Cerebrospinal Fluid Markers in Sporadic Creutzfeldt-Jakob Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. CSF and Patients
2.2. PRNP Codon 129 Genotype, PrPSc Typing and Molecular Classification
2.3. CSF Protein Analysis
2.4. Statistical Analyses
3. Results
3.1. Molecular, Demographic and Clinical Characteristics of sCJD Patients
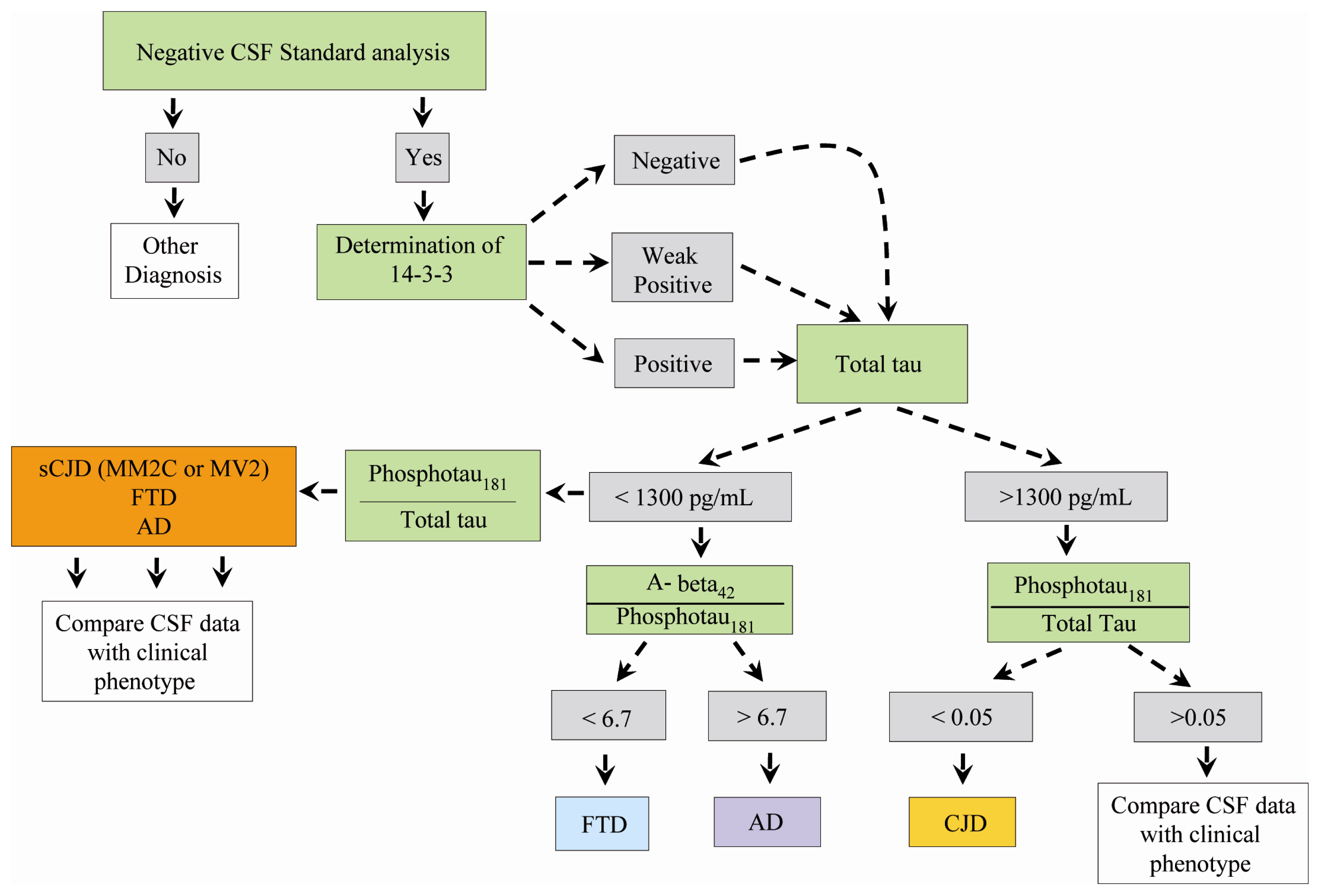
3.2. CSF 14-3-3 Protein Assay and Tau Protein Levels
3.3. CSF 14-3-3 Protein and Tau Protein Levels in Distinct sCJD Subtypes
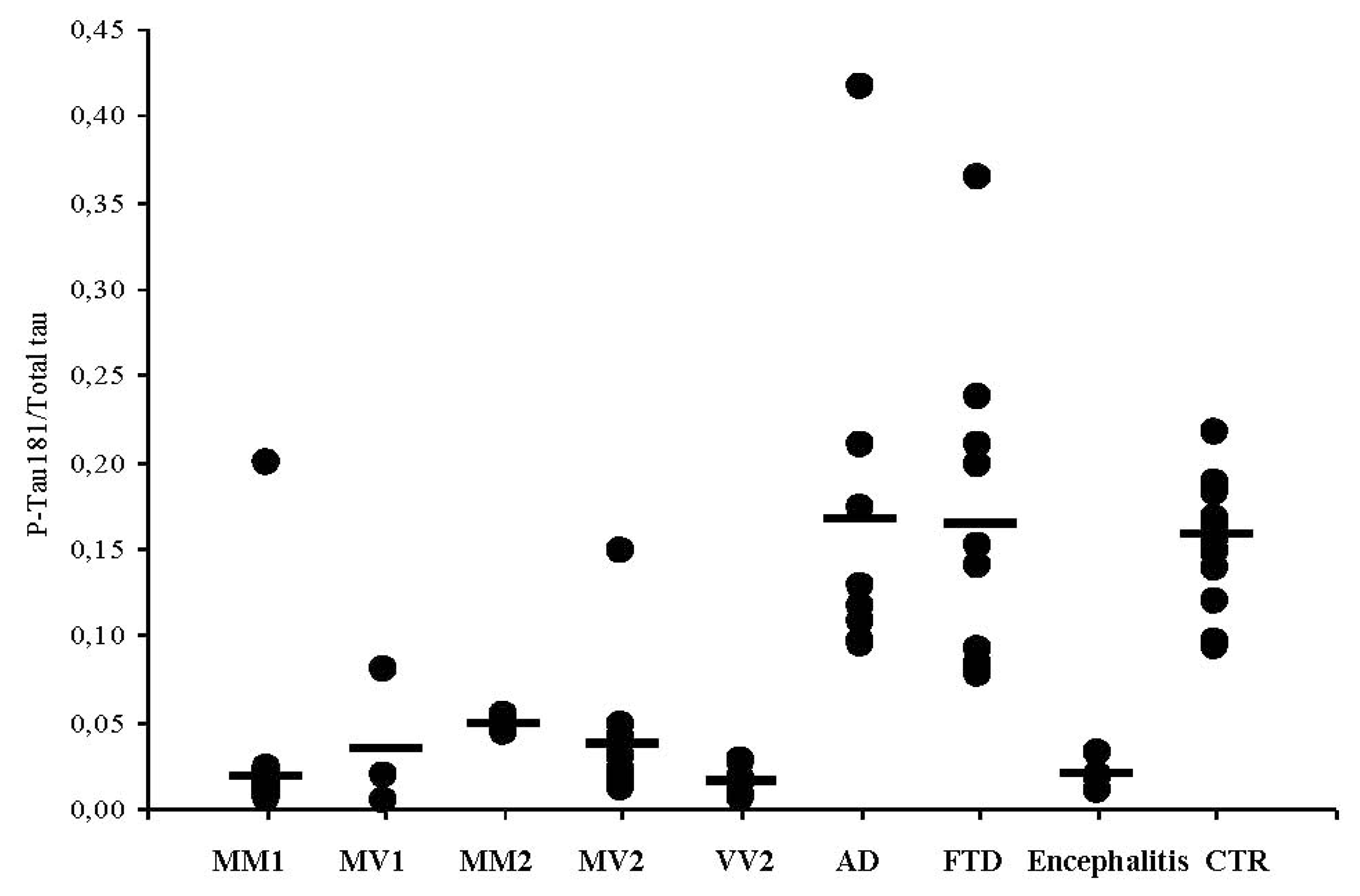
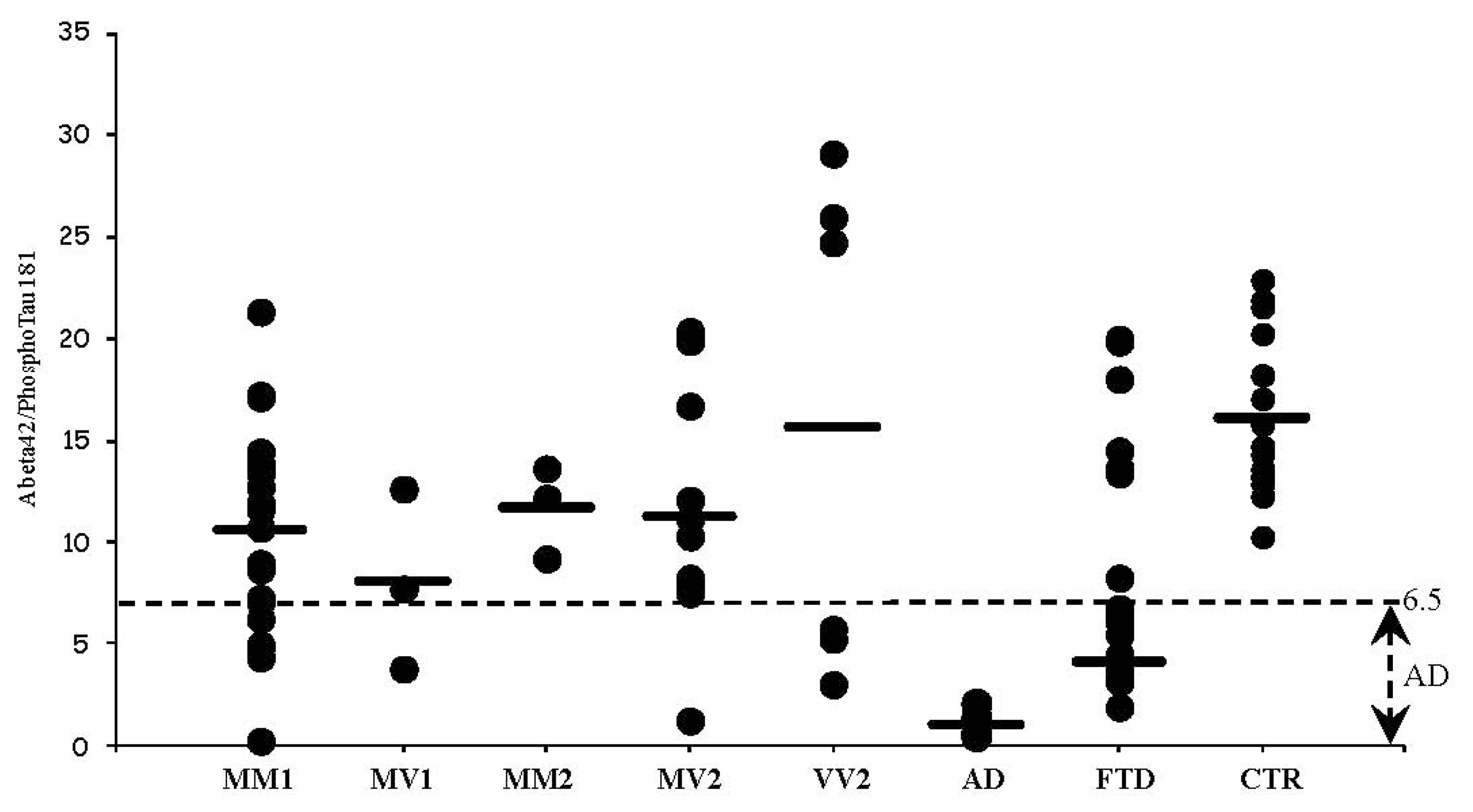
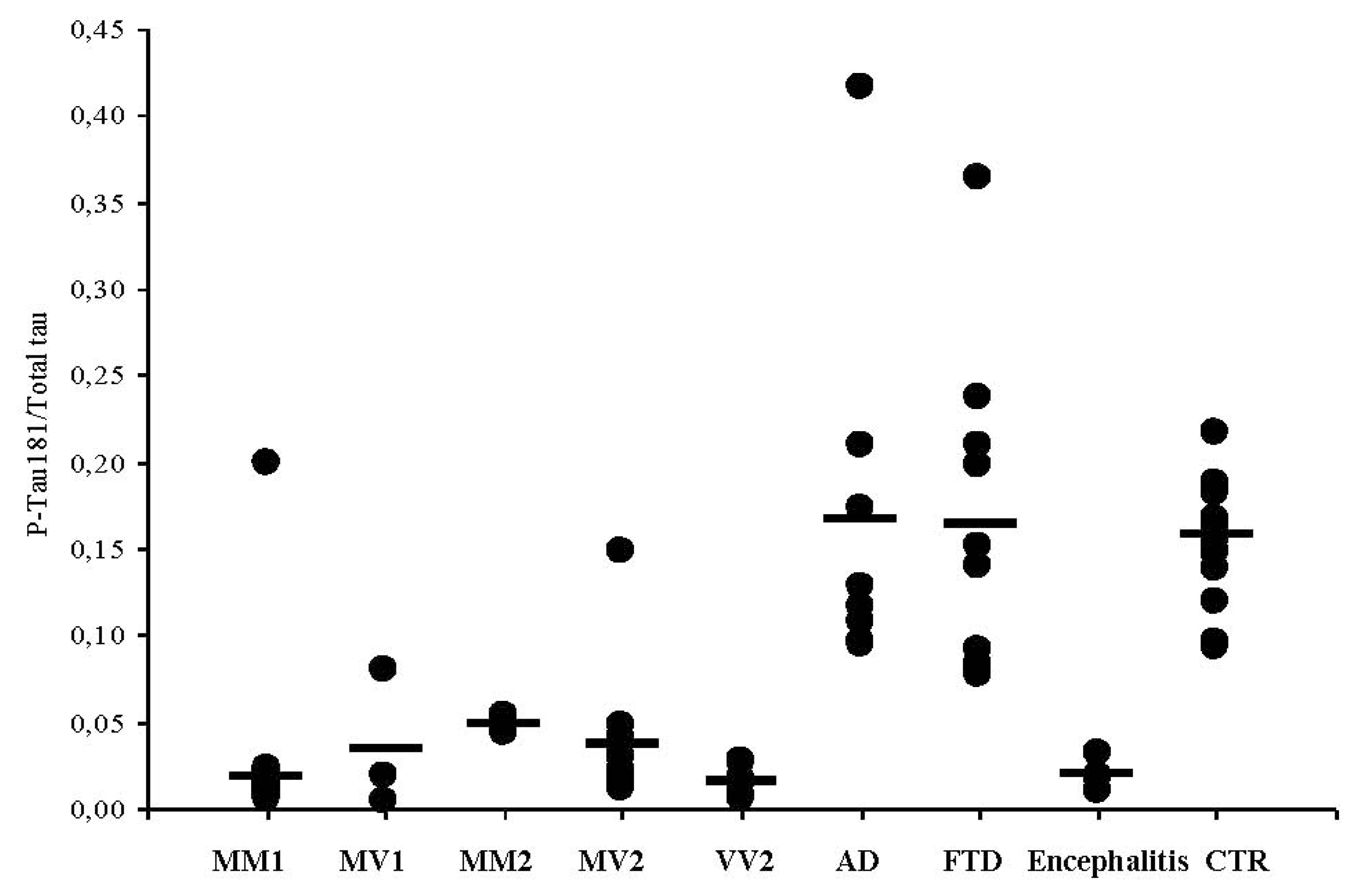
3.4. CSF Phosphotau181 and Aβ1-42 in sCJD Subtypes and Other Neurodegenerative Dementias
4. Conclusions
Acknowledgments
References
- Monaco, S; Zanusso, G; Mazzucco, S; Rizzuto, N. Cerebral amyloidoses: molecular pathways and therapeutic challenges. Curr Med Chem 2006, 13, 1903–1913. [Google Scholar]
- Parchi, P; Castellani, R; Capellari, S; Ghetti, B; Young, K; Chen, SG; Farlow, M; Dickson, DW; Sima, AA; Trojanowski, JQ; Petersen, RB; Gambetti, P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996, 39, 767–778. [Google Scholar]
- Zanusso, G; Farinazzo, A; Prelli, F; Fiorini, M; Gelati, M; Ferrari, S; Righetti, PG; Rizzuto, N; Frangione, B; Monaco, S. Identification of distinct n-terminal truncated forms of prion protein in different Creutzfeldt-Jakob disease subtypes. J Biol Chem 2004, 279, 38936–38942. [Google Scholar]
