Structural Determination of Three Different Series of Compounds as Hsp90 Inhibitors Using 3D-QSAR Modeling, Molecular Docking and Molecular Dynamics Methods

Abstract
:1. Introduction
2. Material and Methods
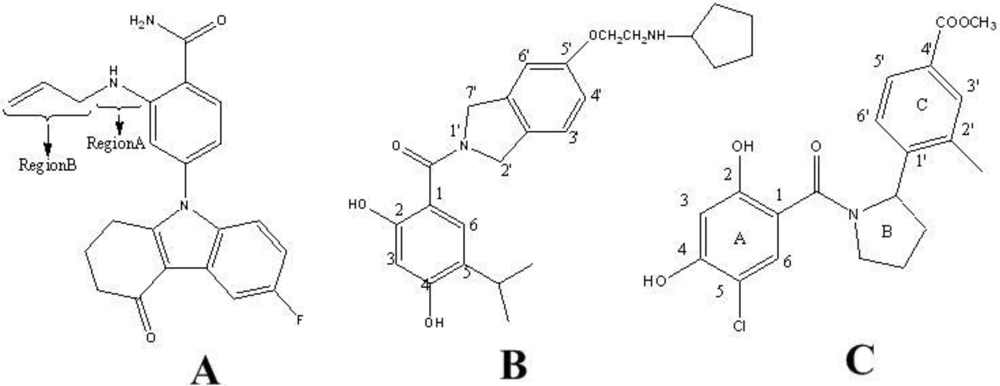
2.1. Data Sets and Biological Activity
2.2. Conformational Sampling and Alignment
2.3. 3D-QSAR Analysis
2.4. Molecular Docking
2.5. Molecular Dynamics Simulations
3. Results and Discussion
3.1. CoMFA and CoMSIA Statistical Results
3.2. Validation of the 3D QSAR Models
3.2.1. BT
3.2.2. AT
3.2.3. DA
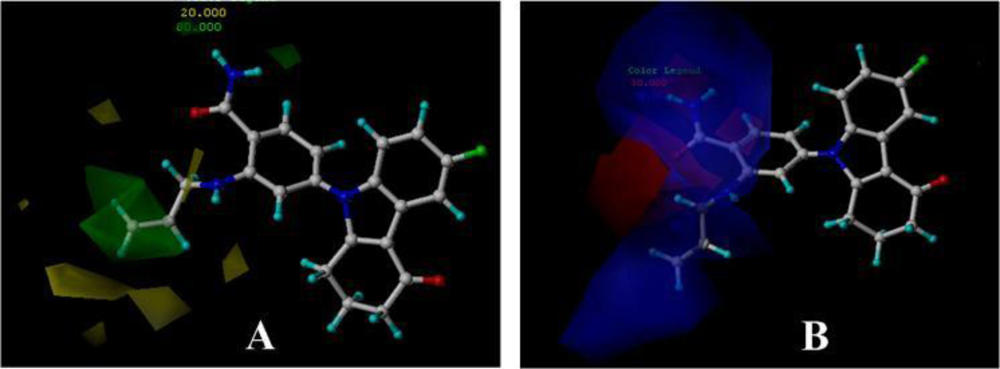
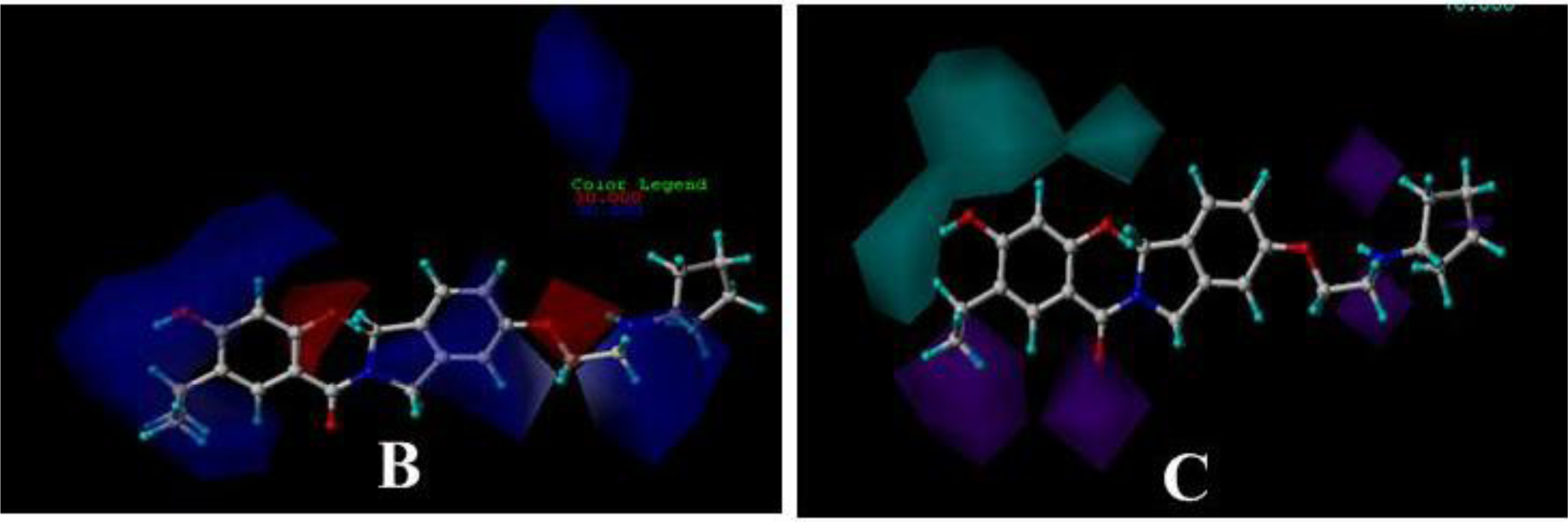
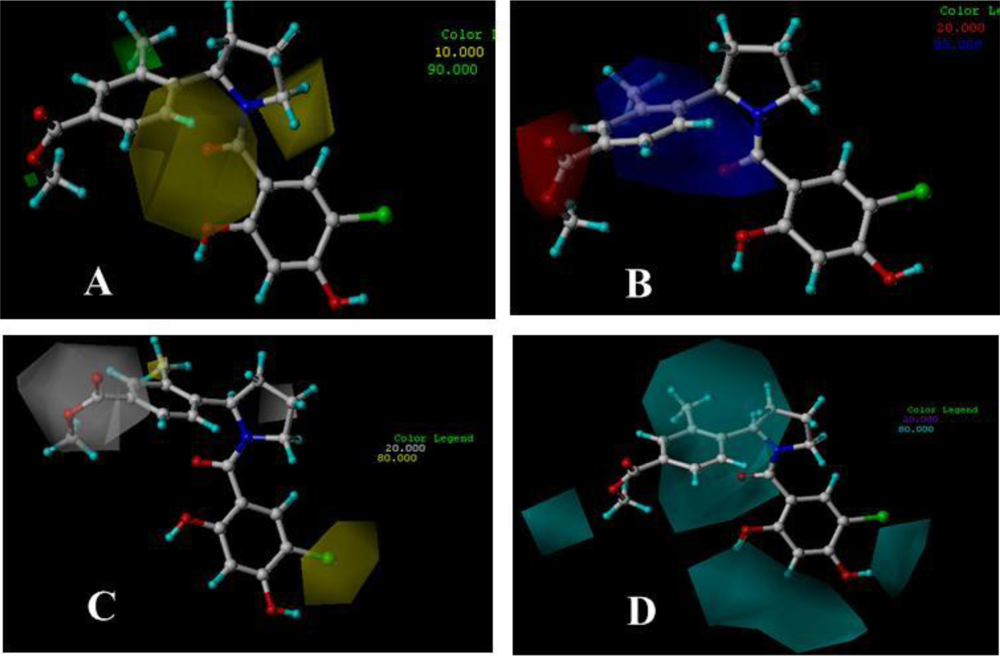
3.3. 3D-QSAR Contour Maps
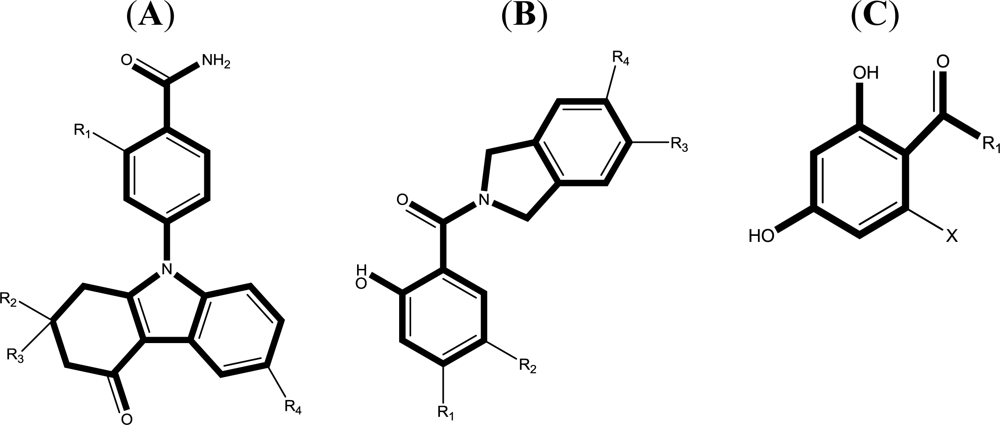
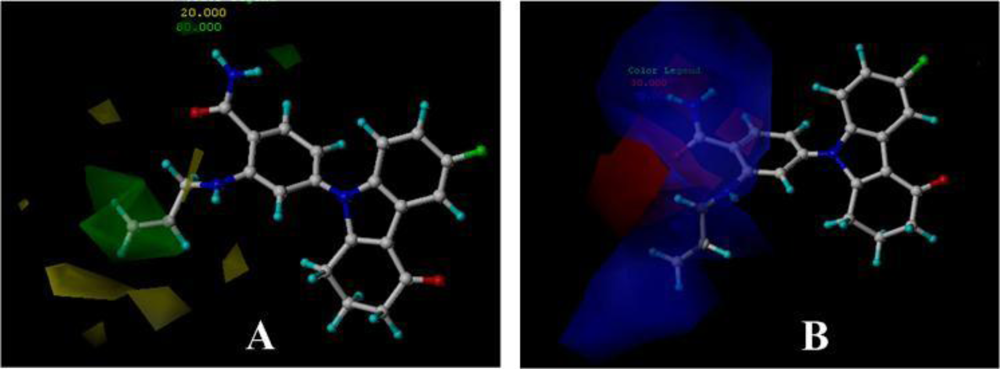
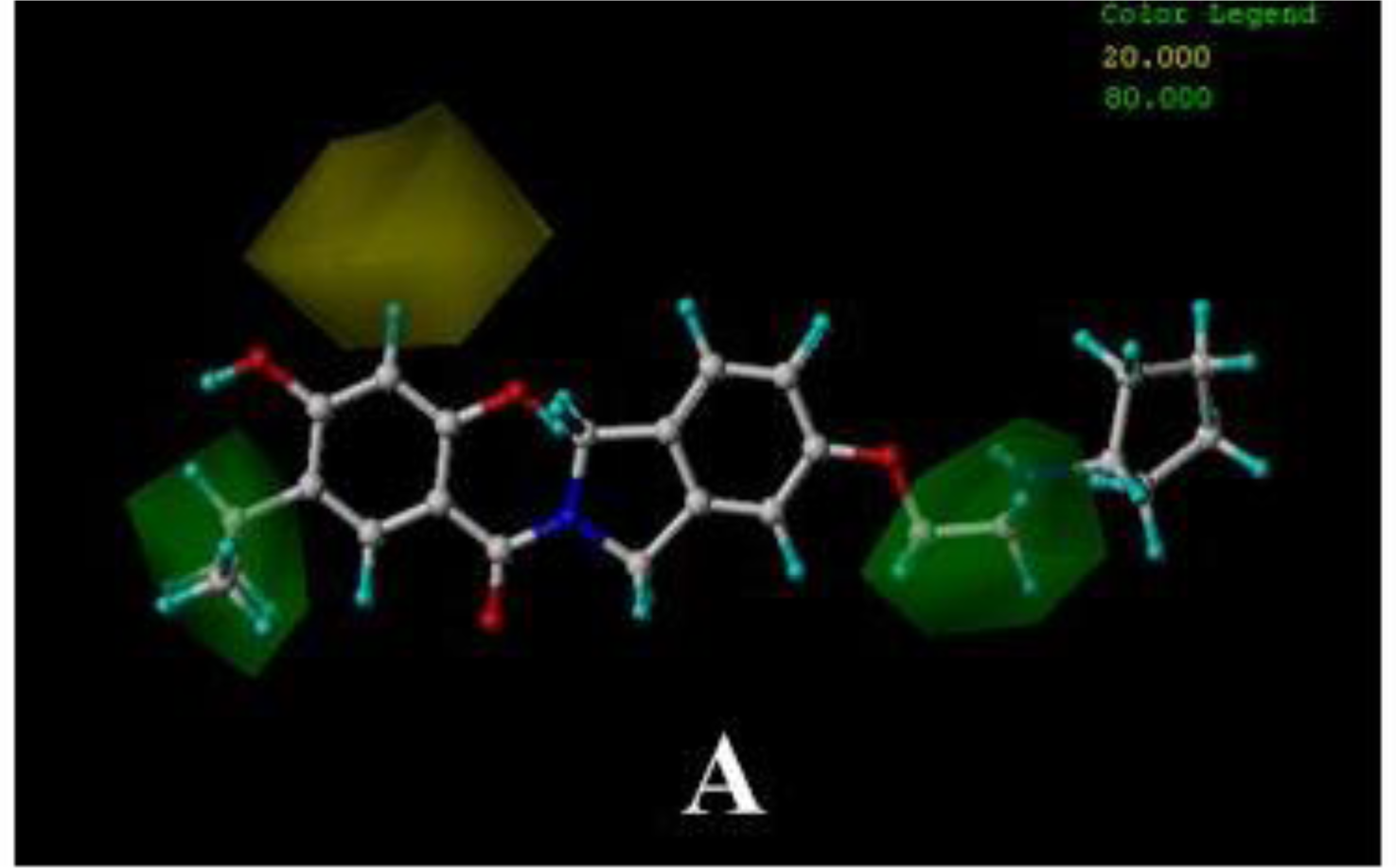
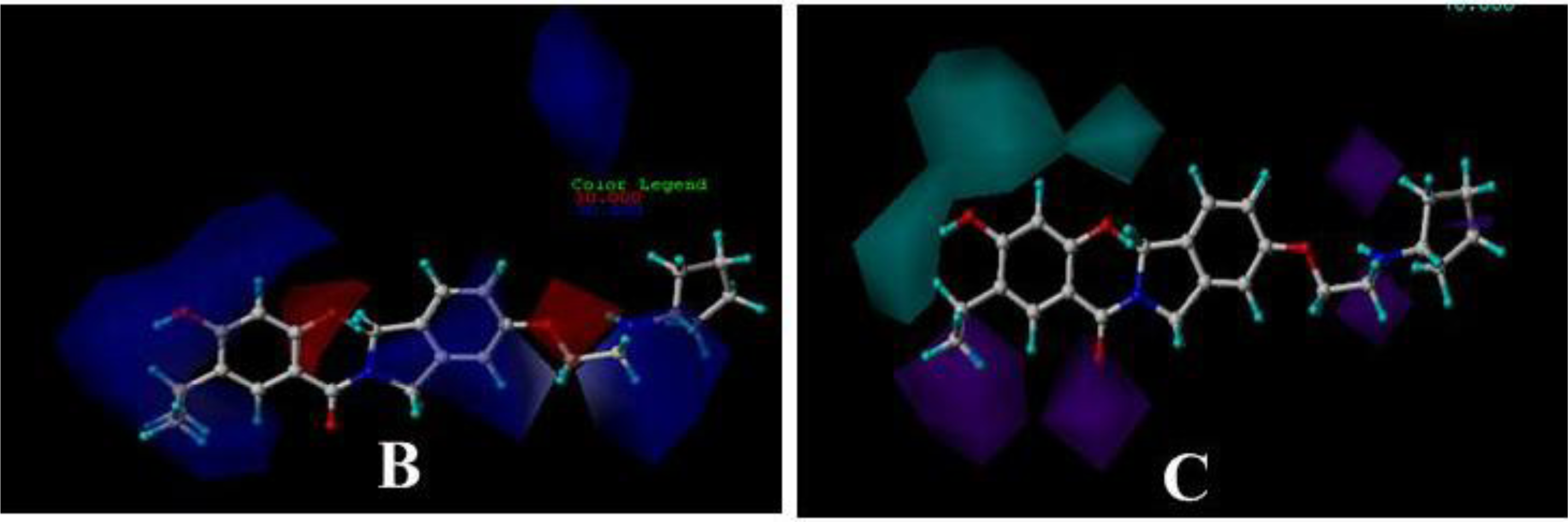
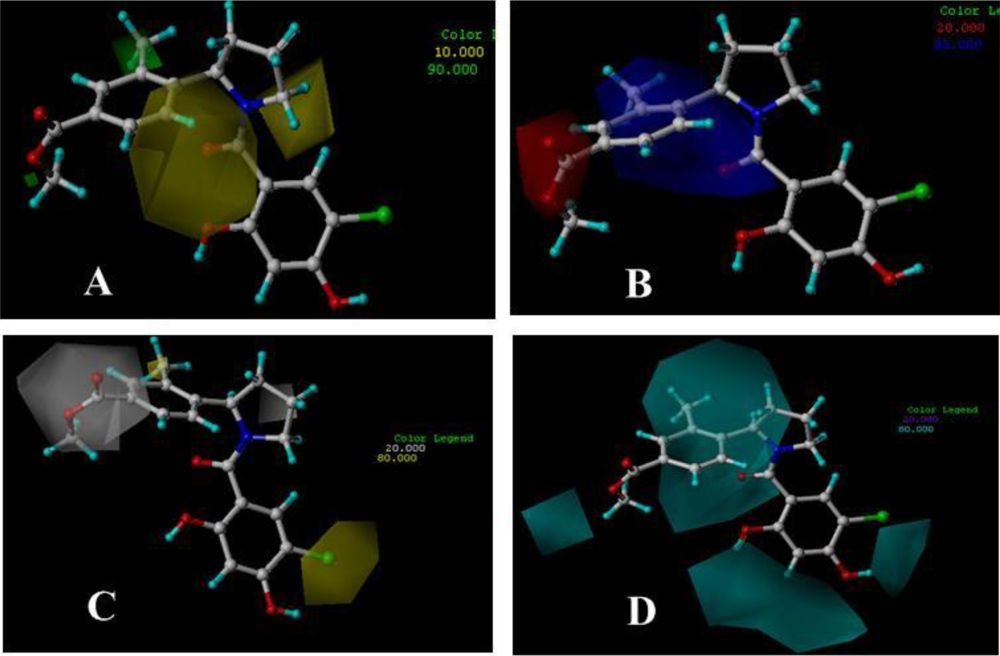
3.3.1. BT
3.3.2. AT
3.3.3. DA
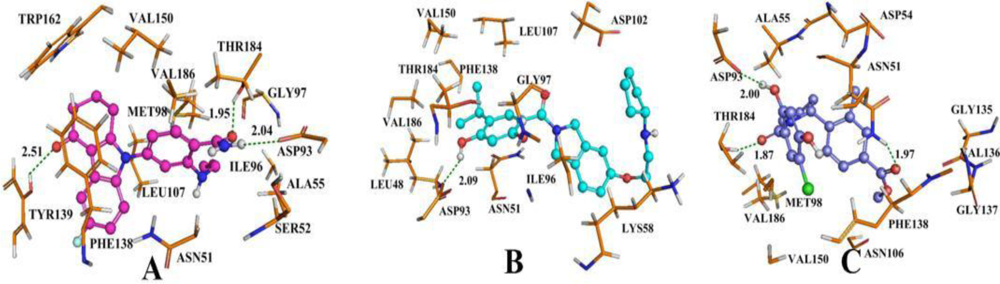
3.4. Docking Analysis and Comparison with 3D Contour Maps
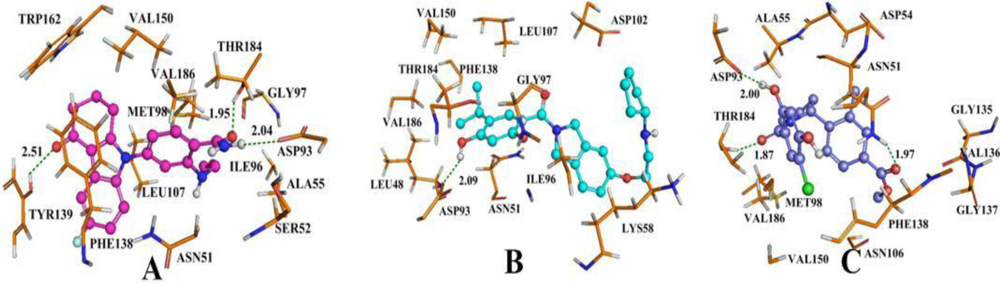
3.4.1. BT
3.4.2. AT
3.4.3. DA
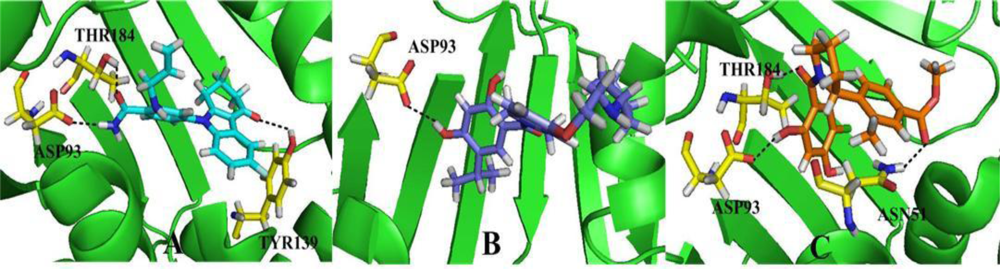
3.5. Comparison of Binding Modes for Each Class
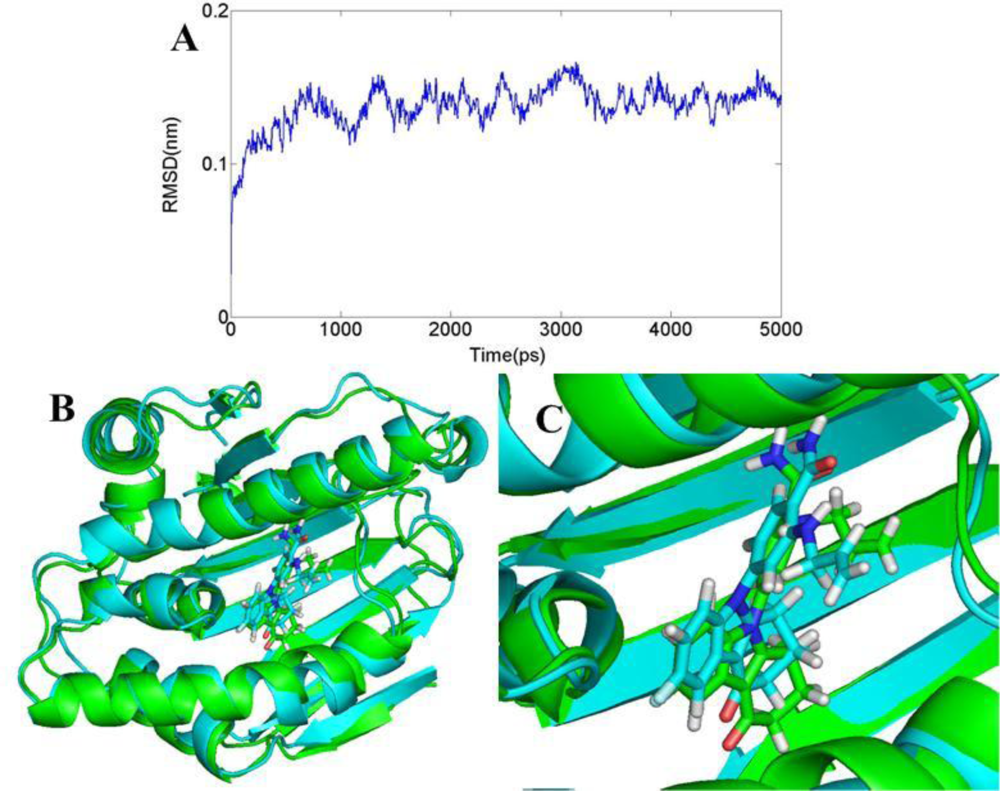
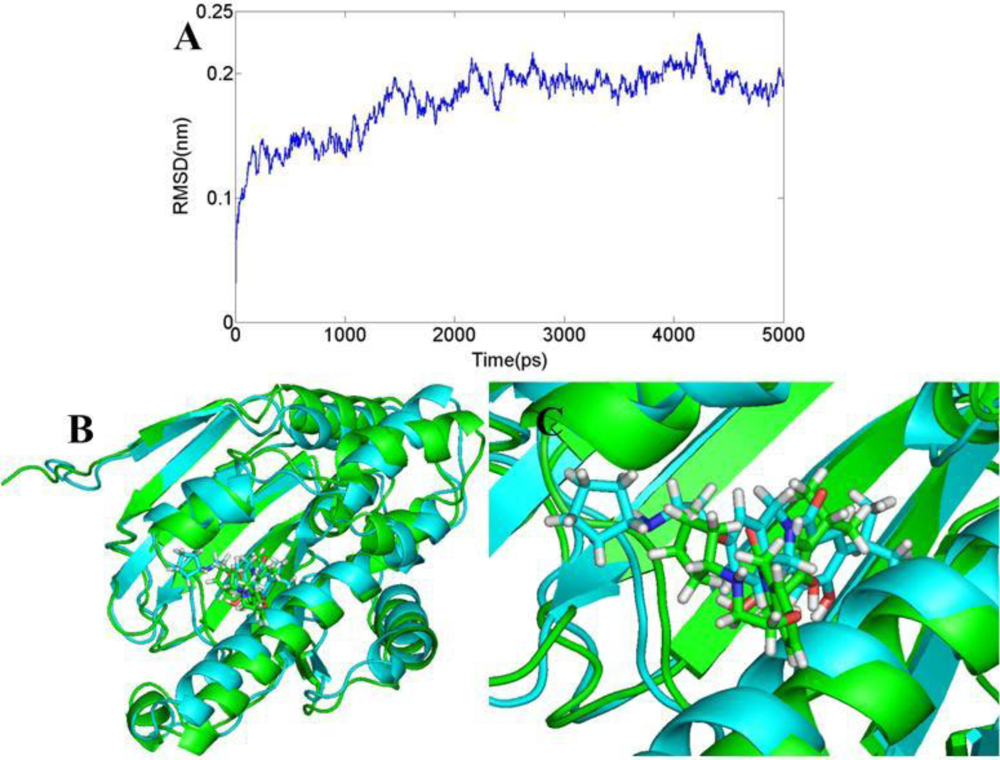
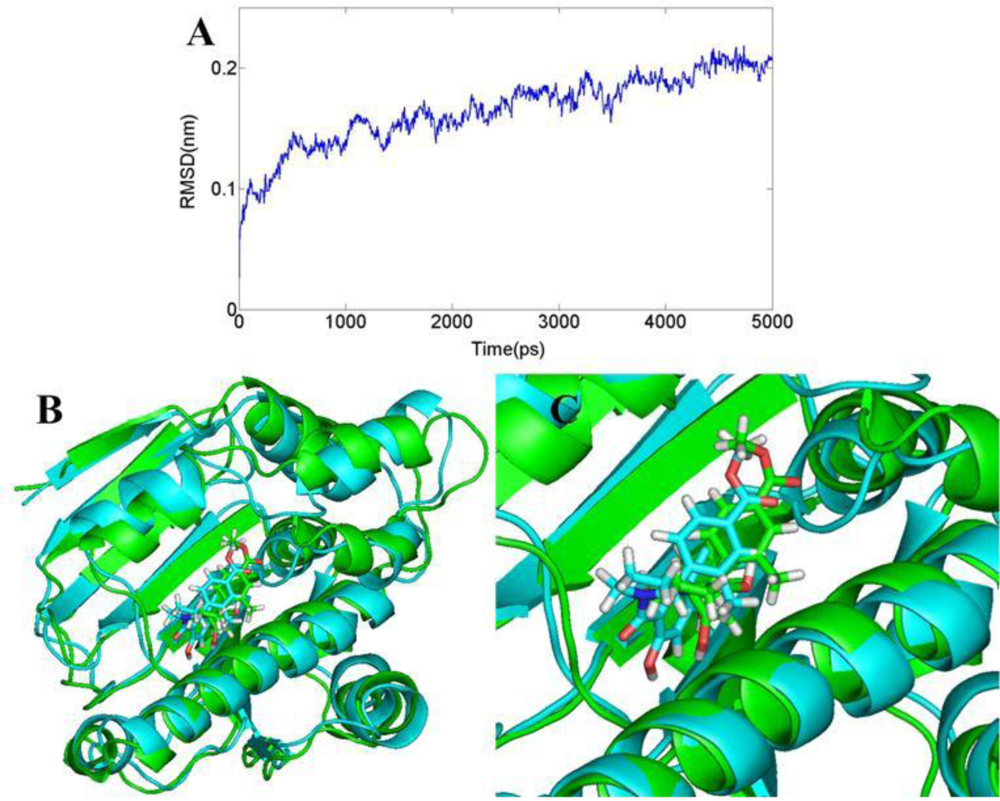
