Amyloidogenic Properties of a D/N Mutated 12 Amino Acid Fragment of the C-Terminal Domain of the Cholesteryl-Ester Transfer Protein (CETP)

Abstract
:1. Introduction
2. Results
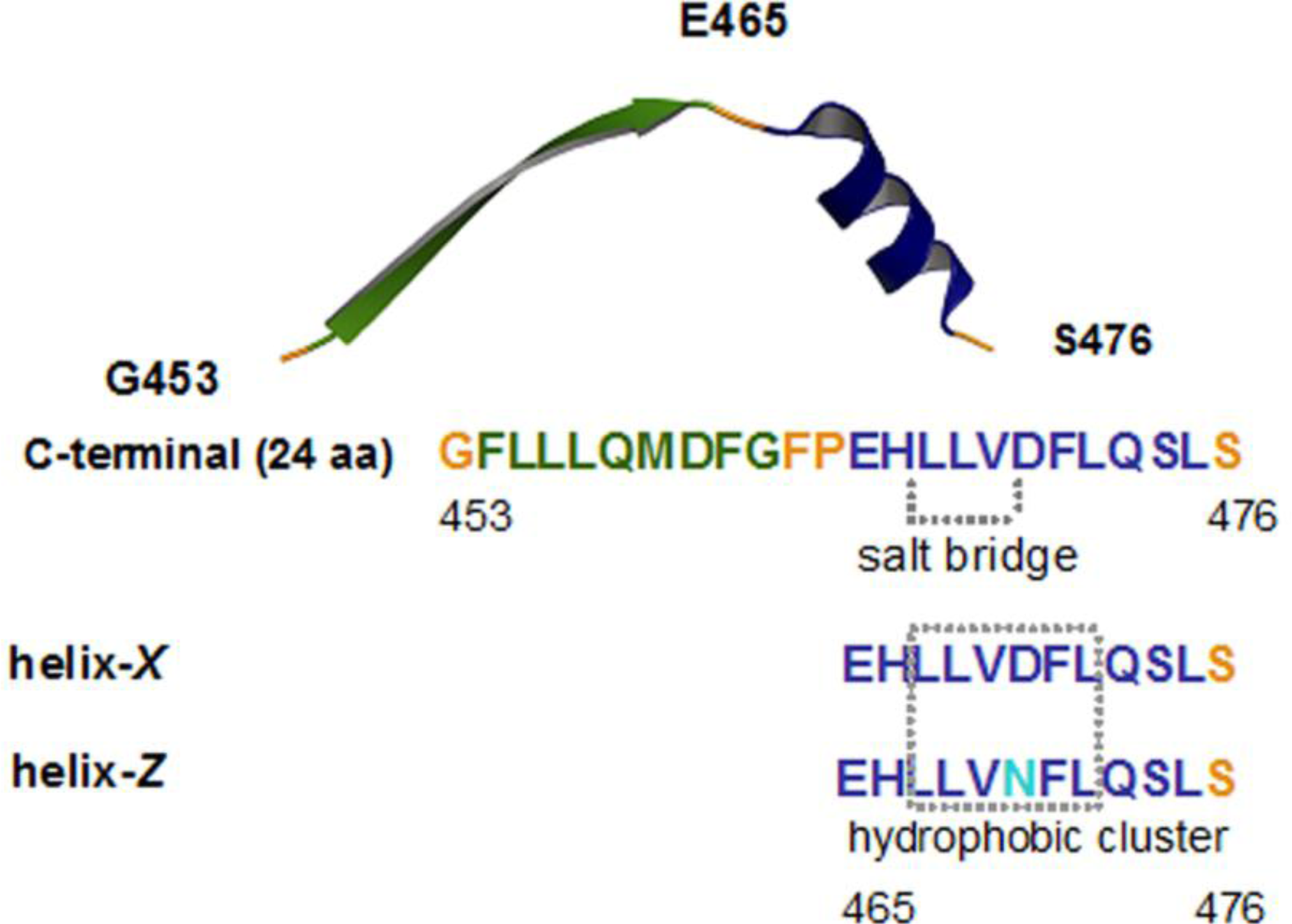
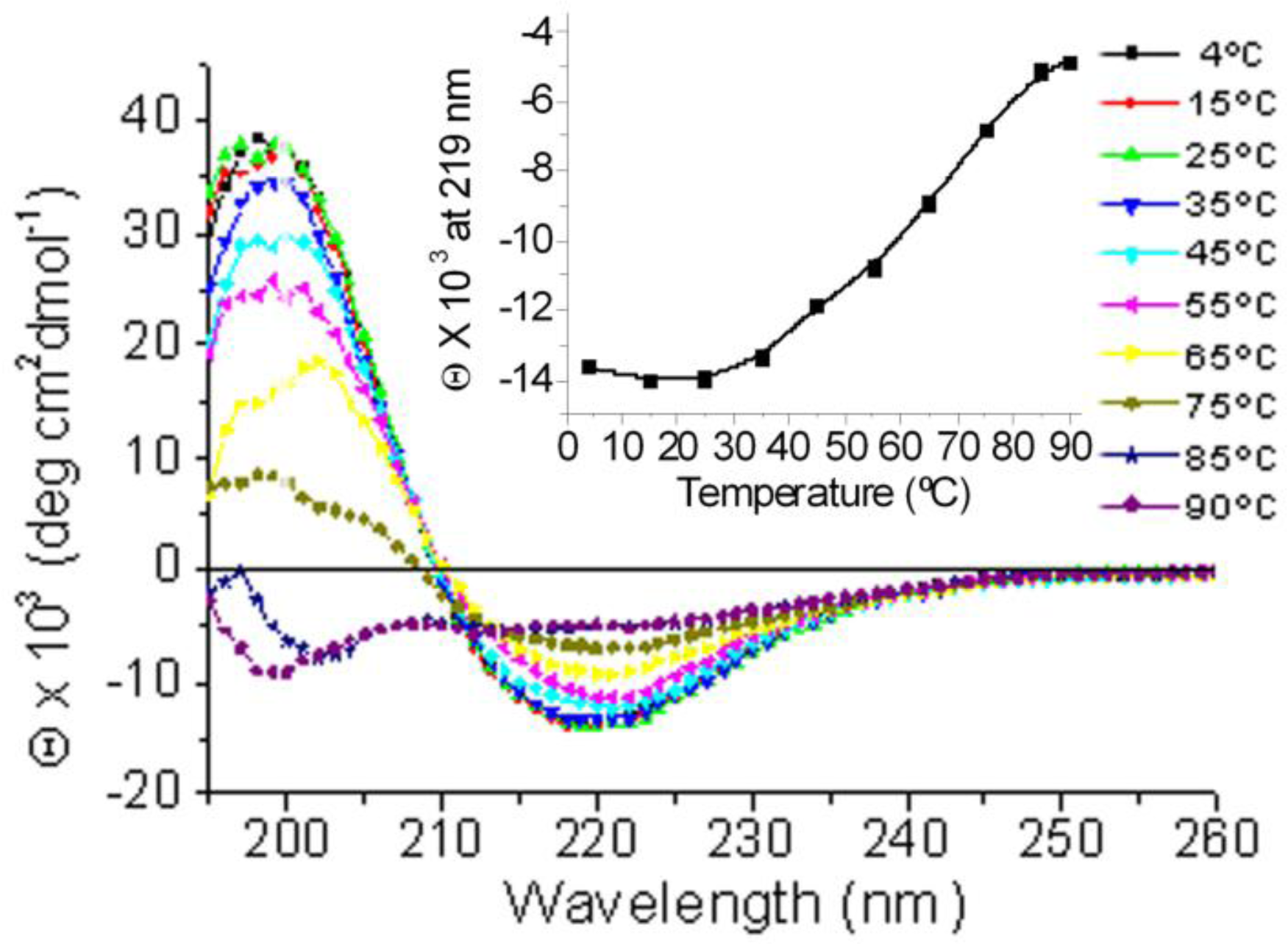
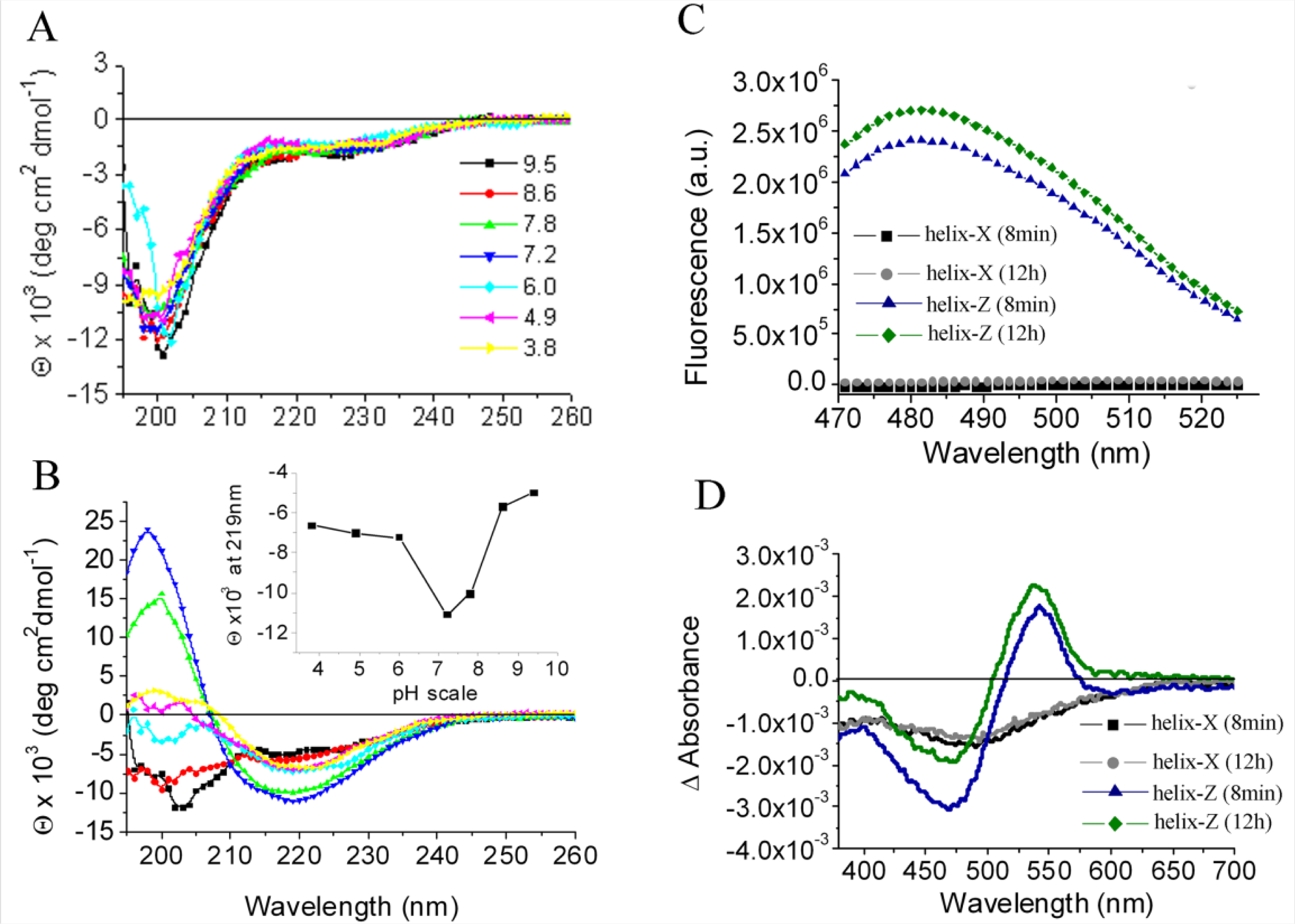
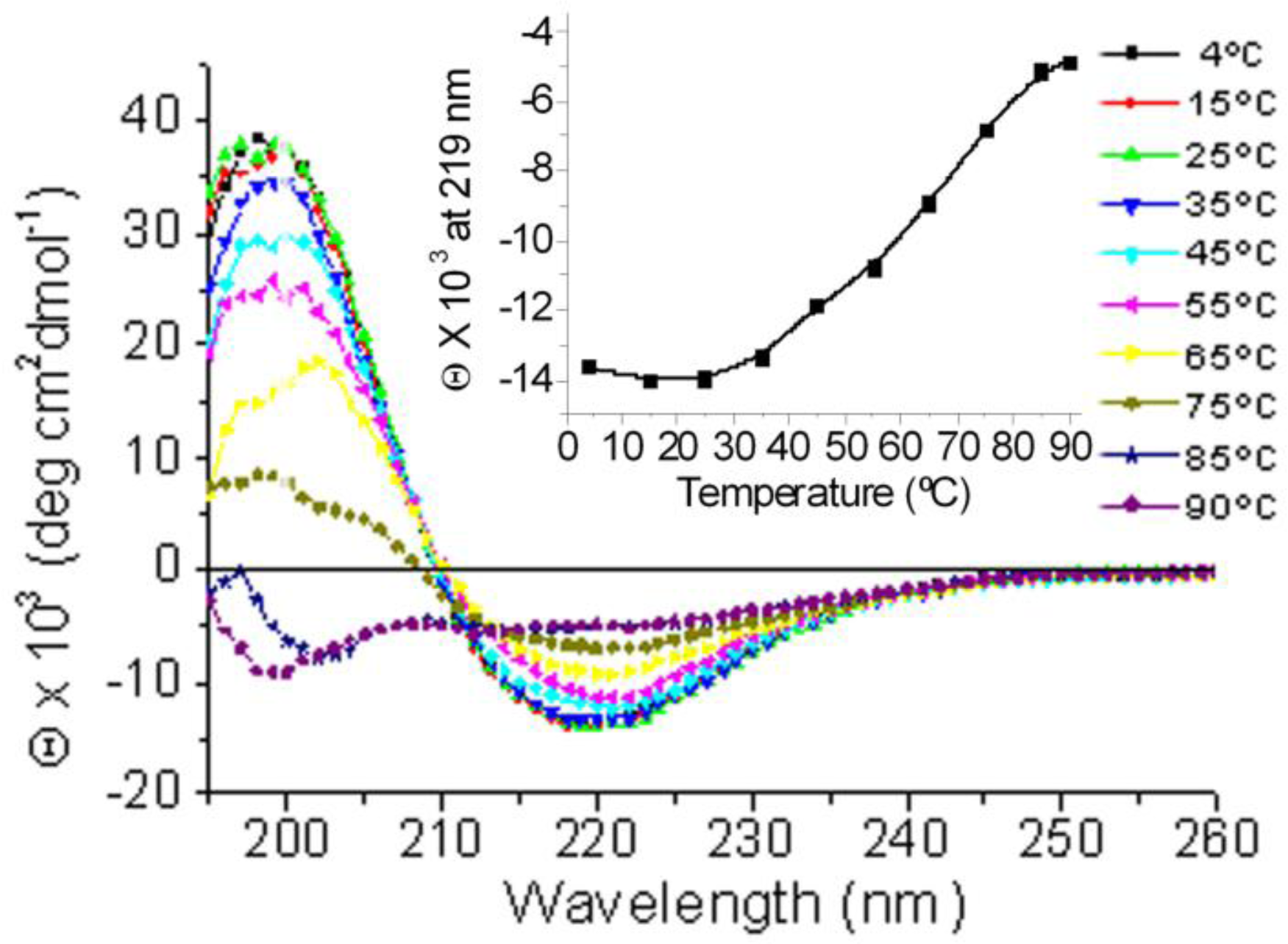
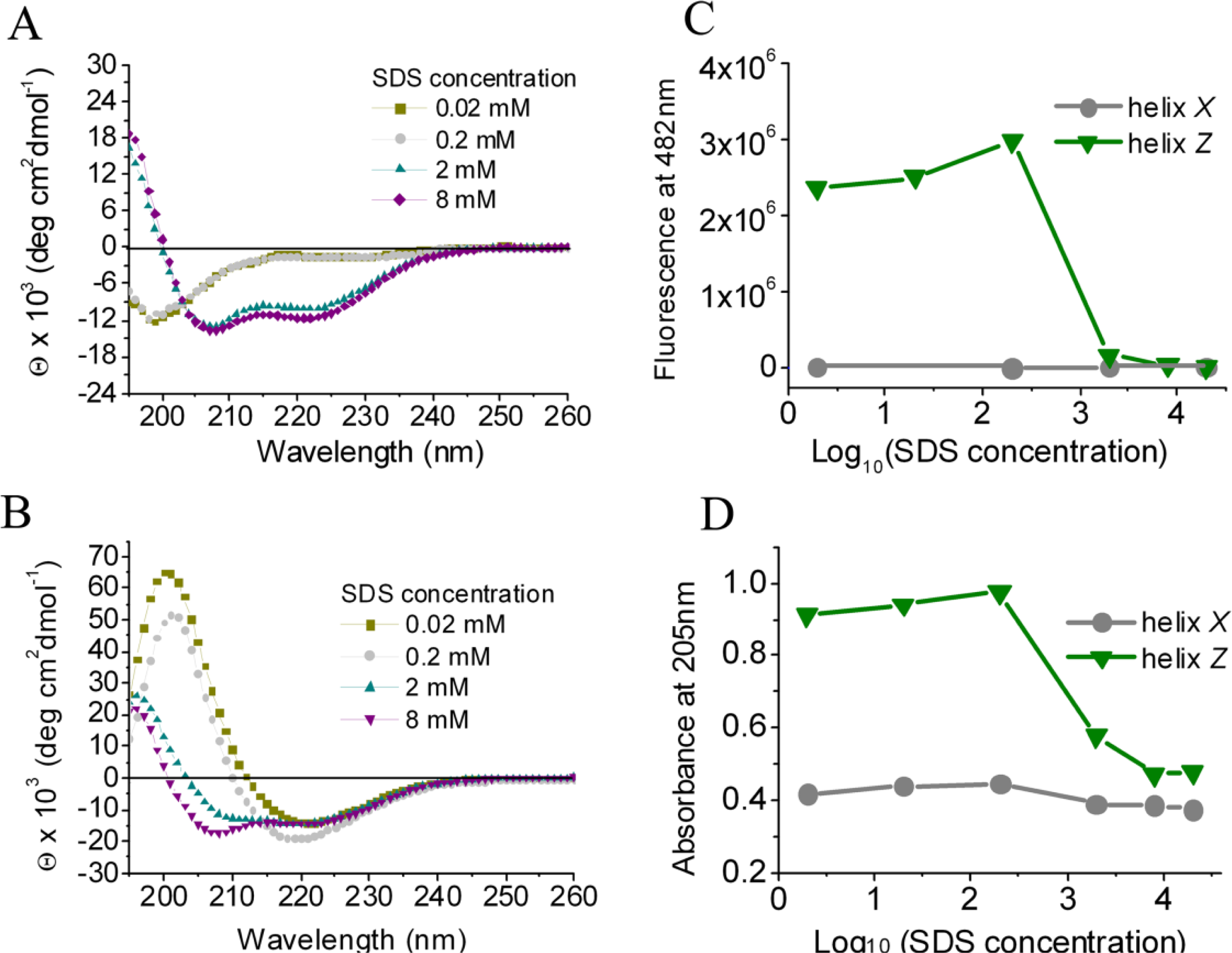
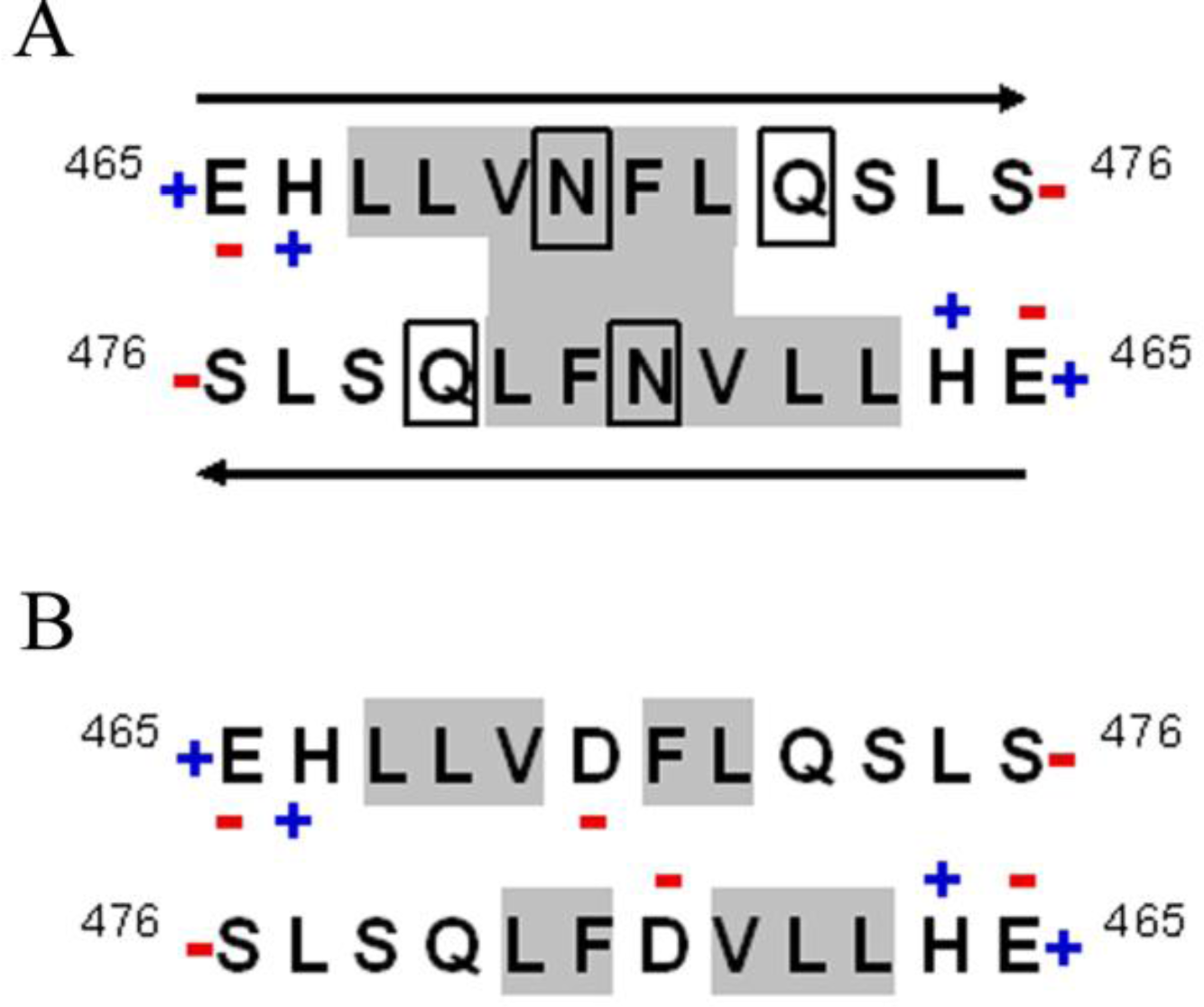
2.1. Conformational Changes in the C-Terminal Domain of CETP
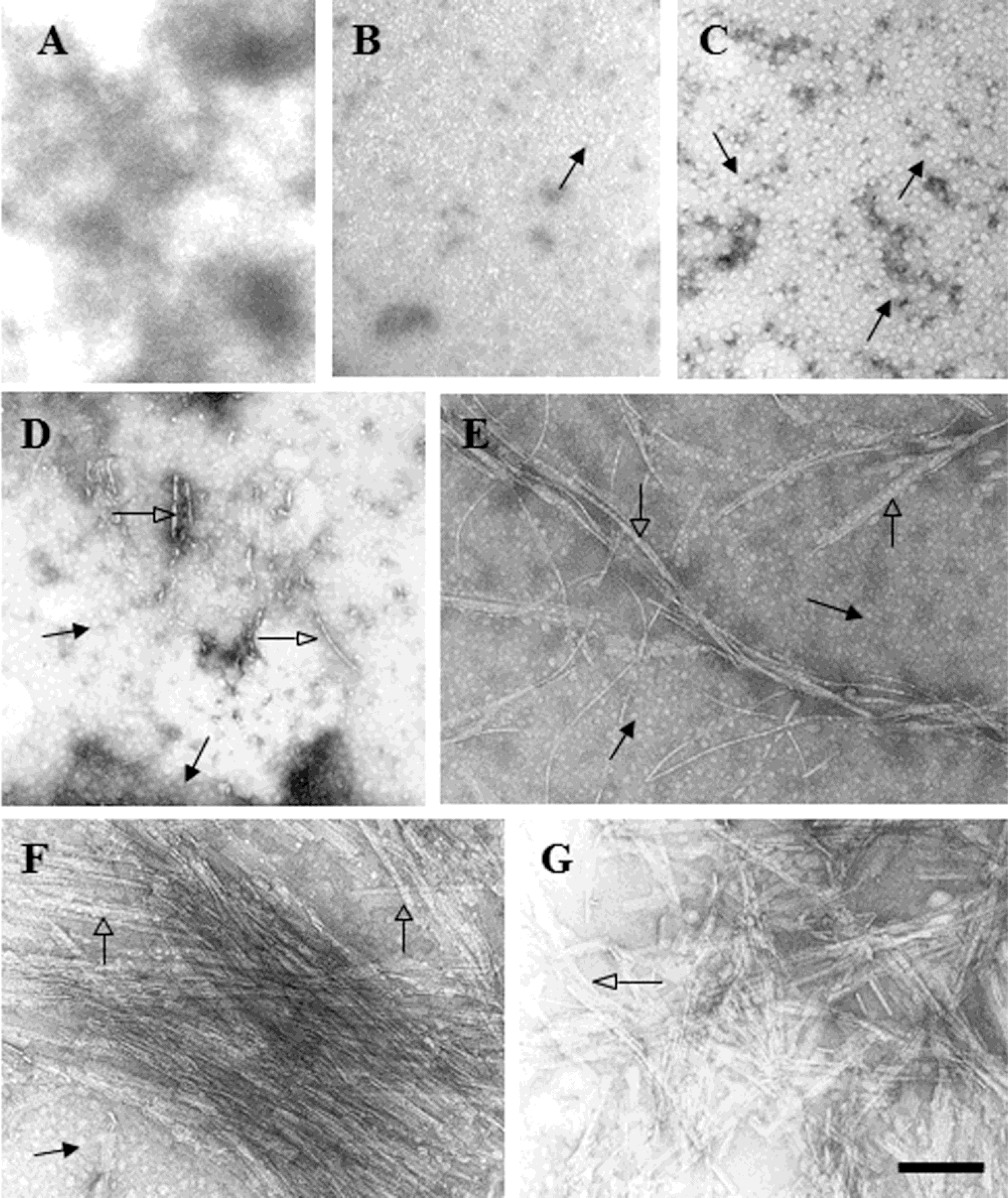
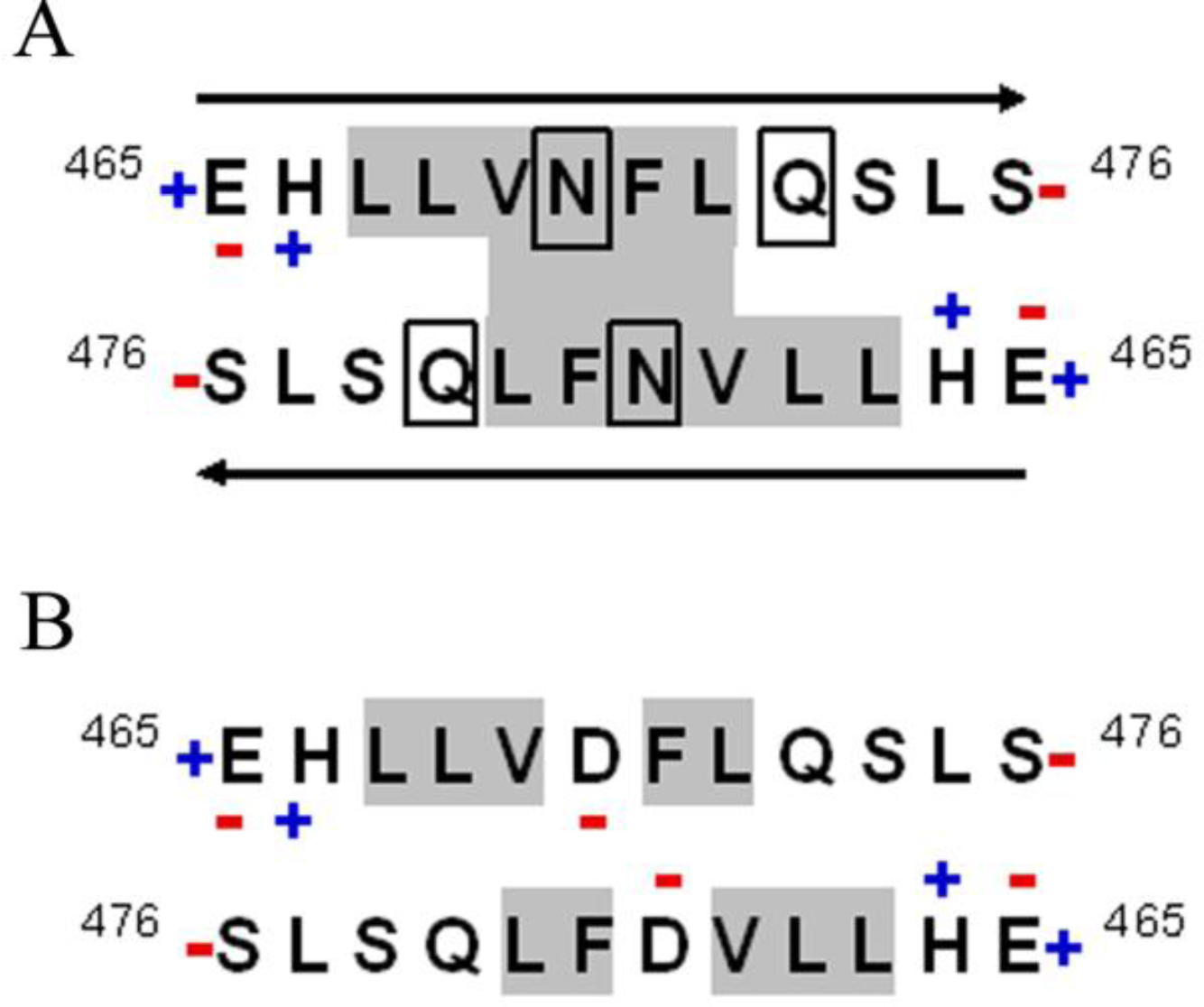
