Structural Modeling and Biochemical Characterization of Recombinant KPN_02809, a Zinc-Dependent Metalloprotease from Klebsiella pneumoniae MGH 78578

Abstract
:1. Introduction
2. Results and Discussion
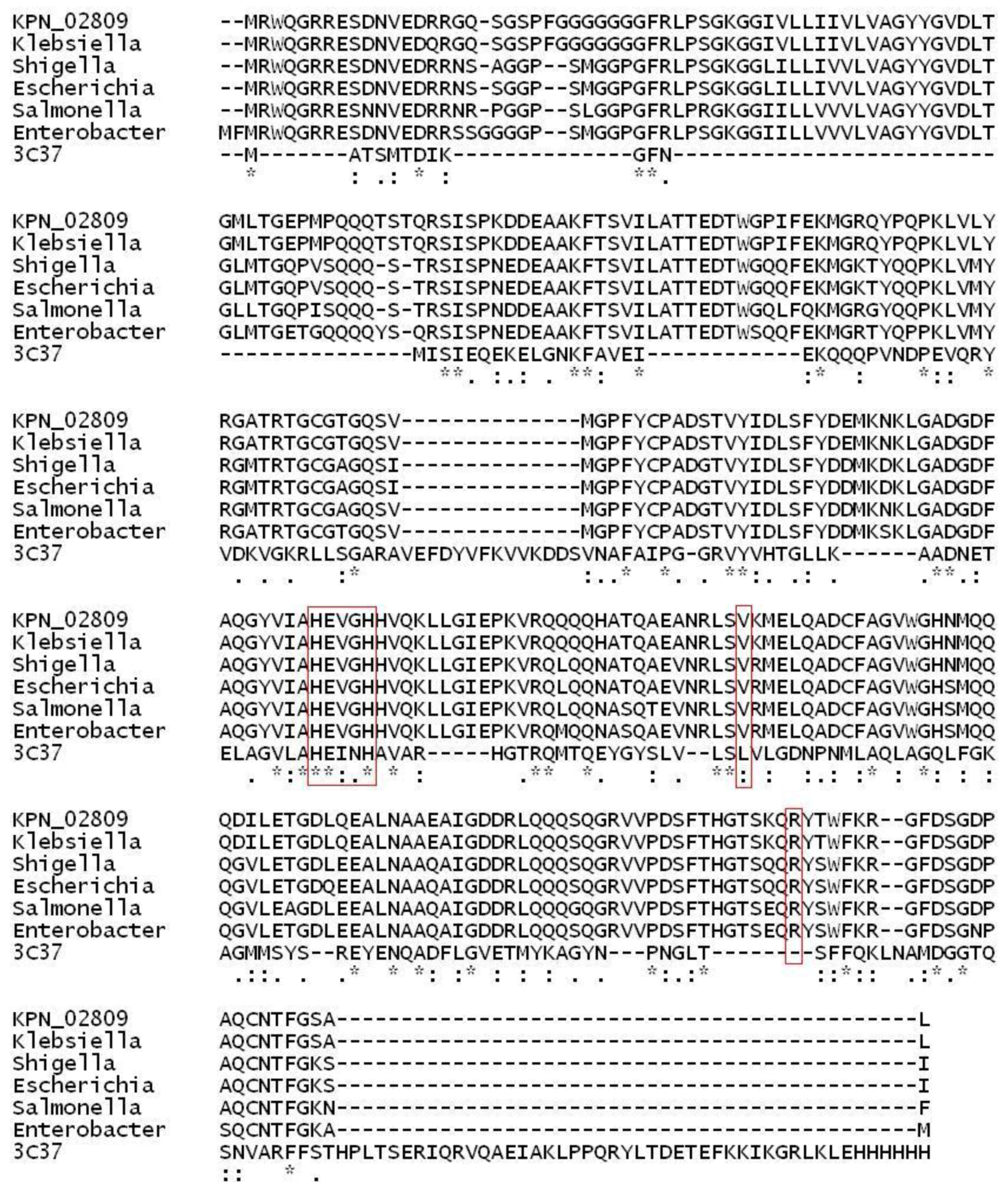
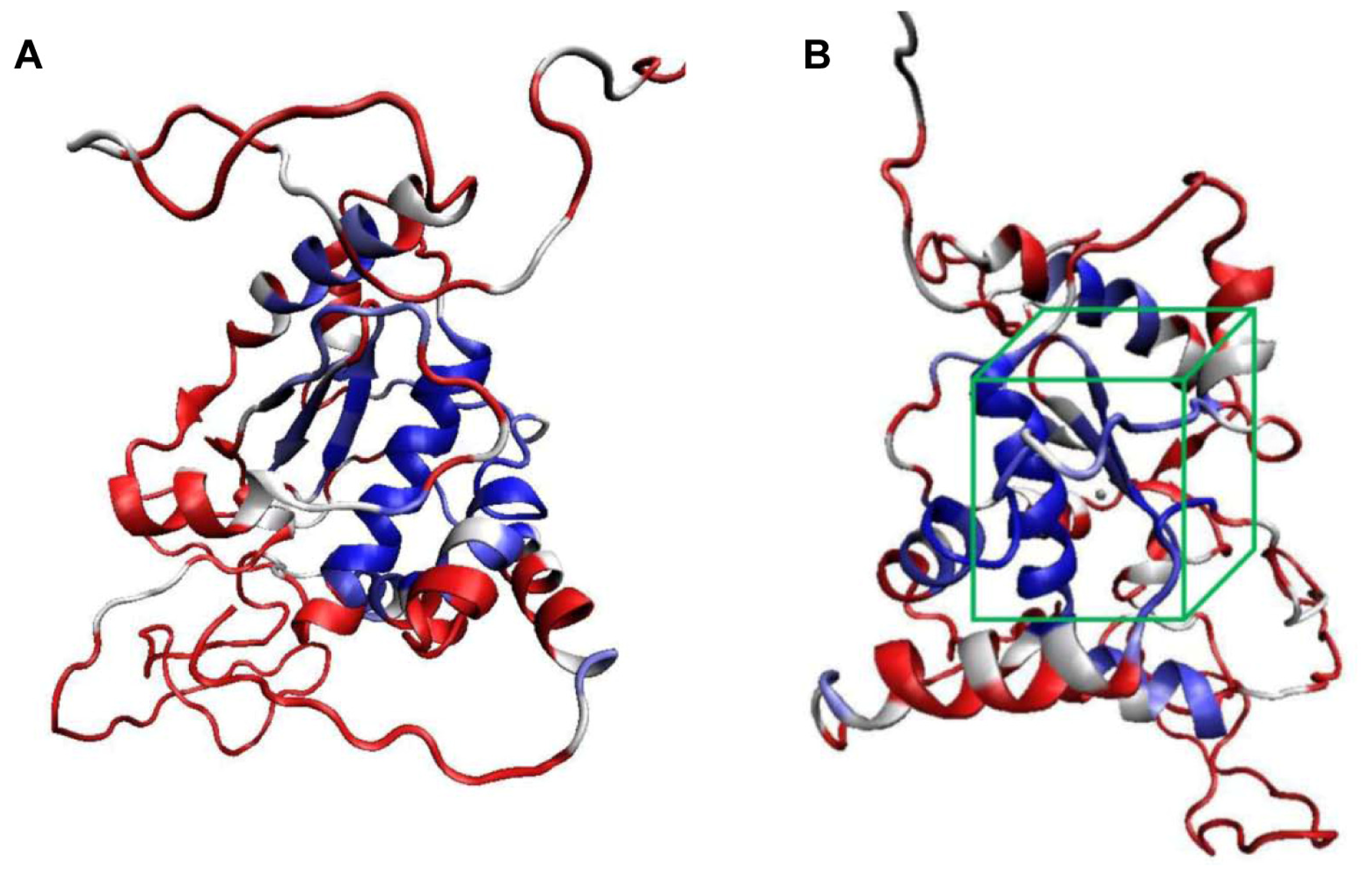
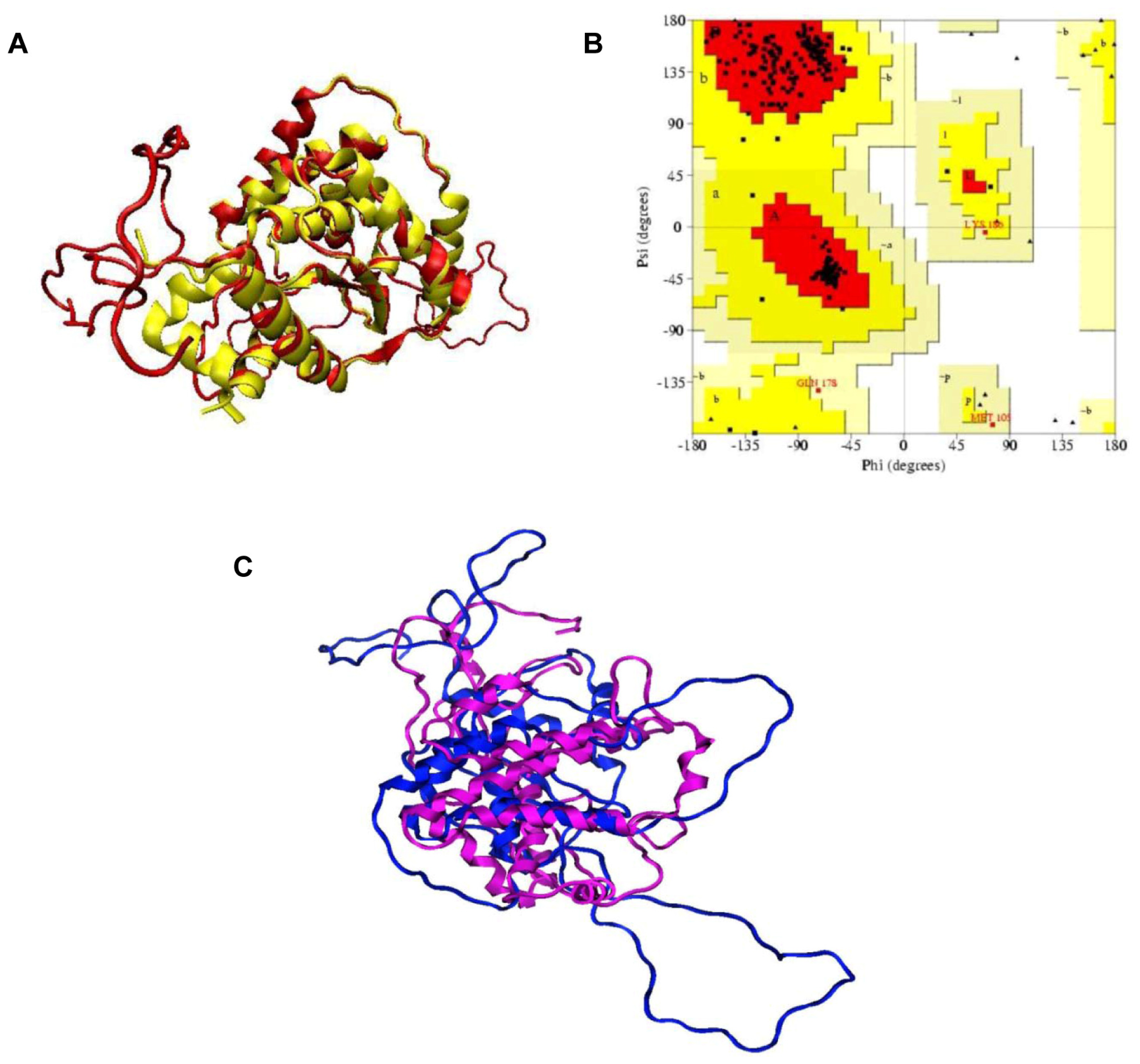
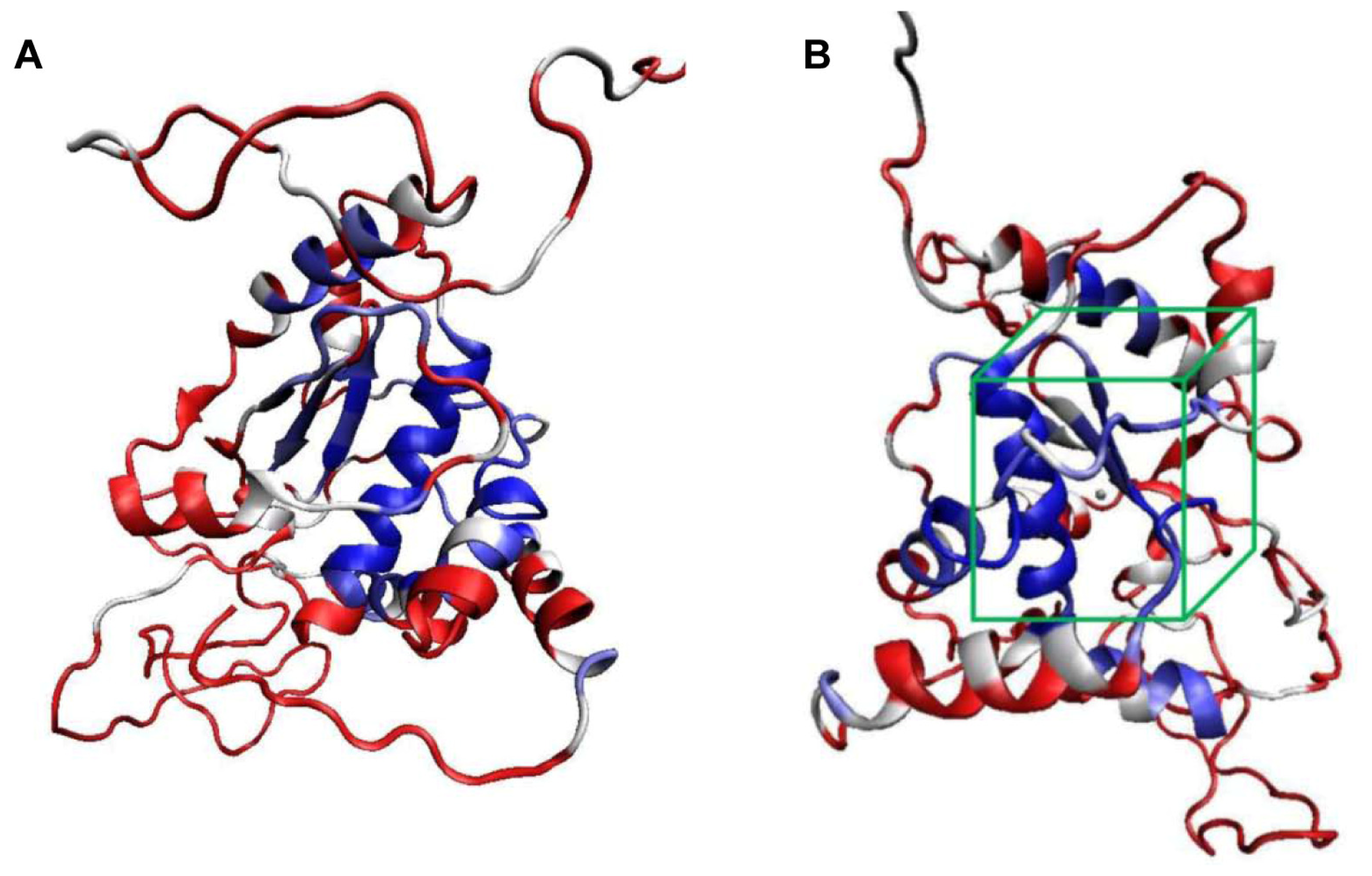
2.1. Structural Modeling and Analysis of KPN_02809
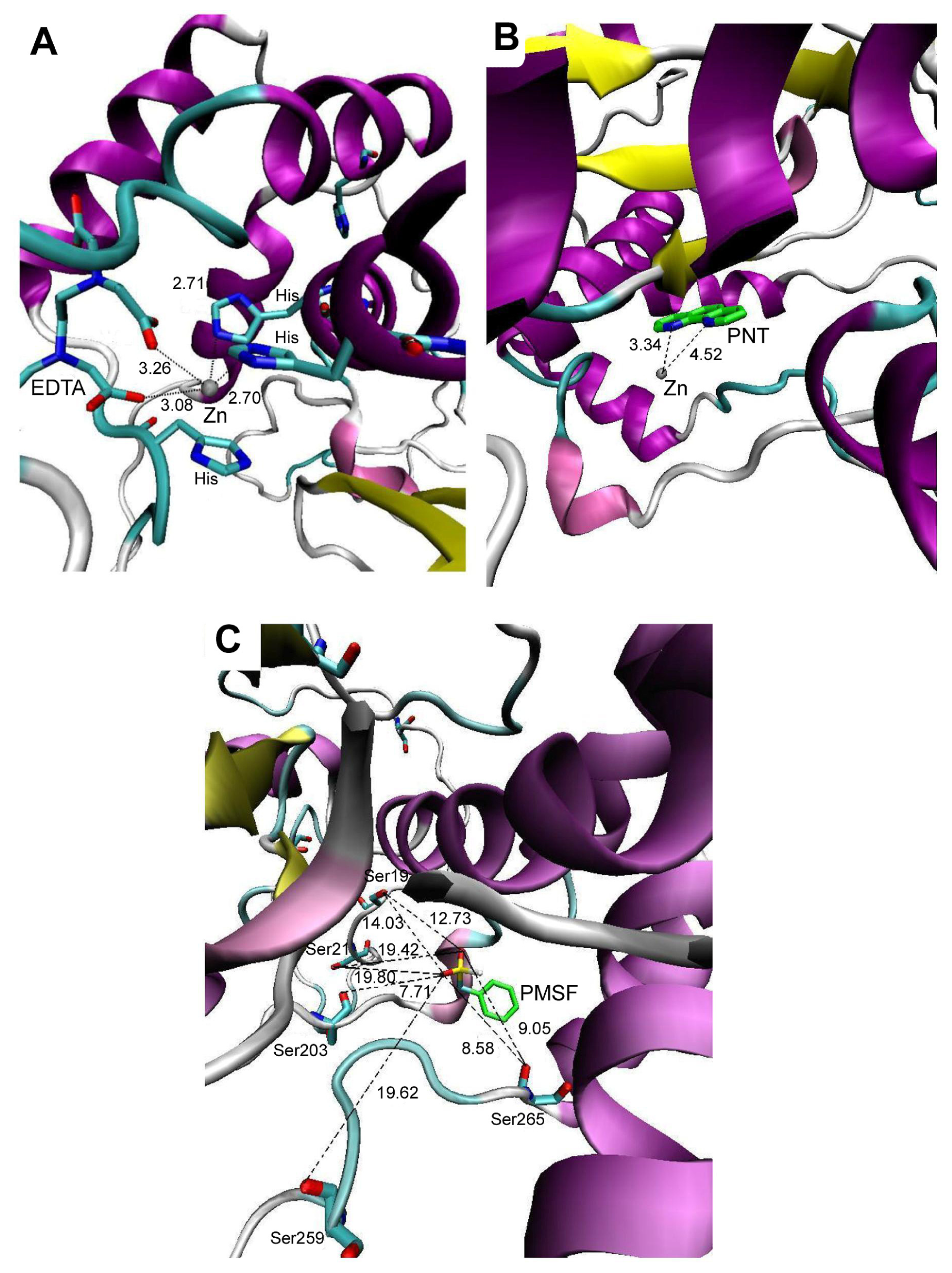
2.2. Molecular Docking Simulation
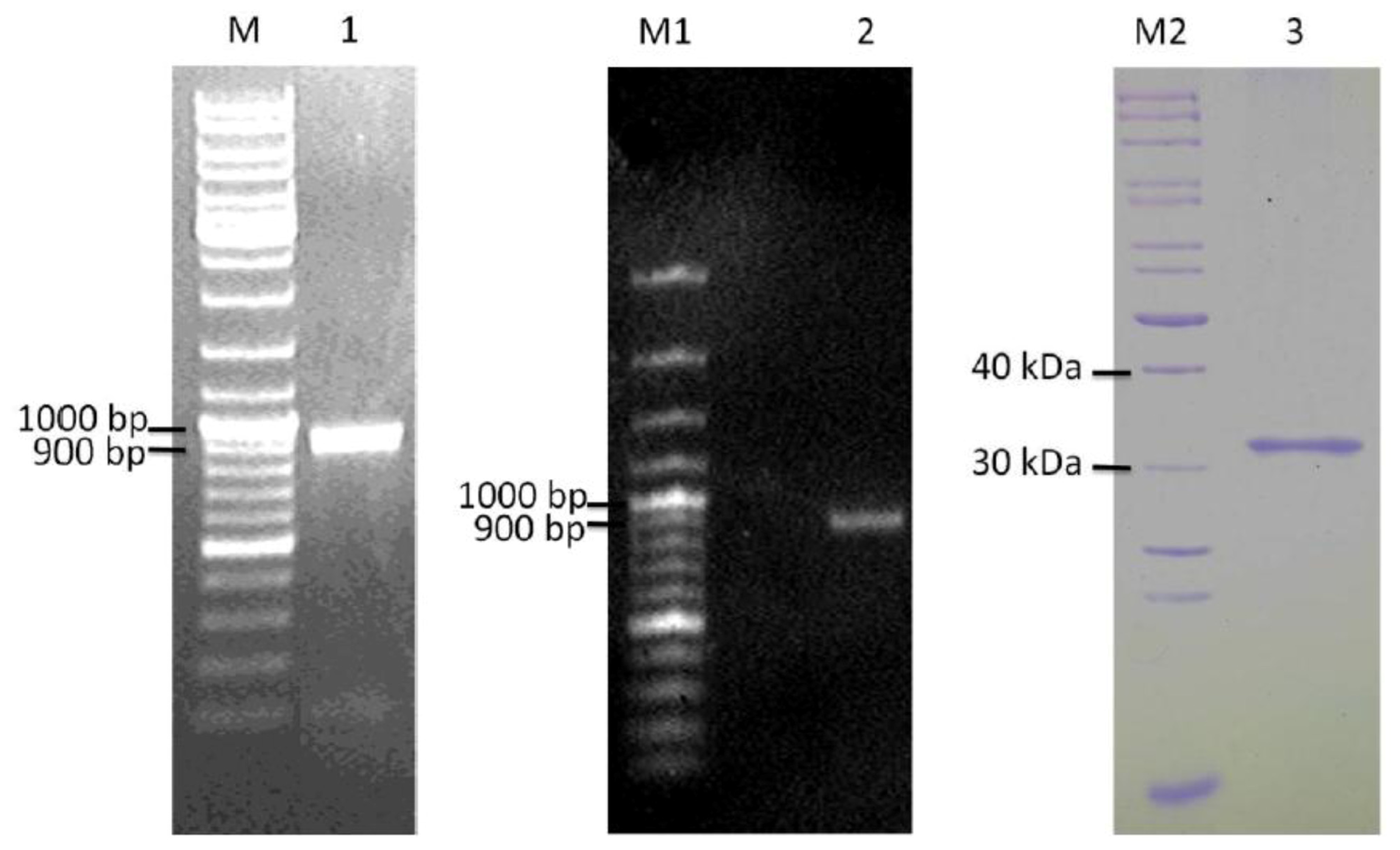
2.3. Detection of ypfJ mRNA Expression in K. pneumoniae MGH 78578
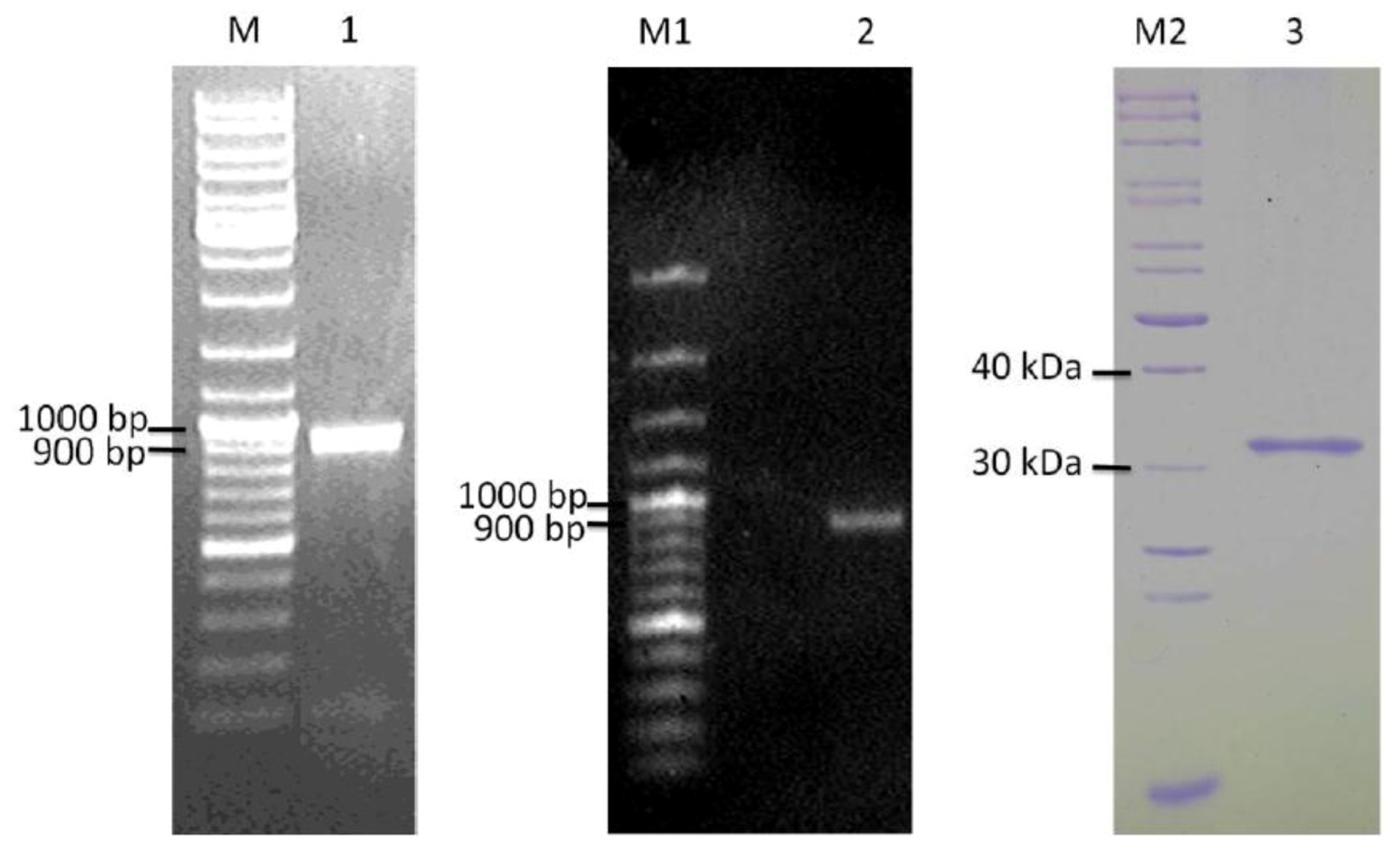
2.4. Cloning and Heterologous Expression of ypfJ Open Reading Frame in E. coli
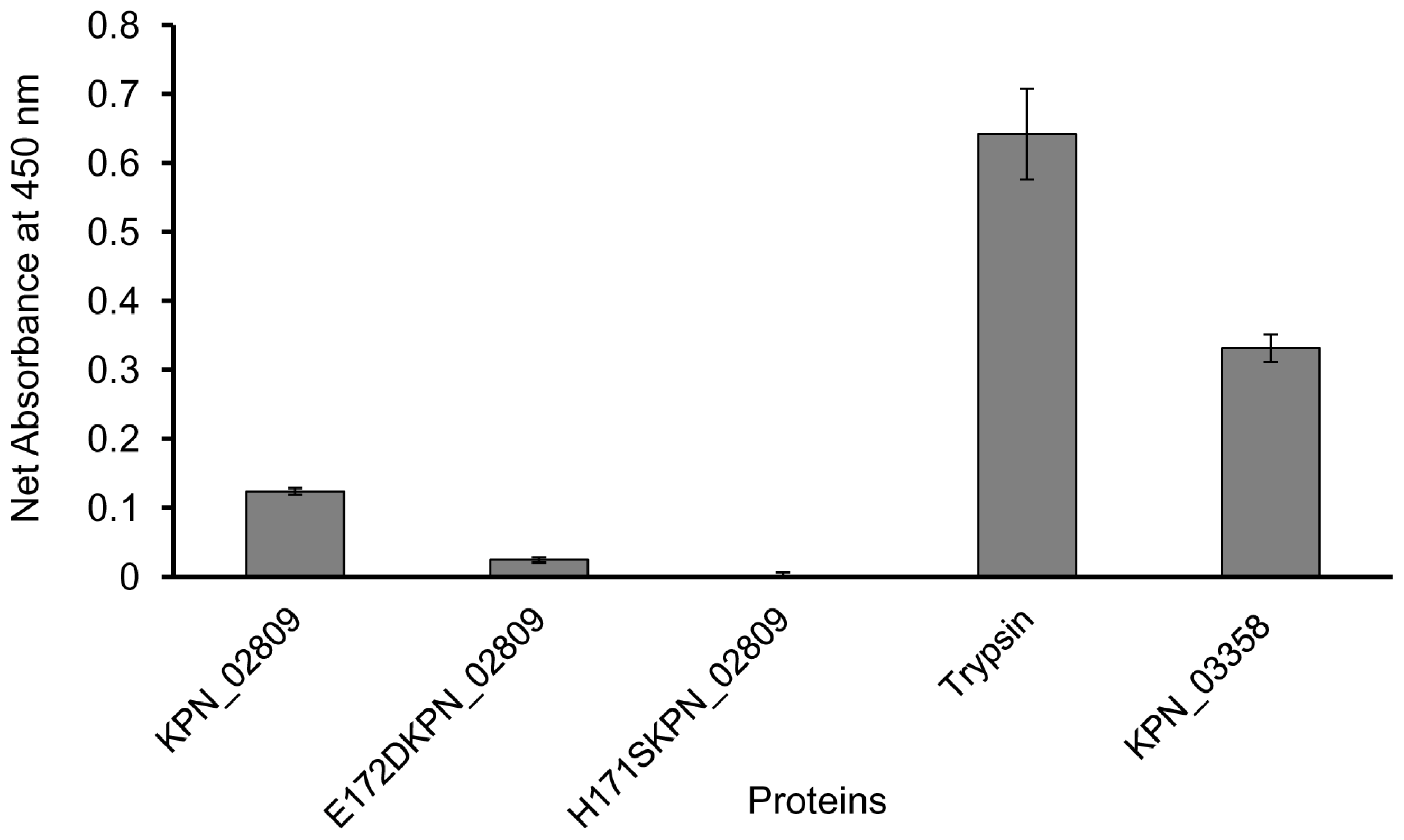
2.5. Proteolytic Activity of Purified Wild-type and Mutants K. pneumoniae KPN_02809
3. Experimental Section
3.1. Homology Modeling of KPN_02809 Protein and Model Assessment
3.2. Molecular Docking Simulation
3.3. Bacterial Strains, Growth and Culture Conditions
