Investigation of the Structure Requirement for 5-HT6 Binding Affinity of Arylsulfonyl Derivatives: A Computational Study

Abstract
:1. Introduction
2. Results and Discussion
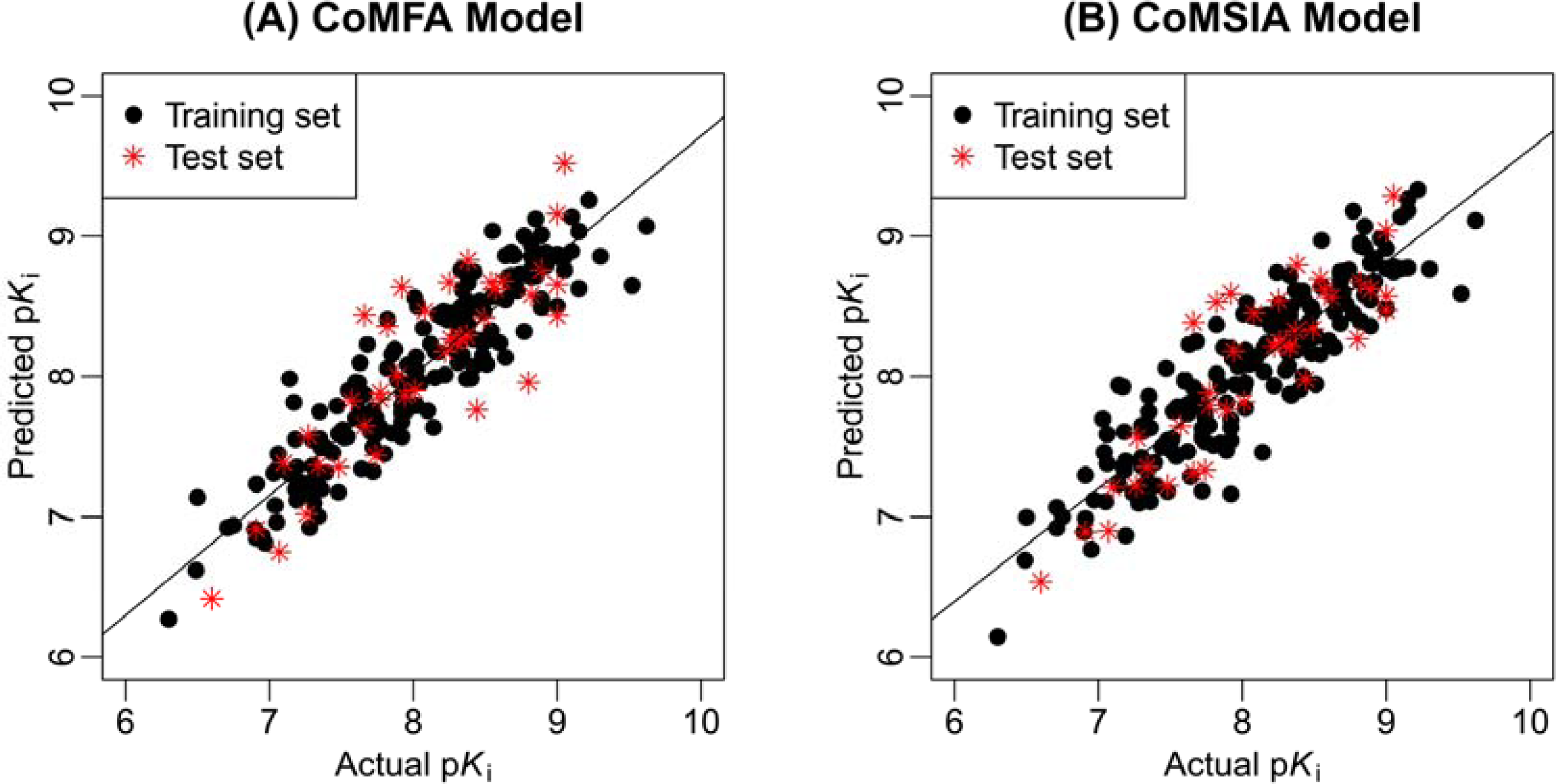
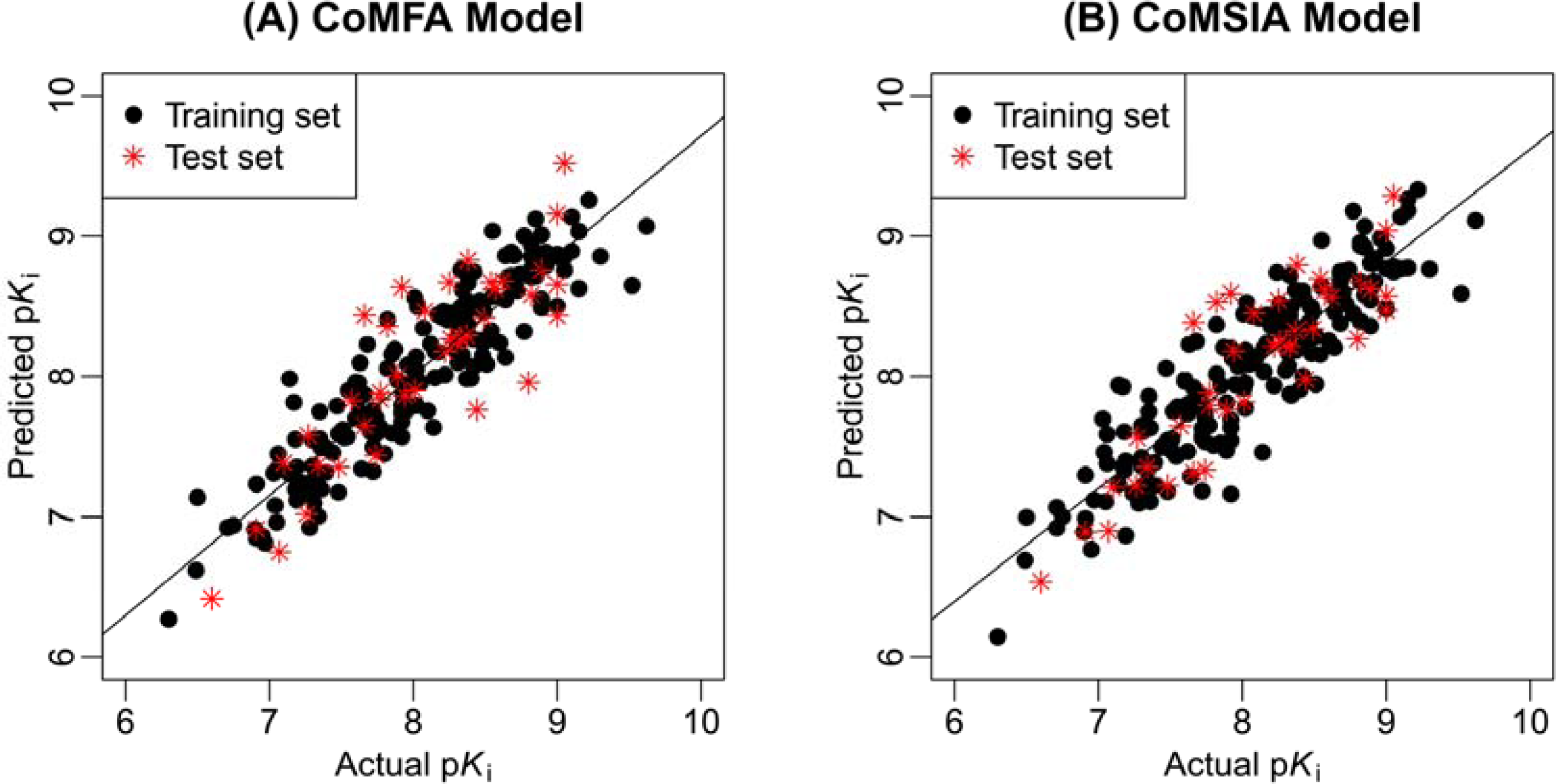
2.1. 3D-QSAR Statistical Results
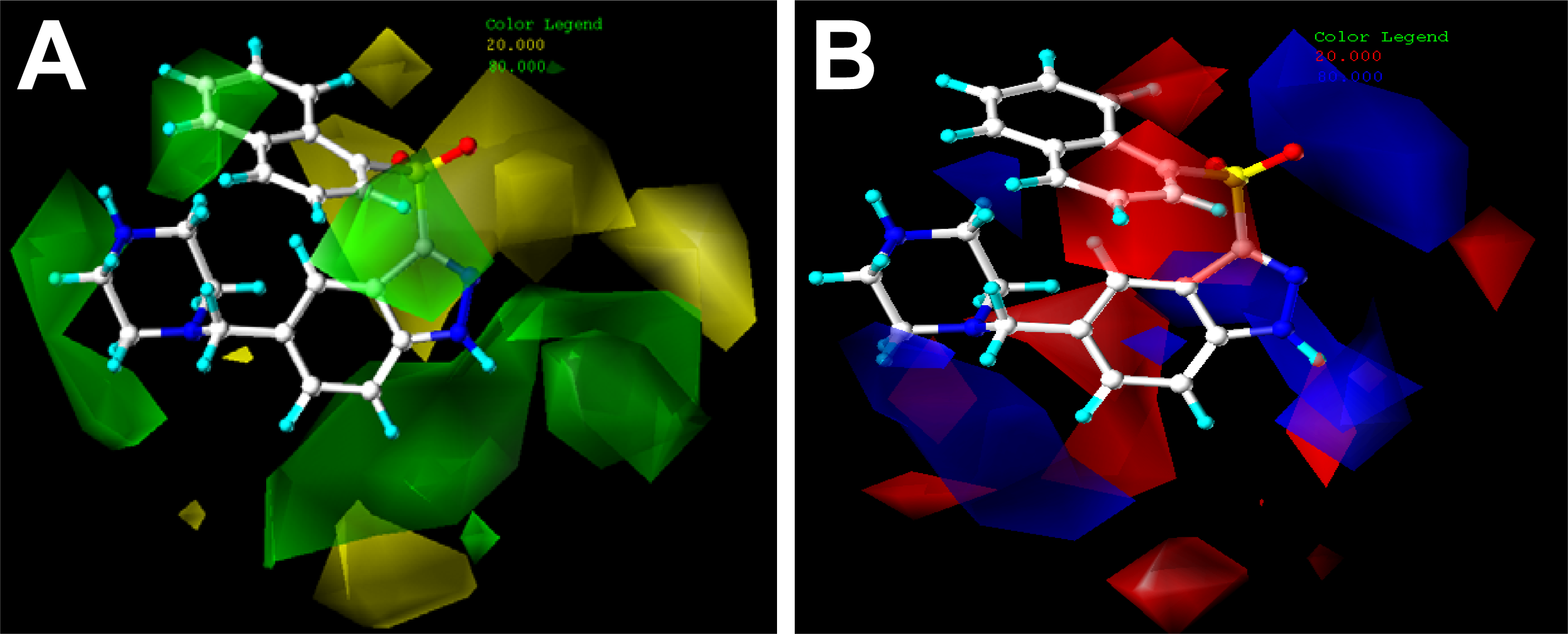
2.2. Contour Maps
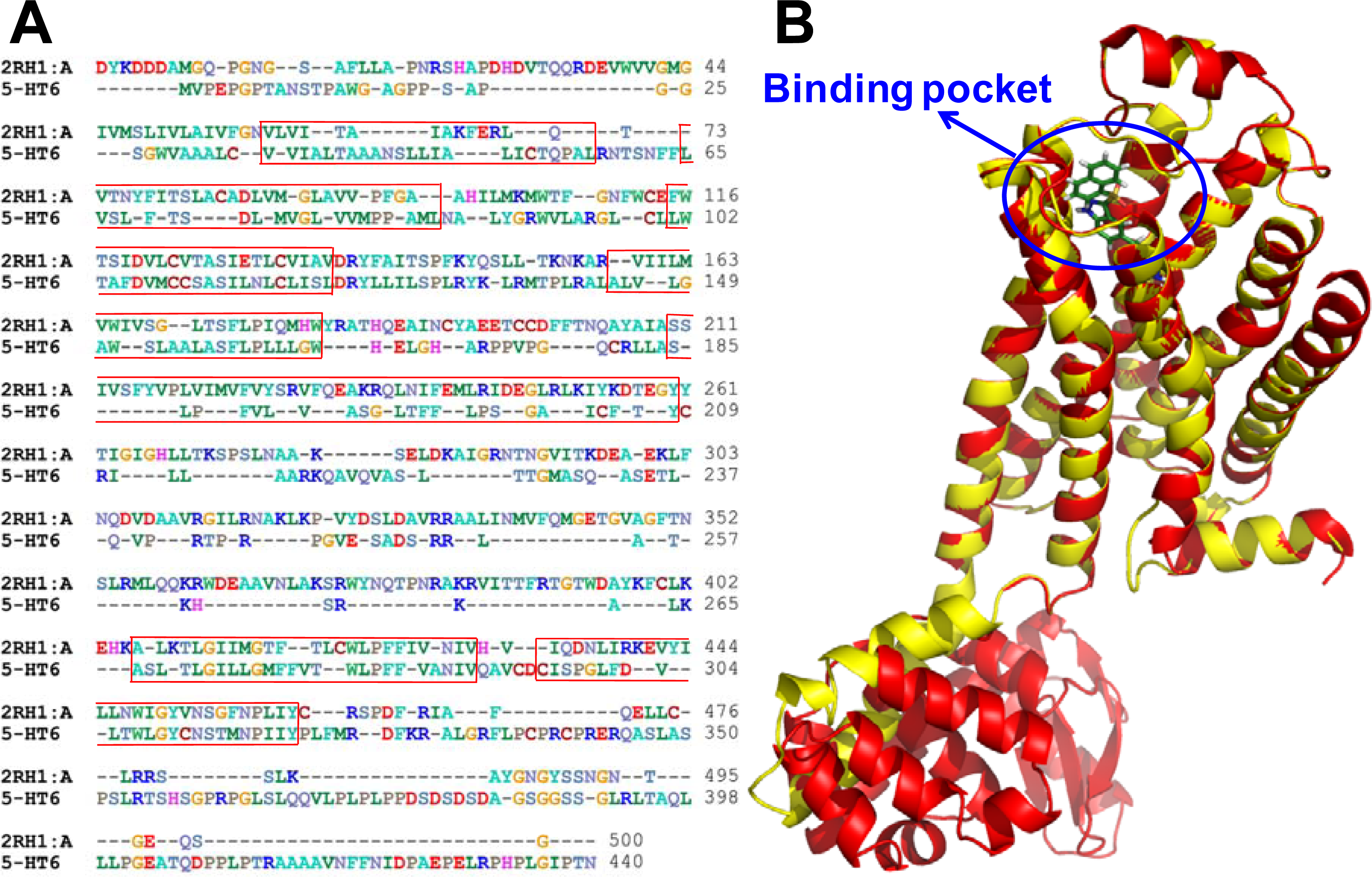
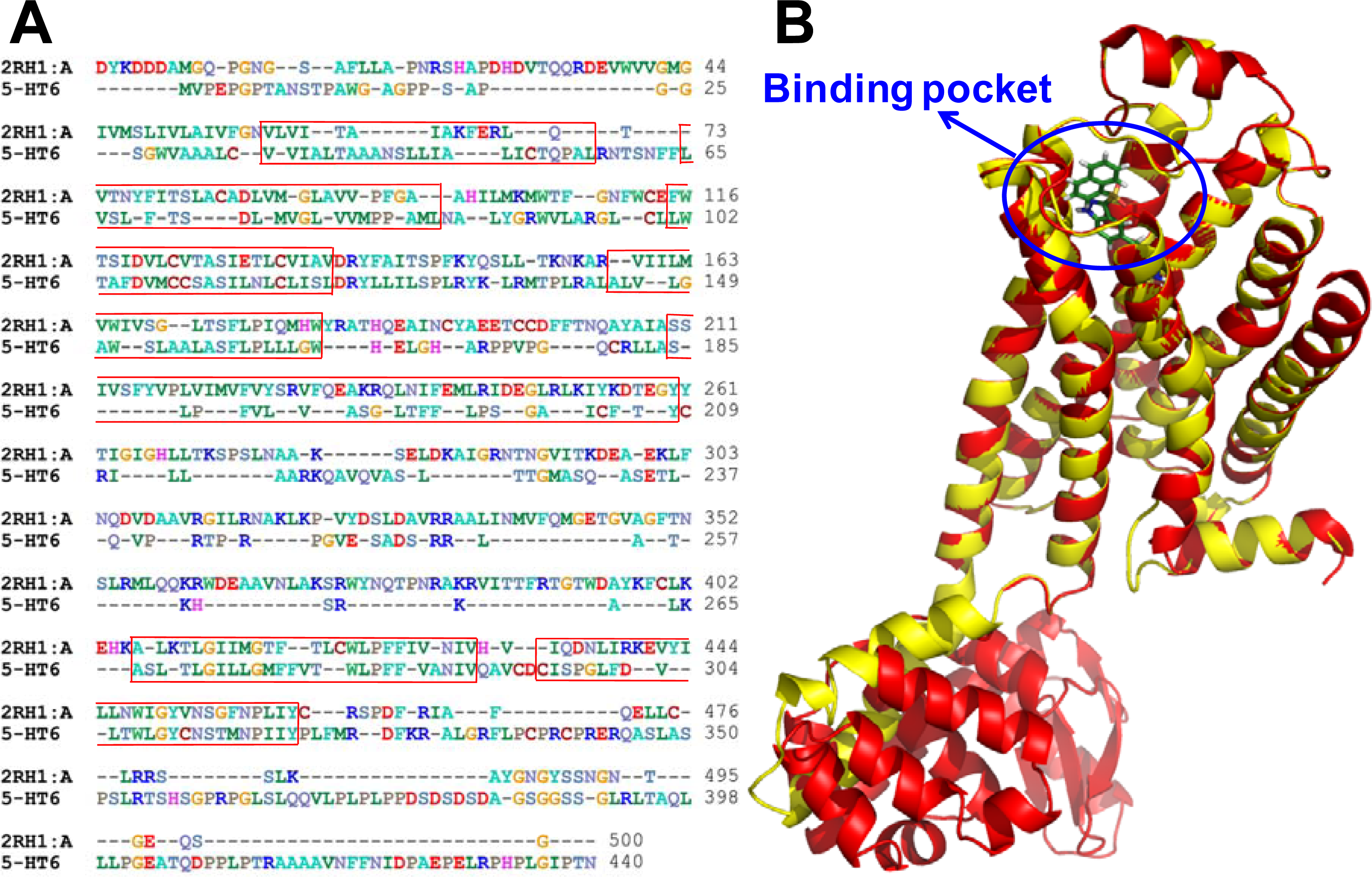
2.3. Homology Modeling Results
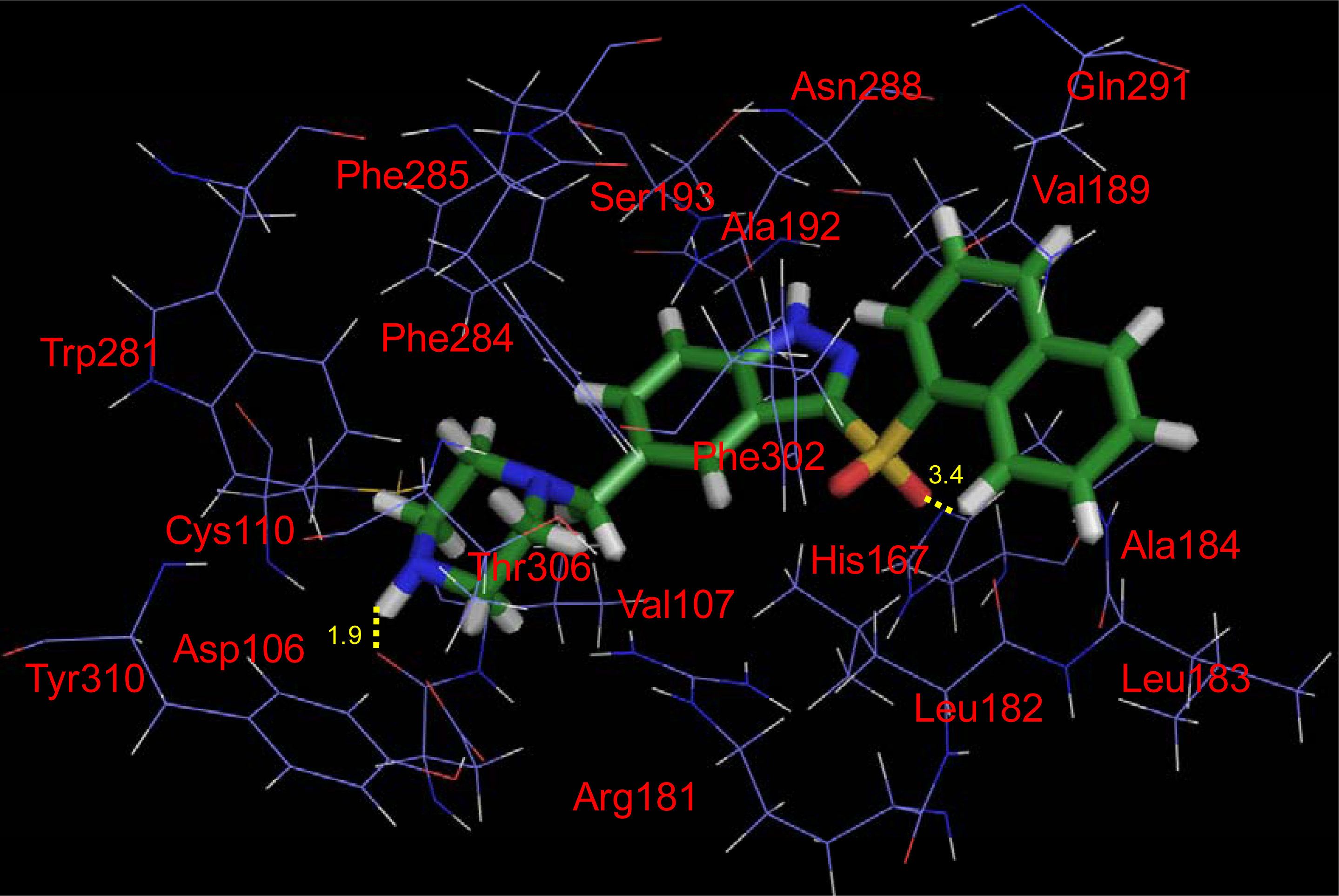
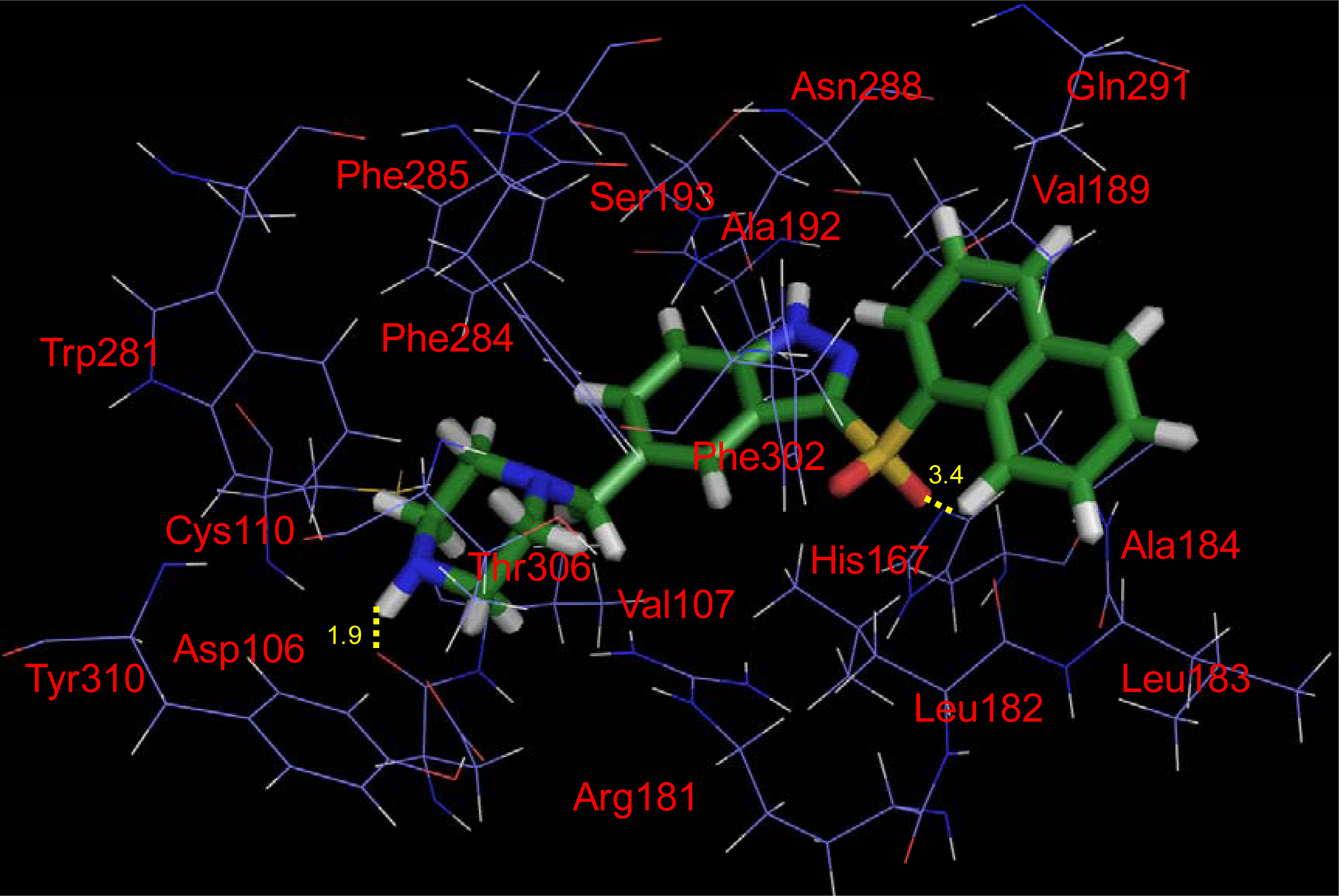
2.4. Docking Results
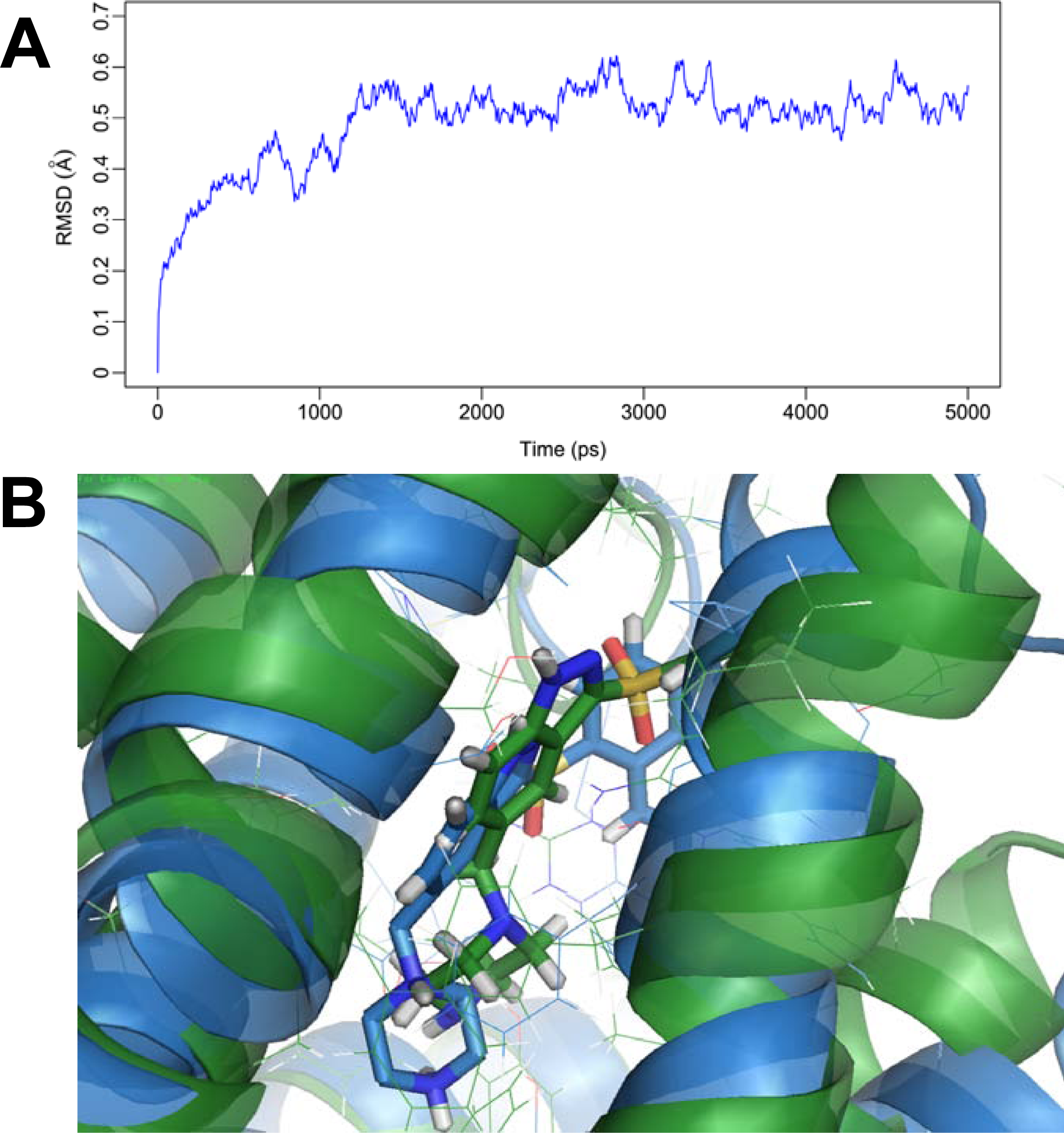
2.5. MD Simulations
3. Material and Experimental Methods
3.1. Dataset and Biological Activity
3.2. Molecular Alignment
3.3. CoMFA and CoMSIA Field Calculation
3.4. 3D-QSAR Model Calculation and Validation
3.5. Homology Modeling
3.6. Molecular Docking
3.7. Molecular Dynamics Simulations
4. Conclusions
Supplementary Information
ijms-12-05011-s001.pdfAcknowledgments
References
- Pullagurla, M; Bondareva, T; Young, R; Glennon, RA. Modulation of the stimulus effects of (+) amphetamine by the 5-HT6 antagonist MS-245. Pharmacol. Biochem. Behav 2004, 78, 263–268. [Google Scholar]
- Bromidge, SM; Brown, AM; Clarke, SE; Dodgson, K; Gager, T; Grassam, HL; Jeffrey, PM; Joiner, GF; King, FD; Middlemiss, DN; et al. 5-chloro-N-(4-methoxy-3-piperazin-1-ylphenyl)-3-methyl-2-benzothiophenesulfon-amide (SB-271046): A potent, selective, and orally bioavailable 5-HT6 receptor antagonist. J. Med. Chem 1999, 42, 202–205. [Google Scholar]