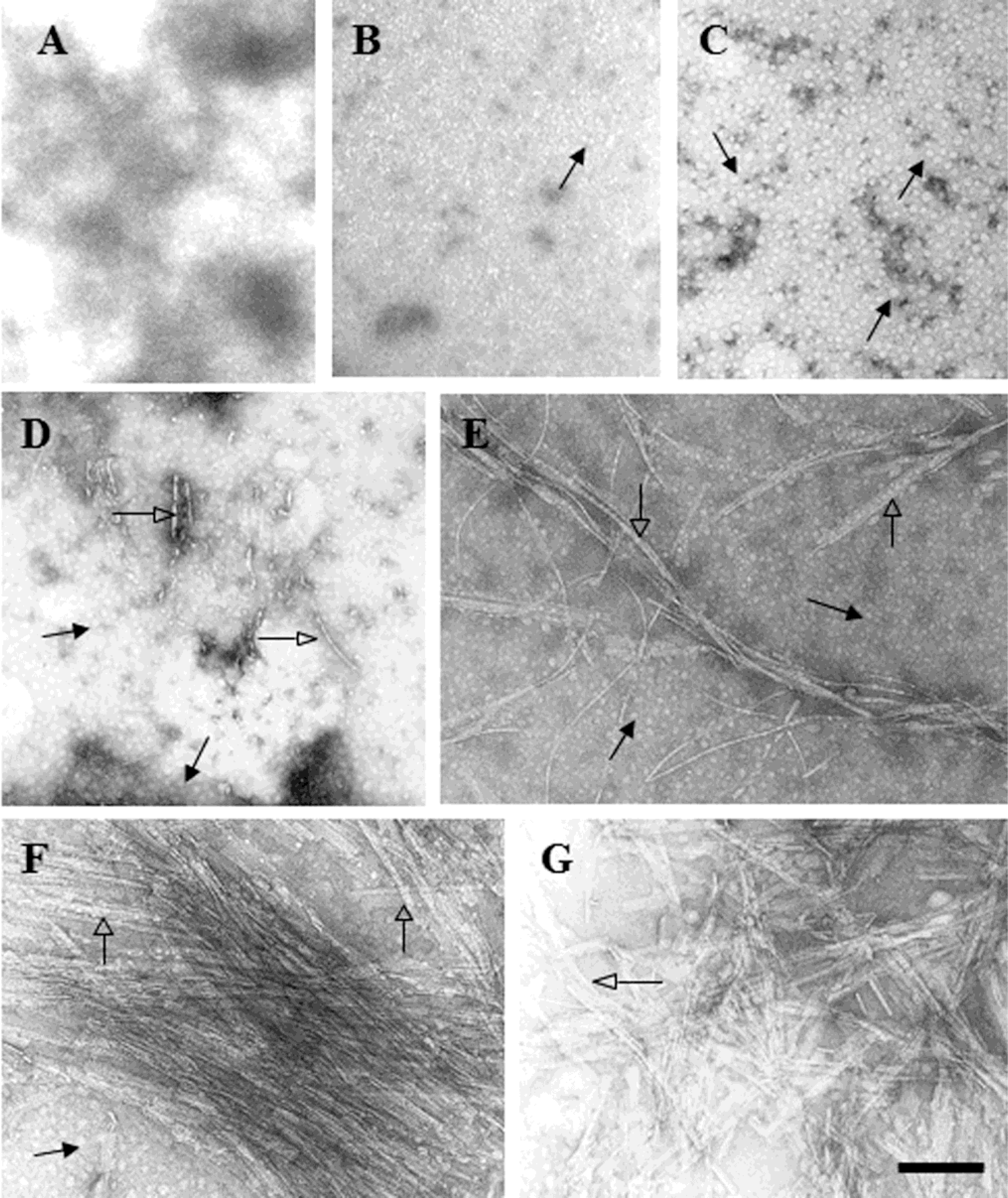
2.2. D470N Mutation in the C-Terminal Domain of CETP Induces Amyloid Fibril Formation
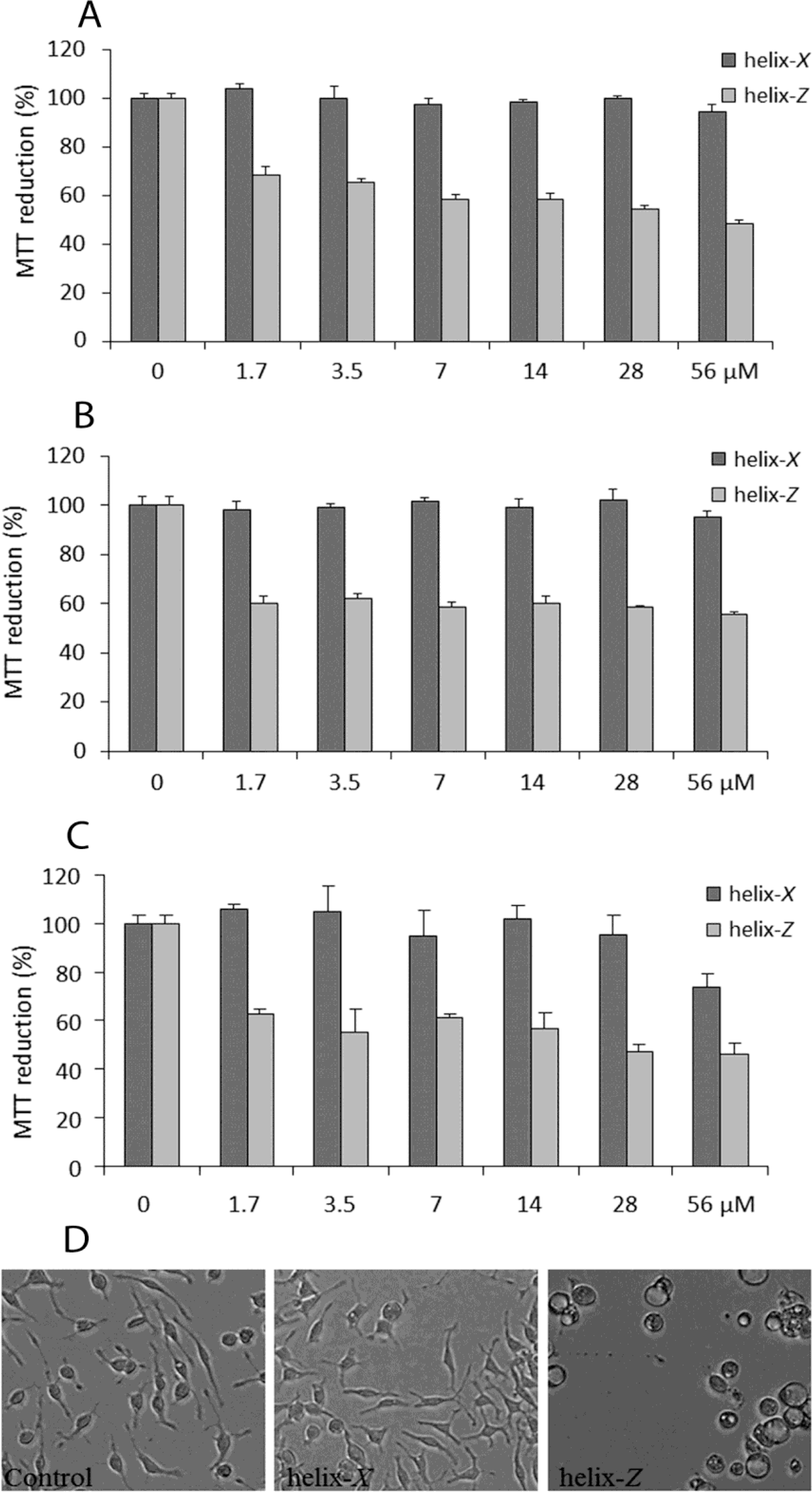
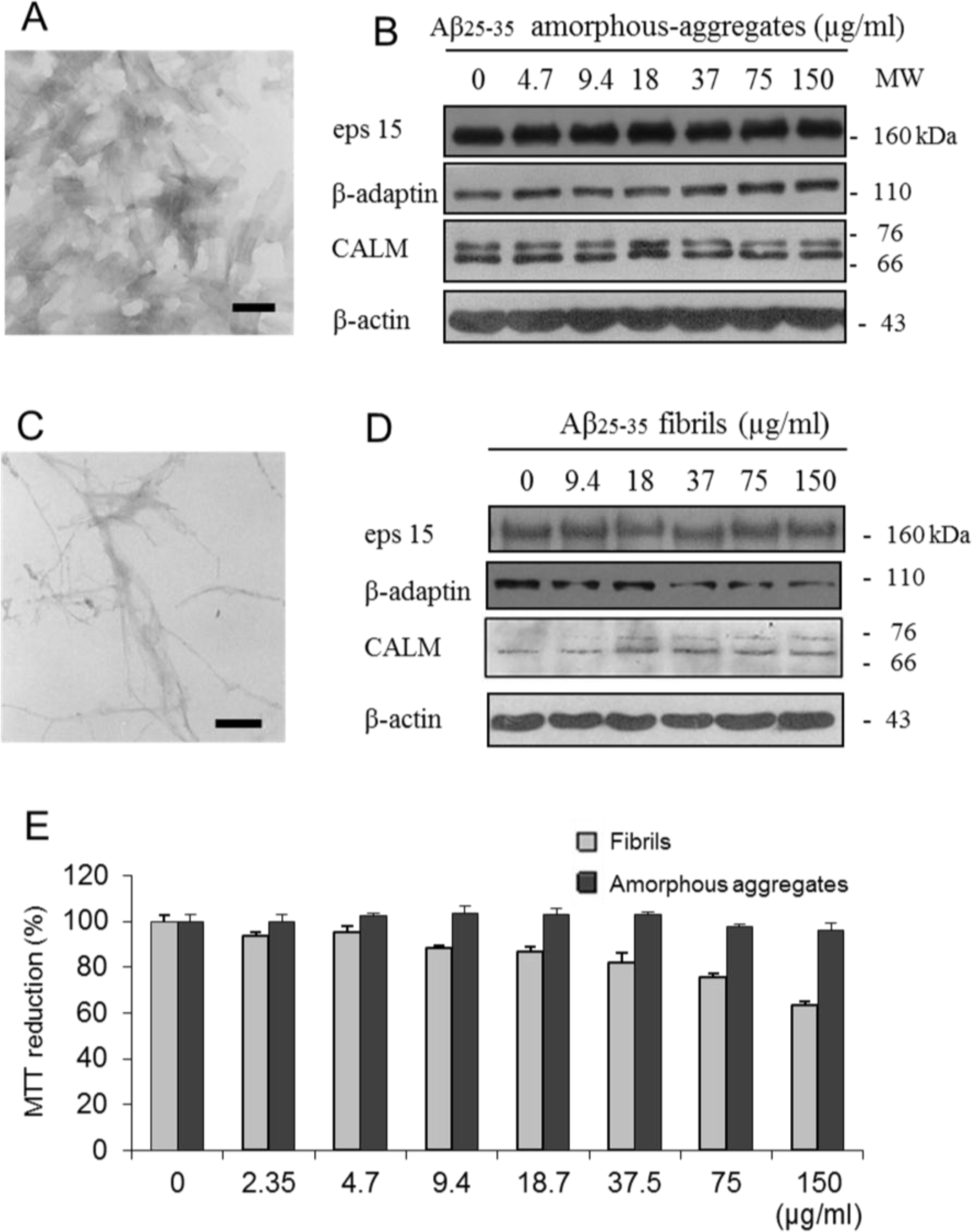
2.3. Cytotoxic Effects Associated with Helix-Z
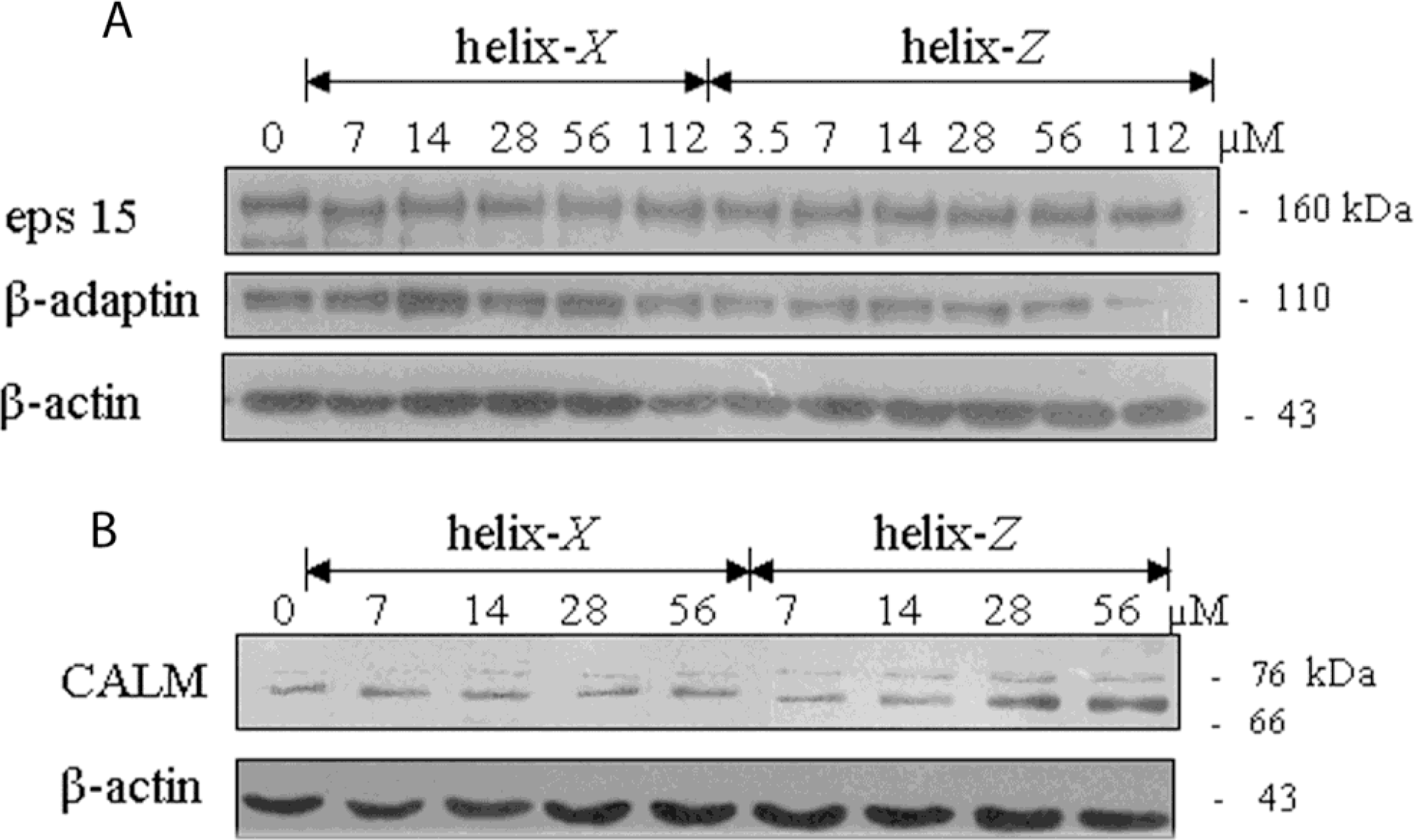
2.4. Endocytic Protein Expression
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Peptide Synthesis and Preparation
4.3. Circular Dichroism Spectroscopy
4.4. Congo Red Spectroscopy and Thioflavin T Fluorescence
4.5. Cell Culture
4.6. Cell Viability Assay
4.7. Western Blot Analysis