- Zanusso, G; Monaco, S. Molecular mechanisms of human prion diseases. Drug Discov Today: Dis Mech 2005, 2, 511–518. [Google Scholar]
- Zerr, I; Pocchiari, M; Collins, S; Brandel, JP; de Pedro Cuesta, J; Knights, RS; Berheimer, H; Cardone, F; Delasnerie-Lauprêtre, N; Cuadrado Corrales, N; et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-jakob disease. Neurology 2000, 55, 811–815. [Google Scholar]
- Zerr, I; Kallenberg, K; Summers, DM; Romero, C; Taratuto, A; Heinemann, U; Breithaupt, M; Varges, D; Meissner, B; Ladogana, A; et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009, 132, 2659–2668. [Google Scholar]
- Otto, M; Wiltfang, J; Tumani, H; Zerr, I; Lantsch, M; Kornhuber, J; Weber, T; Kretzschmar, HA; Poser, S. Elevated levels of tau-protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurosci Lett 1997, 225, 210–212. [Google Scholar]
- Riemenschneider, M; Wagenpfeil, S; Vanderstichele, H; Otto, M; Wiltfang, J; Kretzschmar, H; Vanmechelen, E; Forstl, H; Kurz, A. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol Psychiatr 2003, 8, 343–347. [Google Scholar]
- Sanchez-Juan, P; Green, A; Ladogana, A; Cuadrado-Corrales, N; Saanchez-Valle, R; Mitrova, E; Stoeck, K; Sklaviadis, T; Kulczycki, J; Hess, K; et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2006, 67, 637–643. [Google Scholar]
- Green, A; Sanchez-Juan, P; Ladogana, A; Cuadrado-Corrales, N; Sánchez-Valle, R; Mitrová, E; Stoeck, K; Sklaviadis, T; Kulczycki, J; Heinemann, U; et al. CSF analysis in patients with sporadic CJD and other transmissible spongiform encephalopathies. Eur J Neurol 2007, 14, 121–124. [Google Scholar]