- Meneses, A. Effects of the 5-HT6 receptor antagonist Ro 04-6790 on learning consolidation. Behav. Brain Res 2001, 118, 107–110. [Google Scholar]
- Hamon, M; Doucet, E; Lefèvre, K; Miquel, M-C; Lanfumey, L; Insausti, R; Frechilla, D; Del Rio, J; Vergé, D. Antibodies and antisense oligonucleotide for probing the distribution and putative functions of central 5-HT6 receptors. Neuropsychopharmacology 1999, 21, 68S–76S. [Google Scholar]
- Mitchell, ES; Neumaier, JF. 5-HT6 receptors: A novel target for cognitive enhancement. Pharmacol. Ther 2005, 108, 320–333. [Google Scholar]
- Millan, MJ. The neurobiology and control of anxious states. Prog. Neurobiol 2003, 70, 83–244. [Google Scholar]
- Schechter, LE; Ring, RH; Beyer, CE; Hughes, ZA; Khawaja, X; Malberg, JE; Rosenzweig-Lipson, S. Innovative approaches for the development of antidepressant drugs: Current and future strategies. Neurotherapeutics 2005, 2, 590–611. [Google Scholar]
- Wesolowska, A. Potential role of the 5-HT6 receptor in depression and anxiety: An overview of preclinical data. Pharmacol. Rep 2010, 62, 564–577. [Google Scholar]
- Kohen, R; Metcalf, MA; Khan, N; Druck, T; Huebner, K; Lachowicz, JE; Meltzer, HY; Sibley, DR; Roth, BL; Hamblin, MW. Cloning, characterization, and chromosomal localization of a human 5-HT6 serotonin receptor. J. Neurochem 1996, 66, 47–56. [Google Scholar]
- Monsma, FJ; Shen, Y; Ward, RP; Hamblin, MW; Sibley, DR. Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol. Pharmacol 1993, 43, 320–327. [Google Scholar]