3.4. Total Genomic DNA and RNA Extractions from K. pneumoniae MGH 78578
3.5. Cloning and mutagenesis of K. pneumoniae ypfJ Open Reading Frame
3.6. Expression and Purification of Wild-Type and Mutant KPN_02809 Protein
3.7. Reverse Transcription PCR of ypfJ Gene
3.8. Casein Hydrolysis Assay
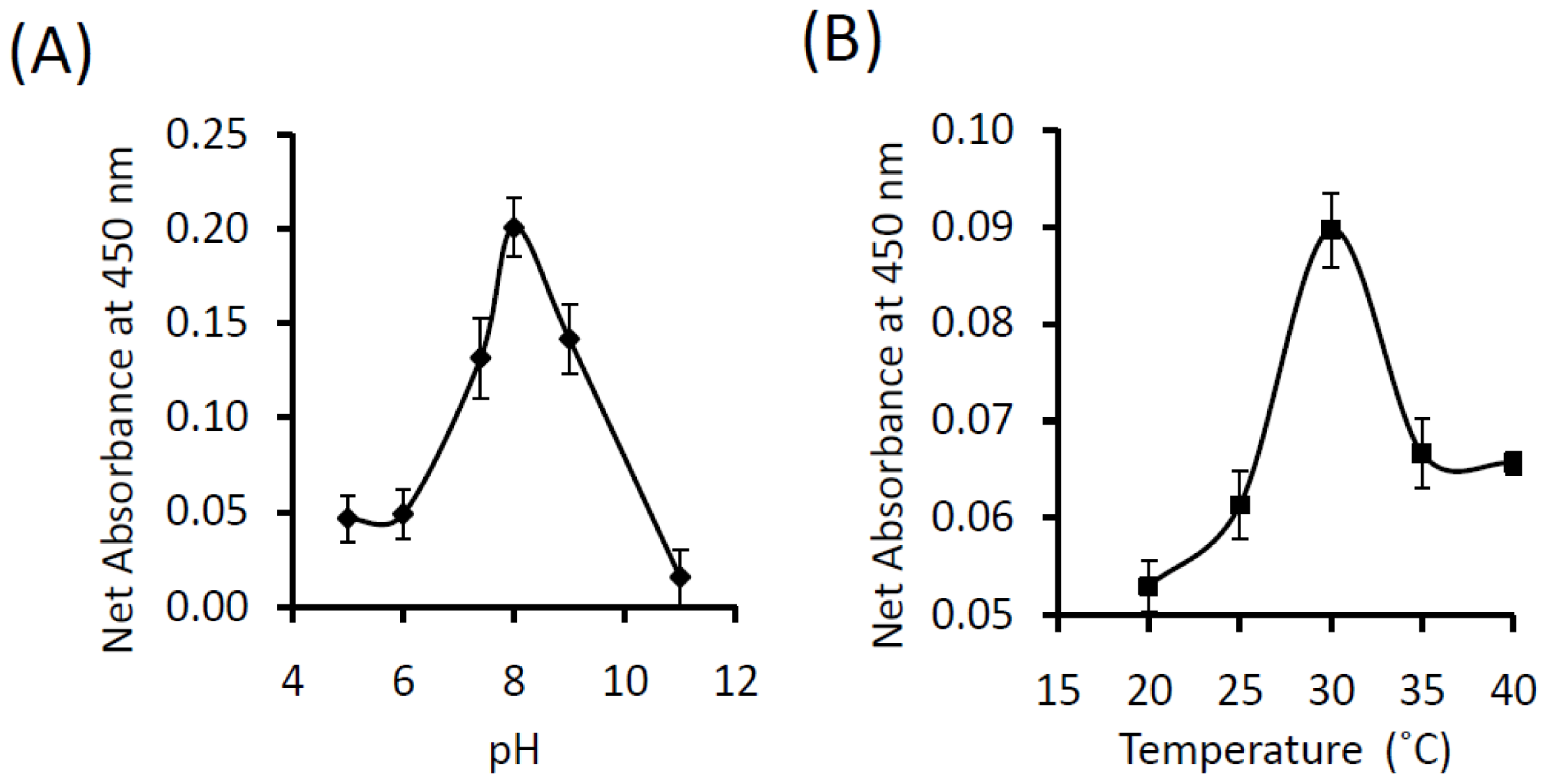
3.9. Determination of pH and Temperature Optima for the Activity of Purified KPN_02809
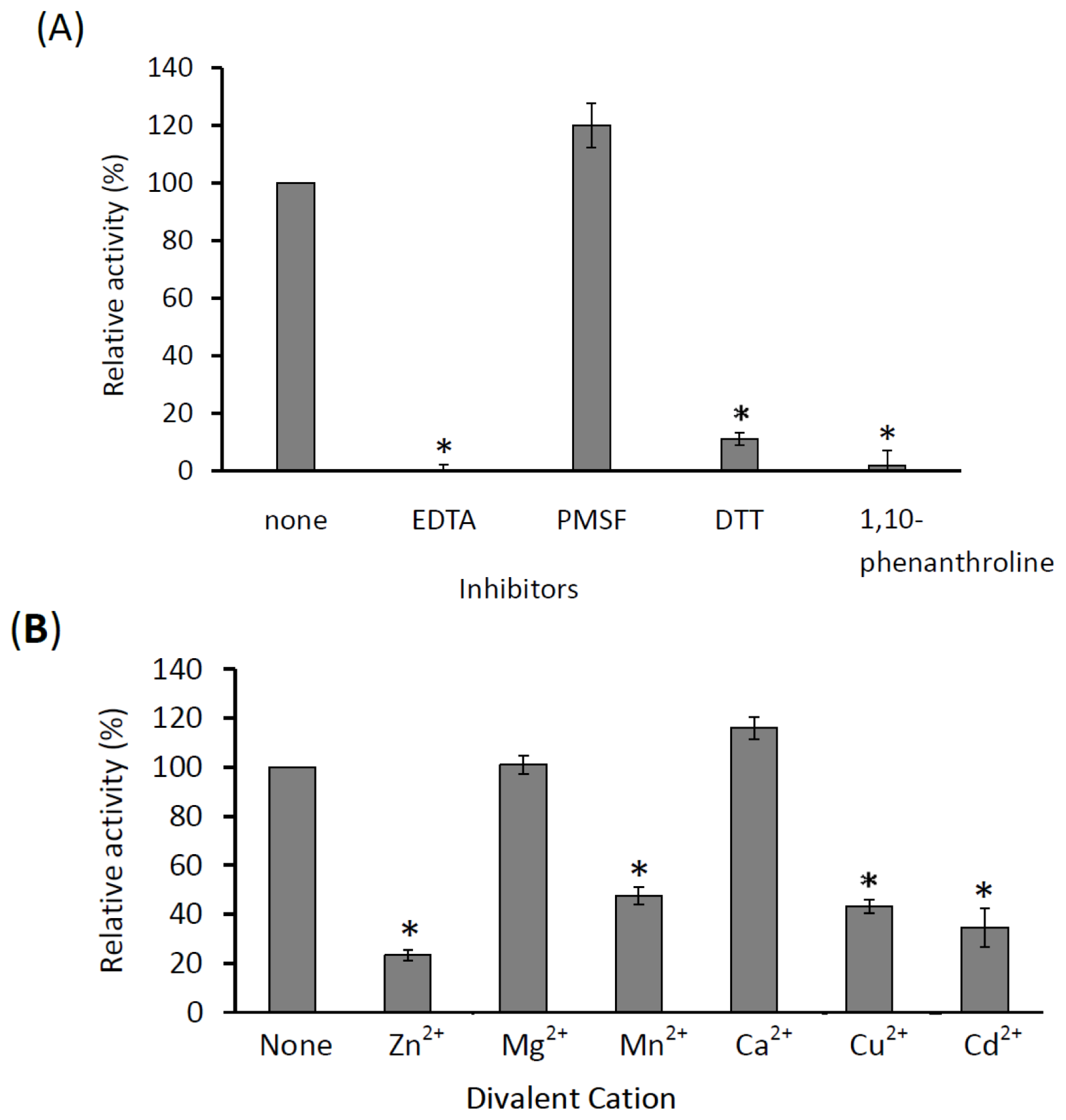
3.10. Effect of Various Inhibitors and Ions on the KPN_02809 Enzymatic Activity
3.11. Quantification of Zinc Ion Content in the KPN_02809 Protein Molecule
4. Conclusions
Acknowledgments
References
- Lawlor, M.S.; Hsu, J.; Rick, P.D.; Miller, V.L. Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol. Microbiol 2005, 58, 1054–1073. [Google Scholar]
- Lau, H.Y.; Clegg, S.; Moore, T.A. Identification of Klebsiella pneumoniae genes uniquely expressed in a strain virulent using a murine model of bacterial pneumonia. Microb. Pathog 2007, 42, 148–155. [Google Scholar]