- Chohan, G; Pennington, C; Mackenzie, JM; Andrews, M; Everington, D; Will, RG; Knight, RS; Green, AJ. The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a 10-year review. J Neurol Neurosurg Psychiat 2010, 81, 1243–1248. [Google Scholar]
- Hsich, G; Kenney, K; Gibbs, CJ; Lee, KH; Harrington, MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996, 335, 924–930. [Google Scholar]
- Zanusso, G; Fiorini, M; Farinazzo, A; Gelati, M; Benedetti, MD; Ferrari, S; Dalla Libera, A; Capaldi, S; Monaco, HL; Rizzuto, N; et al. Phosphorylated 14-3-3zeta protein in the CSF of neuroleptic-treated patients. Neurology 2005, 64, 1618–1620. [Google Scholar]
- Bersano, A; Fiorini, M; Allaria, S; Zanusso, G; Fasoli, E; Gelati, M; Monaco, H; Squintani, G; Monaco, S; Nobile-Orazio, E. Detection of CSF 14-3-3 protein in Guillain-Barré syndrome. Neurology 2006, 67, 2211–2216. [Google Scholar]
- Pennington, C; Chohan, G; Mackenzie, J; Andrews, M; Will, R; Knight, R; Green, A. The role of cerebrospinal fluid proteins as early diagnostic markers for sporadic Creutzfeldt-Jakob disease. Neurosci Lett 2009, 455, 56–59. [Google Scholar]
- Geschwind, MD; Shu, H; Haman, A; Sejvar, JJ; Miller, BL. Rapidly progressive dementia. Ann Neurol 2008, 64, 97–108. [Google Scholar]
- Josephs, KA; Ahlskog, JE; Parisi, JE; Boeve, BF; Crum, BA; Giannini, C; Petersen, RC. Rapidly progressive neurodegenerative dementias. Arch Neurol 2009, 66, 201–207. [Google Scholar]