- Plassat, JL; Amlaiky, N; Hen, R. Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase. Mol. Pharmacol 1993, 44, 229–236. [Google Scholar]
- Glennon, RA; Lee, M; Rangisetty, JB; Dukat, M; Roth, BL; Savage, JE; McBride, A; Rauser, L; Hufeisen, S; Lee, DKH. 2-substituted tryptamines: agents with selectivity for 5-HT6 serotonin receptors. J. Med. Chem 2000, 43, 1011–1018. [Google Scholar]
- Pullagurla, MR; Dukat, M; Setola, V; Roth, B; Glennon, RA. N1-benzenesulfonylgramine and N1-benzenesulfonylskatole: novel 5-HT6 receptor ligand templates. Bioorg. Med. Chem. Lett 2003, 13, 3355–3359. [Google Scholar]
- Elokdah, H; Li, D; McFarlane, G; Bernotas, RC; Robichaud, AJ; Magolda, RL; Zhang, GM; Smith, D; Schechter, LE. Novel 1-(azacyclyl)-3-arylsulfonyl-1H-pyrrolo[2,3-b]pyridines as 5-HT6 agonists and antagonists. Bioorg. Med. Chem 2007, 15, 6208–6226. [Google Scholar]
- Liu, KG; Lo, JR; Comery, TA; Zhang, GM; Zhang, JY; Kowal, DM; Smith, DL; Di, L; Kerns, EH; Schechter, LE; et al. A regiospecific synthesis of a series of 1-sulfonyl azepinoindoles as potent 5-HT6 ligands. Bioorg. Med. Chem. Lett 2008, 18, 3929–3931. [Google Scholar]
- Liu, KG; Lo, JR; Comery, TA; Zhang, GM; Zhang, JY; Kowal, DM; Smith, DL; Di, L; Kerns, EH; Schechter, LE; et al. 1-sulfonylindazoles as potent and selective 5-HT6 ligands. Bioorg. Med. Chem. Lett 2009, 19, 2413–2415. [Google Scholar]
