Different Impacts of Cardiovascular Risk Factors on Oxidative Stress

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. General Characteristics of the Study Population
2.2. Oxidative Stress and Antioxidant Enzyme Activity
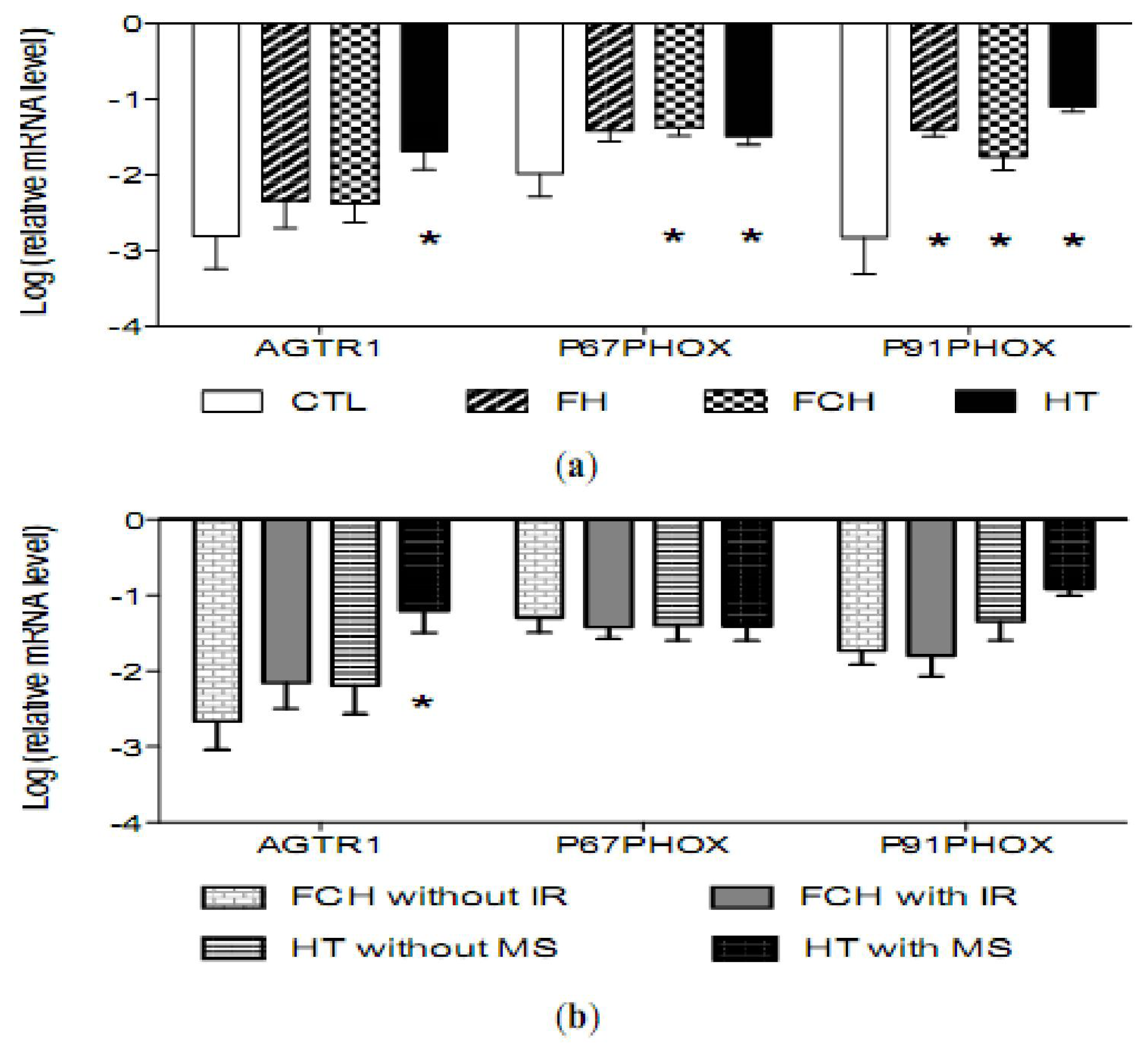
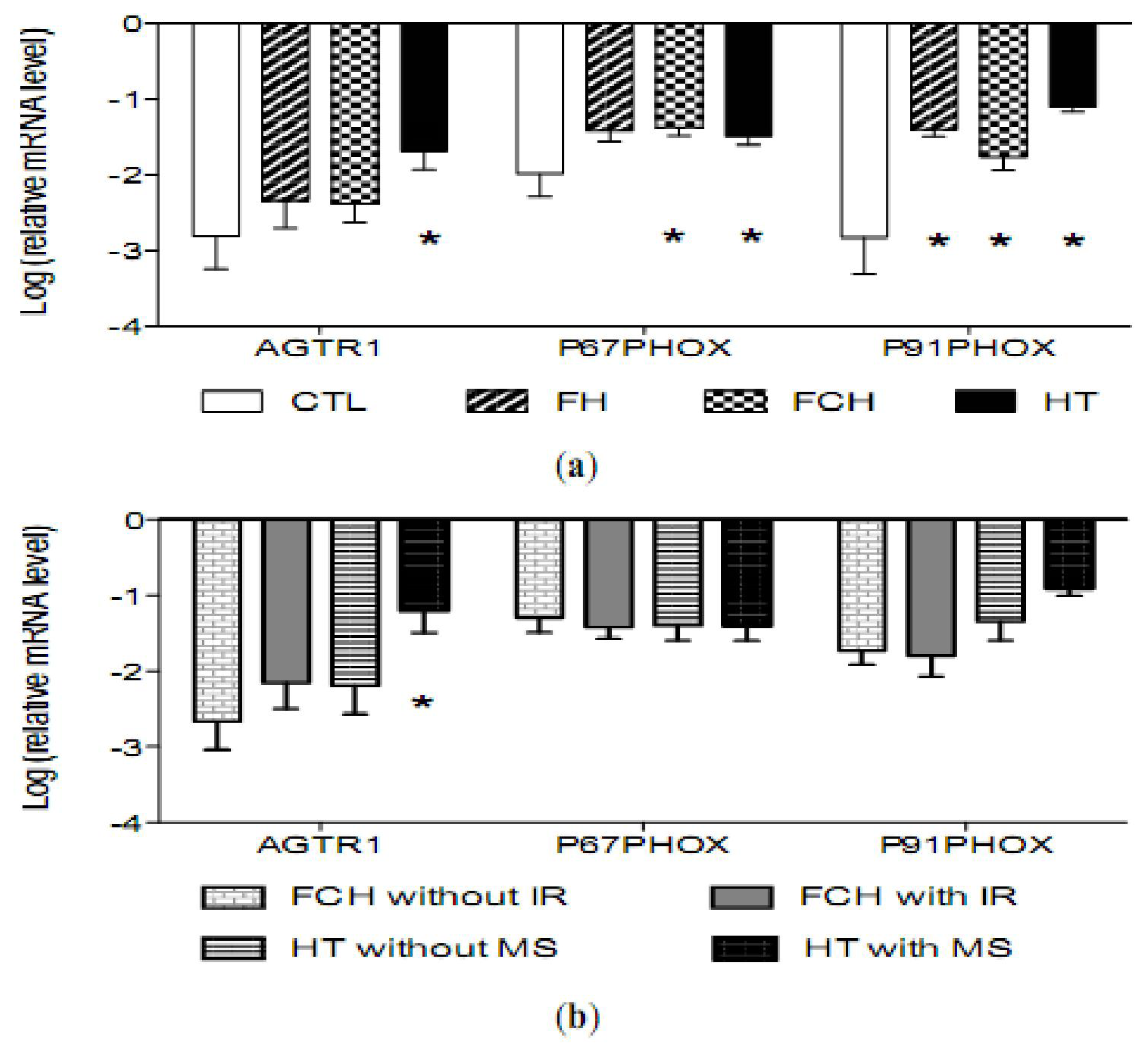
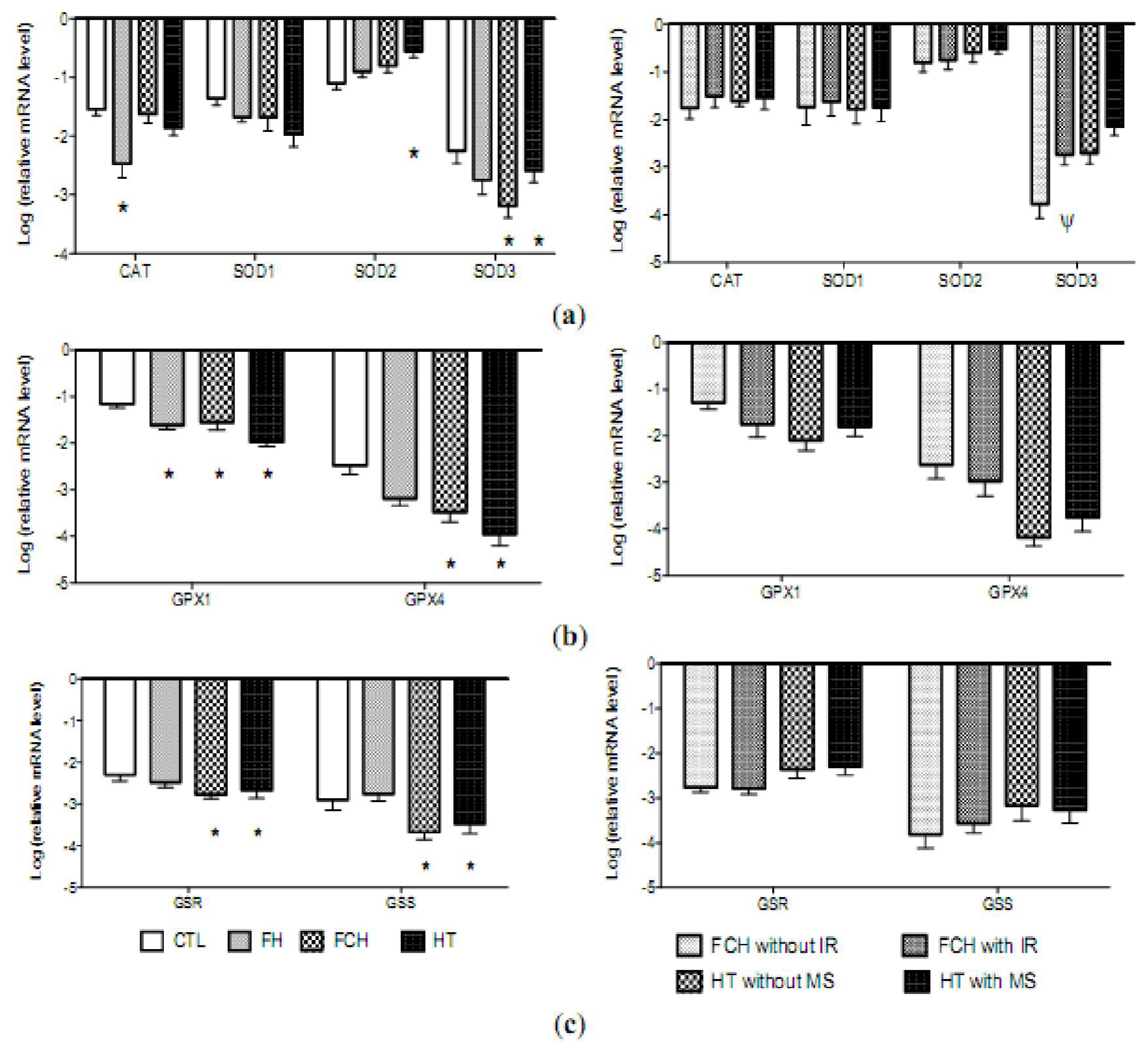
2.3. mRNA Levels of Pro-Oxidant Genes
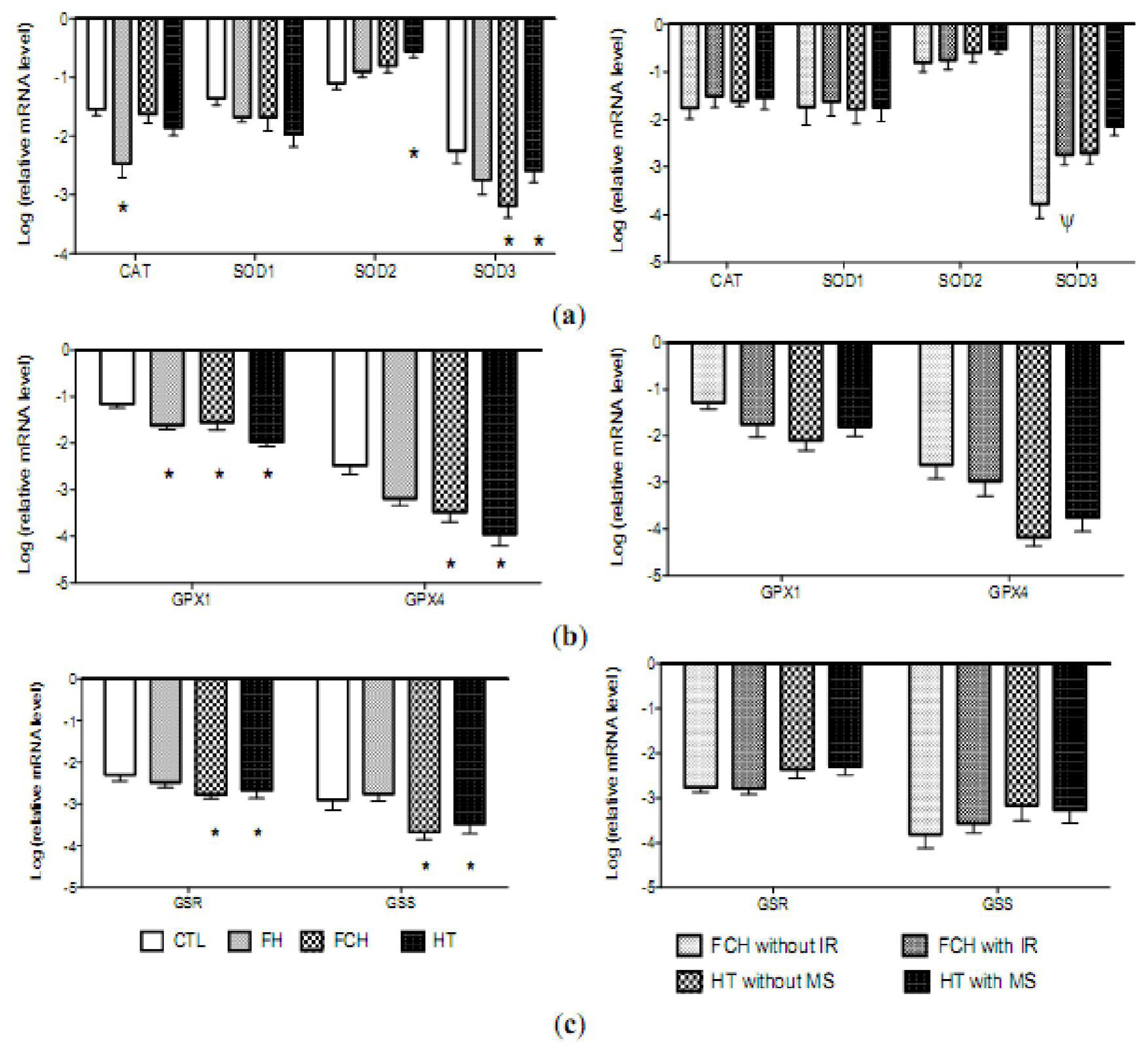
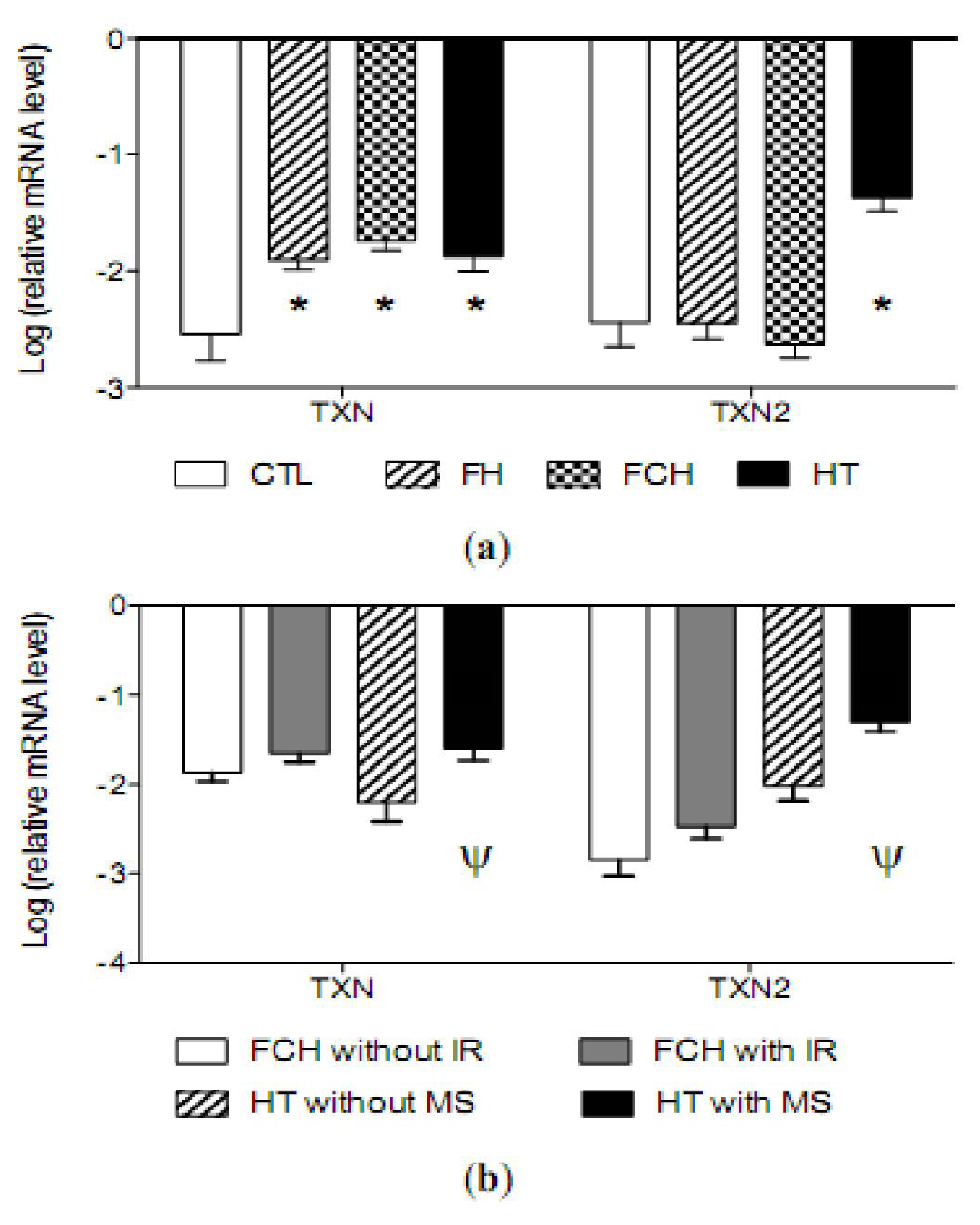
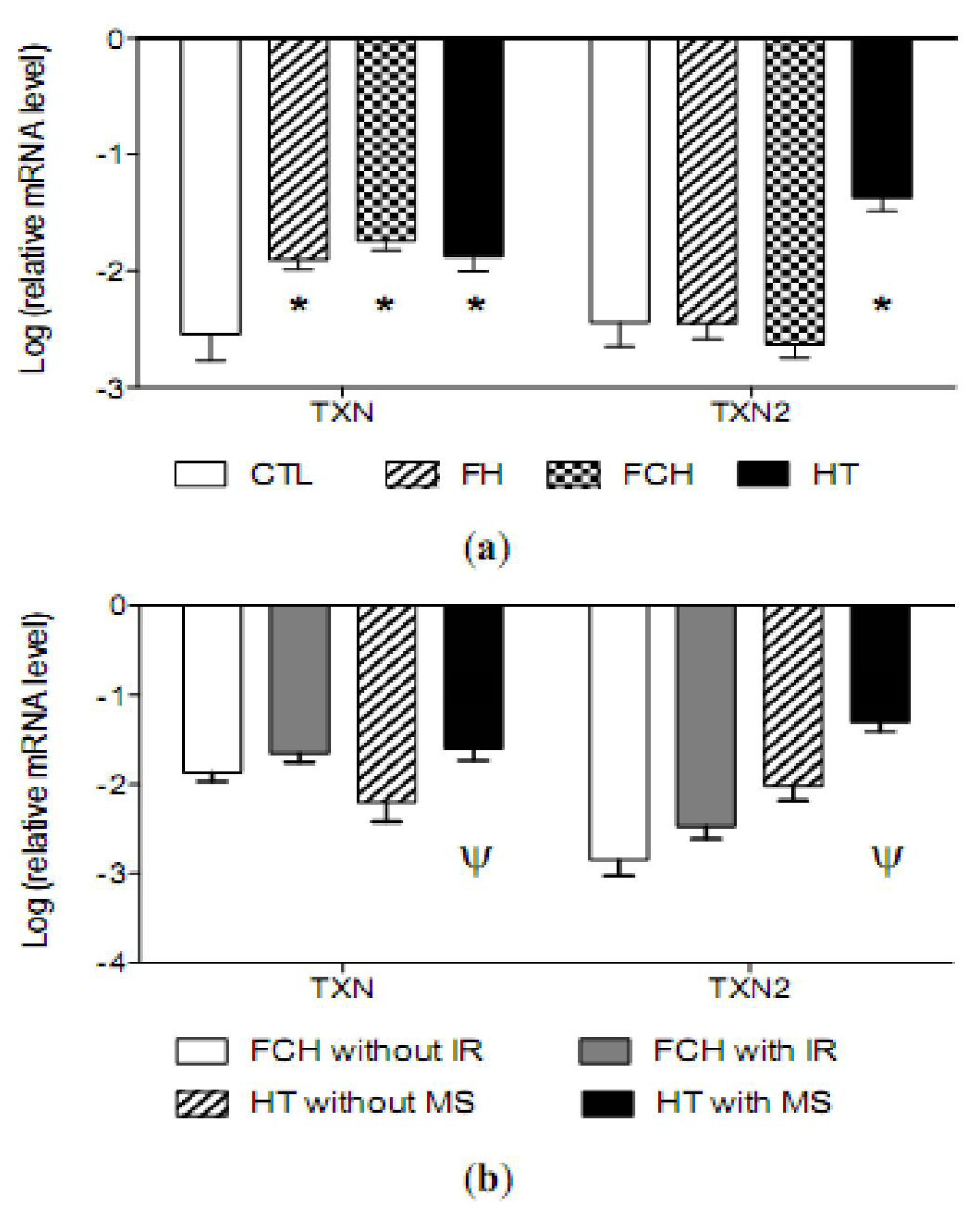
2.4. mRNA Levels of Antioxidant Enzymes
2.5. Discussion
3. Experimental Section
3.1. Selection of Study Participants
3.2. Analytical Procedures
3.3. DNA Extraction and 8-oxo-Deoxyguanosine Measurement
3.4. mRNA Extraction and Measurement
3.5. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-12-06146-s001.pdfAcknowledgments
References
- Raij, L. Nitric oxide in hypertension: Relationship with renal injury and left ventricular hypertrophy. Hypertension 1998, 31, 189–193. [Google Scholar]
- Romero, J; Reckelhoff, J. State-of-the-Art lecture. Role of angiotensin and oxidative stress in essential hypertension. Hypertension 1999, 34, 943–949. [Google Scholar]
- McIntyre, M; Bohr, D; Dominiczak, A. Endothelial function in hypertension: The role of superoxide anion. Hypertension 1999, 34, 539–545. [Google Scholar]
- Orie, NN; Zidek, W; Tepel, M. Reactive oxygen species in essential hypertension and non-insulin-dependent diabetes mellitus. Am J Hypertens 1999, 12, 1169–1174. [Google Scholar]
- Yasunari, K; Maeda, K; Nakamura, M; Yoshikawa, J. Oxidative stress in leukocytes is a possible link between blood pressure, blood glucose, and C-reacting protein. Hypertension 2002, 39, 777–780. [Google Scholar]
- Martinez-Hervas, S; Fandos, M; Real, JT; Espinosa, O; Chaves, FJ; Saez, GT; Salvador, A; Cerdá, C; Carmena, R; Ascaso, JF. Insulin resistance and oxidative stress in familial combined hyperlipidemia. Atherosclerosis 2008, 199, 384–389. [Google Scholar]
- Chaves, FJ; Mansego, ML; Blesa, S; Gonzalez-Albert, V; Jimenez, J; Tormos, MC; Espinosa, O; Giner, V; Iradi, A; Saez, G; Redon, J. Inadequate cytoplasmic antioxidant enzymes response contributes to the oxidative stress in human hypertension. Am J Hypertens 2007, 20, 62–69. [Google Scholar]