3.6. Molecular Dynamics Simulations
3.6.1. BT
3.6.2. AT
3.6.3. DA
4. Conclusions
Supplementary Material
Acknowledgments
References
- Smith, DF; Whitesell, L; Katsanis, E. Molecular chaperones: biology and prospects for pharmacological intervention. Pharmacol. Rev 1998, 50, 493–513. [Google Scholar]
- Smith, DF. Chaperones in signal transduction. In Molecular Chaperones in the Cell; Lund, P, Ed.; Oxford University Press: Oxford, UK, 2001; pp. 165–178. [Google Scholar]
- Jolly, C; Morimoto, RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst 2000, 92, 1564–1572. [Google Scholar]
- Solit, DB; Chiosis, G. Development and application of Hsp90 inhibitors. Drug Discov. Today 2008, 13, 38–43. [Google Scholar]
- Neckers, L. Heat shock protein 90: the cancer chaperone. J. Biosci 2007, 32, 517–530. [Google Scholar]
- Pratt, WB. The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc. Soc. Exp. Biol. Med 1998, 217, 420–434. [Google Scholar]
- Maloney, A; Workman, P. Hsp90 as a new therapeutic target for cancer therapy: The story unfolds. Expert Opin. Biol. Ther 2002, 2, 3–24. [Google Scholar]
- Conroy, SE; Latchman, DS. Do heat shock proteins have a role in breast cancer? Br. J. Cancer 1996, 74, 717–721. [Google Scholar]
