Design of New Competitive Dengue Ns2b/Ns3 Protease Inhibitors—A Computational Approach

Abstract
:1. Introduction
2. Results and Discussion
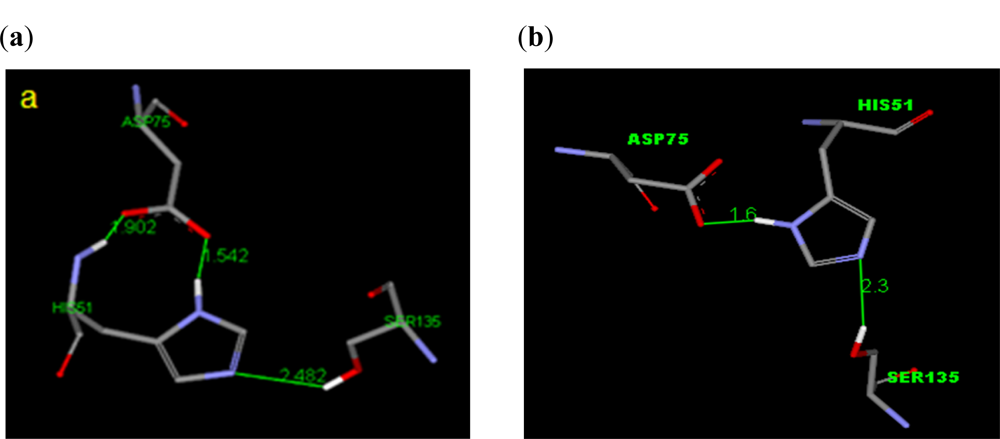
2.1. Spatial Arrangement at the Active Site of the DEN2 NS2B/NS3 Protease
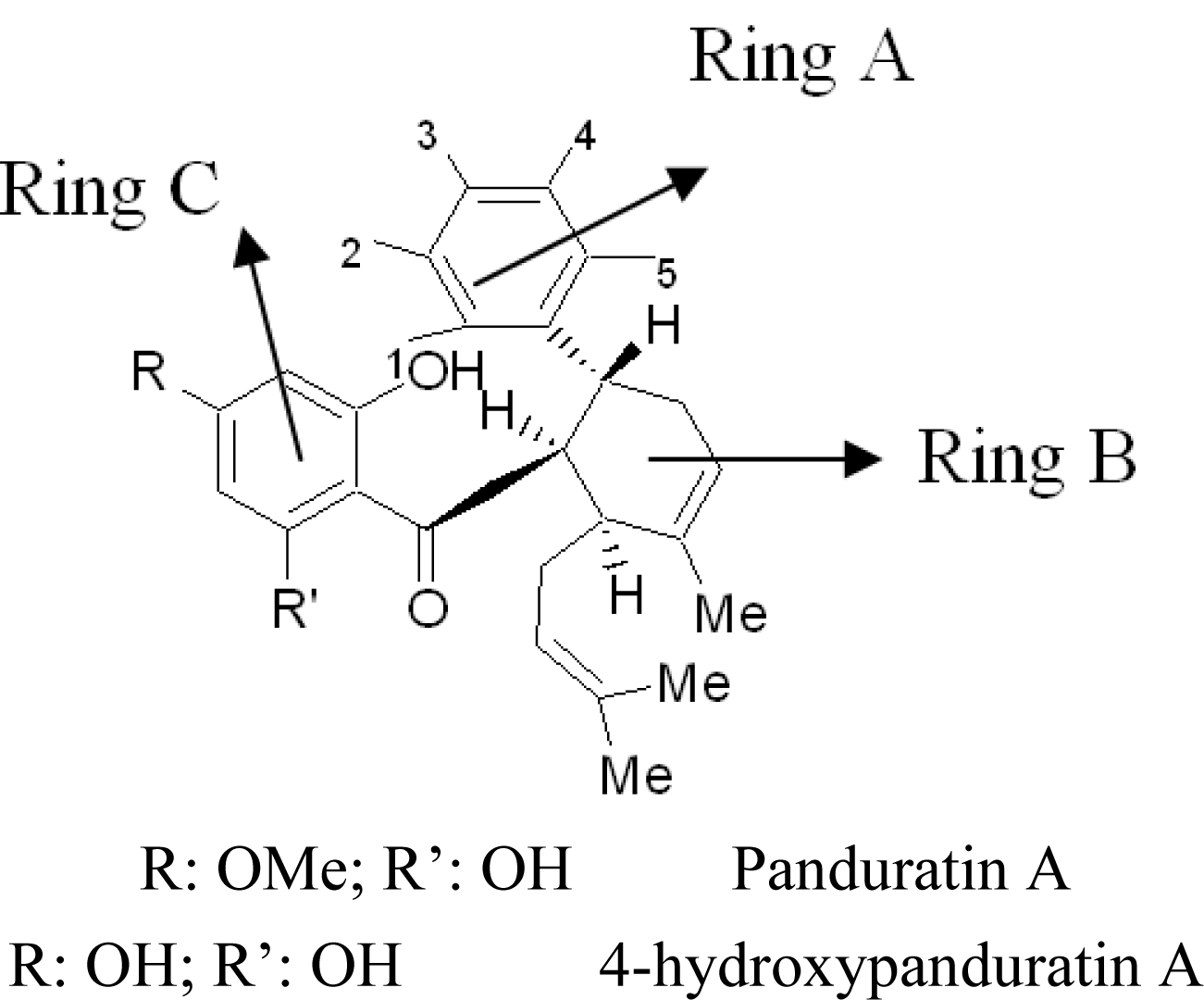
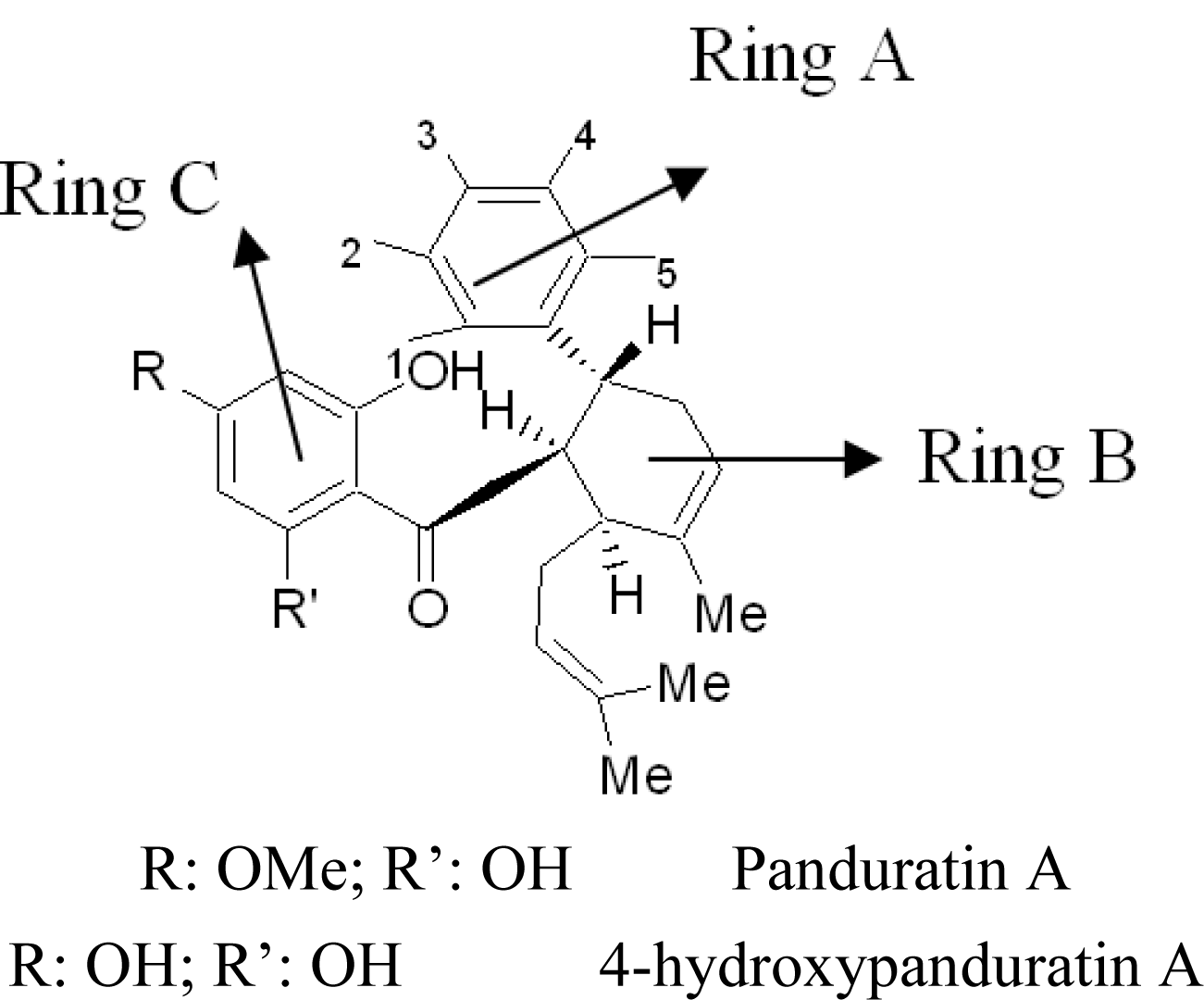
2.2. Competitive Inhibitors Extracted from Boesenbergia Rotunda
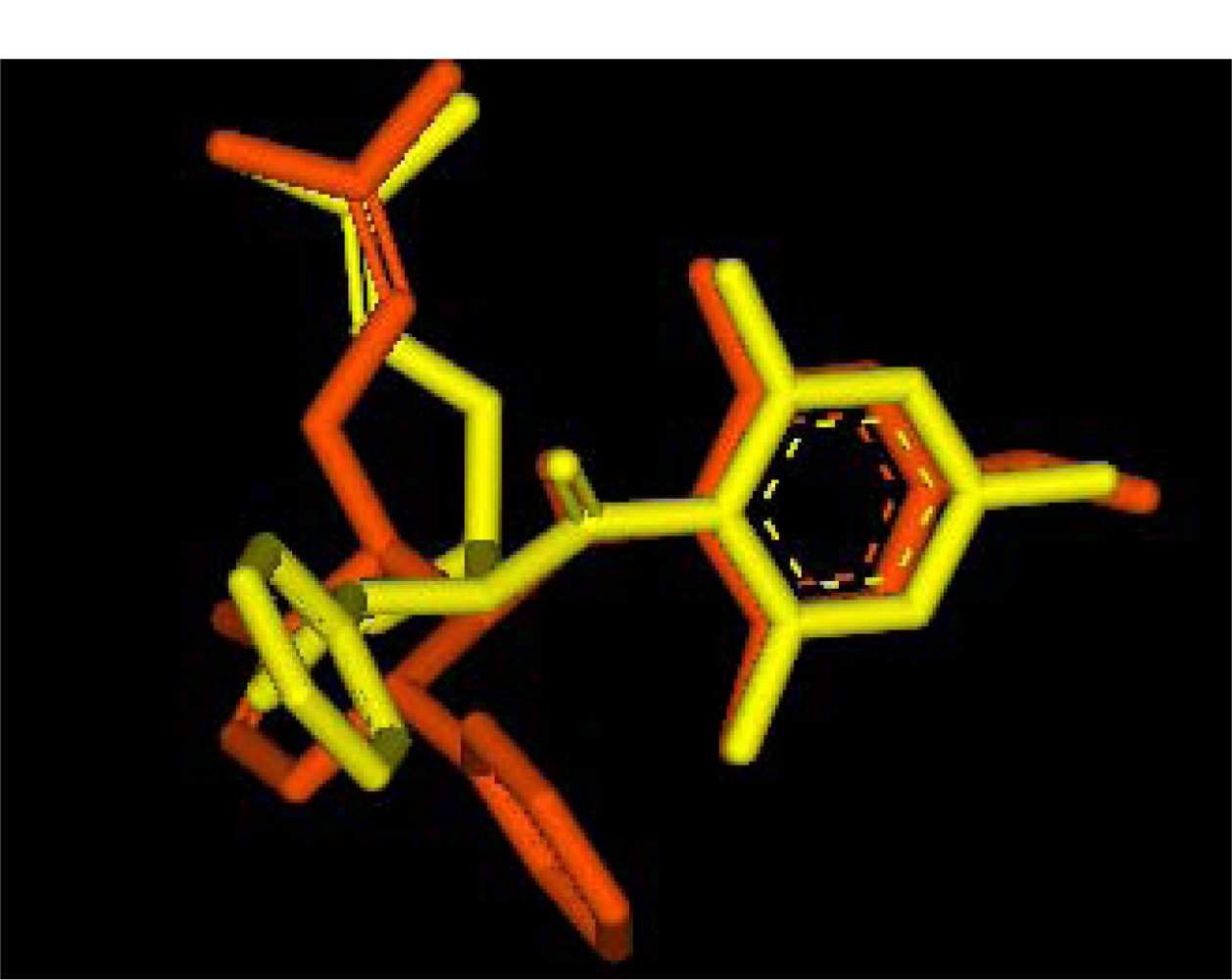
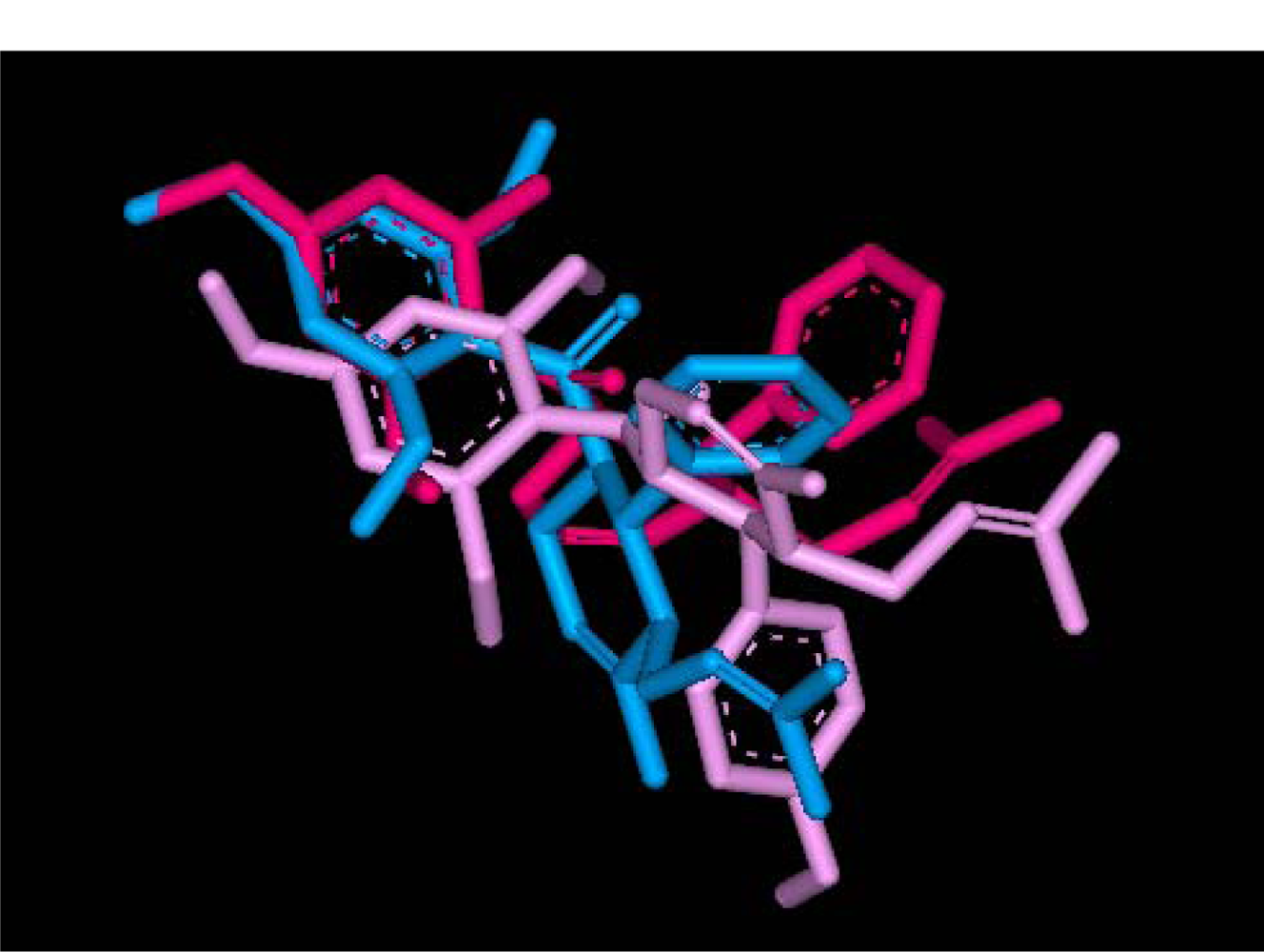
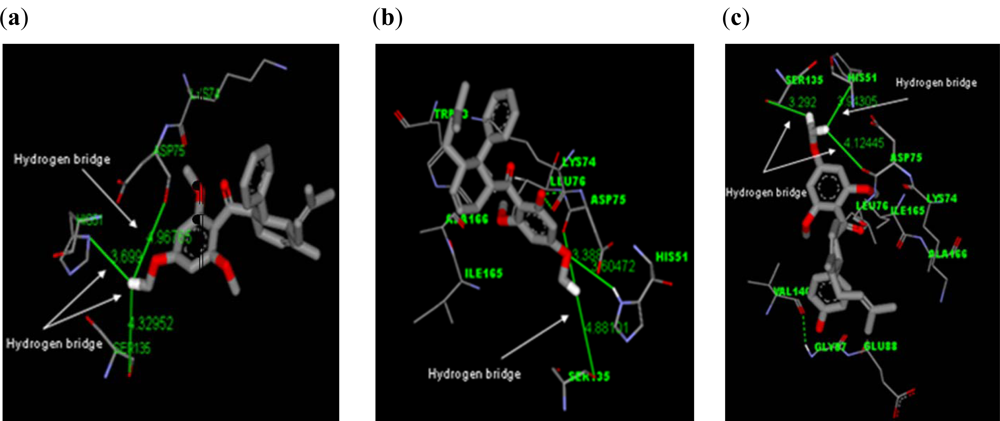
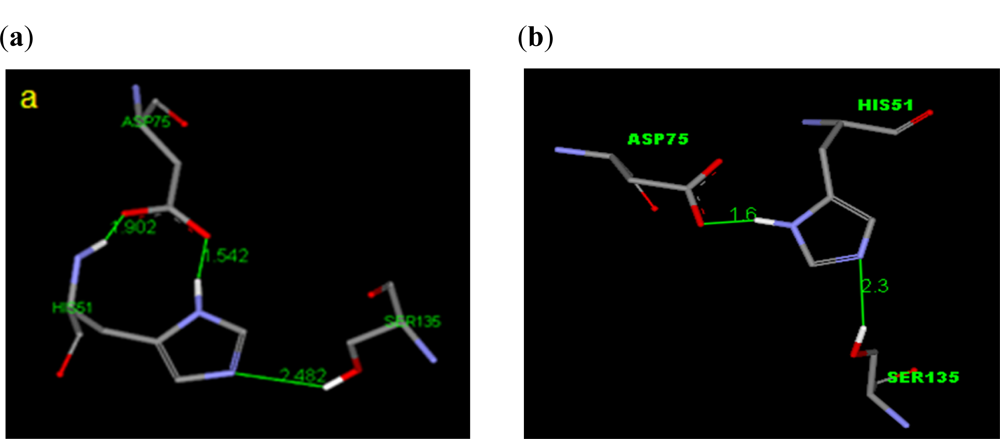
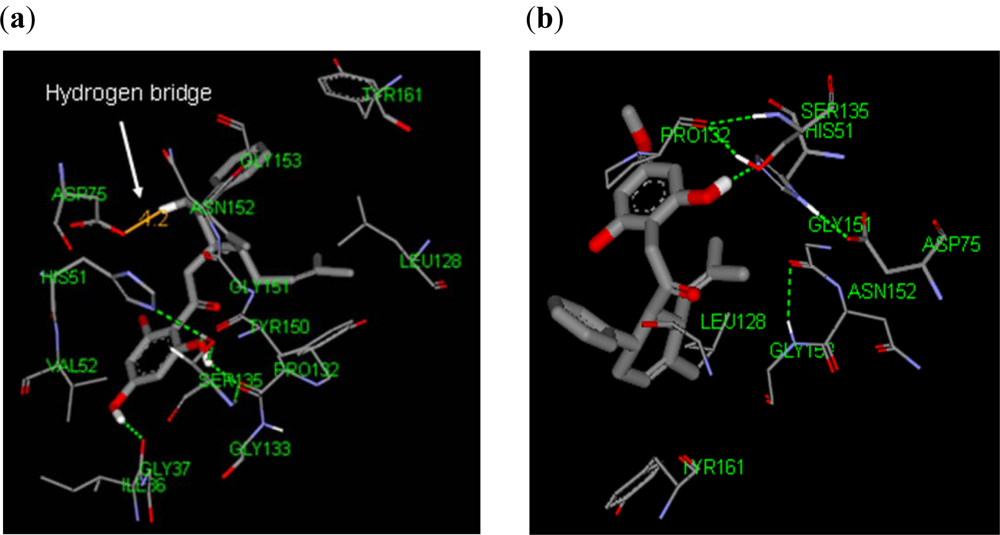
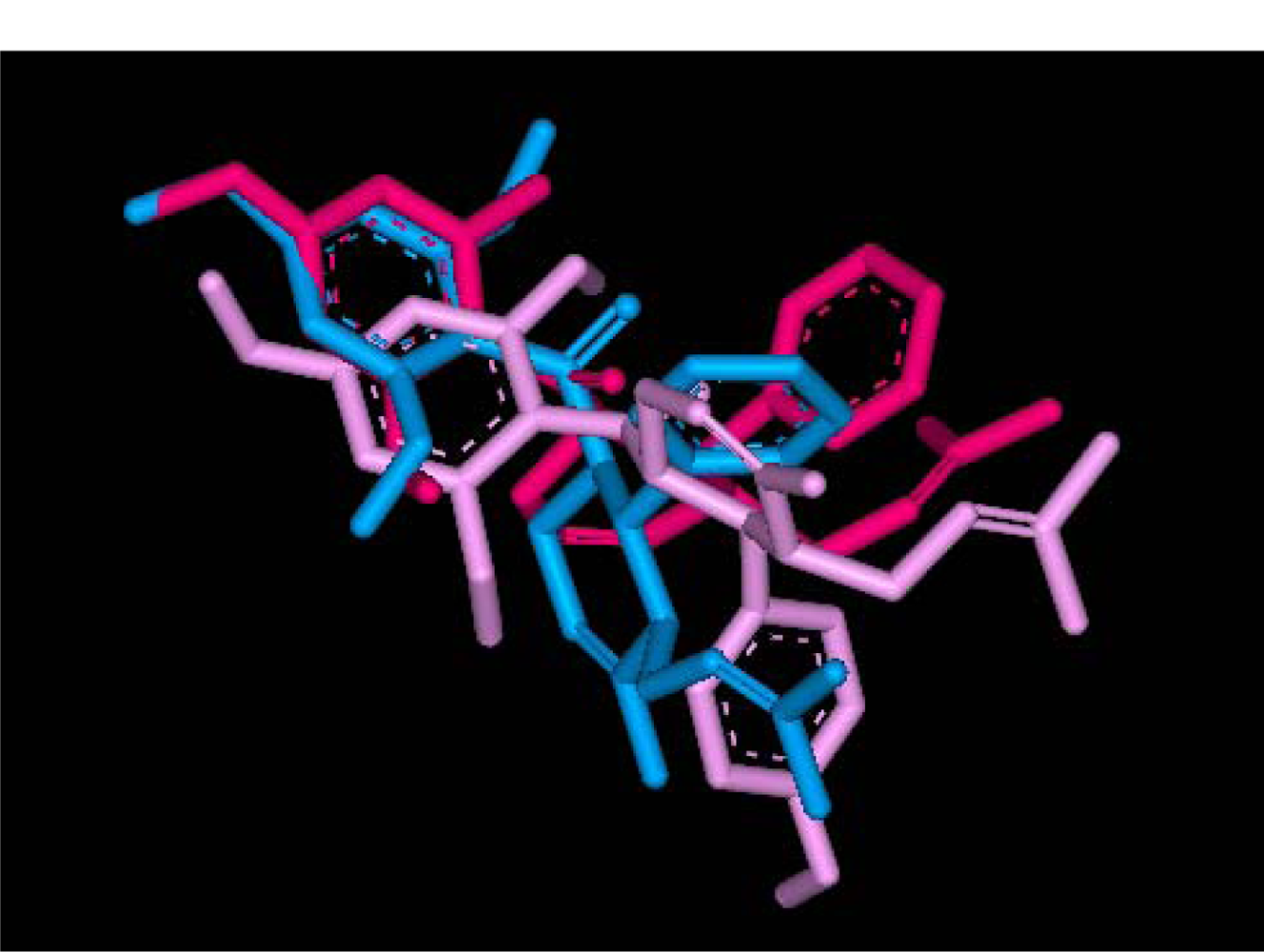
2.2.1. Docking of 4-Hydroxypanduratin A and Panduratin A to DEN2 NS2B/NS3
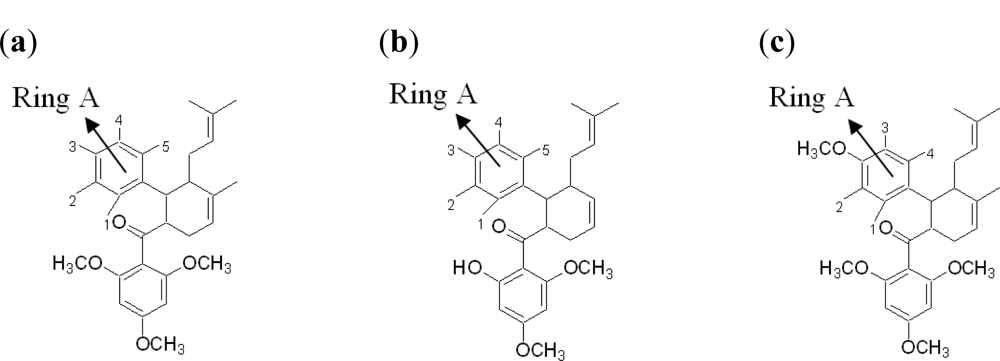
2.2.2. Design of New Dengue Virus Inhibitors Based on 4-Hydroxypanduratin A and Panduratin A
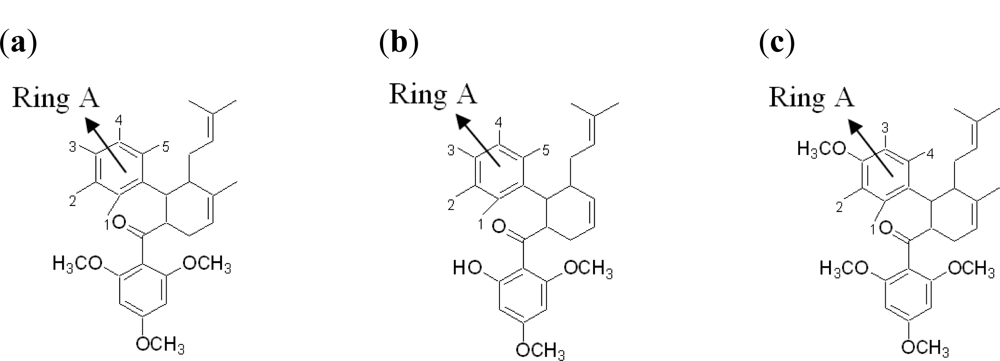
2.3. Synthetic Competitive Dengue Virus Inhibitors