- Liu, KG; Lo, JR; Comery, TA; Zhang, GM; Zhang, JY; Kowal, DM; Smith, DL; Di, L; Kerns, EH; Schechter, LE; et al. Identification of a novel series of 3-piperidinyl-5-sulfonylindazoles as potent 5-HT6 ligands. Bioorg. Med. Chem. Lett 2009, 19, 3214–3216. [Google Scholar]
- Liu, KG; Lo, JR; Comery, TA; Zhang, GM; Zhang, JY; Kowal, DM; Smith, DL; Di, L; Kerns, EH; Schechter, LE; et al. Identification of a series of benzoxazoles as potent 5-HT6 ligands. Bioorg. Med. Chem. Lett 2009, 19, 1115–1117. [Google Scholar]
- Bernotas, RC; Lenicek, S; Antane, S; Cole, DC; Harrison, BL; Robichaud, AJ; Zhang, GM; Smith, D; Platt, B; Lin, Q; et al. Novel 1-aminoethyl-3-arylsulfonyl-1H-pyrrolo[2,3-b] pyridines are potent 5-HT6 agonists. Bioorg. Med. Chem 2009, 17, 5153–5163. [Google Scholar]
- Bernotas, RC; Antane, S; Shenoy, R; Le, V-D; Chen, P; Harrison, BL; Robichaud, AJ; Zhang, GM; Smith, D; Schechter, LE. 3-(arylsulfonyl)-1-(azacyclyl)-1H-indoles are 5-HT6 receptor modulators. Bioorg. Med. Chem. Lett 2010, 20, 1657–1660. [Google Scholar]
- Liu, KG; Robichaud, AJ; Greenfield, AA; Lo, JR; Grosanu, C; Mattes, JF; Cai, Y; Zhang, GM; Zhang, JY; Kowal, DM; et al. Identification of 3-sulfonylindazole derivatives as potent and selective 5-HT6 antagonists. Bioorg. Med. Chem 2011, 19, 650–662. [Google Scholar]
- Sleight, AJ; Boess, FG; Bös, M; Levet-Trafit, B; Riemer, C; Bourson, A. Characterization of Ro 04-6790 and Ro 63-0563: potent and selective antagonists at human and rat 5-HT6 receptors. Br. J. Pharmacol 1998, 124, 556–562. [Google Scholar]