- Ferranini, M; Helta, S; Zocchi, MR; Rugarli, C. Unusual expression and localization of heat shock proteins in human tumour cells. Int. J. Cancer 1992, 16, 613–619. [Google Scholar]
- Kawanishi, K; Shiozaki, H; Doki, Y; Sakita, I; Inoue, M; Yano, M; Tsujinaka, T; Shamma, A; Monden, M. Prognostic significance of heat shock proteins 27 and 70 in patients with squamous cell carcinoma of the esophagus. Cancer 1999, 85, 1649–1657. [Google Scholar]
- Jameel, A; Skilton, RA; Campbell, TA; Chander, SK; Coombes, RC; Luqmani, YA. Clinical and biological significance of Hsp89a in human breast cancer. Int. J. Cancer 1992, 50, 409–415. [Google Scholar]
- Lebeau, J; Le Cholony, C; Prosperi, MT; Goubin, G. Constitutiveoverexpression of 89 kDa heat shock protein gene in the HBL100 mammary cell line converted to a tumorigenic phenotype by the EJ/T24 Harvey-ras oncogene. Oncogene 1991, 6, 1125–1132. [Google Scholar]
- Wataba, K; Saito, T; Fukunaka, K; Nishimura, M; Kudo, R. Overexpression of heat shock proteins in carcinogenic endometrium. Int. J. Cancer 2001, 91, 448–456. [Google Scholar]
- Cornford, PA; Dodson, AR; Parsons, KF; Desmond, AD; Woolfenden, A; Fordham, M; Neoptolemos, JP; Ke, Y; Foster, CS. Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res 2000, 60, 7099–7105. [Google Scholar]
