Oxidant Stress and Signal Transduction in the Nervous System with the PI 3-K, Akt, and mTOR Cascade

Abstract
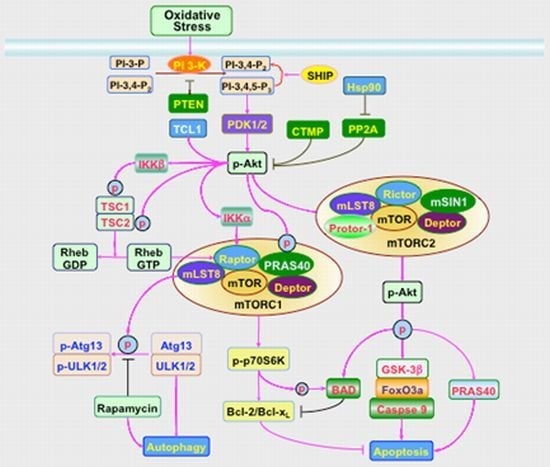
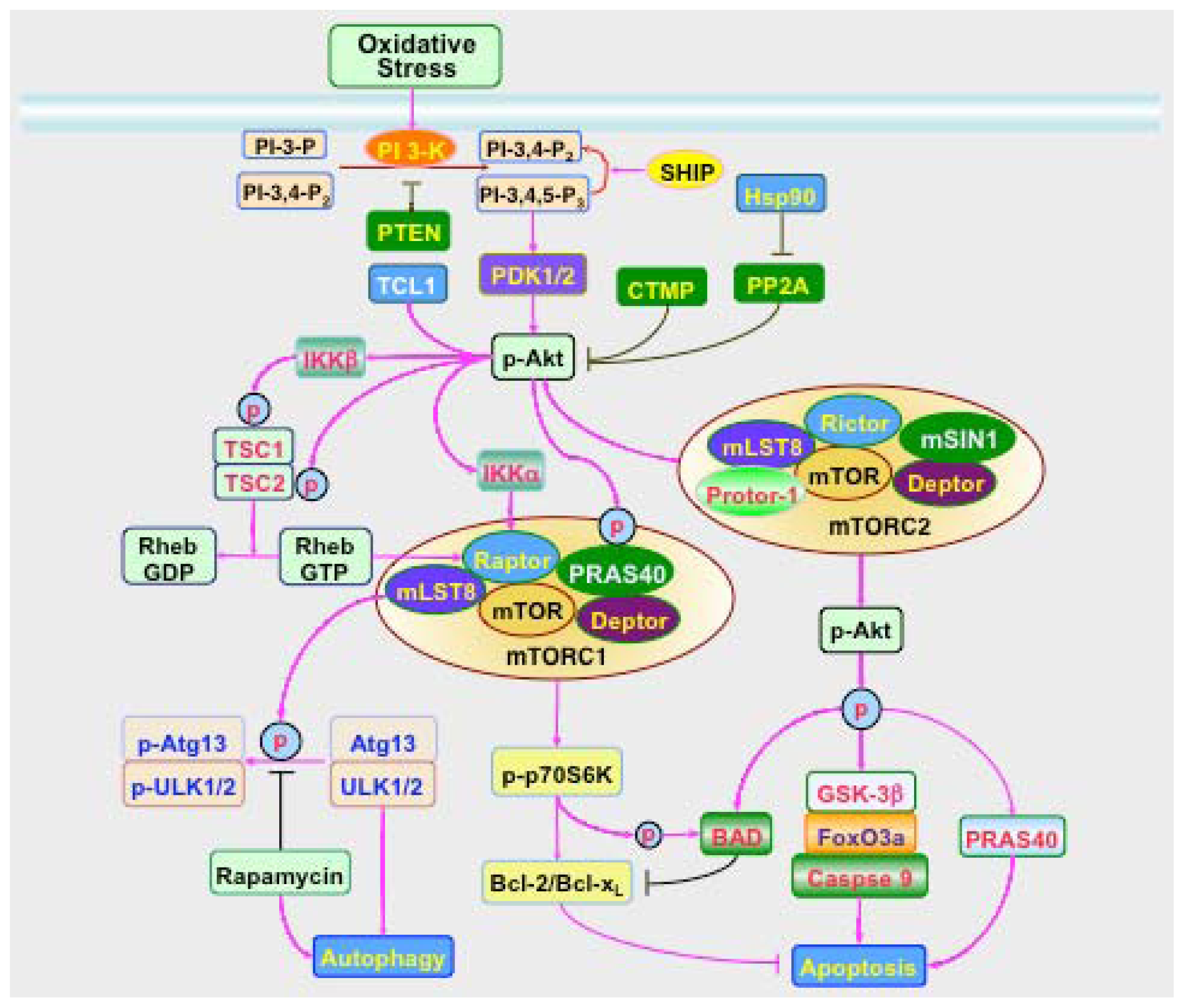
:
{kind=link}
{kind=link}
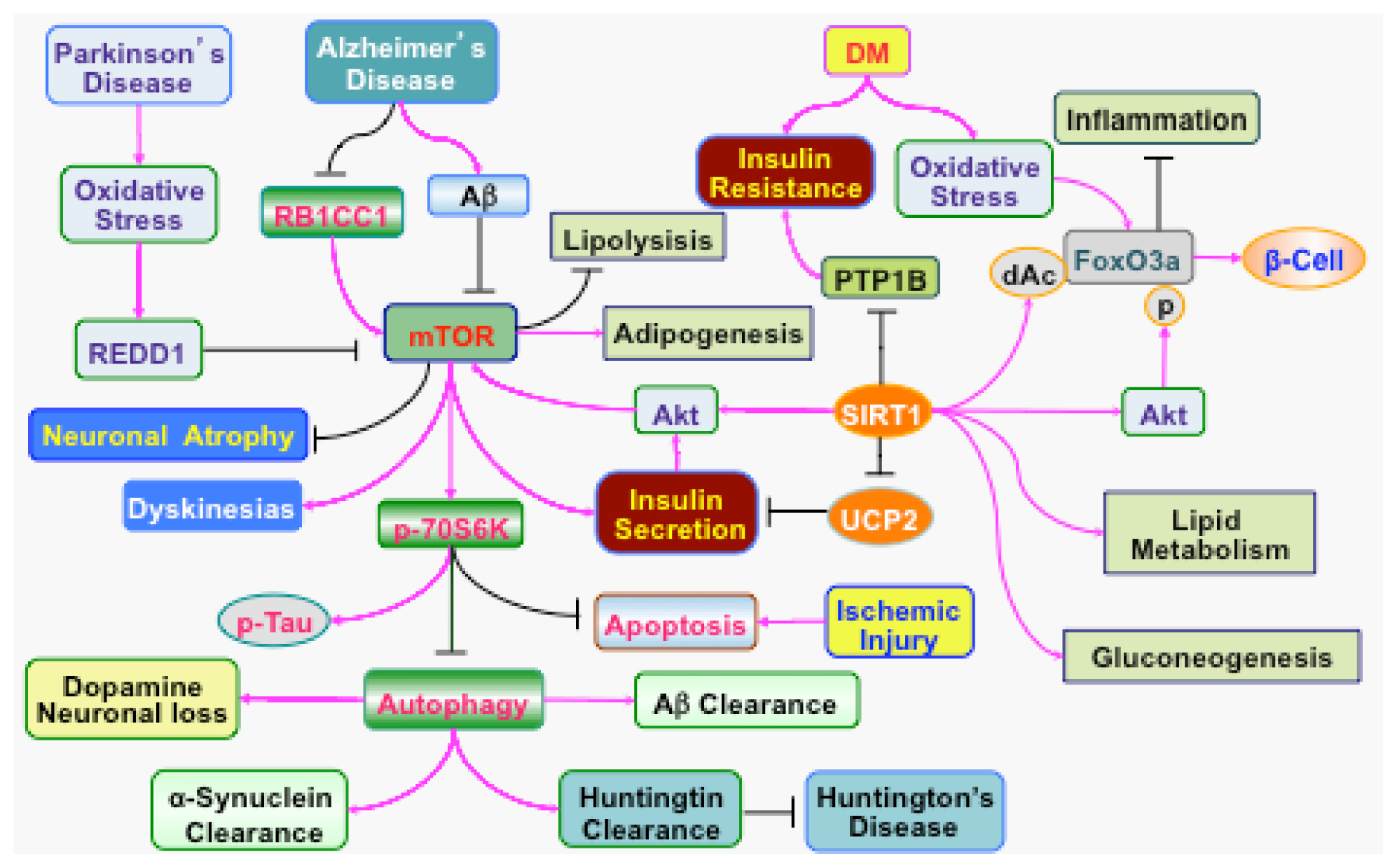{kind=link}
1. Introduction
2. Oxidant Stress and Cellular Injury
2.1. Cellular Survival and Demise during Neurodegeneration
2.2. Apoptotic Early and Late Phases
2.3. The Sub-Constructs of Autophagy
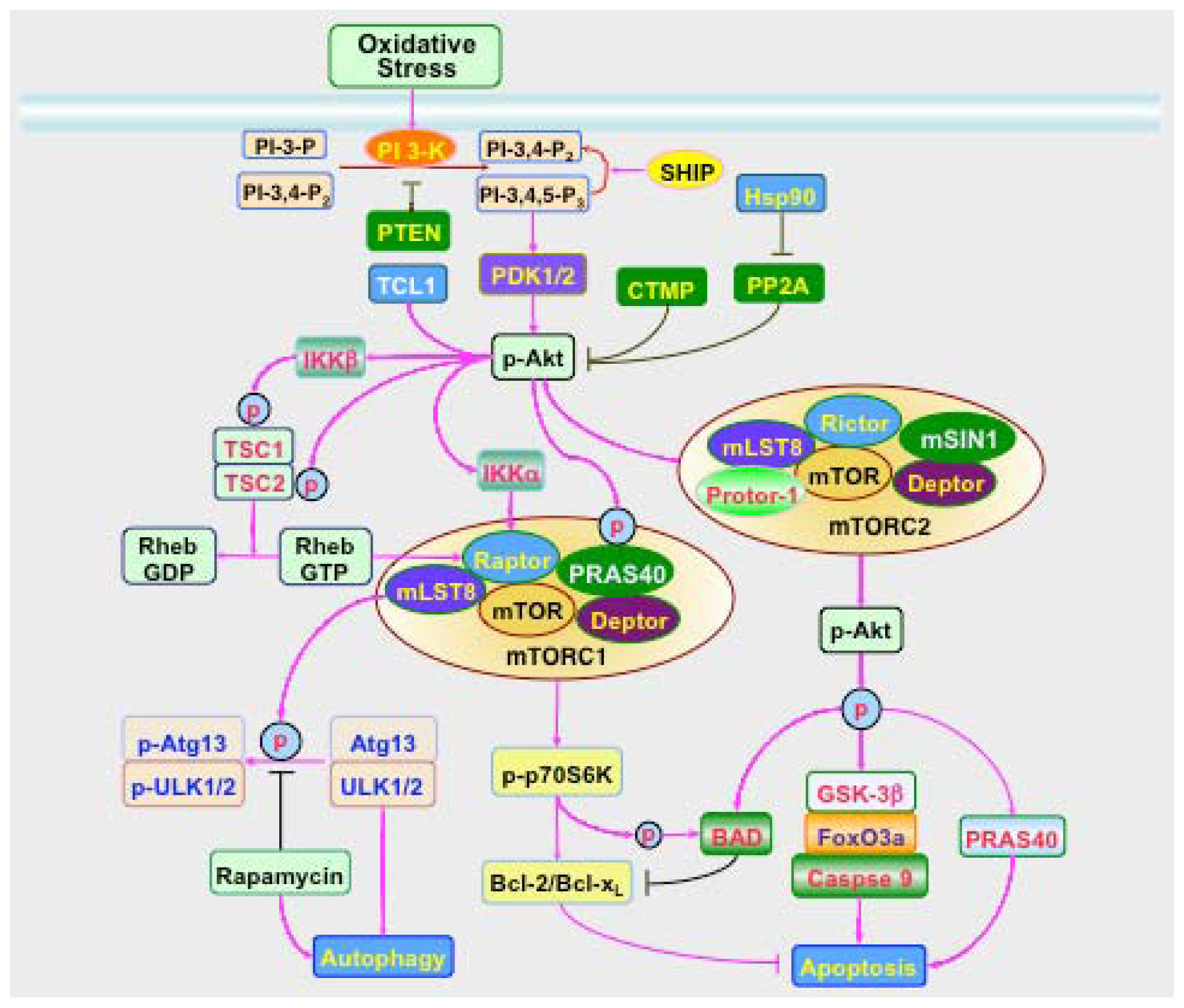