- Haryani, Y.; Noorzaleha, A.S.; Fatimah, A.B.; Noorjahan, B.A.; Patrick, G.B.; Shamsinar, A.T.; Laila, R.A.S.; Son, R. Incidence of Klebsiella pneumonia in street foods sold in Malaysia and their characterization by antibiotic resistance, plasmid profiling, and RAPD-PCR analysis. Food Control 2007, 18, 847–853. [Google Scholar]
- Maltezou, H.C.; Giakkoupi, P.; Maragos, A.; Bolikas, M.; Raftopoulos, V.; Papahatzaki, H.; Vrouhos, G.; Liakou, V.; Vatopoulos, A.C. Outbreak of infections due to KPC-2-producing Klebsiella pneumoniae in a hospital in Crete (Greece). J. Infect 2009, 58, 213–219. [Google Scholar]
- Jongeneel, C.V.; Bouvier, J.; Bairoch, A. A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett 1989, 242, 211–214. [Google Scholar]
- Jain, E.; Bairoch, A.; Duvaud, S.; Phan, I.; Redaschi, N.; Suzek, B.E.; Martin, M.J.; McGarvey, P.; Gasteiger, E. Infrastructure for the life sciences: Design and implementation of the UniProt website. BMC Bioinforma 2009, 10, 136. [Google Scholar]