4.8. Optical Microscopy
4.9. Electron Microscopy
5. Conclusions
Acknowledgments
References
- Uversky, VN; Oldfield, CJ; Midic, U; Xie, H; Xue, B; Vucetic, S; Iakoucheva, LM; Obradovic, Z; Dunker, AK. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics 2009, 10, S7. [Google Scholar]
- Berson, JF; Theos, AC; Harper, DC; Tenza, D; Raposo, G; Marks, MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell. Biol 2003, 161, 521–533. [Google Scholar]
- Dobson, CM. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Xicohtencatl-Cortes, J; Castillo, R; Mas-Oliva, J. In search of new structural states of exchangeable apolipoproteins. Biochem. Biophys. Res. Commun 2004, 324, 467–470. [Google Scholar]
- Ramos, S; Campos-Terán, J; Mas-Oliva, J; Nylander, T; Castillo, R. Forces between hydrophilic surfaces adsorbed with apolipoprotein AII alpha helices. Langmuir 2008, 24, 8568–8575. [Google Scholar]
- Mendoza-Espinosa, P; Moreno, A; Castillo, R; Mas-Oliva, J. Lipid dependant disorder-to-order conformational transitions in apolipoprotein CI derived peptides. Biochem. Biophys. Res. Commun 2008, 365, 8–15. [Google Scholar]
