4 | −30 | 22 | −6.2 | −34 | 27 | −6.2 | −29 | 23 |
| R161D | −6.6 | −5.2 | −1.4 | −8.3 | −4.9 | −3.4 | −5.7 | −7.2 | 1.5 | −5.2 | −38 | 33 |
| S164A | −8.0 | −1.1 | −6.9 | −6.3 | −17 | 11 | −5.4 | −24 | 19 | −5.5 | −40 | 35 |
| S164D | −7.2 | −2.6 | −4.6 | −6.2 | −64 | 58 | −5.4 | −25 | 20 | −5.4 | −15 | 9.6 |
| T165A | −5.7 | −26 | 20 | −6.7 | −18 | 11 | −5.4 | −21 | 16 | −6.1 | −31 | 25 |
| T165D | −7.8 | −37 | 29 | −8.8 | −22 | 13 | −5.4 | −55 | 50 | −6.7 | −6.9 | 0.2 |
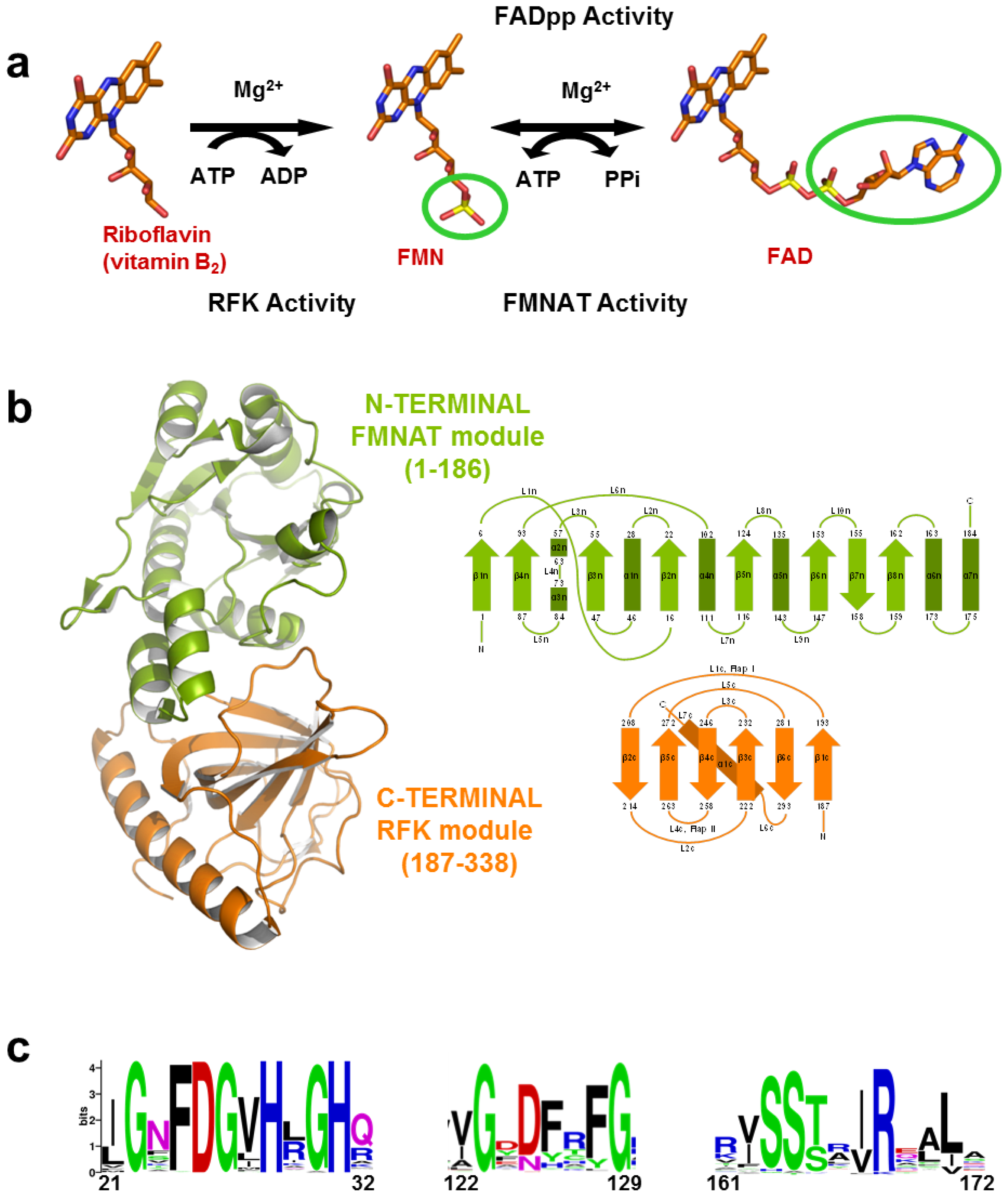
(a) Scheme for the reactions catalysed by the RFK and the FMNAT modules of CaFADS. (b) Cartoon representation and topology of the crystal structure of CaFADS (2X0K). The N-terminal FMNAT and C-terminal RFK modules are coloured in green and orange, respectively. (c) Logo of sequence homology of the motifs putatively involved in the FMNAT catalytic activity in the FADS family. The sequence logo was produced using the server (available online: http://weblogo.berkeley.edu; accessed on15 February 2007).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
References
- Müller, F. Free Flavins: Synthesis, Chemical and Physical Properties. In Chemistry and Biochemistry of Flavoenzymes; Müller, F., Ed.; CRC Press: Boca Raton, FL, USA, 1991; Volume 1, pp. 1–71. [Google Scholar]
- Powers, H.J. Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr 2003, 77, 1352–1360. [Google Scholar]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol 2007, 11, 195–202. [Google Scholar]
- Merrill, A.H., Jr; Lambeth, J.D.; Edmondson, D.E.; McCormick, D.B. Formation and mode of action of flavoproteins. Annu. Rev. Nutr. 1981, 1, 281–317. [Google Scholar]
- Bacher, A.; Eberhardt, S.; Fischer, M.; Kis, K.; Richter, G. Biosynthesis of vitamin b2 (riboflavin). Annu. Rev. Nutr 2000, 20, 153–167. [Google Scholar]
- Roje, S. Vitamin B biosynthesis in plants. Phytochemistry 2007, 68, 1904–1921. [Google Scholar]
- Barile, M.; Brizio, C.; Valenti, D.; De Virgilio, C.; Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem 2000, 267, 4888–4900. [Google Scholar]
- Brizio, C.; Galluccio, M.; Wait, R.; Torchetti, E.M.; Bafunno, V.; Accardi, R.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem. Biophys. Res. Commun 2006, 344, 1008–1016. [Google Scholar]
- Serrano, A.; Ferreira, P.; Martínez-Júlvez, M.; Medina, M. The prokaryotic FAD synthetase family: A potential drug target. Curr. Pharm. Des. 2012, in press. [Google Scholar]