- Stephens, JW; Gable, DR; Hurel, SJ; Miller, GJ; Cooper, JA; Humphries, SE. Increased plasma markers of oxidative stress are associated with coronary heart disease in males with diabetes mellitus and with 10-year risk in a prospective sample of males. Clin Chem 2006, 52, 446–452. [Google Scholar]
- Redon, J; Oliva, MR; Tormos, C; Giner, V; Chaves, J; Iradi, A; Sáez, GT. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar]
- Nakazono, K; Watanabe, N; Matsuno, K; Sasaki, J; Sato, T; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc Natl Acad Sci USA 1991, 88, 10045–10048. [Google Scholar]
- Touyz, RM; Tabet, F; Schiffrin, EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol 2003, 30, 860–866. [Google Scholar]
- Papaharalambus, CA; Griendling, KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med 2007, 17, 48–54. [Google Scholar]
- Heistad, DD. Oxidative stress and vascular disease: 2005 Duff lecture. Arterioscler Thromb Vasc Biol 2006, 26, 689–695. [Google Scholar]
- Tamer, L; Sucu, N; Polat, G; Ercan, B; Aytacoglu, B; Yucebilgic, G; Unlü, A; Dikmengil, M; Atik, U. Decreased serum total antioxidant status and erythrocyte-reduced glutathione levels are associated with increased serum malondialdehyde in atherosclerotic patients. Arch Med Res 2002, 33, 257–260. [Google Scholar]
- Espinosa, O; Jimenez-Almazan, J; Chaves, FJ; Tormos, MC; Clapes, S; Iradi, A; Salvador, A; Fandos, M; Redón, J; Sáez, GT. Urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG), a reliable oxidative stress marker in hypertension. Free Radic Res 2007, 41, 546–554. [Google Scholar]
- Ferri, J; Martinez-Hervas, S; Espinosa, O; Fandos, M; Pedro, T; Real, JT; Chaves, FJ; Cerdá, C; Sáez, G; Ascaso, JF. [8-oxo-7,8-dihydro-2′-deoxyguanosine as a marker of DNA oxidative stress in individuals with combined familiar hyperlipidemia]. Med Clin (Barc) 2008, 131, 1–4. [Google Scholar]
- Abbas, AM; Elsamanoudy, AZ. Effects of 17β-estradiol and antioxidant administration on oxidative stress and insulin resistance in ovariectomized rats. Can J Physiol Pharmacol 2011, 89, 497–504. [Google Scholar]
- Bloch-Damti, A; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal 2005, 7, 1553–1567. [Google Scholar]
- West, IC. Radicals and oxidative stress in diabetes. Diabet Med 2000, 17, 171–180. [Google Scholar]
- Packer, L. Packer, L, Traber, MG, Xin, W, Eds.; Cell Regulation by Thiol Antioxidants: From Glutathione to Lipoate to Anethole Dithiolethione. In Proceedings of the International Symposium on Natural Antioxidants Molecular Mechanisms and Health Effects; AOCS Press: Champaing, IL, USA, 1995; pp. 223–235. [Google Scholar]