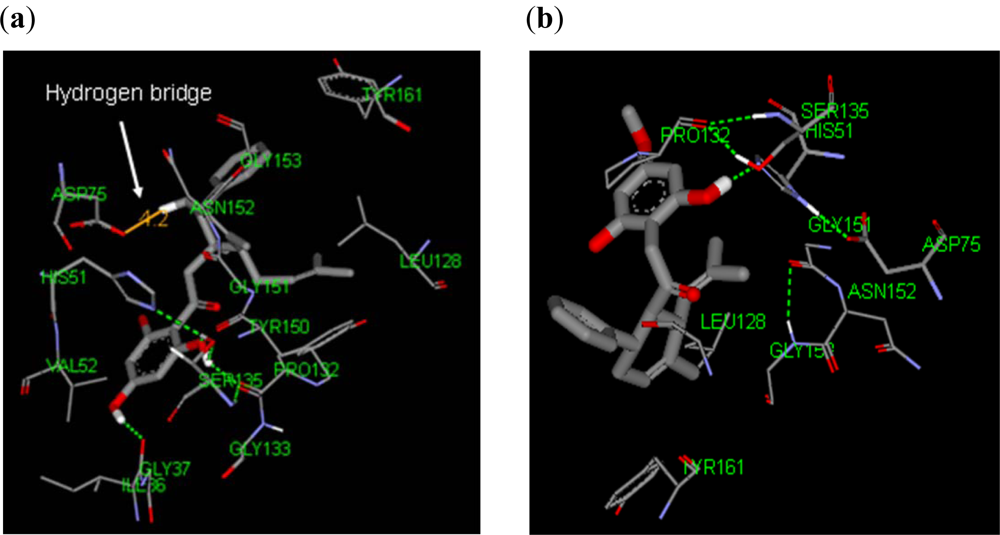
2.3.1. Docking of 246DA, 2446DA and 20H46DA to DEN2 NS2B/NS3
2.3.2. Design of New Dengue Virus Inhibitors Based on 246DA, 2446DA and 20H46DA
3. Experimental Section
3.1. Receptor 3D Structure
3.2. Docking of Ligands (Stage 1)
3.3. Computation of Interaction Energy and Binding Energy (Stage 1)
3.4. Selection of Possible Active Inhibitors (Stage 1)
3.5. Design of New Dengue Virus Inhibitors (Stage 2)
4. Conclusions
Acknowledgments
References
- Morens, D; Fauci, A. Dengue and hemorrhagic fever: A potential threat to public health in United States. J. Am. Med. Assoc 2008, 299, 214–216. [Google Scholar]
- Irie, K; Mohan, PM; Sasaguri, Y; Putnak, R; Padmanabhan, R. Sequence analysis of cloned dengue virus type 2 genome (new guinea-C strain). Gene 1989, 75, 197–211. [Google Scholar]
- Bazan, JR; Fletterick, R. Detection of a trypsin like serine protease domain in flaviviruses and pestiviruses. Virology 1989, 171, 637–639. [Google Scholar]