- Mashhadi, Z.; Xu, H.; Grochowski, L.L.; White, R.H. Archaeal RibL: A new FAD synthetase that is air sensitive. Biochemistry 2010, 49, 8748–8755. [Google Scholar]
- Galluccio, M.; Brizio, C.; Torchetti, E.M.; Ferranti, P.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli, purification and characterization of isoform 2 of human FAD synthetase. Protein Expr. Purif 2007, 52, 175–181. [Google Scholar]
- Mashhadi, Z.; Zhang, H.; Xu, H.; White, R.H. Identification and characterization of an archaeon-specific riboflavin kinase. J. Bacteriol 2008, 190, 2615–2618. [Google Scholar]
- Bonnefond, L.; Frugier, M.; Touze, E.; Lorber, B.; Florentz, C.; Giege, R.; Sauter, C.; Rudinger-Thirion, J. Crystal structure of human mitochondrial tyrosyl-tRNA synthetase reveals common and idiosyncratic features. Structure 2007, 15, 1505–1516. [Google Scholar]
- Giancaspero, T.A.; Locato, V.; de Pinto, M.C.; De Gara, L.; Barile, M. The occurrence of riboflavin kinase and FAD synthetase ensures FAD synthesis in tobacco mitochondria and maintenance of cellular redox status. FEBS J 2009, 276, 219–231. [Google Scholar]
- Manstein, D.J.; Pai, E.F. Purification and characterization of FAD synthetase from Brevibacterium ammoniagenes. J. Biol. Chem 1986, 261, 16169–16173. [Google Scholar]
- Yruela, I.; Arilla-Luna, S.; Medina, M.; Contreras-Moreira, B. Evolutionary divergence of chloroplast FAD synthetase proteins. BMC Evol. Biol 2010, 10, 311. [Google Scholar] [Green Version]
- Frago, S.; Martínez-Júlvez, M.; Serrano, A.; Medina, M. Structural analysis of FAD synthetase from Corynebacterium ammoniagenes. BMC Microbiol 2008, 8, 160. [Google Scholar]
- Herguedas, B.; Martinez-Julvez, M.; Frago, S.; Medina, M.; Hermoso, J.A. Oligomeric state in the crystal structure of modular FAD synthetase provides insights into its sequential catalysis in prokaryotes. J. Mol. Biol 2010, 400, 218–230. [Google Scholar]
- Karthikeyan, S.; Zhou, Q.; Osterman, A.L.; Zhang, H. Ligand binding-induced conformational changes in riboflavin kinase: Structural basis for the ordered mechanism. Biochemistry 2003, 42, 12532–12538. [Google Scholar]
- Serrano, A.; Frago, S.; Herguedas, B.; Martínez-Júlvez, M.; Velázquez-Campoy, A.; Medina, M. Key residues at the riboflavin kinase catalytic site of bifunctional riboflavin kinase/FMN adenylyltransferase from Corynebacterium ammoniagenes. Cell Biochem. Biophys. 2012. [Google Scholar] [CrossRef]