- Cooke, MS; Evans, MD; Herbert, KE; Lunec, J. Urinary 8-oxo-2′-deoxyguanosine—Source, significance and supplements. Free Radic Res 2000, 32, 381–397. [Google Scholar]
- Griffiths, HR; Moller, L; Bartosz, G; Bast, A; Bertoni-Freddari, C; Collins, A; Cooke, M; Coolen, S; Haenen, G; Hoberg, AM; Loft, S; Lunec, J; Olinski, R; Parry, J; Pompella, A; Poulsen, H; Verhagen, H; Astley, SB. Biomarkers. Mol Aspects Med 2002, 23, 101–208. [Google Scholar]
- Nakabeppu, Y; Tsuchimoto, D; Ichinoe, A; Ohno, M; Ide, Y; Hirano, S; Yoshimura, D; Tominaga, Y; Furuichi, M; Sakumi, K. Biological significance of the defense mechanisms against oxidative damage in nucleic acids caused by reactive oxygen species: From mitochondria to nuclei. Ann N Y Acad Sci 2004, 1011, 101–111. [Google Scholar]
- Salo, DC; Pacifici, RE; Lin, SW; Giulivi, C; Davies, KJ. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. J Biol Chem 1990, 265, 11919–11927. [Google Scholar]
- Davies, KJ. Protein damage and degradation by oxygen radicals. I. general aspects. J Biol Chem 1987, 262, 9895–9901. [Google Scholar]
- Pigeolet, E; Corbisier, P; Houbion, A; Lambert, D; Michiels, C; Raes, M; Zachary, MD; Remacle, J. Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech Ageing Dev 1990, 51, 283–297. [Google Scholar]
- Pigeolet, E; Remacle, J. Susceptibility of glutathione peroxidase to proteolysis after oxidative alteration by peroxides and hydroxyl radicals. Free Radic Biol Med 1991, 11, 191–195. [Google Scholar]
- Dalton, TP; Shertzer, HG; Puga, A. Regulation of gene expression by reactive oxygen. Annu Rev Pharmacol Toxicol 1999, 39, 67–101. [Google Scholar]
- Morel, Y; Barouki, R. Repression of gene expression by oxidative stress. Biochem J 1999, 342, 481–496. [Google Scholar]
- Meilhac, O; Zhou, M; Santanam, N; Parthasarathy, S. Lipid peroxides induce expression of catalase in cultured vascular cells. J Lipid Res 2000, 41, 1205–1213. [Google Scholar]
- Zhou, L; Xiang, W; Potts, J; Floyd, M; Sharan, C; Yang, H; Ross, J; Nyanda, AM; Guo, Z. Reduction in extracellular superoxide dismutase activity in African-American patients with hypertension. Free Radic Biol Med 2006, 41, 1384–1391. [Google Scholar]