- Liljas, A.; Piškur, J. Textbook of Structural Biology; World Scientific: Singapore, Singapore, 2009. [Google Scholar]
- Dabonne, S.; Moallic, C.; Sine, J.P.; Niamke, S.; Dion, M.; Colas, B. Cloning, expression and characterization of a 46.5-kDa metallopeptidase from Bacillus halodurans H4 sharing properties with the pitrilysin family. Biochim. Biophys. Acta 2005, 1725, 136–143. [Google Scholar]
- Fujimura-Kamada, K.; Nouvet, F.J.; Michaelis, S. A novel membrane-associated metalloprotease, Ste24p, is required for the first step of NH2-terminal processing of the yeast a-factor precursor. J. Cell Biol 1997, 136, 271–285. [Google Scholar]
- Luciano, P.; Tokatlidis, K.; Chambre, I.; Germanique, J.C.; Geli, V. The mitochondrial processing peptidase behaves as a zinc-metallopeptidase. J. Mol. Biol 1998, 280, 193–199. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of metallopeptidases. Methods Enzymol 1995, 248, 183–228. [Google Scholar]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the web: A case study using the Phyre server. Nat. Protoc 2009, 4, 363–371. [Google Scholar]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res 2009, 37, D211–215. [Google Scholar]
- Hernick, M.; Fierke, C.A. Zinc hydrolases: The mechanisms of zinc-dependent deacetylases. Arch. Biochem. Biophys 2005, 433, 71–84. [Google Scholar]
- Kuan, C.S.; Wong, M.T.; Choi, S.B.; Chang, C.C.; Yee, Y.H.; Wahab, H.A.; Normi, Y.M.; Too, W.C.; Few, L.L. Klebsiella pneumoniae yggG gene product: A zinc-dependent metalloprotease. Int. J. Mol. Sci 2011, 12, 4441–4455. [Google Scholar]
- Kyte, J. Structure in Protein Chemistry, 2nd ed; Garland Science: New York, NY, USA, 2006. [Google Scholar]
- Bell, J.; Ash, D.E.; Snyder, L.M.; Kulathila, R.; Blackburn, N.J.; Merkler, D.J. Structural and functional investigations on the role of zinc in bifunctional rat peptidylglycine alpha-amidating enzyme. Biochemistry 1997, 36, 16239–16246. [Google Scholar]