- Biamonte, MA; Van de Water, R; Joseph, WA; Scannevin, RH; Perret, D; Lee, WC. Heat shock Protein 90: Inhibitors in clinical trials. J. Med. Chem 2010, 53, 3–17. [Google Scholar]
- Porter, JR; Fritz, CC; Depew, KM. Discovery and development of Hsp90 inhibitors: A promising pathway for cancer therapy. Curr. Opin. Chem. Biol 2010, 14, 1–9. [Google Scholar]
- Whitesell, L; Lindquist, SL. Hsp90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar]
- Wandinger, SK; Richter, K; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem 2008, 283, 18473–18477. [Google Scholar]
- BeBoer, C; Dietz, A. The description and antibiotic production of Streptomyces hygroscopicus var, Geldanus. J. Antibiot 1976, 29, 1182–1188. [Google Scholar]
- Supko, JG; Hickman, RL; Grever, MR; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol 1997, 36, 305–315. [Google Scholar]
- Kelland, LR; Sharp, SY; Rogers, PM; Myers, TG; Workman, P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J. Natl. Cancer Inst 1999, 91, 1940–1949. [Google Scholar]
- Chiosis, G; Huezo, H; Rosen, N; Whitesell, E; Mimnaugh, L; Neckers, L. 17AAG: Low target binding affinity and potent cell activity-finding an explanation. Mol. Cancan Ther 2003, 2, 123–129. [Google Scholar]
- Chiosis, G. Discovery and development of purine-scaffold Hsp90 inhibitors. Curr. Top. Med. Chem 2006, 6, 1183–1191. [Google Scholar]