- Frago, S.; Velázquez-Campoy, A.; Medina, M. The puzzle of ligand binding to Corynebacterium ammoniagenes FAD synthetase. J. Biol. Chem 2009, 284, 6610–6619. [Google Scholar]
- Park, Y.S.; Gee, P.; Sanker, S.; Schurter, E.J.; Zuiderweg, E.R.; Kent, C. Identification of functional conserved residues of CTP:glycerol-3-phosphate cytidylyltransferase. Role of histidines in the conserved HXGH in catalysis. J. Biol. Chem 1997, 272, 15161–15166. [Google Scholar]
- Veitch, D.P.; Gilham, D.; Cornell, R.B. The role of histidine residues in the HXGH site of CTP:phosphocholine cytidylyltransferase in CTP binding and catalysis. Eur. J. Biochem 1998, 255, 227–234. [Google Scholar]
- Sanker, S.; Campbell, H.A.; Kent, C. Negative cooperativity of substrate binding but not enzyme activity in wild-type and mutant forms of CTP:glycerol-3-phosphate cytidylyltransferase. J. Biol. Chem 2001, 276, 37922–37928. [Google Scholar]
- Saridakis, V.; Christendat, D.; Kimber, M.S.; Dharamsi, A.; Edwards, A.M.; Pai, E.F. Insights into ligand binding and catalysis of a central step in NAD+ synthesis: Structures of Methanobacterium thermoautotrophicum NMN adenylyltransferase complexes. J. Biol. Chem 2001, 276, 7225–7232. [Google Scholar]
- Saridakis, V.; Pai, E.F. Mutational, structural, and kinetic studies of the ATP-binding site of Methanobacterium thermoautotrophicum nicotinamide mononucleotide adenylyltransferase. J. Biol. Chem 2003, 278, 34356–34363. [Google Scholar]
- Fersht, A.R. Dissection of the structure and activity of the tyrosyl-tRNA synthetase by site-directed mutagenesis. Biochemistry 1987, 26, 8031–8037. [Google Scholar]
- D’Angelo, I.; Raffaelli, N.; Dabusti, V.; Lorenzi, T.; Magni, G.; Rizzi, M. Structure of nicotinamide mononucleotide adenylyltransferase: A key enzyme in NAD(+) biosynthesis. Structure 2000, 8, 993–1004. [Google Scholar]
- Izard, T. The crystal structures of phosphopantetheine adenylyltransferase with bound substrates reveal the enzyme’s catalytic mechanism. J. Mol. Biol 2002, 315, 487–495. [Google Scholar]
- Weber, C.H.; Park, Y.S.; Sanker, S.; Kent, C.; Ludwig, M.L. A prototypical cytidylyltransferase: CTP:glycerol-3-phosphate cytidylyltransferase from Bacillus subtilis. Structure 1999, 7, 1113–1124. [Google Scholar]