3. Signal Transduction and Cell Survival with PI 3-K, Akt, and mTOR
3.1. PI 3-K and PDK
3.2. Akt
3.3. mTOR
3.4. Apoptosis and Autophagy in the PI 3-K, Akt, and mTOR Cascade
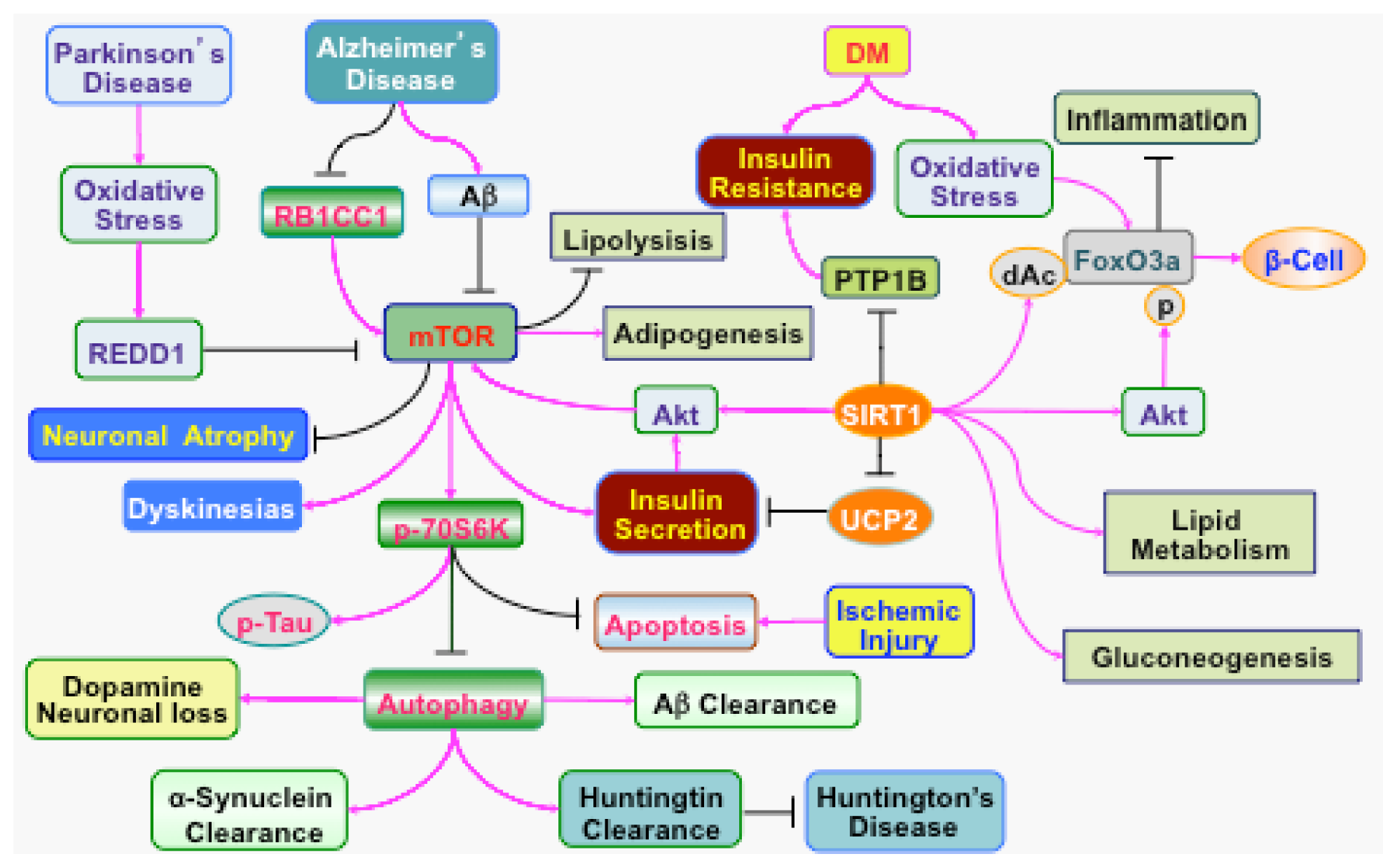
4. Clinical Disorders Modulated by PI 3-K, Akt, and mTOR
4.1 Diabetes Mellitus in the Nervous System
4.2. Acute Injury in the Nervous System
4.3. Chronic Neurodegeneration
5. Conclusions and Perspectives
Acknowledgments
- Conflict of Interest The authors declare no conflict of interest.