- Chambers, TJ; Nestorowicz, A; Amberg, SM; Rice, CM. Mutagenesis of the yellow fever virus NS2B protein: Effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J. Virol 1993, 67, 6797–6807. [Google Scholar]
- Falgout, B; Pethel, M; Zhang, Y; Lai, C. Both non structural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus non structural protein. J. Virol 1991, 65, 2467–2475. [Google Scholar]
- Leyssen, P; de Clercq, E; Neyts, J. Perspective for the treatment of infections with flaviviridae. Clin. Microbiology 2003, 13, 67–82. [Google Scholar]
- Sampath, A; Padmanabhan, R. Molecular targets for flavivirus drug discovery. Antivir. Res 2009, 81, 6–15. [Google Scholar]
- Tan, SK; Pippen, R; Yusof, R; Ibrahim, H; Khalid, N; Rahman, NA. Inhibitory activity of cylohexenyl chalcone derivatives and flavanoids of fingerroot, Boesenbergia rotunda (L.), towards dengue-2 virus NS3 protease. Bioorg. Med. Chem. Lett 2006, 16, 3337–3340. [Google Scholar]
- Yean, KL; Othman, R; Wahab, HA; Yusof, R; Rahman, NA. A revisit into the DEN2 NS2B/NS3 virus protease homology model: strcutural verification and comparison with crystal structure of HCV NS3/4A and DEN2 NS3. Malays J Sci 2006, 25, 15–22. [Google Scholar]
- Yean Kee, L; Tan, SK; Wahab, HA; Yusof, R; Rahman, NA. Non substrate based inhibitors of dengue virus serine protease: A molecular docking approach to study binding interactions between protease and inhibitors. Asia Pacific J. Mol. Biol. Biotechnol 2007, 15, 53–59. [Google Scholar]
- Brinkworth, RI; Fairlie, DP; Young, PR. Homology model of the dengue 2 virus NS3 protease: putative interactions with both substrate and NS2B cofactor. J. Gen. Virol 1999, 80, 1167–1177. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Ki (μM) | Complexation Energy (kcal/mol) |
|---|---|---|
| 4-hydroxypanduratin | 21 ± 8.00 a | −69.8 |
| Panduratin A | 25 ± 6.00 a | −57.8 |
| 4-hydroxypanduratin A | ||||||