- Jung, C.M.; Matsushita, O.; Katayama, S.; Minami, J.; Sakurai, J.; Okabe, A. Identification of metal ligands in the Clostridium histolyticum ColH collagenase. J. Bacteriol 1999, 181, 2816–2822. [Google Scholar]
- Holland, D.R.; Hausrath, A.C.; Juers, D.; Matthews, B.W. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci 1995, 4, 1955–1965. [Google Scholar]
- Gomez-Ortiz, M.; Gomis-Ruth, F.X.; Huber, R.; Aviles, F.X. Inhibition of carboxypeptidase A by excess zinc: Analysis of the structural determinants by X-ray crystallography. FEBS Lett 1997, 400, 336–340. [Google Scholar]
- McCall, K.A.; Huang, C.; Fierke, C.A. Function and mechanism of zinc metalloenzymes. J. Nutr 2000, 130, 1437S–1446S. [Google Scholar]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 2005, 33, 511–518. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc 2010, 5, 725–738. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234, 779–815. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr 1993, 26, 283–291. [Google Scholar]
- Konc, J.; Janezic, D. ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics 2010, 26, 1160–1168. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- Brundiers, R.; Lavie, A.; Veit, T.; Reinstein, J.; Schlichting, I.; Ostermann, N.; Goody, R.S.; Konrad, M. Modifying human thymidylate kinase to potentiate azidothymidine activation. J. Biol. Chem 1999, 274, 35289–35292. [Google Scholar]
- Malito, E.; Sekulic, N.; Too, W.C.; Konrad, M.; Lavie, A. Elucidation of human choline kinase crystal structures in complex with the products ADP or phosphocholine. J. Mol. Biol 2006, 364, 136–151. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Absorbance (500 nm) | Zinc concentration (μM) | Protein concentration (μM) | Ratio of zinc to protein concentration |
|---|---|---|---|---|
| BSA (negative control) | 0.001 | 0 | 15.15 | 0.1 |
| KPN_02809 | 0.456 | 149 | 124.60 | 1.2 |
| E172DKPN_02809 | 0.167 | 61.85 | 62.50 | 1.0 |
| H171SKPN_02809 | 0.073 | 27.04 | 62.50 | 0.4 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wong, M.T.; Choi, S.B.; Kuan, C.S.; Chua, S.L.; Chang, C.H.; Normi, Y.M.; See