- Chiosis, G; Timaul, MN; Lucas, B; Munster, PN; Zheng, FF; Sepp-Lorenzino, L; Rosen, N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol 2001, 8, 289–299. [Google Scholar]
- Radanyi, C; Le Bras, G; Marsaud, V; Peyrat, JF; Messaoudi, S; Catelli, MG; Brion, JD; Alami, M; Renoir, JM. Antiproliferative and apoptotic activities of tosylcyclonovobiocic acids as potent heat shock protein 90 inhibitors in human cancer cells. Cancer Lett 2009, 274, 88–94. [Google Scholar]
- Dymock, B; Barril, X; Beswick, M; Collier, A; Davies, N; Drysdale, M; Fink, A; Fromont, C; Hubbard, ER; Massey, A; Surgenor, A; Wright, L. Adenine derived inhibitors of the molecular chaperone HSP90-SAR explained through multiple X-ray structures. Bioorg. Med. Chem. Lett 2004, 14, 325–328. [Google Scholar]
- Sanam, R; Tajne, S; Gundla, R; Vadivelan, S; Machiraju, PK; Dayam, R; Narasu, L; Jagarlapudi, S; Neamati, N. Combined pharmacophore and structure-guided studies to identify diverse HSP90 inhibitors. J. Mol. Graph. Model 2010, 28, 472–477. [Google Scholar]
- Jadhav, VD; Duerfeldt, AS; Blagg, BSJ. Design, synthesis, and biological activity of bicyclic radester analogues. Bioorg. Med. Chem. Lett 2009, 19, 6845–6850. [Google Scholar]