- Di Massimo, C; Lo Presti, R; Corbacelli, C; Pompei, A; Scarpelli, P; de Amicis, D; Caimi, G; Tozzi Ciancarelli, MG. Impairment of plasma nitric oxide availability in senescent healthy individuals: Apparent involvement of extracellular superoxide dismutase activity. Clin Hemorheol Microcirc 2006, 35, 231–237. [Google Scholar]
- Horiuchi, M; Tsutsui, M; Tasaki, H; Morishita, T; Suda, O; Nakata, S; Nihei, S; Miyamoto, M; Kouzuma, R; Okazaki, M; Yanagihara, N; Adachi, T; Nakashima, Y. Upregulation of vascular extracellular superoxide dismutase in patients with acute coronary syndromes. Arterioscler Thromb Vasc Biol 2004, 24, 106–111. [Google Scholar]
- Azevedo-Martins, AK; Curi, R. Fatty acids decrease catalase activity in human leukaemia cell lines. Cell Biochem Funct 2008, 26, 87–94. [Google Scholar]
- Drummond, GR; Cai, H; Davis, ME; Ramasamy, S; Harrison, DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res 2000, 86, 347–354. [Google Scholar]
- Li, WG; Miller, FJ, Jr; Zhang, HJ; Spitz, DR; Oberley, LW; Weintraub, NL. H(2)O(2)-induced O(2) production by a non-phagocytic NAD(P)H oxidase causes oxidant injury. J Biol Chem 2001, 276, 29251–29256. [Google Scholar]
- Wu, L; Juurlink, BH. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension 2002, 39, 809–814. [Google Scholar]
- Dobrian, AD; Davies, MJ; Schriver, SD; Lauterio, TJ; Prewitt, RL. Oxidative stress in a rat model of obesity-induced hypertension. Hypertension 2001, 37, 554–560. [Google Scholar]
- Reckelhoff, JF; Romero, JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol 2003, 284, R893–R912. [Google Scholar]
- Martino, F; Pignatelli, P; Martino, E; Morrone, F; Carnevale, R; di Santo, S; Buchetti, B; Loffredo, L; Violi, F. Early increase of oxidative stress and soluble CD40L in children with hypercholesterolemia. J Am Coll Cardiol 2007, 49, 1974–1981. [Google Scholar]
- Real, JT; Martínez-Hervás, S; Tormos, MC; Domenech, E; Pallardó, FV; Sáez-Tormo, G; Redon, J; Carmena, R; Chaves, FJ; Ascaso, JF; García-García, AB. Increased oxidative stress levels and normal antioxidant enzyme activity in circulating mononuclear cells from patients of familial hypercholesterolemia. Metabolism 2010, 59, 293–298. [Google Scholar]
- Martinez-Hervas, S; Real, JT; Ivorra, C; Priego, A; Chaves, FJ; Pallardo, FV; Viña, JR; Redon, J; Carmena, R; Ascaso, JF. Increased plasma xanthine oxidase activity is related to nuclear factor kappa beta activation and inflammatory markers in familial combined hyperlipidemia. Nutr Metab Cardiovasc Dis 2010, 20, 734–739. [Google Scholar]