- Olland, A.M.; Underwood, K.W.; Czerwinski, R.M.; Lo, M.C.; Aulabaugh, A.; Bard, J.; Stahl, M.L.; Somers, W.S.; Sullivan, F.X.; Chopra, R. Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J. Biol. Chem 2002, 277, 3698–3707. [Google Scholar]
- Efimov, I.; Kuusk, V.; Zhang, X.; McIntire, W.S. Proposed steady-state kinetic mechanism for Corynebacterium ammoniagenes FAD synthetase produced by Escherichia coli. Biochemistry 1998, 37, 9716–9723. [Google Scholar]
- Izard, T.; Geerlof, A. The crystal structure of a novel bacterial adenylyltransferase reveals half of sites reactivity. EMBO J 1999, 18, 2021–2030. [Google Scholar]
- Perona, J.J.; Rould, M.A.; Steitz, T.A. Structural basis for transfer RNA aminoacylation by Escherichia coli glutaminyl-tRNA synthetase. Biochemistry 1993, 32, 8758–8771. [Google Scholar]
- Lowe, G.; Tansley, G. The stereochemical course of nucleotidyl transfer catalysed by NAD pyrophosphorylase. Eur. J. Biochem 1983, 132, 117–120. [Google Scholar]
- Leskovac, V. Comprehensive Enzyme Kinetics; Kluwer Adacemic/Plenum Publishers: New York, NY, USA; p. 2003.
- Torchetti, E.M.; Bonomi, F.; Galluccio, M.; Gianazza, E.; Giancaspero, T.A.; Lametti, S.; Indiveri, C.; Barile, M. Human FAD synthase (isoform 2): A component of the machinery that delivers FAD to apo-flavoproteins. FEBS J 2011, 278, 4434–4449. [Google Scholar]
- Huerta, C.; Borek, D.; Machius, M.; Grishin, N.V.; Zhang, H. Structure and mechanism of a eukaryotic FMN adenylyltransferase. J. Mol. Biol 2009, 389, 388–400. [Google Scholar]
- Gerdes, S.Y.; Scholle, M.D.; D’Souza, M.; Bernal, A.; Baev, M.V.; Farrell, M.; Kurnasov, O.V.; Daugherty, M.D.; Mseeh, F.; Polanuyer, B.M.; et al. From genetic footprinting to antimicrobial drug targets: Examples in cofactor biosynthetic pathways. J. Bacteriol 2002, 184, 4555–4572. [Google Scholar]


| kcat (min−1) | KmFMN (μM) | KmATP (μM) | kcat/KmFMN (min−1 μM−1) | kcat/KmATP (min−1 μM−1) | |
|---|---|---|---|---|---|
| WT a | 17 | 1.2 | 36 | 14 | 0.48 |
| H28A | <0.1 | - | - | - | - |
| H28D | <0.1 | - | - | - | - |
| H31A | <0.1 | - | - | - | - |
| H31D | <0.1 | - | - | - | - |
| N125A | <0.1 | - | - | - | - |
| N125D | <0.1 | - | - | - | - |
| R161A | 15 | 6.1 | 1.0 | 2.4 | 15 |
| R161D | 3.9 | 12.0 | 8.3 | 0.32 | 0.47 |
| S164A | 17 | 20 | 86 | 0.84 | 0.20 |
| S164D | <0.1 | - | - | - | - |
| T165A | 8.1 | 2.1 | 2.3 | 3.8 | 3.4 |
| T165D | <0.1 | - | - | - | - |