- Chen, CY; Chen, CYC. Insights into designing the dual-targeted HER2/HSP90 inhibitors. J. Mol. Graph. Model 2010, 29, 21–31. [Google Scholar]
- Liu, HC; Lyu, PC; Leong, MK; Tsai, KC; Hsiue, GH. 3D-QSAR studies on PU3 analogues by comparative molecular field analysis. Bioorg. Med. Chem. Lett 2004, 14, 731–734. [Google Scholar]
- Roy, KK; Singh, S; Saxena, AK. Integration-mediated prediction enrichment of quantitative model for Hsp90 inhibitors as anti-cancer agents: 3D-QSAR study. Mol Divers 2010. [CrossRef]
- Barta, TE; Veal, JM; Rice, JW; Partridge, JM; Fadden, RP; Ma, W; Jenks, M; Geng, LF; Hanson, GJ; Huang, KH; et al. Discovery of benzamide tetrahydro-4H-carbazol-4-ones as novel small molecule inhibitors of Hsp90. Bioorg. Med. Chem. Lett 2008, 18, 3517–3521. [Google Scholar]
- Woodhead, AJ; Angove, H; Carr, MG; Chessari, G; Congreve, M; Coyle, JE; Cosme, J; Graham, B; Day, PJ; Downham, R; et al. Discovery of (2,4-Dihydroxy-5-isopropylphenyl) [5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl] methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem 2010, 53, 5956–5969. [Google Scholar]
- Kung, PP; Funk, L; Meng, J; Collins, M; Zhou, JZX; Johnson, MC; Ekker, A; Wang, J; Mehta, P; Yin, MJ; et al. Dihydroxylphenyl amides as inhibitors of the Hsp90 molecular chaperone. Bioorg. Med. Chem. Lett 2008, 18, 6273–6278. [Google Scholar]