References
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. SIRT1: New avenues of discovery for disorders of oxidative stress. Expert Opin. Ther. Targets 2012, 16, 167–178. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. Oxidative stress: Biomarkers and novel therapeutic pathways. Exp. Gerontol 2010, 45, 217–234. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. Translating cell survival and cell longevity into treatment strategies with SIRT1. Rom. J. Morphol. Embryol 2011, 52, 1173–1185. [Google Scholar]
- Jayaram, H.N.; Kusumanchi, P.; Yalowitz, J.A. NMNAT expression and its relation to NAD metabolism. Curr. Med. Chem 2011, 18, 1962–1972. [Google Scholar]
- Yang, H.; Jin, X.; Kei Lam, C.W.; Yan, S.K. Oxidative stress and diabetes mellitus. Clin. Chem. Lab. Med 2011, 49, 1773–1782. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. The vitamin nicotinamide: Translating nutrition into clinical care. Molecules 2009, 14, 3446–3485. [Google Scholar]
- Maiese, K.; Hou, J.; Chong, Z.Z.; Shang, Y.C. Erythropoietin, forkhead proteins, and oxidative injury: biomarkers and biology. TheScientificWorldJournal 2009, 9, 1072–1104. [Google Scholar]
- Muley, M.M.; Thakare, V.N.; Patil, R.R.; Kshirsagar, A.D.; Naik, S.R. Silymarin improves the behavioural, biochemical and histoarchitecture alterations in focal ischemic rats: A comparative evaluation with piracetam and protocatachuic acid. Pharmacol. Biochem. Behav 2012, 102, 286–293. [Google Scholar]
- Sun, L.; Gu, L.; Wang, S.; Yuan, J.; Yang, H.; Zhu, J.; Zhang, H. N-acetylcysteine protects against apoptosis through modulation of group I metabotropic glutamate receptor activity. PLoS One 2012, 7, e32503. [Google Scholar]
- Suzen, S.; Cihaner, S.S.; Coban, T. Synthesis and comparison of antioxidant properties of indole-based melatonin analogue indole amino acid derivatives. Chem. Biol. Drug Des 2012, 79, 76–83. [Google Scholar]
- Tupe, R.S.; Tupe, S.G.; Agte, V.V. Dietary nicotinic acid supplementation improves hepatic zinc uptake and offers hepatoprotection against oxidative damage. Br. J. Nutr 2011, 25, 1–9. [Google Scholar]
- Wong, D.Z.; Kadir, H.A.; Lee, C.L.; Goh, B.H. Neuroprotective properties of Loranthus parasiticus aqueous fraction against oxidative stress-induced damage in NG108–15 cells. J. Nat. Med 2012, 66, 544–551. [Google Scholar]
- Zhang, G.; Zhao, Z.; Gao, L.; Deng, J.; Wang, B.; Xu, D.; Liu, B.; Qu, Y.; Yu, J.; Li, J.; et al. Gypenoside attenuates white matter lesions induced by chronic cerebral hypoperfusion in rats. Pharmacol. Biochem. Behav 2011, 99, 42–51. [Google Scholar]
- Cacciatore, I.; Baldassarre, L.; Fornasari, E.; Mollica, A.; Pinnen, F. Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid. Med. Cell. Longevity 2012, 2012, 240146. [Google Scholar]
- Escobar, J.; Pereda, J.; Lopez-Rodas, G.; Sastre, J. Redox signaling and histone acetylation in acute pancreatitis. Free Radic. Biol. Med 2012, 52, 819–837. [Google Scholar]
- Lange, C.A.; Bainbridge, J.W. Oxygen sensing in retinal health and disease. Ophthalmologica 2012, 227, 115–131. [Google Scholar]
- Wang, J.; Sun, P.; Bao, Y.; Dou, B.; Song, D.; Li, Y. Vitamin E renders protection to PC12 cells against oxidative damage and apoptosis induced by single-walled carbon nanotubes. Toxicol. In Vitro 2012, 26, 32–41. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol 2005, 75, 207–246. [Google Scholar]
- Li, R.P.; Wang, Z.Z.; Sun, M.X.; Hou, X.L.; Sun, Y.; Deng, Z.F.; Xiao, K. Polydatin protects learning and memory impairments in a rat model of vascular dementia. Phytomedicine 2012, 19, 677–681. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. New strategies for Alzheimer’s disease and cognitive impairment. Oxid. Med. Cell. Longevity 2009, 2, 279–289. [Google Scholar]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med. Chem 2011, 18, 4949–4975. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. Stress in the brain: Novel cellular mechanisms of injury linked to Alzheimer’s disease. Brain Res. Brain Res. Rev 2005, 49, 1–21. [Google Scholar]
- Maiese, K.; Chong, Z.; Li, F. Reducing Oxidative Stress and Enhancing Neurovascular Longevity during Diabetes Mellitus. In Neurovascular Medicine: Pursuing Cellular Longevity for Healthy Aging; Maiese, K., Ed.; Oxford University Press: New York, NY, USA, 2009; pp. 540–564. [Google Scholar]
- Srivastava, S.; Haigis, M.C. Role of sirtuins and calorie restriction in neuroprotection: Implications in Alzheimer’s and Parkinson’s diseases. Curr. Pharm. Des 2011, 17, 3418–3433. [Google Scholar]
- Chong, Z.Z.; Shang, Y.C.; Zhang, L.; Wang, S.; Maiese, K. Mammalian target of rapamycin: Hitting the bull’s-eye for neurological disorders. Oxid. Med. Cell. Longevity 2010, 3, 374–391. [Google Scholar]
- Enz, R. Metabotropic glutamate receptors and interacting proteins: Evolving drug targets. Curr. Drug Targets 2012, 13, 145–156. [Google Scholar]
- Hyrskyluoto, A.; Reijonen, S.; Kivinen, J.; Lindholm, D.; Korhonen, L. GADD34 mediates cytoprotective autophagy in mutant huntingtin expressing cells via the mTOR pathway. Exp. Cell Res 2012, 318, 33–42. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Li, F. Driving cellular plasticity and survival through the signal transduction pathways of metabotropic glutamate receptors. Curr. Neurovasc. Res 2005, 2, 425–446. [Google Scholar]
- Nagley, P.; Higgins, G.C.; Atkin, J.D.; Beart, P.M. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim. Biophys. Acta 2010, 1802, 167–185. [Google Scholar]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet 2012, 21, 2245–2262. [Google Scholar]
- Yang, D.; Wang, C.E.; Zhao, B.; Li, W.; Ouyang, Z.; Liu, Z.; Yang, H.; Fan, P.; O’Neill, A.; Gu, W.; et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum. Mol. Genet 2010, 19, 3983–3994. [Google Scholar]
- Al Sweidi, S.; Sanchez, M.G.; Bourque, M.; Morissette, M.; Dluzen, D.; Di Paolo, T. Oestrogen receptors and signalling pathways: Implications for neuroprotective effects of sex steroids in Parkinson’s disease. J. Neuroendocrinol 2012, 24, 48–61. [Google Scholar]
- Asaithambi, A.; Kanthasamy, A.; Saminathan, H.; Anantharam, V.; Kanthasamy, A.G. Protein kinase D1 (PKD1) activation mediates a compensatory protective response during early stages of oxidative stress-induced neuronal degeneration. Mol. Neurodegener. 2011, 6. [Google Scholar] [CrossRef]
- Das, F.; Dey, N.; Venkatesan, B.; Kasinath, B.S.; Ghosh-Choudhury, N.; Choudhury, G.G. High glucose upregulation of early-onset Parkinson’s disease protein DJ-1 integrates the PRAS40/TORC1 axis to mesangial cell hypertrophy. Cell. Signal 2011, 23, 1311–1319. [Google Scholar]
- Khan, M.M.; Ahmad, A.; Ishrat, T.; Khan, M.B.; Hoda, M.N.; Khuwaja, G.; Raza, S.S.; Khan, A.; Javed, H.; Vaibhav, K.; et al. Resveratrol attenuates 6-hydroxydopamine-induced oxidative damage and dopamine depletion in rat model of Parkinson’s disease. Brain Res 2010, 1328, 139–151. [Google Scholar]
- Kook, Y.H.; Ka, M.; Um, M. Neuroprotective cytokines repress PUMA induction in the 1-methyl-4-phenylpyridinium (MPP(+)) model of Parkinson’s disease. Biochem. Biophys. Res. Commun 2011, 411, 370–374. [Google Scholar]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Deleidi, M.; Serapide, M.F.; Pluchino, S.; Marchetti, B. Plasticity of subventricular zone neuroprogenitors in MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) mouse model of Parkinson’s disease involves cross talk between inflammatory and Wnt/beta-Catenin signaling pathways: Functional consequences for neuroprotection and repair. J. Neurosci 2012, 32, 2062–2085. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Therapeutic promise and principles: Metabotropic glutamate receptors. Oxid. Med. Cell. Longevity 2008, 1, 1–14. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. Targeting disease through novel pathways of apoptosis and autophagy. Expert Opin. Ther. Targets 2012, in press. [Google Scholar]
- Banach, M.; Piskorska, B.; Czuczwar, S.; Borowicz, K. Nitric oxide, epileptic seizures, and action of antiepileptic drugs. CNS Neurol. Disord. Drug Targets 2011, 10, 808–819. [Google Scholar]
- O’Dell, C.M.; Das, A.; Wallace, G.t.; Ray, S.K.; Banik, N.L. Understanding the basic mechanisms underlying seizures in mesial temporal lobe epilepsy and possible therapeutic targets: A review. J. Neurosci. Res 2012, 90, 913–924. [Google Scholar]
- Sensi, S.L.; Paoletti, P.; Koh, J.Y.; Aizenman, E.; Bush, A.I.; Hershfinkel, M. The neurophysiology and pathology of brain zinc. J. Neurosci 2011, 31, 16076–16085. [Google Scholar]
- Talos, D.M.; Sun, H.; Zhou, X.; Fitzgerald, E.C.; Jackson, M.C.; Klein, P.M.; Lan, V.J.; Joseph, A.; Jensen, F.E. The Interaction between early life epilepsy and autistic-like behavioral consequences: A role for the mammalian target of rapamycin (mTOR) pathway. PLoS One 2012, 7, e35885. [Google Scholar]
- Beard, R.S., Jr; Reynolds, J.J.; Bearden, S.E. Metabotropic glutamate receptor 5 mediates phosphorylation of vascular endothelial cadherin and nuclear localization of beta-catenin in response to homocysteine. Vascul. Pharmacol 2012, 56, 159–167. [Google Scholar]