Too, W.C.; Wahab, H.A.; Few, L.L. Structural Modeling and Biochemical Characterization of Recombinant KPN_02809, a Zinc-Dependent Metalloprotease from Klebsiella pneumoniae MGH 78578. Int. J. Mol. Sci. 2012, 13, 901-917. https://doi.org/10.3390/ijms13010901
Wong MT, Choi SB, Kuan CS, Chua SL, Chang CH, Normi YM, See Too WC, Wahab HA, Few LL. Structural Modeling and Biochemical Characterization of Recombinant KPN_02809, a Zinc-Dependent Metalloprotease from Klebsiella pneumoniae MGH 78578. International Journal of Molecular Sciences. 2012; 13(1):901-917. https://doi.org/10.3390/ijms13010901
Chicago/Turabian StyleWong, Mun Teng, Sy Bing Choi, Chee Sian Kuan, Siang Ling Chua, Chiat Han Chang, Yahaya Mohd Normi, Wei Cun See Too, Habibah A. Wahab, and Ling Ling Few. 2012. "Structural Modeling and Biochemical Characterization of Recombinant KPN_02809, a Zinc-Dependent Metalloprotease from Klebsiella pneumoniae MGH 78578" International Journal of Molecular Sciences 13, no. 1: 901-917. https://doi.org/10.3390/ijms13010901