- McNally, JS; Davis, ME; Giddens, DP; Saha, A; Hwang, J; Dikalov, S; Jo, H; Harrison, DG. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol 2003, 285, H2290–2297. [Google Scholar]
- Cabelli, DE; Allen, D; Bielski, BH; Holcman, J. The interaction between Cu(I) superoxide dismutase and hydrogen peroxide. J Biol Chem 1989, 264, 9967–9971. [Google Scholar]
- Grune, T; Blasig, IE; Sitte, N; Roloff, B; Haseloff, R; Davies, KJ. Peroxynitrite increases the degradation of aconitase and other cellular proteins by proteasome. J Biol Chem 1998, 273, 10857–10862. [Google Scholar]
- Kunsch, C; Medford, RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 1999, 85, 753–766. [Google Scholar]
- Karamouzis, I; Pervanidou, P; Berardelli, R; Iliadis, S; Papassotiriou, I; Karamouzis, M; Chrousos, GP; Kanaka-Gantenbein, C. Enhanced oxidative stress and platelet activation combined with reduced antioxidant capacity in obese prepubertal and adolescent girls with full or partial metabolic syndrome. Horm Metab Res 2011, 43, 607–613. [Google Scholar]
- Chaves, FJ; Corella, D; Blesa, S; Mansego, ML; Marin, P; Portoles, O; Sorlí, JV; González-Albert, V; Tormos, MC; García-García, AB; Sáez, G; Redon, J. Xanthine oxidoreductase polymorphisms: Influence in blood pressure and oxidative stress levels. Pharmacogenet Genomics 2007, 17, 589–596. [Google Scholar]
- Vaziri, ND; Dicus, M; Ho, ND; Boroujerdi-Rad, L; Sindhu, RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int 2003, 63, 179–185. [Google Scholar]
- Miller, JD; Weiss, RM; Serrano, KM; Brooks, RM; Berry, CJ, II; Zimmerman, K; Young, SG; Heistad, DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation 2009, 119, 2693–2701. [Google Scholar]
- Ruiz, MC; Moreno, JM; Ruiz, N; Vargas, F; Asensio, C; Osuna, A. Effect of statin treatment on oxidative stress and renal function in renal transplantation. Transplant Proc 2006, 38, 2431–2433. [Google Scholar]
- Rosenson, RS. Fenofibrate: Treatment of hyperlipidemia and beyond. Expert Rev Cardiovasc Ther 2008, 6, 1319–1330. [Google Scholar]
- von Haehling, S; Anker, SD; Bassenge, E. Statins and the role of nitric oxide in chronic heart failure. Heart Fail Rev 2003, 8, 99–106. [Google Scholar]
- Rudich, A; Kozlovsky, N; Potashnik, R; Bashan, N. Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. Am J Physiol 1997, 272, E935–E940. [Google Scholar]