- Bonilla, C.; Zurita, M.; Otero, L.; Aguayo, C.; Rico, M.A.; Vaquero, J. The severity of brain damage determines bone marrow stromal cell therapy efficacy in a traumatic brain injury model. J. Trauma Acute Care Surg 2012, 72, 1203–1212. [Google Scholar]
- Duris, K.; Manaenko, A.; Suzuki, H.; Rolland, W.B.; Krafft, P.R.; Zhang, J.H. α7 Nicotinic acetylcholine receptor agonist PNU-282987 attenuates early brain injury in a perforation model of subarachnoid hemorrhage in rats. Stroke 2011, 42, 3530–3536. [Google Scholar]
- Goffus, A.M.; Anderson, G.D.; Hoane, M. Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid. Med. Cell. Longevity 2010, 3, 145–152. [Google Scholar]
- Guo, J.M.; Dong, W.Z.; Liu, A.J.; Cheng, M.H.; Su, D.F. Nicotinamide postpones stroke in stroke-prone spontaneously hypertensive rats. CNS Neurosci. Ther 2012, 18, 267–268. [Google Scholar]
- Lanfranconi, S.; Locatelli, F.; Corti, S.; Candelise, L.; Comi, G.P.; Baron, P.L.; Strazzer, S.; Bresolin, N.; Bersano, A. Growth factors in ischemic stroke. J. Cell. Mol. Med 2011, 15, 1645–1687. [Google Scholar]
- Ma, Y.; Qu, Y.; Fei, Z. Vascular endothelial growth factor in cerebral ischemia. J. Neurosci. Res 2011, 89, 969–978. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. Erythropoietin and oxidative stress. Curr. Neurovasc. Res 2008, 5, 125–142. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Li, F.; Shang, Y.C. Erythropoietin: Elucidating new cellular targets that broaden therapeutic strategies. Prog. Neurobiol 2008, 85, 194–213. [Google Scholar]
- Raza, S.S.; Khan, M.M.; Ashafaq, M.; Ahmad, A.; Khuwaja, G.; Khan, A.; Siddiqui, M.S.; Safhi, M.M.; Islam, F. Silymarin protects neurons from oxidative stress associated damages in focal cerebral ischemia: A behavioral, biochemical and immunohistological study in Wistar rats. J. Neurol. Sci 2011, 309, 45–54. [Google Scholar]
- Zhao, L.D.; Wang, J.H.; Jin, G.R.; Zhao, Y.; Zhang, H.J. Neuroprotective effect of Buyang Huanwu decoction against focal cerebral ischemia/reperfusion injury in rats—Time window and mechanism. J. Ethnopharmacol 2012, 140, 339–344. [Google Scholar]
- Chong, Z.Z.; Maiese, K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: Diversified control of cell growth, inflammation, and injury. Histol. Histopathol 2007, 22, 1251–1267. [Google Scholar]
- Kato, S.; Aoyama, M.; Kakita, H.; Hida, H.; Kato, I.; Ito, T.; Goto, T.; Hussein, M.H.; Sawamoto, K.; Togari, H.; et al. Endogenous erythropoietin from astrocyte protects the oligodendrocyte precursor cell against hypoxic and reoxygenation injury. J. Neurosci. Res 2011, 89, 1566–1574. [Google Scholar]
- Kigerl, K.A.; Ankeny, D.P.; Garg, S.K.; Wei, P.; Guan, Z.; Lai, W.; McTigue, D.M.; Banerjee, R.; Popovich, P.G. System x(c)(−) regulates microglia and macrophage glutamate excitotoxicity in vivo. Exp. Neurol 2012, 233, 333–341. [Google Scholar]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.; et al. Reactive astrocytes and Wnt/beta-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol. Dis 2011, 41, 508–527. [Google Scholar]
- Shang, Y.C.; Chong, Z.Z.; Hou, J.; Maiese, K. FoxO3a governs early microglial proliferation and employs mitochondrial depolarization with caspase 3, 8, and 9 cleavage during oxidant induced apoptosis. Curr. Neurovasc. Res 2009, 6, 223–238. [Google Scholar]
- Shang, Y.C.; Chong, Z.Z.; Hou, J.; Maiese, K. Wnt1, FoxO3a, and NF-kappaB oversee microglial integrity and activation during oxidant stress. Cell. Signal 2010, 22, 1317–1329. [Google Scholar]
- Jiang, Y.L.; Ning, Y.; Ma, X.L.; Liu, Y.Y.; Wang, Y.; Zhang, Z.; Shan, C.X.; Xu, Y.D.; Yin, L.M.; Yang, Y.Q. Alteration of the proteome profile of the pancreas in diabetic rats induced by streptozotocin. Int. J. Mol. Med 2011, 28, 153–160. [Google Scholar]
- John, C.M.; Ramasamy, R.; Al Naqeeb, G.; Dhiab Al-Nuaimi, A.H.; Adam, A. Enhanced CD4+CD25+ regulatory T cells with splenic proliferation and protection against oxidative stress by nicotinamide in gestational diabetes. Curr. Med. Chem. 2012, in press. [Google Scholar]
- Kurban, S.; Mehmetoglu, I.; Yerlikaya, H.F.; Gonen, S.; Erdem, S. Effect of chronic regular exercise on serum ischemia-modified albumin levels and oxidative stress in type 2 diabetes mellitus. Endocr. Res 2011, 36, 116–123. [Google Scholar]
- Lee, Y.; Hong, Y.; Lee, S.R.; Chang, K.T. Autophagy contributes to retardation of cardiac growth in diabetic rats. Lab. Anim. Res 2012, 28, 99–107. [Google Scholar]
- Liu, Z.; Stanojevic, V.; Brindamour, L.J.; Habener, J.F. GLP1–derived nonapeptide GLP1(28–36)amide protects pancreatic beta-cells from glucolipotoxicity. J. Endocrinol 2012, 213, 143–154. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Novel avenues of drug discovery and biomarkers for diabetes mellitus. J. Clin. Pharmacol 2011, 51, 128–152. [Google Scholar]
- Maiese, K.; Morhan, S.D.; Chong, Z.Z. Oxidative stress biology and cell injury during type 1 and type 2 diabetes mellitus. Curr. Neurovasc. Res 2007, 4, 63–71. [Google Scholar]
- Maiese, K.; Shang, Y.C.; Chong, Z.Z.; Hou, J. Diabetes mellitus: Channeling care through cellular discovery. Curr. Neurovasc. Res 2010, 7, 59–64. [Google Scholar]
- Pang, J.; Xi, C.; Dai, Y.; Gong, H.; Zhang, T.M. Altered expression of base excision repair genes in response to high glucose-induced oxidative stress in HepG2 hepatocytes. Med. Sci. Monit. 2012, 18, BR281–285. [Google Scholar]
- Tang, L.; Zhang, Y.; Jiang, Y.; Willard, L.; Ortiz, E.; Wark, L.; Medeiros, D.; Lin, D. Dietary wolfberry ameliorates retinal structure abnormalities in db/db mice at the early stage of diabetes. Exp. Biol. Med. (Maywood) 2011, 236, 1051–1063. [Google Scholar]
- Zengi, A.; Ercan, G.; Caglayan, O.; Tamsel, S.; Karadeniz, M.; Simsir, I.; Harman, E.; Kahraman, C.; Orman, M.; Cetinkalp, S.; et al. Increased oxidative DNA damage in lean normoglycemic offspring of type 2 diabetic patients. Exp. Clin. Endocrinol. Diabetes 2011, 119, 467–471. [Google Scholar]
- Acquaah-Mensah, G.K.; Taylor, R.C.; Bhave, S.V. PACAP interactions in the mouse brain: Implications for behavioral and other disorders. Gene 2012, 491, 224–231. [Google Scholar]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov 2011, 10, 868–880. [Google Scholar]
- Hwang, S.K.; Kim, H.H. The functions of mTOR in ischemic diseases. BMB Rep 2011, 44, 506–511. [Google Scholar]
- Maiese, K. The many facets of cell injury: Angiogenesis to autophagy. Curr. Neurovasc. Res 2012, 9, 83–84. [Google Scholar]
- Broe, M.; Shepherd, C.E.; Milward, E.A.; Halliday, G.M. Relationship between DNA fragmentation, morphological changes and neuronal loss in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 2001, 101, 616–624. [Google Scholar]
- Louneva, N.; Cohen, J.W.; Han, L.Y.; Talbot, K.; Wilson, R.S.; Bennett, D.A.; Trojanowski, J.Q.; Arnold, S.E. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am. J. Pathol 2008, 173, 1488–1495. [Google Scholar]
- Grammas, P.; Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J. Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int. J. Clin. Exp. Pathol 2011, 4, 616–627. [Google Scholar]
- Tatton, N.A. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson’s disease. Exp. Neurol 2000, 166, 29–43. [Google Scholar]
- Shang, Y.C.; Chong, Z.Z.; Wang, S.; Maiese, K. Erythropoietin and Wnt1 govern pathways of mTOR, Apaf-1, and XIAP in inflammatory microglia. Curr. Neurovasc. Res 2011, 8, 270–285. [Google Scholar]
- Zhou, X.; Wang, L.; Wang, M.; Xu, L.; Yu, L.; Fang, T.; Wu, M. Emodin-induced microglial apoptosis is associated with TRB3 induction. Immunopharmacol. Immunotoxicol 2011, 33, 594–602. [Google Scholar]
- Qin, A.P.; Liu, C.F.; Qin, Y.Y.; Hong, L.Z.; Xu, M.; Yang, L.; Liu, J.; Qin, Z.H.; Zhang, H.L. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 2010, 6, 738–753. [Google Scholar]
- Wang, J.Y.; Xia, Q.; Chu, K.T.; Pan, J.; Sun, L.N.; Zeng, B.; Zhu, Y.J.; Wang, Q.; Wang, K.; Luo, B.Y. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: A widely used inhibitor of autophagy. J. Neuropathol. Exp. Neurol 2011, 70, 314–322. [Google Scholar]
- Baba, H.; Sakurai, M.; Abe, K.; Tominaga, R. Autophagy-mediated stress response in motor neuron after transient ischemia in rabbits. J. Vasc. Surg 2009, 50, 381–387. [Google Scholar]
- Canu, N.; Tufi, R.; Serafino, A.L.; Amadoro, G.; Ciotti, M.T.; Calissano, P. Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J. Neurochem 2005, 92, 1228–1242. [Google Scholar] [Green Version]
- Xue, L.; Fletcher, G.C.; Tolkovsky, A.M. Autophagy is activated by apoptotic signalling in sympathetic neurons: An alternative mechanism of death execution. Mol. Cell. Neurosci 1999, 14, 180–198. [Google Scholar]
- Chong, Z.Z.; Maiese, K. Mammalian target of rapamycin signaling in diabetic cardiovascular disease. Cardiovasc. Diabetol. 2012, 11. [Google Scholar] [CrossRef]
- Troy, C.M.; Akpan, N.; Jean, Y.Y. Regulation of caspases in the nervous system implications for functions in health and disease. Prog. Mol. Biol. Transl. Sci 2011, 99, 265–305. [Google Scholar]