- Boess, FG; Riemer, C; Bös, M; Bentley, J; Bourson, A; Sleight, AJ. The 5-hydroxytryptamine6 receptor-selective radioligand [3H]Ro 63–0563 labels 5-hydroxytryptamine receptor binding sites in rat and porcine striatum. Mol. Pharmacol 1998, 54, 577–583. [Google Scholar]
- Holenz, J; Pauwels, PJ; Díaz, JL; Mercè, R; Codony, X; Buschmann, H. Medicinal chemistry strategies to 5-HT6 receptor ligands as potential cognitive enhancers and antiobesity agents. Drug Discov. Today 2006, 11, 283–299. [Google Scholar]
- Russell, MGN; Dias, R. Memories are made of this (perhaps): A review of serotonin 5-HT6 receptor ligands and their biological functions. Curr. Top. Med. Chem 2002, 2, 643–654. [Google Scholar]
- Muthas, D; Sabnis, YA; Lundborg, M; Karlén, A. Is it possible to increase hit rates in structure-based virtual screening by pharmacophore filtering? An investigation of the advantages and pitfalls of post-filtering. J. Mol. Graph. Model 2008, 26, 1237–1251. [Google Scholar]
- Xie, H-Z; Li, L-L; Ren, J-X; Zou, J; Yang, L; Wei, Y-Q; Yang, S-Y. Pharmacophore modeling study based on known spleen tyrosine kinase inhibitors together with virtual screening for identifying novel inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 1944–1949. [Google Scholar]
- Alzate-Morales, JH; Vergara-Jaque, A; Caballero, J. Computational study on the interaction of N1 substituted pyrazole derivatives with B-Raf kinase: An unusual water wire hydrogen-bond network and novel interactions at the entrance of the active site. J. Chem. Inf. Model 2010, 50, 1101–1112. [Google Scholar]