| Position in benzyl ring A | Complexation energy of derivatives (kcal/mol) | |||||
| –OH | –COO− | –NO2 | –NH3+ | –Cl | –CH3 | |
| 1 | −37.1 | 58.1 | −1.3 | −50.1 | −43.1 | −38.6 |
| 2 | −50.4 | 31.1 | 240.6 | −35.5 | −50.7 | −44.0 |
| 3 | −27.1 | 65.2 | −49.3 | −63.0 | −53.8 | −43.9 |
| 4 | −40.7 | −22.5 | −58.4 | −58.9 | −41.4 | −38.5 |
| 5 | −36.1 | −91.4 | −43.4 | −93.3 | −51.6 | −54.1 |
| Panduratin A | ||||||
| Position in benzyl ring A | Complexation energy of derivatives (kcal/mol) | |||||
| –OH | –COO− | –NO2 | –NH3+ | –Cl | –CH3 | |
| 1 | 70.9 | 13.8 | 59.9 | 120.9 | 54.8 | 64.4 |
| 2 | 68.4 | 36.8 | −24.3 | −14.4 | −31.6 | −37.9 |
| 3 | −24.7 | 5.3 | −48.2 | −10.7 | −62.2 | −62.6 |
| 4 | −51.4 | 27.2 | −40.6 | −42.7 | −69.1 | −53.7 |
| 5 | −60.8 | −44.7 | −68.0 | −64.5 | −55.2 | −54.0 |
| No | Molecule | R | R’ | Complexation energy (kcal/mol) |
|---|---|---|---|---|
| 1 | 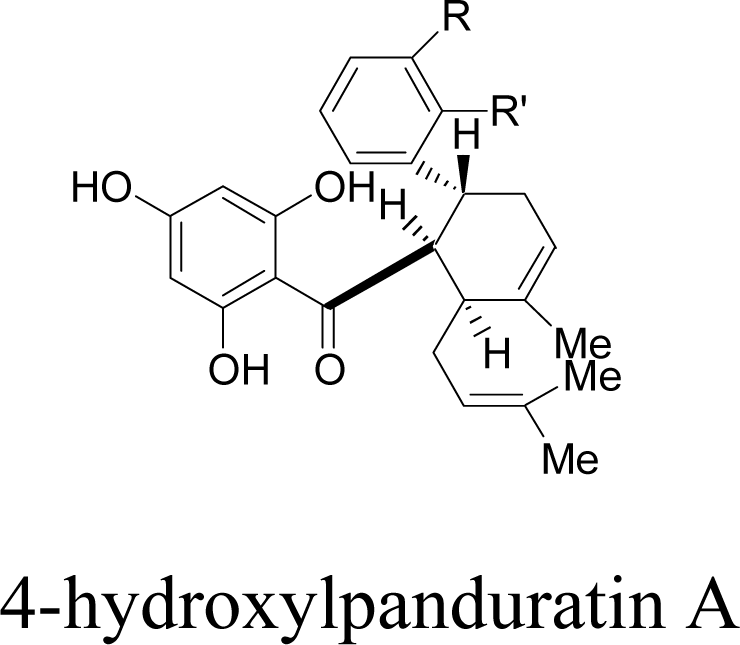 | R = NO2 | R’ = H | −68.7 |