- Ceriello, A. Oxidative stress and glycemic regulation. Metabolism 2000, 49, 27–29. [Google Scholar]
- Yaworsky, K; Somwar, R; Klip, A. Packer, L, Rösen, P, Tritschler, H, King, G, Eds.; Interrelationship between Oxidative Stress and Insulin Resistance. In Antioxidants in Diabetes Management; Marcel Dekker: New York, NY, USA, 2000; pp. 275–302. [Google Scholar]
- Maddux, BA; See, W; Lawrence, JC, Jr; Goldfine, AL; Goldfine, ID; Evans, JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes 2001, 50, 404–410. [Google Scholar]
- Rudich, A; Tirosh, A; Potashnik, R; Khamaisi, M; Bashan, N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia 1999, 42, 949–957. [Google Scholar]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal 2000, 2, 811–820. [Google Scholar]
- Park, YS; Fujiwara, N; Koh, YH; Miyamoto, Y; Suzuki, K; Honke, K; Taniguchi, N. Induction of thioredoxin reductase gene expression by peroxynitrite in human umbilical vein endothelial cells. Biol Chem 2002, 383, 683–691. [Google Scholar]
- Chiueh, CC; Andoh, T; Chock, PB. Induction of thioredoxin and mitochondrial survival proteins mediates preconditioning-induced cardioprotection and neuroprotection. Ann N Y Acad Sci 2005, 1042, 403–418. [Google Scholar]
- Zelko, IN; Mariani, TJ; Folz, RJ. Superoxide dismutase multigene family: A comparison for the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution and expression. Free Radic Biol Med 2002, 33, 337–349. [Google Scholar]
- Third Report of the National Cholesterol Education Program (NCEP). Expert Panel on Detection Evaluation Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 106, 3143–3421.
- Chaves, FJ; Real, JT; Garcia-Garcia, AB; Civera, M; Armengod, ME; Ascaso, JF; Carmena, R. Genetic diagnosis of familial hypercholesterolemia in a South European outbreed population: Influence of low-density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL, and high-density lipoprotein cholesterol. J Clin Endocrinol Metab 2001, 86, 4926–4932. [Google Scholar]
- Castro Cabezas, M; de Bruin, TW; Erkelens, DW. Familial combined hyperlipidaemia: 1973–1991. Neth J Med 1992, 40, 83–95. [Google Scholar]
- Ascaso, JF; Romero, P; Real, JT; Priego, A; Valdecabres, C; Carmena, R. Insulin resistance quantification by fasting insulin plasma values and HOMA index in a non-diabetic population. Med Clin (Barc) 2001, 117, 530–533. [Google Scholar]