- Nopparat, C.; Porter, J.E.; Ebadi, M.; Govitrapong, P. The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J. Pineal. Res 2010, 49, 382–389. [Google Scholar]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1–dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ 2010, 17, 268–277. [Google Scholar]
- Wang, S.; Chong, Z.Z.; Shang, Y.C.; Maiese, K. WISP1 (CCN4) autoregulates its expression and nuclear trafficking of beta-catenin during oxidant stress with limited effects upon neuronal autophagy. Curr. Neurovasc. Res 2012, 9, 89–99. [Google Scholar]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci 2009, 29, 13578–13588. [Google Scholar]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 2010, 5, e9979. [Google Scholar]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 2009, 4, e5515. [Google Scholar]
- Liu, Y.; Shi, S.; Gu, Z.; Du, Y.; Liu, M.; Yan, S.; Gao, J.; Li, J.; Shao, Y.; Zhong, W.; et al. Impaired autophagic function in rat islets with aging. Age (Dordr) 2012. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar]
- Hu, P.; Lai, D.; Lu, P.; Gao, J.; He, H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int. J. Mol. Med 2012, 29, 613–618. [Google Scholar]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; De Tata, V. Palmitate activates autophagy in INS-1E beta-cells and in isolated rat and human pancreatic islets. PLoS One 2012, 7, e36188. [Google Scholar]
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog. Neurobiol. 2012, in press. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. Erythropoietin: New directions for the nervous system. Int. J. Mol. Sci 2012, 13, 11102–11129. [Google Scholar]
- Aksu, U.; Demirci, C.; Ince, C. The pathogenesis of acute kidney injury and the toxic triangle of oxygen, reactive oxygen species and nitric oxide. Contrib. Nephrol 2011, 174, 119–128. [Google Scholar]
- Balan, V.; Miller, G.S.; Kaplun, L.; Balan, K.; Chong, Z.Z.; Li, F.; Kaplun, A.; VanBerkum, M.F.; Arking, R.; Freeman, D.C.; et al. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem 2008, 283, 27810–27819. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell. Signal 2007, 19, 1150–1162. [Google Scholar]
- Bailey, T.J.; Fossum, S.L.; Fimbel, S.M.; Montgomery, J.E.; Hyde, D.R. The inhibitor of phagocytosis, O-phospho-l-serine, suppresses Muller glia proliferation and cone cell regeneration in the light-damaged zebrafish retina. Exp. Eye Res 2010, 91, 601–612. [Google Scholar]
- Chong, Z.Z.; Kang, J.; Li, F.; Maiese, K. mGluRI targets microglial activation and selectively prevents neuronal cell engulfment through Akt and Caspase dependent pathways. Curr. Neurovasc. Res 2005, 2, 197–211. [Google Scholar]
- Kang, J.Q.; Chong, Z.Z.; Maiese, K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol 2003, 64, 557–569. [Google Scholar]
- Kang, J.Q.; Chong, Z.Z.; Maiese, K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res 2003, 74, 37–51. [Google Scholar]
- Shang, Y.C.; Chong, Z.Z.; Hou, J.; Maiese, K. The forkhead transcription factor FoxO3a controls microglial inflammatory activation and eventual apoptotic injury through caspase 3. Curr. Neurovasc. Res 2009, 6, 20–31. [Google Scholar]
- Koh, P.O. Nicotinamide attenuates the decrease of astrocytic phosphoprotein PEA-15 in focal cerebral ischemic injury. J. Vet. Med. Sci 2012, 74, 377–380. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. “Sly as a FOXO”: New paths with Forkhead signaling in the brain. Curr. Neurovasc. Res 2007, 4, 295–302. [Google Scholar]
- Balduini, W.; Carloni, S.; Buonocore, G. Autophagy in hypoxia-ischemia induced brain injury. J. Matern. Fetal Neonatal Med 2012, 25, 30–34. [Google Scholar]
- Deretic, V.; Jiang, S.; Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol 2012, 22, 397–406. [Google Scholar]
- Silva, D.F.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondria: The common upstream driver of amyloid-beta and tau pathology in Alzheimer’s disease. Curr. Alzheimer Res 2011, 8, 563–572. [Google Scholar]
- Scott, R.C.; Juhasz, G.; Neufeld, T.P. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol 2007, 17, 1–11. [Google Scholar]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000, 150, 1507–1513. [Google Scholar]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1–arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar]
- Kuroyanagi, H.; Yan, J.; Seki, N.; Yamanouchi, Y.; Suzuki, Y.; Takano, T.; Muramatsu, M.; Shirasawa, T. Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignment. Genomics 1998, 51, 76–85. [Google Scholar]
- Yan, J.; Kuroyanagi, H.; Kuroiwa, A.; Matsuda, Y.; Tokumitsu, H.; Tomoda, T.; Shirasawa, T.; Muramatsu, M. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem. Biophys. Res. Commun 1998, 246, 222–227. [Google Scholar]
- Yan, J.; Kuroyanagi, H.; Tomemori, T.; Okazaki, N.; Asato, K.; Matsuda, Y.; Suzuki, Y.; Ohshima, Y.; Mitani, S.; Masuho, Y.; et al. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene 1999, 18, 5850–5859. [Google Scholar]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar]
- Ding, Z.; Liang, J.; Li, J.; Lu, Y.; Ariyaratna, V.; Lu, Z.; Davies, M.A.; Westwick, J.K.; Mills, G.B. Physical association of PDK1 with AKT1 is sufficient for pathway activation independent of membrane localization and phosphatidylinositol 3 kinase. PLoS One 2010, 5, e9910. [Google Scholar]
- Glidden, E.J.; Gray, L.G.; Vemuru, S.; Li, D.; Harris, T.E.; Mayo, M.W. Multiple site acetylation of rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem 2012, 287, 581–588. [Google Scholar]
- Chen, J.X.; Tuo, Q.; Liao, D.F.; Zeng, H. Inhibition of protein tyrosine phosphatase improves angiogenesis via enhancing Ang-1/Tie-2 signaling in diabetes. Exp. Diabetes Res 2012, 2012, 836759. [Google Scholar]
- Deblon, N.; Bourgoin, L.; Veyrat-Durebex, C.; Peyrou, M.; Vinciguerra, M.; Caillon, A.; Maeder, C.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; et al. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br. J. Pharmacol 2012, 165, 2325–2340. [Google Scholar]
- Hou, J.; Chong, Z.Z.; Shang, Y.C.; Maiese, K. Early apoptotic vascular signaling is determined by Sirt1 through nuclear shuttling, forkhead trafficking, bad, and mitochondrial caspase activation. Curr. Neurovasc. Res 2010, 7, 95–112. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. Mechanistic insights into diabetes mellitus and oxidative stress. Curr. Med. Chem 2007, 14, 1729–1738. [Google Scholar]
- Saha, A.K.; Xu, X.J.; Lawson, E.; Deoliveira, R.; Brandon, A.E.; Kraegen, E.W.; Ruderman, N.B. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes 2010, 59, 2426–2434. [Google Scholar]
- Lu, M.J.; Chen, Y.S.; Huang, H.S.; Ma, M.C. Erythropoietin alleviates post-ischemic injury of rat hearts by attenuating nitrosative stress. Life Sci 2012, 90, 776–784. [Google Scholar]
- Koshimizu, T.; Kawai, M.; Kondou, H.; Tachikawa, K.; Sakai, N.; Ozono, K.; Michigami, T. Vinculin functions as regulator of chondrogenesis. J. Biol. Chem 2012, 287, 15760–15775. [Google Scholar]
- Chong, Z.Z.; Shang, Y.C.; Maiese, K. Cardiovascular disease and mTOR signaling. Trends Cardiovasc. Med 2011, 21, 151–155. [Google Scholar]
- James, M.F.; Stivison, E.; Beauchamp, R.; Han, S.; Li, H.; Wallace, M.R.; Gusella, J.F.; Stemmer-Rachamimov, A.O.; Ramesh, V. Regulation of mTOR complex 2 signaling in neurofibromatosis 2-deficient target cell types. Mol. Cancer Res 2012, 10, 649–659. [Google Scholar]
- Weber, J.D.; Gutmann, D.H. Deconvoluting mTOR biology. Cell Cycle 2012, 11, 236–248. [Google Scholar]
- Chong, Z.Z.; Wang, S.; Shang, Y.C.; Maiese, K. Targeting cardiovascular disease with novel SIRT1 pathways. Future Cardiol 2012, 8, 89–100. [Google Scholar]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol 2004, 24, 200–216. [Google Scholar]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 2007, 25, 209–226. [Google Scholar]
- Gingras, A.C.; Kennedy, S.G.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 1998, 12, 502–513. [Google Scholar]
- Bhandari, B.K.; Feliers, D.; Duraisamy, S.; Stewart, J.L.; Gingras, A.C.; Abboud, H.E.; Choudhury, G.G.; Sonenberg, N.; Kasinath, B.S. Insulin regulation of protein translation repressor 4E-BP1, an eIF4E-binding protein, in renal epithelial cells. Kidney Int 2001, 59, 866–875. [Google Scholar]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol 2002, 4, 648–657. [Google Scholar]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem 2009, 284, 12783–12791. [Google Scholar]
- Cai, S.L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol 2006, 173, 279–289. [Google Scholar]
- Oshiro, N.; Takahashi, R.; Yoshino, K.; Tanimura, K.; Nakashima, A.; Eguchi, S.; Miyamoto, T.; Hara, K.; Takehana, K.; Avruch, J.; et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem 2007, 282, 20329–20339. [Google Scholar]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem 2008, 283, 15619–15627. [Google Scholar]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar]
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. PRAS40 is an integral regulatory component of erythropoietin mTOR signaling and cytoprotection. PLoS One 2012, 7, e45456. [Google Scholar]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J. Biol. Chem 2003, 278, 10189–10194. [Google Scholar]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol 2007, 9, 316–323. [Google Scholar]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem 2007, 282, 20036–20044. [Google Scholar]