- Villacañas, O; Pérez, JJ; Rubio-Martínez, J. Structural analysis of the inhibition of Cdk4 and Cdk6 by p16 (INK4a) through molecular dynamics simulations. J. Biomol. Struct. Dyn 2002, 20, 347–358. [Google Scholar]
- Doddareddy, MR; Cho, YS; Koh, HY; Pae, AN. CoMFA and CoMSIA 3D QSAR analysis on N1-arylsulfonylindole compounds as 5-HT6 antagonists. Bioorg. Med. Chem 2004, 12, 3977–3985. [Google Scholar]
- López-Rodríguez, ML; Benhamú, B; de la Fuente, T; Sanz, A; Pardo, L; Campillo, M. A three-dimensional pharmacophore model for 5-hydroxytryptamine6 (5-HT6) receptor antagonists. J. Med. Chem 2005, 48, 4216–4219. [Google Scholar]
- Goodarzi, M; Freitas, MP; Ghasemi, N. QSAR studies of bioactivities of 1-(azacyclyl)-3-arylsulfonyl-1H-pyrrolo[2,3-b]pyridines as 5-HT6 receptor ligands using physicochemical descriptors and MLR and ANN-modeling. Eur. J. Med. Chem 2010, 45, 3911–3915. [Google Scholar]
- Sharma, BK; Singh, P; Sarbhai, K; Prabhakar, YS. A quantitative structure-activity relationship study on serotonin 5-HT6 receptor ligands: indolyl and piperidinyl sulphonamides. SAR QSAR Environ. Res 2010, 21, 369–388. [Google Scholar]
- Durdagi, S; Papadopoulos, MG; Zoumpoulakis, PG; Koukoulitsa, C; Mavromoustakos, T. A computational study on cannabinoid receptors and potent bioactive cannabinoid ligands: Homology modeling, docking, de novo drug design and molecular dynamics analysis. Mol. Divers 2010, 14, 257–276. [Google Scholar]
- SWISS-MODEL. Available online: http://swissmodel.expasy.org/ (accessed on 18 July 2011).