| R = NH3+ | R’ = H | −68.9 | ||
| R = CH2NH3+ | R’ = H | −69.7 | ||
| R = NH2+CH3 | R’ = H | −68.9 | ||
| R = H | R’ = COO− | −91.4 | ||
| R = H | R’ = NH3+ | −93.3 | ||
| 2 | 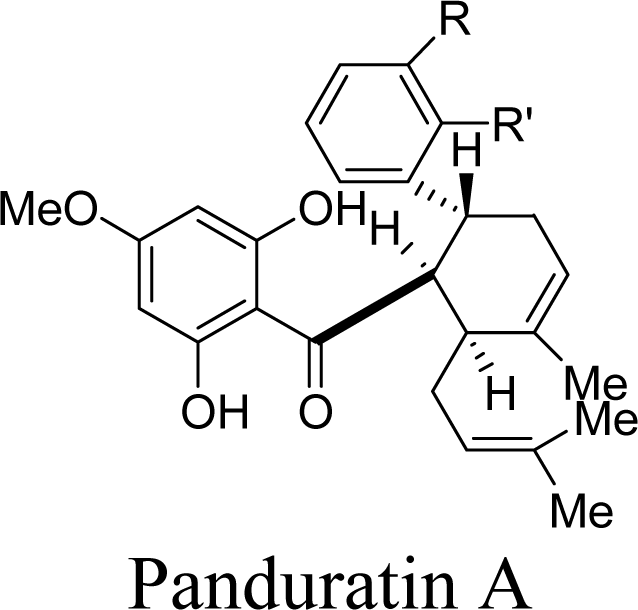 | R = Cl | R’ = H | −69.1 |
| R = OCH3 | R’ = H | −70.2 | ||
| R = SH | R’ = H | −58.8 | ||
| R = NH2+CH3 | R’ = H | −64.8 | ||
| R = H | R’ = OH | −60.8 | ||
| R = H | R’ = NO2 | −58.0 | ||
| R = H | R’ = NH3+ | −64.5 | ||
| R = H | R’ = Cl | −55.2 | ||
| R = H | R’ = CH2OH | −66.0 | ||
| R = H | R’ = SH | −63.3 | ||
| R = H | R’= CH2NH3+ | −58.6 |
| Molecule | Ki (μM) | Complexation Energy (kcal/mol) |
|---|---|---|
| 246DA | 19.84 | −76.8 |
| 20H46DA | 24.36 | −61.0 |
| 2446DA | 39.68 | −43.5 |
| Name | Molecule | R | R’ | Complexation Energy (kcal/mol) |
|---|---|---|---|---|
| 246DA | 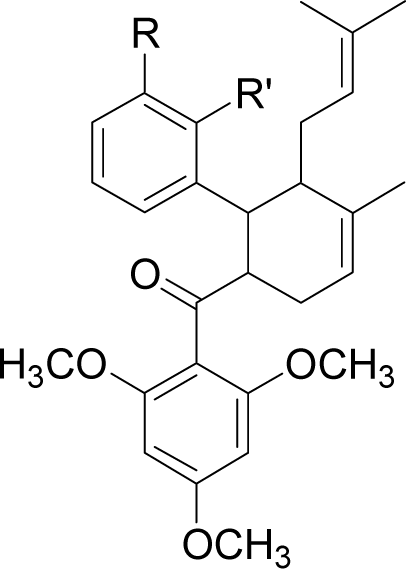 | R = OH | R’ = H | −70.2 |
| R = COO− | R’ = H | −117.9 | ||
| R = NH3+ | R’ = H | −76.8 | ||
| R = OCH3 | R’ = H | −81.2 | ||