- Klerkx, AH; El Harchaoui, K; van der Steeg, WA; Boekholdt, SM; Stroes, ES; Kastelein, JJ; Kuivenhoven, JA. Cholesteryl ester transfer protein (CETP) inhibition beyond raising high-density lipoprotein cholesterol levels: pathways by which modulation of CETP activity may alter atherogenesis. Arterioscler. Thromb. Vasc. Biol 2006, 26, 706–715. [Google Scholar]
- Bolaños-García, VM; Soriano-García, M; Mas-Oliva, J. Stability of the C-terminal peptide of CETP mediated through an (i, i + 4) array. Biochim. Biophys. Acta 1998, 1384, 7–15. [Google Scholar]
- Wang, S; Wang, X; Deng, L; Rassart, E; Milne, RS; Tall, AR. Point mutagenesis of carboxyl-terminal amino acids of cholesteryl ester transfer protein. J. Biol. Chem 1993, 66, 1955–1959. [Google Scholar]
- Wang, S; Kussie, P; Deng, L; Tall, A. Defective binding of neutral lipids by a carboxyl-terminal deletion mutant of cholesteryl ester transfer protein. Evidence for a carboxyl-terminal cholesteryl ester binding site essential for neutral lipid transfer activity. J. Biol. Chem 1995, 270, 612–618. [Google Scholar]
- Qiu, X; Mistry, A; Ammirati, MJ; Chrunyk, BA; Clark, RW; Cong, Y; Culp, JS; Danley, DE; Freeman, TB; Geoghegan, KF; Griffor, MC; Hawrylik, SJ; Hayward, CM; Hensley, P; Hoth, LR; Karam, GA; Lira, ME; Lloyd, DB; McGrath, KM; Stutzman-Engwall, KJ; Subashi, AK; Subashi, TA; Thompson, JF; Wang, IK; Zhao, H; Seddon, AP. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat. Struct. Mol. Biol 2007, 14, 106–113. [Google Scholar]
- García-González, V; Mas-Oliva, J. Structural Arrangement that Supports Lipid Transfer in the cholesteryl-ester transfer protein (CETP). USA-México Workshop in Biological Chemistry: Multidisciplinary Approaches to Protein Folding, Mexico City, Mexico, 25–27 March 2009.