- Cramer, RD, III; Bunce, JD; Patterson, DE. Cross validation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies. Quant. Struct. Act. Relat 1988, 7, 18–25. [Google Scholar]
- Klebe, G; Abraham, U; Mietzner, T. Molecular similarity in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem 1994, 37, 4130–4146. [Google Scholar]
- Martin, YC. 3D-QSAR: Current state, scope, and limitations. Perspect. Drug Disc. Des 1998, 12, 3–23. [Google Scholar]
- Matthew, C; Cramer, RD, III; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem 1989, 10, 982–1012. [Google Scholar]
- Zaheer-ul, H; Uddin, R; Yuan, H; Petukhov, PA; Choudhary, MI; Madura, JD. Receptor-based modeling and 3D-QSAR for a quantitative production of the Butyrylcholinesterase inhibitors based on genetic algorithm. J. Chem. Inf. Model 2008, 48, 1092–1103. [Google Scholar]
- Wold, S; Ruhe, A; Wold, H; Dunn, WJ, III. The collinearity problemin linear regression. The partial least squares (PLS) approach to generalized inverses SIAM. J. Sci. Stat. Comput 1984, 5, 735–743. [Google Scholar]
- Kirkpatrick, P. Virtual screening: Gliding to success. Nat. Rev. Drug Discov 2004, 3, 299. [Google Scholar]
- Homepage of RCSB Protein Data Bank. Available online: http://www.pdb.org (accessed on 27 January 2011).