- O’Brien, E; Asmar, R; Beilin, L; Imai, Y; Mallion, J; Mancia, G; Mengden, T; Myers, M; Padfield, P; Palatini, P; Parati, G; Pickering, T; Redon, J; Staessen, J; Stergiou, G; Verdecchia, P. European Society of Hypertension Working Group on Blood Pressure Monitoring. European Society of Hypertension recommendations for conventional, ambulatory and home blood pressure measurement. J Hypertens 2003, 21, 821–848. [Google Scholar]
- Oltra, AM; Carbonell, F; Tormos, C; Irati, A; Sáez, GT. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic Biol Med 2001, 30, 1286–1292. [Google Scholar]
- Navarro, J; Obrador, E; Pellicer, JA; Aseni, M; Viña, J; Estrela, JM. Blood glutathione as an index of radiation-induced oxidative stress in mice and humans. Free Radic Biol Med 1997, 22, 1203–1209. [Google Scholar]
- Wong, SH; Knight, JA; Hopfer, SM; Zaharia, O; Leach, CN, Jr; Sunderman, FW, Jr. Lipoperoxides in plasma as measured by liquid-chromatographic separation of malondialdehyde-thiobarbituric acid adduct. Clin Chem 1987, 33, 214–220. [Google Scholar]
- Muñiz, P; Valls, V; Perez-Broseta, C; Iradi, A; Climent, JV; Oliva, MR; Sáez, GT. The role of 8-hydroxy-2′-deoxyguanosine in rifamycin-induced DNA damage. Free Radic Biol Med 1995, 18, 747–755. [Google Scholar]
- Nebot, C; Moutet, M; Huet, P; Xu, JZ; Yadan, JC; Chaudiere, J. Spectrophotometric assay of superoxide dismutase activity based on the activated autoxidation of a tetracyclic catechol. Anal Biochem 1993, 214, 442–451. [Google Scholar]
- Aebi, H. Catalase. in vitro Methods Enzymol 1984, 105, 121–126. [Google Scholar]
- Paglia, DE; Valentine, WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 1967, 70, 158–169. [Google Scholar]
- Chomczynski, P; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat Protoc 2006, 1, 581–585. [Google Scholar]
- Rozen, S; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000, 132, 365–386. [Google Scholar]
- Vandesompele, J; de Preter, K; Pattyn, F; Poppe, B; van Roy, N; de Paepe, A; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002, 3, RESEARCH0034. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Variables | Controls (n = 20) | FH (n = 17) | FCH (n = 30) | HT (n = 43) |
|---|---|---|---|---|
| Age (year) | 39.9(14.3) | 42.6(13.3) | 45.6(8.5) | 46.3(9.6) |
| Gender (M/F) | 12/8 | 3/14 | 20/10 + | 26/17 + |