- Alonso, AL; Zentella-Dehesa, A; Mas-Oliva, J. Characterization of a naturally occurring new version of the cholesterol ester transfer protein (CETP) from small intestine. Mol. Cell. Biochem 2003, 245, 173–182. [Google Scholar]
- Mendoza-Espinosa, P; García-González, V; Moreno, A; Castillo, R; Mas-Oliva, J. Disorder-to-order conformational transitions in protein structure and its relationship to disease. Mol. Cell. Biochem 2009, 330, 105–120. [Google Scholar]
- El Khoury, J; Luster, AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends. Pharmacol. Sci 2008, 29, 626–632. [Google Scholar]
- Hickman, SE; Allison, EK; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci 2008, 28, 8354–8360. [Google Scholar]
- Manzano-León, N; Delgado-Coello, NB; Guaderrama-Díaz, M; Mas-Oliva, J. Beta-adaptin: Key molecule for microglial scavenger receptor function under oxidative stress. Biochem. Biophys. Res. Commun 2006, 351, 588–594. [Google Scholar]
- Abe, K; Saito, H. Both oxidative stress-dependent and independent effects of amyloid beta protein are detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. Brain Res 1999, 830, 146–154. [Google Scholar]
- Liu, Y; Peterson, DA; Schubert, D. Amyloid beta peptide alters intracellular vesicle trafficking and cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 1998, 95, 13266–13271. [Google Scholar]
- Broome, BM; Hecht, MH. Nature disfavors sequences of alternating polar and non-polar amino acids: implications for amyloidogenesis. J. Mol. Biol 2000, 296, 961–968. [Google Scholar]
- Schwartz, R; Istrail, S; King, J. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci 2001, 10, 1023–1031. [Google Scholar]
- Chiti, F; Calamai, M; Taddei, N; Stefani, M; Ramponi, G; Dobson, CM. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA 2002, 99, 16419–16426. [Google Scholar]
- López De La Paz, M; Goldie, K; Zurdo, J; Lacroix, E; Dobson, CM; Hoenger, A; Serrano, L. De novo designed peptide-based amyloid fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 16052–16057. [Google Scholar]
- Monsellier, E; Chiti, F. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep 2007, 8, 737–742. [Google Scholar]
- Uversky, VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci 2002, 11, 739–756. [Google Scholar]
- Wetzel, R. Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res 2006, 39, 671–679. [Google Scholar]
- Nelson, R; Sawaya, MR; Balbirnie, M; Madsen, AØ; Riekel, C; Grothe, R; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar]
- Radivojac, P; Obradovic, Z; Smith, DK; Zhu, G; Vucetic, S; Brown, CJ; Lawson, JD; Dunker, AK. Protein flexibility and intrinsic disorder. Protein Sci 2004, 13, 71–80. [Google Scholar]
- Ladokhin, AS; White, SH. Folding of amphipathic alpha-helices on membranes: energetics of helix formation by melittin. J. Mol. Biol 1999, 285, 1363–1369. [Google Scholar]
- White, SH; Wimley, WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta 1998, 1376, 339–352. [Google Scholar]
- Aguilar-Gaytan, R; Mas-Oliva, J. Oxidative stress impairs endocytosis of the scavenger receptor class A. Biochem. Biophys. Res. Commun 2003, 305, 510–517. [Google Scholar]
- Mousavi, SA; Malerød, L; Berg, T; Kjeken, R. Clathrin-dependent endocytosis. Biochem. J 2004, 377, 1–16. [Google Scholar]
- Ford, MG; Pearse, BM; Higgins, MK; Vallis, Y; Owen, DJ; Gibson, A; Hopkins, CR; Evans, PR; McMahon, HT. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 2001, 291, 1051–1055. [Google Scholar]
- Legandre-Guillemin, V; Wasiak, S; Hussain, NK; Angers, A; McPherson, PS. ENTH/ANTH proteins and clatrin-mediated membrane budding. J. Cell. Sci 2004, 117, 9–18. [Google Scholar]
- Klunk, WE; Jacob, RF; Mason, RP. Quantifying amyloid beta-peptide (Abeta) aggregation using the Congo red-Abeta (CR-abeta) spectrophotometric assay. Anal. Biochem 1999, 266, 66–76. [Google Scholar]
- Knowles, TP; Fitzpatrick, AW; Meehan, S; Mott, HR; Vendruscolo, M; Dobson, CM; Welland, ME. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar]
Supplementary Data





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | helix-X | helix-Z | Aβ1–42 | Aβ25–35 |
|---|---|---|---|---|
| MW (Da) | 1399.8 | 1399.6 | 4514 | 1060.3 |
| Isoelectric point | 4.17 | 5.13 | 5.21 | 8.75 |
| Hydrophobicity (kcal/mol) | 0.27 | 0.28 | 0.21 | 0.37 |
| μH (kcal/mol) | 0.41 | 0.41 | 0.08 | 0.03 |
| pH | helix-X | helix-Z | Aβ1–42 | Aβ25–35 |
|---|---|---|---|---|
| 4 | 0.07 | 0.65 | 3.17 | 1.02 |
| 4.5 | −0.47 | 0.34 | 1.55 | 1.01 |
| 5 | −0.87 | 0.06 | 0.38 | 1.00 |
| 5.5 | −1.16 | −0.18 | −0.49 | 1.00 |
| 6 | −1.47 | −0.48 | −1.42 | 0.99 |
| 6.5 | −1.75 | −0.75 | −2.25 | 0.99 |
| 7 | −1.91 | −0.91 | −2.72 | 0.99 |
| 7.5 | −1.97 | −0.97 | −2.92 | 0.99 |
| 8 | −2.00 | −1.01 | −3.01 | 0.97 |
| 8.5 | −2.06 | −1.06 | −3.1 | 0.93 |
| 9 | −2.16 | −1.17 | −3.32 | 0.79 |
| 9.5 | −2.39 | −1.39 | −3.81 | 0.52 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
García-González, V.; Mas-Oliva, J. Amyloidogenic Properties of a D/N Mutated 12 Amino Acid Fragment of the C-Terminal Domain of the Cholesteryl-Ester Transfer Protein (CETP). Int. J. Mol. Sci. 2011, 12, 2019-2035. https://doi.org/10.3390/ijms12032019
García-González V, Mas-Oliva J. Amyloidogenic Properties of a D/N Mutated 12 Amino Acid Fragment of the C-Terminal Domain of the Cholesteryl-Ester Transfer Protein (CETP). International Journal of Molecular Sciences. 2011; 12(3):2019-2035. https://doi.org/10.3390/ijms12032019
Chicago/Turabian StyleGarcía-González, Victor, and Jaime Mas-Oliva. 2011. "Amyloidogenic Properties of a D/N Mutated 12 Amino Acid Fragment of the C-Terminal Domain of the Cholesteryl-Ester Transfer Protein (CETP)" International Journal of Molecular Sciences 12, no. 3: 2019-2035. https://doi.org/10.3390/ijms12032019