- Welch, W; Ruppert, J; Jain, AN. Hammerhead: Fast, fully automated docking of flexible ligands to protein binding sites. Chem. Biol 1996, 3, 449–462. [Google Scholar]
- Lindahl, E; Hess, B; Van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model 2001, 7, 306–317. [Google Scholar]
- Van der Spoel, D; Van Buuren, AR; Tieleman, DP; Berendsen, HJ. Molecular dynamics simulations of peptides from BPTI: A closer look at amide-aromatic interactions. J. Biomol. NMR 1996, 8, 229–238. [Google Scholar]
- Darden, T; York, D; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Berendsen, HJC; Grigerra, JR; Straatsma, TPJ. The missing term in effective pair potential. Phys. Chem 1987, 91, 6269–6271. [Google Scholar]
- Hess, B; Bekker, H; Berendsen, HJC. Fraaije JGEM Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Wang, GX; Li, Y; Liu, XL; Wang, YH. Understanding the aquatic toxicity of pesticide: Structure-activity relationship and molecular descriptors to distinguish the ratings of toxicity. QSAR Comb. Sci 2009, 28, 1418–1431. [Google Scholar]
- Bringmann, G; Rummey, CJ. 3D-QSAR Investigations on antimalarial naphthylisoquinoline alkaloids, by Comparative Molecular Similarity Indices Analysis (CoMSIA), based on different alignment approaches. J. Chem. Inf. Comput. Sci 2003, 43, 304–316. [Google Scholar]
- Bohm, M; St rzebecher, J; Klebe, G. Three-dimensional quantitative structure activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem 1999, 42, 458–477. [Google Scholar]
- Du, J; Qin, J; Liu, HX; Yao, XJ. 3D-QSAR and molecular docking studies of selective agonists for the thyroidhormone receptor β. J. Mol. Graph. Model 2008, 27, 95–104. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Benzamide tetrahydro-4H-carbazol-4-one analogs | AT13387 | Dihydroxylphenyl amides |
|---|---|---|---|
| CoMFA | CoMSIA | CoMSIA | |
| R2cv | 0.482 | 0.715 | 0.645 |
| R2ncv | 0.903 | 0.892 | 0.858 |
| SEE | 0.22 | 0.304 | 0.478 |
| F | 78.818 | 86.941 | 60.608 |
| R2pred | 0.5747 | 0.7013 | 0.7177 |
| SEP | 0.507 | 0.494 | 0.757 |
| Nc | 2 | 2 | 2 |
| Field Contribution | |||
| S | 0.825 | 0.179 | 0.153 |
| E | 0.175 | 0.322 | 0.285 |
| H | - | - | 0.29 |
| D | - | 0.499 | 0.273 |
| A | - | - | - |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Wang, F.; Ma, Z.; Wang, X.; Wang, Y. Structural Determination of Three Different Series of Compounds as Hsp90 Inhibitors Using 3D-QSAR Modeling, Molecular Docking and Molecular Dynamics Methods. Int. J. Mol. Sci. 2011, 12, 946-970. https://doi.org/10.3390/ijms12020946
Liu J, Wang F, Ma Z, Wang X, Wang Y. Structural Determination of Three Different Series of Compounds as Hsp90 Inhibitors Using 3D-QSAR Modeling, Molecular Docking and Molecular Dynamics Methods. International Journal of Molecular Sciences. 2011; 12(2):946-970. https://doi.org/10.3390/ijms12020946
Chicago/Turabian StyleLiu, Jianling, Fangfang Wang, Zhi Ma, Xia Wang, and Yonghua Wang. 2011. "Structural Determination of Three Different Series of Compounds as Hsp90 Inhibitors Using 3D-QSAR Modeling, Molecular Docking and Molecular Dynamics Methods" International Journal of Molecular Sciences 12, no. 2: 946-970. https://doi.org/10.3390/ijms12020946