- Baldeiras, IE; Ribeiro, MH; Pacheco, P; Machado, A; Santana, I; Cunha, L; Oliveira, CR. Diagnostic value of CSF protein profile in a Portuguese population of sCJD patients. J Neurol 2009, 256, 1540–1550. [Google Scholar]
- Wang, GR; Gao, C; Shi, Q; Zhou, W; Chen, JM; Dong, CF; Shi, S; Wang, X; Wei, Y; Jiang, HY; et al. Elevated levels of tau protein in cerebrospinal fluid of patients with probable Creutzfeldt-Jakob disease. Am J Med Sci 2010, 340, 291–295. [Google Scholar]
- Meiner, Z; Kahana, E; Baitcher, F; Korczyn, AD; Chapman, J; Cohen, OS; Milo, R; Aharon-Perez, J; Abramsky, O; Gabizon, R; Rosenmann, H. Tau and 14-3-3 of genetic and sporadic Creutzfeldt-Jakob disease patients in Israel. J Neurol 2011, 258, 255–262. [Google Scholar]
- Otto, M; Wiltfang, J; Cepek, L; Neumann, M; Mollenhauer, B; Steinacker, P; Ciesielczyk, B; Schulz-Schaeffer, W; Kretzschmar, HA; Poser, S. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2002, 58, 192–197. [Google Scholar]
- Hansson, O; Zetterberg, H; Buchhave, P; Londos, E; Blennow, K; Minthon, L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 2006, 5, 228–234. [Google Scholar]
- Deisenhammer, F; Egg, R; Giovannoni, G; Hemmer, B; Petzold, A; Sellebjerg, F; Teunissen, C; Tumani, H. EFNS guidelines on disease-specific CSF investigations. Eur J Neurol 2009, 16, 760–770. [Google Scholar]
- Van Everbroeck, B; Quoilin, S; Boons, J; Martin, JJ; Cras, P. A prospective study of CSF markers in 250 patients with possible Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiat 2003, 74, 1210–1214. [Google Scholar]