| R = CH2NH3+ | R’ = H | −74.1 | ||
| R = H | R’ = NH3+ | −91.9 | ||
| R = H | R’ = CH2OH | −90.4 | ||
| R = H | R’ = CH2NH3+ | −87.6 | ||
| 20H46DA | 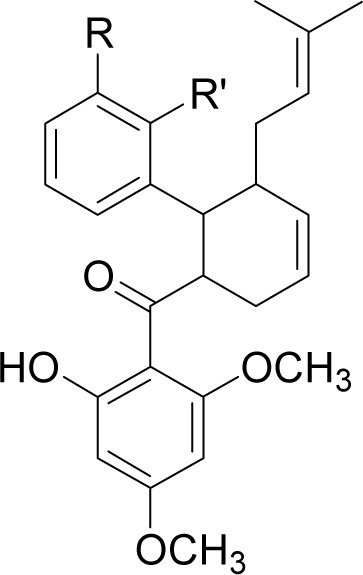 | R = OH | R’ = H | −61.6 |
| R = COO− | R’ = H | −61.8 | ||
| R = NH3+ | R’ = H | −164.7 | ||
| R = CH3 | R’ = H | −61.0 | ||
| R = CH2OH | R’ = H | −77.4 | ||
| R = H | R’ = COO− | −83.5 | ||
| R = H | R’ = NO2 | −64.3 | ||
| R = H | R’ = NH3+ | −68.3 | ||
| R = H | R’ = OCH3 | −64.7 | ||
| R = H | R’ = CH2NH3+ | −129.1 | ||
| 2446DA | 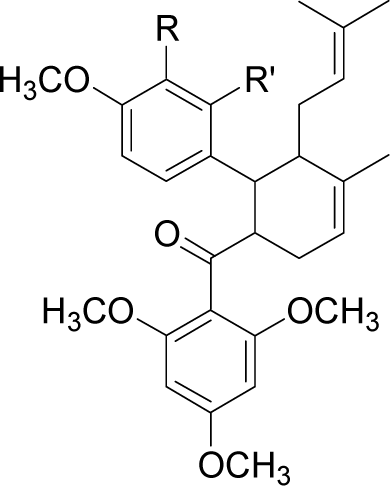 | R = H | R’ = OH | −95.3 |
| R = H | R’ = COO− | −108.9 | ||
| R = H | R’ = NO2 | −84.7 | ||
| R = H | R’ = NH3+ | −88.9 | ||
| R = H | R’ = Cl | −98.7 | ||
| R = H | R’ = CH3 | −87.2 | ||
| R = H | R’ = CH2OH | −95.7 | ||
| R = H | R’ = OCH3 | −83.8 | ||
| R = H | R’ = SH | −73.5 | ||
| R = H | R’ = CH2NH3+ | −116.7 | ||
| R = OH | R’ = H | −88.9 | ||
| R = NO2 | R’ = H | −84.3 | ||
| R = NH3+ | R’ = H | −116.5 | ||
| R = Cl | R’ = H | −79.1 | ||
| R = CH3 | R’ = H | −76.4 | ||
| R = OCH3 | R’ = H | −81.6 | ||
| R = SH | R’ = H | −68.1 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Frimayanti, N.; Chee, C.F.; Zain, S.M.; Rahman, N.A. Design of New Competitive Dengue Ns2b/Ns3 Protease Inhibitors—A Computational Approach. Int. J. Mol. Sci. 2011, 12, 1089-1100. https://doi.org/10.3390/ijms12021089
Frimayanti N, Chee CF, Zain SM, Rahman NA. Design of New Competitive Dengue Ns2b/Ns3 Protease Inhibitors—A Computational Approach. International Journal of Molecular Sciences. 2011; 12(2):1089-1100. https://doi.org/10.3390/ijms12021089
Chicago/Turabian StyleFrimayanti, Neni, Chin Fei Chee, Sharifuddin M. Zain, and Noorsaadah Abd. Rahman. 2011. "Design of New Competitive Dengue Ns2b/Ns3 Protease Inhibitors—A Computational Approach" International Journal of Molecular Sciences 12, no. 2: 1089-1100. https://doi.org/10.3390/ijms12021089