| kcat (min−1) | KmFAD (μM) | KmPPi (μM) | kcat/KmFAD (min−1 μM−1) | kcat/KmPPi (min−1 μM−1) | |
|---|---|---|---|---|---|
| WT | 13 | 1.4 | 114 | 8.6 | 0.110 |
| H28A | <0.1 | - | - | - | - |
| H28D | <0.1 | - | - | - | - |
| H31A | <0.1 | - | - | - | - |
| H31D | <0.1 | - | - | - | - |
| N125A | <0.5 | - | - | - | - |
| N125D | 2.1 | 15 | 320 | 0.14 | 0.007 |
| R161A | 6.9 | 1.7 | 215 | 4.4 | 0.032 |
| R161D | 2.1 | 17 | 483 | 0.12 | 0.004 |
| S164A | 1.7 | 17 | 229 | 0.10 | 0.007 |
| S164D | <0.1 | - | - | - | - |
| T165A | 1.7 | 1.3 | 537 | 1.2 | 0.003 |
| T165D | <0.1 | - | - | - | - |
| Kd (μM) | ||||
|---|---|---|---|---|
| FADS:FMN 10 mM Mg2+ | FADS:FAD 10 mM Mg2+ | FADS:ATP 10 mM Mg2+ | FADS:ATP 0 mM Mg2+ | |
| WT | 7.8 | 0.7 | 30 a | 5.5 |
| H28A | 13 | 48 | >650 a | very weak |
| H28D | 7.9 | 1.8 | 130 a | 40 |
| H31A | 18 | 18 | 43 a | very weak |
| H31D | 6.1 | 12 | >50 a | very weak |
| N125A | 5.6 | 2.7 | 123 a | 6.4 |
| N125D | 38 | 30 | 70 a | 91 |
| R161A | 18 | 0.8 | 27 a | 27 |
| R161D | 14 | 1.0 | 61 a | 164 |
| S164A | 1.4 | 26 | 111 a | 86 |
| S164D | 5.3 | 27 | 104 a | 114 |
| T165A | 65 | 11 | 118 a | 32 |
| T165D | 2.0 | 0.4 | 103 a | 11 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Serrano, A.; Frago, S.; Velázquez-Campoy, A.; Medina, M. Role of Key Residues at the Flavin Mononucleotide (FMN):Adenylyltransferase Catalytic Site of the Bifunctional Riboflavin Kinase/Flavin Adenine Dinucleotide (FAD) Synthetase from Corynebacterium ammoniagenes. Int. J. Mol. Sci. 2012, 13, 14492-14517. https://doi.org/10.3390/ijms131114492
Serrano A, Frago S, Velázquez-Campoy A, Medina M. Role of Key Residues at the Flavin Mononucleotide (FMN):Adenylyltransferase Catalytic Site of the Bifunctional Riboflavin Kinase/Flavin Adenine Dinucleotide (FAD) Synthetase from Corynebacterium ammoniagenes. International Journal of Molecular Sciences. 2012; 13(11):14492-14517. https://doi.org/10.3390/ijms131114492
Chicago/Turabian StyleSerrano, Ana, Susana Frago, Adrián Velázquez-Campoy, and Milagros Medina. 2012. "Role of Key Residues at the Flavin Mononucleotide (FMN):Adenylyltransferase Catalytic Site of the Bifunctional Riboflavin Kinase/Flavin Adenine Dinucleotide (FAD) Synthetase from Corynebacterium ammoniagenes" International Journal of Molecular Sciences 13, no. 11: 14492-14517. https://doi.org/10.3390/ijms131114492