- Gmitterová, K; Heinemann, U; Bodemer, M; Krasnianski, A; Meissner, B; Kretzschmar, HA; Zerr, I. 14-3-3 CSF levels in sporadic Creutzfeldt-Jakob disease differ across molecular subtypes. Neurobiol Aging 2009, 30, 1842–1850. [Google Scholar]
- Castellani, RJ; Colucci, M; Xie, Z; Zou, W; Li, C; Parchi, P; Capellari, S; Pastore, M; Rahbar, MH; Chen, SG; Gambetti, P. Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology 2004, 63, 436–442. [Google Scholar]
- Grossman, M; Farmer, J; Leight, S; Work, M; Moore, P; van Deerlin, V; Pratico, D; Clark, CM; Coslett, HB; Chatterjee, A; et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol 2005, 57, 721–729. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Molecular subtype | Relative% | Gender F/M | Mean age (years) | Time from disease onset to lumbar puncture (months) | Disease duration (months) |
|---|---|---|---|---|---|
| MM-1 (n = 76) | 59 | 33/43 | 70 ± 8 (53–99) | 2.5 ± 2.9 (0.5–18) | 6 ± 5 (0.5–27) |
| MV-1 (n = 7) | 6 | 5/2 | 70 ± 7 (52–76) | 4.1 ± 2.7 (1–9) | 10.5 ± 4 (6–17) |
| MM-2 (n = 4) | 3 | 2/2 | 60 ± 4 (54–62) | 5.8 ± 3 (4–11) | 11 ± 9 (4–18) |
| MV-2 (n = 20) | 16 | 4/16 | 67 ± 8 (53–81) | 7 ± 5 (2–24) | 17.4 ± 10 (7–48) |
| VV-2 (n = 20) | 16 | 11/9 | 65 ± 8 (54–79) | 4.2 ± 3.2 (1–13) | 7.3 ± 5.5 (3–25) |
| fCJD (n = 5) | - | 3/2 | 57 ± 10 (47–73) | 2 ± 1 | 5.8 ± 1.4 (4–8) |