| Body mass index (kg/m2) | 25.9(3.0) | 25.5(3.6) | 26.9(3.9) | 30.6(5.1) *,+,Ω |
| Office SBP (mmHg) | 121.6(11.0) | 120.2(10.7) | 139.5(5.9) *,+ | 157.8(21.3) *,+, Ω |
| Office DBP (mmHg) | 76.8(5.8) | 73.3(9.1) | 87.3(4.0) *,+ | 99.7(12.7) *,+, Ω |
| 24-h SBP (mmHg) | - | - | - | 142.0(16.9) |
| 24-h DBP (mmHg) | - | - | - | 90.5(10.5) |
| Baseline glucose (mg/dL) | 89.7(6.4) | 88.4(8.9) | 101.3(16.4) *,+ | 104.3(20.4) *,+ |
| Total-cholesterol (mg/dL) | 189.7(35.9) | 304.8(65.7) * | 271.9(57.5) * | 208.5(33.7) +, Ω |
| HDL-cholesterol (mg/dL) | 47.6(9.7) | 58.1(13.4) * | 40.1(9.9) *,+ | 44.8(8.9) +, Ω. |
| Triglycerides (mg/dL) | 102.2(48.6) | 127.5(51.9) | 294.4(175.1) *,+ | 146.2(71.6) Ω |
| Variables | Controls (n = 20) | FH (n = 17) | FCH (n = 30) | HT (n = 43) |
|---|---|---|---|---|
| GSH | 22.9 ± 0.9 | 18.3 ± 1.1 * | 18.7 ± 0.6 * | 15.7 ± 0.6 *, Ω |
| GSSG | 0.28 ± 0.04 | 0.43 ± 0.04 * | 0.32 ± 0.02 + | 1.13 ± 0.05 *,+, Ω |
| GSSG/GSH | 1.3 ± 0.2 | 2.5 ± 0.3 * | 1.8 ± 0.2 * | 7.8 ± 0.4 +, Ω |
| MDA | 0.23 ± 0.02 | 0.30 ± 0.04 | 0.26 ± 0.02 | 0.92 ± 0.29 *,+, Ω |
| 8-oxo-dG | 4.8 ± 0.3 | 5.5 ± 0.3 | 5.7 ± 0.2 * | 6.8 ± 0.2 *,+, Ω |
| Catalase | 217.0 ± 10.1 | 140.2 ± 11.4 * | 173.3 ± 10.3 * | 110.9 ± 4.5 *,+, Ω |
| GPX1 | 58.5 ± 1.8 | 54.1 ± 2.1 | 51.7 ± 1.2 * | 32.8 ± 0.9 *,+, Ω |
| SOD | 6.9 ± 0.6 | 5.1 ± 0.7 | 5.6 ± 0.7 | 3.8 ± 0.3 *,+, Ω |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mansego, M.L.; Redon, J.; Martinez-Hervas, S.; Real, J.T.; Martinez, F.; Blesa, S.; Gonzalez-Albert, V.; Saez, G.T.; Carmena, R.; Chaves, F.J. Different Impacts of Cardiovascular Risk Factors on Oxidative Stress. Int. J. Mol. Sci. 2011, 12, 6146-6163. https://doi.org/10.3390/ijms12096146
Mansego ML, Redon J, Martinez-Hervas S, Real JT, Martinez F, Blesa S, Gonzalez-Albert V, Saez GT, Carmena R, Chaves FJ. Different Impacts of Cardiovascular Risk Factors on Oxidative Stress. International Journal of Molecular Sciences. 2011; 12(9):6146-6163. https://doi.org/10.3390/ijms12096146
Chicago/Turabian StyleMansego, Maria L., Josep Redon, Sergio Martinez-Hervas, Jose T. Real, Fernando Martinez, Sebastian Blesa, Veronica Gonzalez-Albert, Guillermo T. Saez, Rafael Carmena, and Felipe J. Chaves. 2011. "Different Impacts of Cardiovascular Risk Factors on Oxidative Stress" International Journal of Molecular Sciences 12, no. 9: 6146-6163. https://doi.org/10.3390/ijms12096146