- Dan, H.C.; Adli, M.; Baldwin, A.S. Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by I kappa B kinase alpha. Cancer Res 2007, 67, 6263–6269. [Google Scholar]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 1997, 91, 243–252. [Google Scholar]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar]
- Yen, C.J.; Izzo, J.G.; Lee, D.F.; Guha, S.; Wei, Y.; Wu, T.T.; Chen, C.T.; Kuo, H.P.; Hsu, J.M.; Sun, H.L.; et al. Bile acid exposure up-regulates tuberous sclerosis complex 1/mammalian target of rapamycin pathway in Barrett’s-associated esophageal adenocarcinoma. Cancer Res 2008, 68, 2632–2640. [Google Scholar]
- Chong, Z.Z.; Hou, J.; Shang, Y.C.; Wang, S.; Maiese, K. EPO relies upon novel signaling of Wnt1 that requires Akt1, FoxO3a, GSK-3beta, and beta-Catenin to foster vascular integrity during experimental diabetes. Curr. Neurovasc. Res 2011, 8, 103–120. [Google Scholar]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation 2002, 106, 2973–2979. [Google Scholar]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. AKT1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-xL and caspase 1, 3, and 9. Exp. Cell Res 2004, 296, 196–207. [Google Scholar]
- Hou, J.; Chong, Z.Z.; Shang, Y.C.; Maiese, K. FoxO3a governs early and late apoptotic endothelial programs during elevated glucose through mitochondrial and caspase signaling. Mol. Cell. Endocrinol 2010, 321, 194–206. [Google Scholar]
- Hou, J.; Wang, S.; Shang, Y.C.; Chong, Z.Z.; Maiese, K. Erythropoietin employs cell longevity pathways of SIRT1 to foster endothelial vascular integrity during oxidant stress. Curr. Neurovasc. Res 2011, 8, 220–235. [Google Scholar]
- Mannell, H.K.; Pircher, J.; Chaudhry, D.I.; Alig, S.K.; Koch, E.G.; Mettler, R.; Pohl, U.; Krotz, F. ARNO regulates VEGF-dependent tissue responses by stabilizing endothelial VEGFR-2 surface expression. Cardiovasc. Res 2012, 93, 111–119. [Google Scholar]
- Su, K.H.; Shyue, S.K.; Kou, Y.R.; Ching, L.C.; Chiang, A.N.; Yu, Y.B.; Chen, C.Y.; Pan, C.C.; Lee, T.S. β Common receptor integrates the erythropoietin signaling in activation of endothelial nitric oxide synthase. J. Cell. Physiol 2011, 226, 3330–3339. [Google Scholar]
- Chen, T.; Zhang, L.; Qu, Y.; Huo, K.; Jiang, X.; Fei, Z. The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int. J. Mol. Med 2012, 29, 630–636. [Google Scholar]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow Metab 2004, 24, 728–743. [Google Scholar]
- Chong, Z.Z.; Shang, Y.C.; Hou, J.; Maiese, K. Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways. Oxid. Med. Cell. Longevity 2010, 3, 153–165. [Google Scholar]
- Komandirov, M.A.; Knyazeva, E.A.; Fedorenko, Y.P.; Rudkovskii, M.V.; Stetsurin, D.A.; Uzdensky, A.B. On the role of phosphatidylinositol 3-kinase, protein kinase b/Akt, and glycogen synthase kinase-3β in photodynamic injury of crayfish neurons and glial cells. J. Mol. Neurosci 2011, 45, 229–235. [Google Scholar]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci 2010, 30, 1166–1175. [Google Scholar]
- Shen, J.; Wu, Y.; Xu, J.Y.; Zhang, J.; Sinclair, S.H.; Yanoff, M.; Xu, G.; Li, W.; Xu, G.T. ERKand Akt-dependent neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via modulation of Bcl-xL, Bax, and BAD. Invest. Ophthalmol. Vis. Sci 2010, 51, 35–46. [Google Scholar]
- Zeng, K.W.; Wang, X.M.; Ko, H.; Kwon, H.C.; Cha, J.W.; Yang, H.O. Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid beta-protein via the PI3K/Akt/Bad/Bcl(XL)-regulated mitochondrial apoptotic pathway. Eur. J. Pharmacol 2011, 672, 45–55. [Google Scholar]
- Li, F.; Chong, Z.Z.; Maiese, K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr. Neurovasc. Res 2006, 3, 187–201. [Google Scholar]
- Pineda, D.; Ampurdanes, C.; Medina, M.G.; Serratosa, J.; Tusell, J.M.; Saura, J.; Planas, A.M.; Navarro, P. Tissue plasminogen activator induces microglial inflammation via a noncatalytic molecular mechanism involving activation of mitogen-activated protein kinases and Akt signaling pathways and AnnexinA2 and Galectin-1 receptors. Glia 2012, 60, 526–540. [Google Scholar]
- Perez-Garcia, M.J.; Cena, V.; de Pablo, Y.; Llovera, M.; Comella, J.X.; Soler, R.M. Glial cell line-derived neurotrophic factor increases intracellular calcium concentration. Role of calcium/calmodulin in the activation of the phosphatidylinositol 3–kinase pathway. J. Biol. Chem 2004, 279, 6132–6142. [Google Scholar]
- Xu, J.; Zhang, Q.G.; Li, C.; Zhang, G.Y. Subtoxic N-methyl-d-aspartate delayed neuronal death in ischemic brain injury through TrkB receptor- and calmodulin-mediated PI-3K/Akt pathway activation. Hippocampus 2007, 17, 525–537. [Google Scholar]
- Maiese, K. The challenges for drug development: Cytokines, genes, and stem cells. Curr. Neurovasc. Res 2012, 9, 231–232. [Google Scholar]
- Chattopadhyay, M.; Walter, C.; Mata, M.; Fink, D.J. Neuroprotective effect of herpes simplex virus-mediated gene transfer of erythropoietin in hyperglycemic dorsal root ganglion neurons. Brain 2009, 132, 879–888. [Google Scholar]
- Chong, Z.Z.; Maiese, K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br. J. Pharmacol 2007, 150, 839–850. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. A “FOXO” in sight: Targeting FoxO proteins from conception to cancer. Med. Res. Rev 2009, 29, 395–418. [Google Scholar]
- Storz, P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signaling 2011, 14, 593–605. [Google Scholar]
- Kousteni, S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone 2012, 50, 437–443. [Google Scholar]
- Lam, E.W.; Shah, K.; Brosens, J.J. The diversity of sex steroid action: The role of micro-RNAs and FOXO transcription factors in cycling endometrium and cancer. J. Endocrinol 2012, 212, 13–25. [Google Scholar]
- Lappas, M.; Permezel, M. The anti-inflammatory and antioxidative effects of nicotinamide, a vitamin B(3) derivative, are elicited by FoxO3 in human gestational tissues: Implications for preterm birth. J. Nutr. Biochem 2011, 22, 1195–1201. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. OutFOXOing disease and disability: The therapeutic potential of targeting FoxO proteins. Trends Mol. Med 2008, 14, 219–227. [Google Scholar]
- Xie, Z.; Chen, F.; Wu, X.; Zhuang, C.; Zhu, J.; Wang, J.; Ji, H.; Wang, Y.; Hua, X. Effects of supplemental erythropoietin on its receptor expression and signal transduction pathways in rat model of retinal detachment. Curr. Eye Res 2012, 37, 138–144. [Google Scholar]
- Ma, R.; Xiong, N.; Huang, C.; Tang, Q.; Hu, B.; Xiang, J.; Li, G. Erythropoietin protects PC12 cells from beta-amyloid(25–35)-induced apoptosis via PI3K/Akt signaling pathway. Neuropharmacology 2009, 56, 1027–1034. [Google Scholar]
- Shang, Y.C.; Chong, Z.Z.; Wang, S.; Maiese, K. Prevention of beta-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany NY) 2012, 4, 187–201. [Google Scholar]
- Sun, Z.K.; Yang, H.Q.; Pan, J.; Zhen, H.; Wang, Z.Q.; Chen, S.D.; Ding, J.Q. Protective effects of erythropoietin on tau phosphorylation induced by beta-amyloid. J. Neurosci. Res 2008, 86, 3018–3027. [Google Scholar]
- Kilic, E.; Kilic, U.; Soliz, J.; Bassetti, C.L.; Gassmann, M.; Hermann, D.M. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J 2005, 19, 2026–2028. [Google Scholar]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol 2003, 138, 1107–1118. [Google Scholar]
- Dzietko, M.; Felderhoff-Mueser, U.; Sifringer, M.; Krutz, B.; Bittigau, P.; Thor, F.; Heumann, R.; Buhrer, C.; Ikonomidou, C.; Hansen, H.H. Erythropoietin protects the developing brain against N-methyl-d-aspartate receptor antagonist neurotoxicity. Neurobiol. Dis 2004, 15, 177–187. [Google Scholar]
- Um, M.; Lodish, H.F. Antiapoptotic effects of erythropoietin in differentiated neuroblastoma SH-SY5Y cells require activation of both the STAT5 and AKT signaling pathways. J. Biol. Chem 2006, 281, 5648–5656. [Google Scholar]
- Chong, Z.Z.; Lin, S.H.; Li, F.; Maiese, K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through Akt, Bad, PARP, and mitochondrial associated “anti-apoptotic” pathways. Curr. Neurovasc. Res 2005, 2, 271–285. [Google Scholar]
- Li, F.; Chong, Z.Z.; Maiese, K. Cell life versus cell longevity: The mysteries surrounding the NAD(+) precursor nicotinamide. Curr. Med. Chem 2006, 13, 883–895. [Google Scholar]
- Maiese, K.; Li, F.; Chong, Z.Z. Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends Pharmacol. Sci 2004, 25, 577–583. [Google Scholar]
- Wang, S.; Chong, Z.Z.; Shang, Y.C.; Maiese, K. Wnt1 inducible signaling pathway protein 1 (WISP1) blocks neurodegeneration through phosphoinositide 3 kinase/Akt1 and apoptotic mitochondrial signaling involving Bad, Bax, Bim, and Bcl-xL. Curr. Neurovasc. Res 2012, 9, 20–31. [Google Scholar]
- Koh, P.O. Nicotinamide attenuates the ischemic brain injury-induced decrease of Akt activation and Bad phosphorylation. Neurosci. Lett 2011, 498, 105–109. [Google Scholar]