| Subjects | 14-3-3 Protein positive | Tau protein levels (pg/mL) mean ± SD | Subjects with tau protein >4000 pg/mL |
|---|---|---|---|
| sCJD (n = 127) | 122 (96%) | 3305 ± 1096 | 79 |
| fCJD (n = 5) | 5 (100%) | 4000 | 5 |
| FTD (n = 15) | 2 (13%) | 287 ± 194 | - |
| AD (n = 7) | 2 (28%) | 898 ± 522 | - |
| CTR (n = 15) | - | 188 ± 46 | - |
| Molecular type | 14-3-3 Positivity | Tau protein > 4000 pg/mL | Tau protein 1300< >4000 | Tau protein < 1300 pg/mL |
|---|---|---|---|---|
| MM-1 (n = 76) | 75 (98.5%) | 59 (78%) | 15 (20%) (Mean: 2546 ± 870) (Range: 1443–3864) | 2 (2.6%) (230,1272) |
| MV-1 (n = 7) | 7 (100%) | 2 (28%) | 4 (57%) (Mean: 2270 ± 1120) (Range: 1478–3062) | 1 (14%) (650) |
| MM-2 (n = 4) | 2 (50%) | 1 (25%) | 1 (25%) | 2 (50%) (609,445) |
| MV-2 (n = 20) | 18 (90%) | 4 (20%) | 11 (55%) (Mean: 2211 ± 1068) (Range: 1628–3792) | 5 (25%) (Mean 892 ± 264) |
| VV-2 (n = 20) | 20 (100%) | 13 (65%) | 7 (35%) (Mean: 3433 ± 818) (Range: 1628–3897) | - |
| fCJD (n = 5) | 5 (100%) | 5 | - | - |
| AD (n = 7) | 2 (30%) | - | 1507,1663 | 529 ± 336 |
| FTD (n = 15) | 2 (13%) | - | - | 287 ± 194 |
| CTR (n = 15) | - | - | - | 188 ± 46 |
| Molecular subtype | First spinal tap | Lag between spinal taps (months) | Second spinal tap | ||
|---|---|---|---|---|---|
| 14-3-3 Protein | Tau protein (pg/mL) | 14-3-3 Protein | Tau protein (pg/mL) | ||
| MM-2 | +/− | 609 | 2 | + | 767 |
| MM-2 | + | 445 | 4 | + | 2560 |
| MV-2 | + | 507 | 15 | + | 3792 |
| MV-2 | + | 1040 | 3 | + | 1925 |
| MV-2 | + | 676 | 1 | + | 2659 |