- Pullagurla, MR; Westkaemper, RB; Glennon, RA. Possible differences in modes of agonist and antagonist binding at human 5-HT6 receptors. Bioorg. Med. Chem. Lett 2004, 14, 4569–4573. [Google Scholar]
- Hirst, WD; Abrahamsen, B; Blaney, FE; Calver, AR; Aloj, L; Price, GW; Medhurst, AD. Differences in the central nervous system distribution and pharmacology of the mouse 5-hydroxytryptamine-6 receptor compared with rat and human receptors investigated by radioligand binding, site-directed mutagenesis, and molecular modeling. Mol. Pharmacol 2003, 64, 1295–1308. [Google Scholar]
- Dukat, M; Mosier, PD; Kolanos, R; Roth, BL; Glennon, RA. Binding of serotonin and N1-benzenesulfonyltryptamine-related analogs at human 5-HT6 serotonin receptors: Receptor modeling studies. J. Med. Chem 2008, 51, 603–611. [Google Scholar]
- Hillisch, A; Pineda, LF; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar]
- AbdulHameed, MDM; Hamza, A; Liu, JJ; Zhan, CG. Combined 3D-QSAR modeling and molecular docking study on indolinone derivatives as inhibitors of 3-phosphoinositide-dependent protein kinase-1. J. Chem. Inf. Model 2008, 48, 1760–1772. [Google Scholar]
- Böhm, M; Stürzebecher, J; Klebe, G. Three-dimensional quantitative structure-activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem 1999, 42, 458–477. [Google Scholar]
- NCBI. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 18 July 2011).
- RCSB PDB. Available online: http://www.rcsb.org/pdb/home/home.do/ (accessed on 18 July 2011).