- Pastor, M.D.; Garcia-Yebenes, I.; Fradejas, N.; Perez-Ortiz, J.M.; Mora-Lee, S.; Tranque, P.; Moro, M.A.; Pende, M.; Calvo, S. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J. Biol. Chem 2009, 284, 22067–22078. [Google Scholar]
- Wu, X.; Reiter, C.E.; Antonetti, D.A.; Kimball, S.R.; Jefferson, L.S.; Gardner, T.W. Insulin promotes rat retinal neuronal cell survival in a p70S6K-dependent manner. J. Biol. Chem 2004, 279, 9167–9175. [Google Scholar]
- Maiese, K.; Li, F.; Chong, Z.Z. New avenues of exploration for erythropoietin. JAMA 2005, 293, 90–95. [Google Scholar]
- Galan-Moya, E.M.; Le Guelte, A.; Lima Fernandes, E.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.O.; Scott, M.G.; Junier, M.P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep 2011, 12, 470–476. [Google Scholar]
- Marfia, G.; Madaschi, L.; Marra, F.; Menarini, M.; Bottai, D.; Formenti, A.; Bellardita, C.; Di Giulio, A.M.; Carelli, S.; Gorio, A. Adult neural precursors isolated from post mortem brain yield mostly neurons: an erythropoietin-dependent process. Neurobiol. Dis 2011, 43, 86–98. [Google Scholar]
- Sanghera, K.P.; Mathalone, N.; Baigi, R.; Panov, E.; Wang, D.; Zhao, X.; Hsu, H.; Wang, H.; Tropepe, V.; Ward, M.; et al. The PI3K/Akt/mTOR pathway mediates retinal progenitor cell survival under hypoxic and superoxide stress. Mol. Cell. Neurosci 2011, 47, 145–153. [Google Scholar]
- Hou, G.; Xue, L.; Lu, Z.; Fan, T.; Tian, F.; Xue, Y. An activated mTOR/p70S6K signaling pathway in esophageal squamous cell carcinoma cell lines and inhibition of the pathway by rapamycin and siRNA against mTOR. Cancer Lett 2007, 253, 236–248. [Google Scholar]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest 2010, 120, 2805–2816. [Google Scholar]
- Dormond, O.; Madsen, J.C.; Briscoe, D.M. The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem 2007, 282, 23679–23686. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. The pro-survival pathways of mTOR and protein kinase B target glycogen synthase kinase-3beta and nuclear factor-κB to foster endogenous microglial cell protection. Int. J. Mol. Med 2007, 19, 263–272. [Google Scholar]
- Choi, K.C.; Kim, S.H.; Ha, J.Y.; Kim, S.T.; Son, J.H. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J. Neurochem 2010, 112, 366–376. [Google Scholar]
- Shang, Y.C.; Chong, Z.Z.; Wang, S.; Maiese, K. WNT1 inducible signaling pathway protein 1 (WISP1) targets PRAS40 to govern β-amyloid apoptotic injury of microglia. Curr. Neurovasc. Res 2012, 9, 239–249. [Google Scholar]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jeno, P.; Arrieumerlou, C.; Hall, M.N. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2007, 2, e1217. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. Attempted cell cycle induction in post-mitotic neurons occurs in early and late apoptotic programs through Rb, E2F1, and Caspase 3. Curr. Neurovasc. Res 2006, 3, 25–39. [Google Scholar]
- Yu, Y.; Ren, Q.G.; Zhang, Z.H.; Zhou, K.; Yu, Z.Y.; Luo, X.; Wang, W. Phospho-Rb mediating cell cycle reentry induces early apoptosis following oxygen-glucose deprivation in rat cortical neurons. Neurochem. Res 2012, 37, 503–511. [Google Scholar]
- Bhaskar, K.; Miller, M.; Chludzinski, A.; Herrup, K.; Zagorski, M.; Lamb, B.T. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol. Neurodegener 2009, 4, 14. [Google Scholar]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar]
- Abdullah, A.; Wolfe, R.; Mannan, H.; Stoelwinder, J.U.; Stevenson, C.; Peeters, A. Epidemiologic merit of obese-years, the combination of degree and duration of obesity. Am. J. Epidemiol 2012, 176, 99–107. [Google Scholar]
- Reagan, L.P. Diabetes as a chronic metabolic stressor: Causes, consequences and clinical complications. Exp. Neurol 2012, 233, 68–78. [Google Scholar]
- Kelly, G.S. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: Part 2. Altern. Med. Rev 2010, 15, 313–328. [Google Scholar]
- Lai, C.S.; Tsai, M.L.; Badmaev, V.; Jimenez, M.; Ho, C.T.; Pan, M.H. Xanthigen suppresses preadipocyte differentiation and adipogenesis through down-regulation of PPARgamma and C/EBPs and modulation of SIRT-1, AMPK, and FoxO pathways. J. Agric. Food Chem 2012, 60, 1094–1101. [Google Scholar]
- Chong, Z.Z.; Maiese, K. Enhanced tolerance against early and late apoptotic oxidative stress in mammalian neurons through nicotinamidase and sirtuin mediated pathways. Curr. Neurovasc. Res 2008, 5, 159–170. [Google Scholar]
- Sun, C.; Zhang, F.; Ge, X.; Yan, T.; Chen, X.; Shi, X.; Zhai, Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab 2007, 6, 307–319. [Google Scholar]
- Li, Y.; Xu, S.; Giles, A.; Nakamura, K.; Lee, J.W.; Hou, X.; Donmez, G.; Li, J.; Luo, Z.; Walsh, K.; et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J 2011, 25, 1664–1679. [Google Scholar]
- Frojdo, S.; Durand, C.; Molin, L.; Carey, A.L.; El-Osta, A.; Kingwell, B.A.; Febbraio, M.A.; Solari, F.; Vidal, H.; Pirola, L. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol. Cell. Endocrinol 2011, 335, 166–176. [Google Scholar]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 2006, 4, e31. [Google Scholar]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4. [Google Scholar] [CrossRef]
- Wang, F.; Chan, C.H.; Chen, K.; Guan, X.; Lin, H.K.; Tong, Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 2012, 31, 1546–1557. [Google Scholar]
- Wang, R.H.; Kim, H.S.; Xiao, C.; Xu, X.; Gavrilova, O.; Deng, C.X. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J. Clin. Invest 2011, 121, 4477–4490. [Google Scholar]
- Guo, W.; Qian, L.; Zhang, J.; Zhang, W.; Morrison, A.; Hayes, P.; Wilson, S.; Chen, T.; Zhao, J. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res 2011, 89, 1723–1736. [Google Scholar]
- Hamada, S.; Hara, K.; Hamada, T.; Yasuda, H.; Moriyama, H.; Nakayama, R.; Nagata, M.; Yokono, K. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes 2009, 58, 1321–1332. [Google Scholar]
- Treins, C.; Alliouachene, S.; Hassouna, R.; Xie, Y.; Birnbaum, M.J.; Pende, M. The combined deletion of S6K1 and Akt2 deteriorates glycaemic control in high fat diet. Mol. Cell. Biol 2012, 32, 4004–4011. [Google Scholar]
- Fraenkel, M.; Ketzinel-Gilad, M.; Ariav, Y.; Pappo, O.; Karaca, M.; Castel, J.; Berthault, M.F.; Magnan, C.; Cerasi, E.; Kaiser, N.; et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008, 57, 945–957. [Google Scholar]
- Cho, H.J.; Park, J.; Lee, H.W.; Lee, Y.S.; Kim, J.B. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem. Biophys. Res. Commun 2004, 321, 942–948. [Google Scholar]
- Yeh, W.C.; Bierer, B.E.; McKnight, S.L. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc. Natl. Acad. Sci. USA 1995, 92, 11086–11090. [Google Scholar]
- Chakrabarti, P.; English, T.; Shi, J.; Smas, C.M.; Kandror, K.V. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 2010, 59, 775–781. [Google Scholar]
- Rovira, J.; Marcelo Arellano, E.; Burke, J.T.; Brault, Y.; Moya-Rull, D.; Banon-Maneus, E.; Ramirez-Bajo, M.J.; Gutierrez-Dalmau, A.; Revuelta, I.; Quintana, L.F.; et al. Effect of mTOR inhibitor on body weight: From an experimental rat model to human transplant patients. Transpl. Int 2008, 21, 992–998. [Google Scholar]
- Phan, J.; Peterfy, M.; Reue, K. Lipin expression preceding peroxisome proliferator-activated receptor-γ is critical for adipogenesis in vivo and in vitro. J. Biol. Chem 2004, 279, 29558–29564. [Google Scholar]
- Peterfy, M.; Phan, J.; Xu, P.; Reue, K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet 2001, 27, 121–124. [Google Scholar]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C., Jr; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab 2006, 4, 199–210. [Google Scholar]
- Huffman, T.A.; Mothe-Satney, I.; Lawrence, J.C., Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. USA 2002, 99, 1047–1052. [Google Scholar]
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar]
- Cota, D.; Matter, E.K.; Woods, S.C.; Seeley, R.J. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J. Neurosci 2008, 28, 7202–7208. [Google Scholar]
- Chakrabarti, P.; Anno, T.; Manning, B.D.; Luo, Z.; Kandror, K.V. The mammalian target of rapamycin complex 1 regulates leptin biosynthesis in adipocytes at the level of translation: The role of the 5′-untranslated region in the expression of leptin messenger ribonucleic acid. Mol. Endocrinol 2008, 22, 2260–2267. [Google Scholar]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 form the critical elements for cerebral vascular protection by erythropoietin. J. Cereb. Blood Flow Metab 2003, 23, 320–330. [Google Scholar]
- Toba, H.; Kojima, Y.; Wang, J.; Noda, K.; Tian, W.; Kobara, M.; Nakata, T. Erythropoietin attenuated vascular dysfunction and inflammation by inhibiting NADPH oxidase-derived superoxide production in nitric oxide synthase-inhibited hypertensive rat aorta. Eur. J. Pharmacol 2012, 691, 190–197. [Google Scholar]
- Sheng, B.; Liu, J.; Li, G.H. Metformin preconditioning protects Daphnia pulex from lethal hypoxic insult involving AMPK, HIF and mTOR signaling. Comp. Biochem. Physiol. B Biochem. Mol. Biol 2012, 163, 51–58. [Google Scholar]