| Subjects | Tau protein pg/mL mean ± SD | PhosphoTau 181 pg/mL mean ± SD | PhosphoTau 181/Tau |
|---|---|---|---|
| All sCJD (n = 127) | 3305 ± 1096 | 46 ± 22 | 0.014 |
| MM-1 (n = 76) | 3456 ± 972 | 42 ± 17 | 0.019 |
| MV-1 (n = 7) | 2772 ± 1394 | 48 ± 27 | 0.034 |
| MM-2 (n = 4) | 1455 ± 1701 | 28 ± 4.4 | 0.049 |
| MV-2 (n = 20) | 2211 ± 1068 | 53 ± 17 | 0.038 |
| VV-2 (n = 20) | 3433 ± 817 | 56 ± 40 | 0.016 |
| AD (n = 7) | 898 ± 522 | 105 ± 47 | 0.17 |
| FTD (n = 15) | 287 ± 194 | 37 ± 17 | 0.16 |
| CTR (n = 15) | 188 ± 46 | 29 ± 8 | 0.16 |
| Encephalitis (n = 6) | 1814 ± 163 | 35 ± 11 | 0.019 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zanusso, G.; Fiorini, M.; Ferrari, S.; Gajofatto, A.; Cagnin, A.; Galassi, A.; Richelli, S.; Monaco, S. Cerebrospinal Fluid Markers in Sporadic Creutzfeldt-Jakob Disease. Int. J. Mol. Sci. 2011, 12, 6281-6292. https://doi.org/10.3390/ijms12096281
Zanusso G, Fiorini M, Ferrari S, Gajofatto A, Cagnin A, Galassi A, Richelli S, Monaco S. Cerebrospinal Fluid Markers in Sporadic Creutzfeldt-Jakob Disease. International Journal of Molecular Sciences. 2011; 12(9):6281-6292. https://doi.org/10.3390/ijms12096281
Chicago/Turabian StyleZanusso, Gianluigi, Michele Fiorini, Sergio Ferrari, Alberto Gajofatto, Annachiara Cagnin, Andrea Galassi, Silvia Richelli, and Salvatore Monaco. 2011. "Cerebrospinal Fluid Markers in Sporadic Creutzfeldt-Jakob Disease" International Journal of Molecular Sciences 12, no. 9: 6281-6292. https://doi.org/10.3390/ijms12096281