- Cherezov, V; Rosenbaum, DM; Hanson, MA; Rasmussen, SGF; Thian, FS; Kobilka, TS; Choi, H-J; Kuhn, P; Weis, WI; Kobilka, BK; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar]
- Berendsen, HJC; van der Spoel, D; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 1995, 91, 43–56. [Google Scholar]
- Lindahl, E; Hess, B; van der Spoel, D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model 2001, 7, 306–317. [Google Scholar]
- Aalten, DMF; Bywater, R; Findlay, JBC; Hendlich, M; Hooft, RWW; Vriend, G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des 1996, 10, 255–262. [Google Scholar]
- Schuttelkopf, AW; van Aalten, DMF. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr 2004, D60, 1355–1363. [Google Scholar]
- Berendsen, HJC; Postma, JPM; van Gunsteren, WF; Hermans, J. Intermolecular Forces; Pullman, B, Ed.; Reidel publishing company: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Parrinello, M; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 1981, 52, 7182–7190. [Google Scholar]
- Essmann, U; Perera, L; Berkowitz, M; Darden, T; Lee, H; Pedersen, L. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Lin, JH; Perryman, AL; Schames, JR; McCammon, JA. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc 2002, 124, 5632–5633. [Google Scholar]




| PLS Statistics | Ligand-based model | Receptor-based model | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| q2 | 0.50 | 0.53 | 0.45 | 0.47 |
| OPN | 7 | 7 | 5 | 5 |
| r2ncv | 0.85 | 0.81 | 0.78 | 0.75 |
| SEE | 0.27 | 0.30 | 0.33 | 0.35 |
| F | 127.90 | 104.23 | 123.45 | 104.26 |
| r2pred | 0.71 | 0.76 | 0.59 | 0.62 |
| Contribution Steric | 0.42 | 0.18 | 0.42 | 0.16 |
| Electrostatic | 0.46 | 0.33 | 0.42 | 0.26 |
| Hydrophobic | - | 0.24 | 0.25 | |
| H-bond acceptor | - | 0.12 | 0.16 | |
| CIC2 | 0.07 | 0.07 | 0.11 | 0.12 |
| BEHv2 | 0.05 | 0.06 | 0.05 | 0.05 |
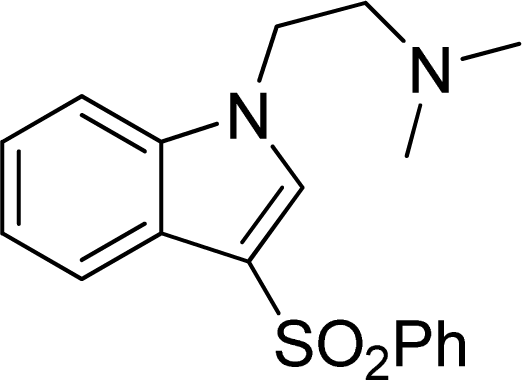
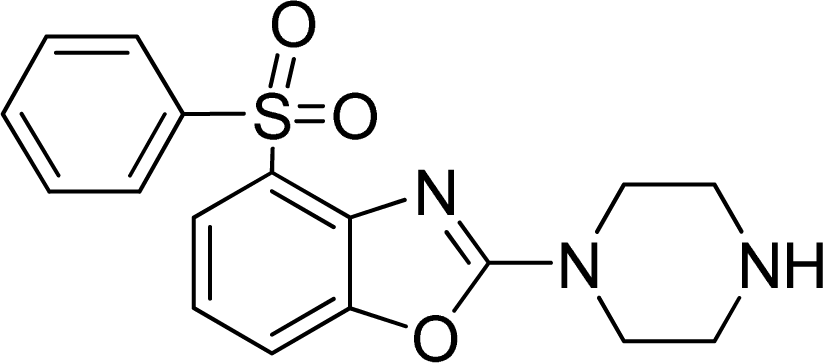
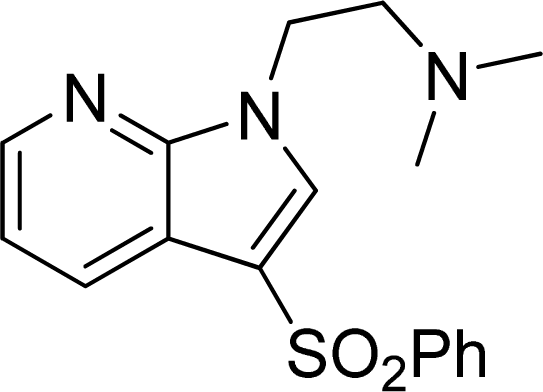
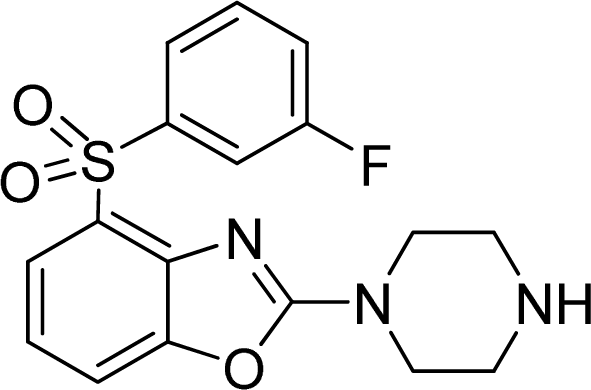



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hao, M.; Li, Y.; Li, H.; Zhang, S. Investigation of the Structure Requirement for 5-HT6 Binding Affinity of Arylsulfonyl Derivatives: A Computational Study. Int. J. Mol. Sci. 2011, 12, 5011-5030. https://doi.org/10.3390/ijms12085011
Hao M, Li Y, Li H, Zhang S. Investigation of the Structure Requirement for 5-HT6 Binding Affinity of Arylsulfonyl Derivatives: A Computational Study. International Journal of Molecular Sciences. 2011; 12(8):5011-5030. https://doi.org/10.3390/ijms12085011
Chicago/Turabian StyleHao, Ming, Yan Li, Hanqing Li, and Shuwei Zhang. 2011. "Investigation of the Structure Requirement for 5-HT6 Binding Affinity of Arylsulfonyl Derivatives: A Computational Study" International Journal of Molecular Sciences 12, no. 8: 5011-5030. https://doi.org/10.3390/ijms12085011