- Koh, P.O. Melatonin prevents ischemic brain injury through activation of the mTOR/p70S6 kinase signaling pathway. Neurosci. Lett 2008, 444, 74–78. [Google Scholar]
- Liu, G.; Detloff, M.R.; Miller, K.N.; Santi, L.; Houle, J.D. Exercise modulates microRNAs that affect the PTEN/mTOR pathway in rats after spinal cord injury. Exp. Neurol 2012, 233, 447–456. [Google Scholar]
- Walker, C.L.; Walker, M.J.; Liu, N.K.; Risberg, E.C.; Gao, X.; Chen, J.; Xu, X.M. Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS One 2012, 7, e30012. [Google Scholar]
- Hu, L.Y.; Sun, Z.G.; Wen, Y.M.; Cheng, G.Z.; Wang, S.L.; Zhao, H.B.; Zhang, X.R. ATP-mediated protein kinase B Akt/mammalian target of rapamycin mTOR/p70 ribosomal S6 protein p70S6 kinase signaling pathway activation promotes improvement of locomotor function after spinal cord injury in rats. Neuroscience 2010, 169, 1046–1062. [Google Scholar]
- Sun, F.; Park, K.K.; Belin, S.; Wang, D.; Lu, T.; Chen, G.; Zhang, K.; Yeung, C.; Feng, G.; Yankner, B.A.; et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 2011, 480, 372–375. [Google Scholar] [Green Version]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci 2010, 13, 1075–1081. [Google Scholar]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar]
- Erlich, S.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis 2007, 26, 86–93. [Google Scholar]
- Sekiguchi, A.; Kanno, H.; Ozawa, H.; Yamaya, S.; Itoi, E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J. Neurotrauma 2012, 29, 946–956. [Google Scholar]
- Shi, G.D.; OuYang, Y.P.; Shi, J.G.; Liu, Y.; Yuan, W.; Jia, L.S. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem. Biophys. Res. Commun 2011, 404, 941–945. [Google Scholar]
- Zhang, W.; Khatibi, N.H.; Yamaguchi-Okada, M.; Yan, J.; Chen, C.; Hu, Q.; Meng, H.; Han, H.; Liu, S.; Zhou, C. Mammalian target of rapamycin (mTOR) inhibition reduces cerebral vasospasm following a subarachnoid hemorrhage injury in canines. Exp. Neurol 2012, 233, 799–806. [Google Scholar]
- Zeng, L.H.; Rensing, N.R.; Wong, M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J. Neurosci 2009, 29, 6964–6972. [Google Scholar]
- Buckmaster, P.S.; Ingram, E.A.; Wen, X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J. Neurosci 2009, 29, 8259–8269. [Google Scholar]
- Holmes, G.L.; Stafstrom, C.E. Tuberous sclerosis complex and epilepsy: Recent developments and future challenges. Epilepsia 2007, 48, 617–630. [Google Scholar]
- Waltereit, R.; Welzl, H.; Dichgans, J.; Lipp, H.P.; Schmidt, W.J.; Weller, M. Enhanced episodic-like memory and kindling epilepsy in a rat model of tuberous sclerosis. J. Neurochem 2006, 96, 407–413. [Google Scholar]
- Zeng, L.H.; Xu, L.; Gutmann, D.H.; Wong, M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol 2008, 63, 444–453. [Google Scholar]
- Lee, S.T.; Chu, K.; Park, J.E.; Jung, K.H.; Jeon, D.; Lim, J.Y.; Lee, S.K.; Kim, M.; Roh, J.K. Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-beta burden in Alzheimer’s disease models. J. Neurochem 2012, 120, 115–124. [Google Scholar]
- Burgos-Ramos, E.; Martos-Moreno, G.A.; Lopez, M.G.; Herranz, R.; Aguado-Llera, D.; Egea, J.; Frechilla, D.; Cenarruzabeitia, E.; Leon, R.; Arilla-Ferreiro, E.; et al. The N-terminal tripeptide of insulin-like growth factor-I protects against beta-amyloid-induced somatostatin depletion by calcium and glycogen synthase kinase 3 beta modulation. J. Neurochem 2009, 109, 360–370. [Google Scholar]
- Echeverria, V.; Zeitlin, R.; Burgess, S.; Patel, S.; Barman, A.; Thakur, G.; Mamcarz, M.; Wang, L.; Sattelle, D.B.; Kirschner, D.A.; et al. Cotinine reduces amyloid-beta aggregation and improves memory in Alzheimer’s disease mice. J. Alzheimers Dis 2011, 24, 817–835. [Google Scholar]
- Li, L.; Xu, B.; Zhu, Y.; Chen, L.; Sokabe, M. DHEA prevents Abeta25-35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology 2010, 59, 323–333. [Google Scholar]
- Chano, T.; Okabe, H.; Hulette, C.M. RB1CC1 insufficiency causes neuronal atrophy through mTOR signaling alteration and involved in the pathology of Alzheimer’s diseases. Brain Res 2007, 1168, 97–105. [Google Scholar]
- Paccalin, M.; Pain-Barc, S.; Pluchon, C.; Paul, C.; Besson, M.N.; Carret-Rebillat, A.S.; Rioux-Bilan, A.; Gil, R.; Hugon, J. Activated mTOR and PKR kinases in lymphocytes correlate with memory and cognitive decline in Alzheimer’s disease. Dementia Geriatr. Cognit. Disord 2006, 22, 320–326. [Google Scholar]
- Slipczuk, L.; Bekinschtein, P.; Katche, C.; Cammarota, M.; Izquierdo, I.; Medina, J.H. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One 2009, 4, e6007. [Google Scholar]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; Yu, F.; Wong, H.; Lin, M.T.; Tampellini, D.; Klann, E.; Blitzer, R.D.; Gouras, G.K. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One 2010, 5, e12845. [Google Scholar]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Aβ exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem 2005, 94, 215–225. [Google Scholar]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem 2005, 93, 105–117. [Google Scholar]
- Esteras, N.; Munoz, U.; Alquezar, C.; Bartolome, F.; Bermejo-Pareja, F.; Martin-Requero, A. Altered calmodulin degradation and signaling in non-neuronal cells from Alzheimer’s disease patients. Curr. Alzheimer Res 2012, 9, 267–277. [Google Scholar]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol 2003, 163, 591–607. [Google Scholar]
- Sarkar, S.; Ravikumar, B.; Rubinsztein, D.C. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol 2009, 453, 83–110. [Google Scholar]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet 2006, 15, 433–442. [Google Scholar]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet 2004, 36, 585–595. [Google Scholar]
- Floto, R.A.; Sarkar, S.; Perlstein, E.O.; Kampmann, B.; Schreiber, S.L.; Rubinsztein, D.C. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 2007, 3, 620–622. [Google Scholar]
- Roscic, A.; Baldo, B.; Crochemore, C.; Marcellin, D.; Paganetti, P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J. Neurochem 2011, 119, 398–407. [Google Scholar]
- Fox, J.H.; Connor, T.; Chopra, V.; Dorsey, K.; Kama, J.A.; Bleckmann, D.; Betschart, C.; Hoyer, D.; Frentzel, S.; Difiglia, M.; et al. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington’s disease. Mol. Neurodegener 2010, 5, 26. [Google Scholar]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010, 5, e9313. [Google Scholar]
- Tain, L.S.; Mortiboys, H.; Tao, R.N.; Ziviani, E.; Bandmann, O.; Whitworth, A.J. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci 2009, 12, 1129–1135. [Google Scholar]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1–mediated 14-3-3 shuttling. Genes Dev 2008, 22, 239–251. [Google Scholar]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci 2006, 26, 9996–10005. [Google Scholar]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 2008, 27, 2432–2443. [Google Scholar]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef]
- Fokas, E.; Yoshimura, M.; Prevo, R.; Higgins, G.; Hackl, W.; Maira, S.M.; Bernhard, E.J.; McKenna, W.G.; Muschel, R.J. NVP-BEZ235 and NVP-BGT226, dual phosphatidylinositol 3-kinase/Mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat. Oncol 2012, 7, 48. [Google Scholar]
- Maiese, K.; Hou, J.; Chong, Z.Z.; Shang, Y.C. A fork in the path: Developing therapeutic inroads with FoxO proteins. Oxid. Med. Cell. Longev 2009, 2, 119–129. [Google Scholar]
- Musteanu, M.; Blaas, L.; Zenz, R.; Svinka, J.; Hoffmann, T.; Grabner, B.; Schramek, D.; Kantner, H.P.; Muller, M.; Kolbe, T.; et al. A mouse model to identify cooperating signaling pathways in cancer. Nat. Methods 2012, 9, 897–900. [Google Scholar]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Horsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar]
- Baryawno, N.; Sveinbjornsson, B.; Eksborg, S.; Chen, C.S.; Kogner, P.; Johnsen, J.I. Small-molecule inhibitors of phosphatidylinositol 3-kinase/Akt signaling inhibit Wnt/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res 2010, 70, 266–276. [Google Scholar]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 2011, 6, e25416. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar]
- Carayol, N.; Vakana, E.; Sassano, A.; Kaur, S.; Goussetis, D.J.; Glaser, H.; Druker, B.J.; Donato, N.J.; Altman, J.K.; Barr, S.; et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12469–12474. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maiese, K.; Chong, Z.Z.; Wang, S.; Shang, Y.C. Oxidant Stress and Signal Transduction in the Nervous System with the PI 3-K, Akt, and mTOR Cascade. Int. J. Mol. Sci. 2012, 13, 13830-13866. https://doi.org/10.3390/ijms131113830
Maiese K, Chong ZZ, Wang S, Shang YC. Oxidant Stress and Signal Transduction in the Nervous System with the PI 3-K, Akt, and mTOR Cascade. International Journal of Molecular Sciences. 2012; 13(11):13830-13866. https://doi.org/10.3390/ijms131113830
Chicago/Turabian StyleMaiese, Kenneth, Zhao Zhong Chong, Shaohui Wang, and Yan Chen Shang. 2012. "Oxidant Stress and Signal Transduction in the Nervous System with the PI 3-K, Akt, and mTOR Cascade" International Journal of Molecular Sciences 13, no. 11: 13830-13866. https://doi.org/10.3390/ijms131113830