Inference of Gene Regulation via miRNAs During ES Cell Differentiation Using MiRaGE Method



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Development of MiRaGE Method
2.2. miRNAs Underrepresented in ES Cells after the Differentiation to Neuronal Cells
2.3. miRNAs May Be Overrepresented in ES Cells after the Differentiation to Neuronal Cells
3. Materials and Methods
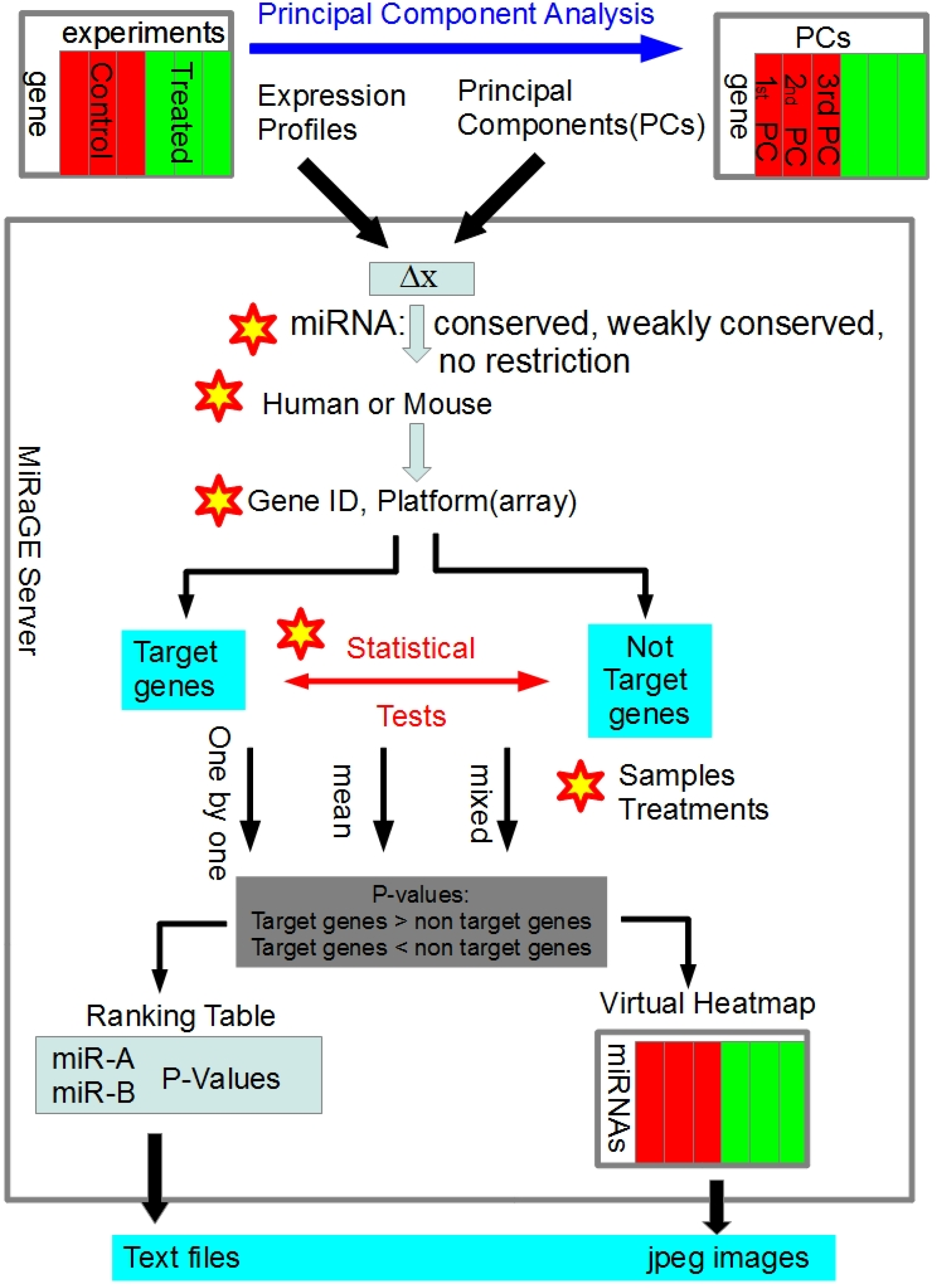
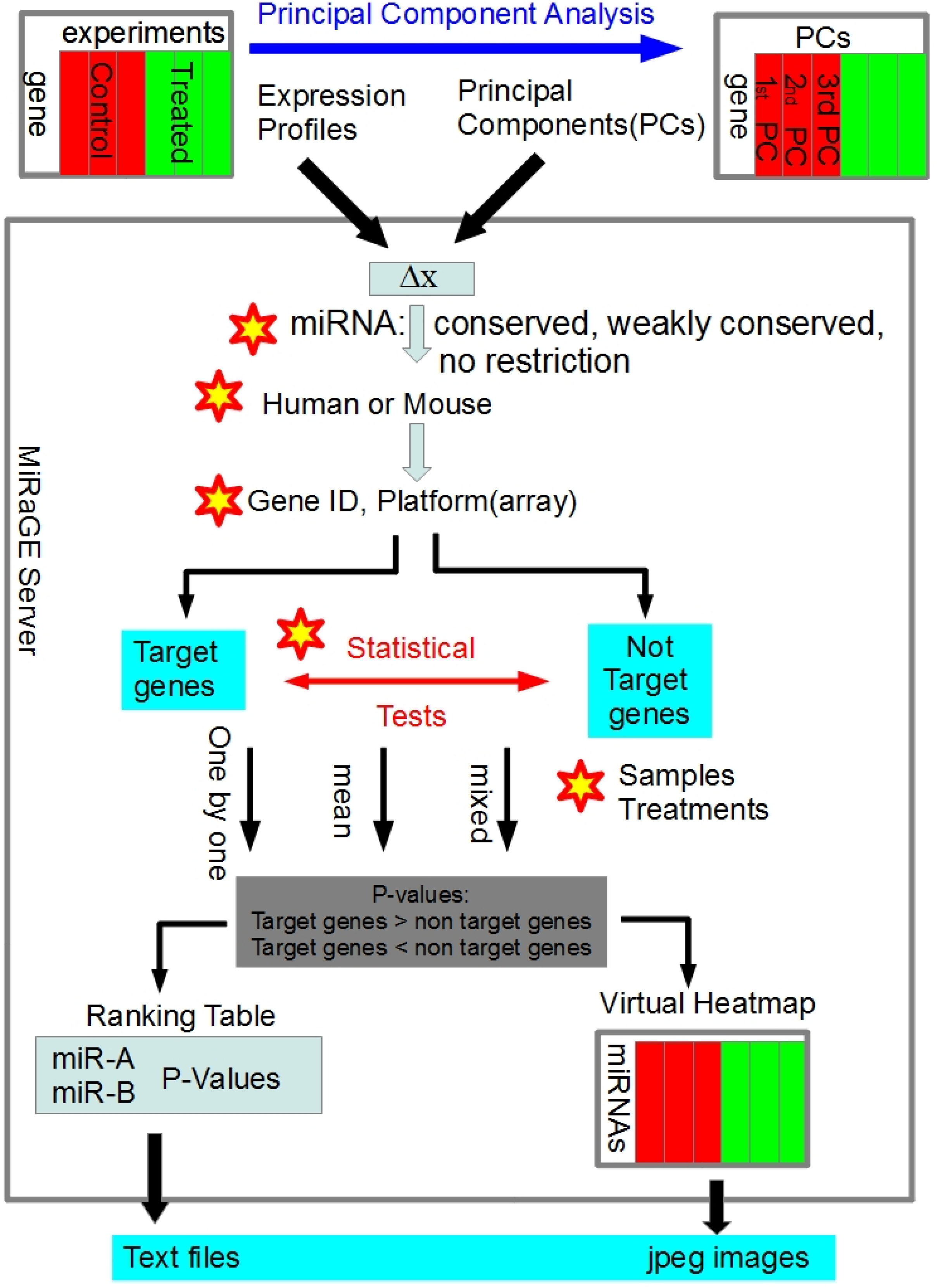
3.1. Detailed Method of MiRaGE
3.2. Inference by MiRaGE Server for the Differentiation from ES Cell
3.3. Principal Component Analysis (PCA)
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009, 19, 92–105. [Google Scholar]
- Taguchi, Y.-h.; Yasuda, J. MiRaGE: Inference of Gene Expression Regulation via microRNA Transfection II. Proceedings of the 7th International Conference on Intelligent Computing, ICIC2011, Zhengzhou, China, 11–14 August 2011, Huang, D.-S., Gan, Y., Premaratne, P., Han, K., Eds.; Springer: New York, NY, USA, 2011; pp. 129–135. [Google Scholar]
- Taguchi, Y.-h.; Yasuda, J. Inference of Gene Expression Regulation via microRNA Transfection. Proceedings of the 6th International Conference on Intelligent Computing, ICIC2010, Changsha, China, 18–21 August 2010, Huang, D.-S., Zhao, Z., Bevilacqua, V., Figueroa, J.C., Eds.; Springer: New York, NY, USA, 2010; pp. 672–679. [Google Scholar]
- Volinia, S.; Visone, R.; Galasso, M.; Rossi, E.; Croce, C.M. Identification of microRNA activity by Targets’ Reverse EXpression. Bioinformatics 2010, 26, 91–97. [Google Scholar]
- van Dongen, S.; Abreu-Goodger, C.; Enright, A.J. Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods 2008, 5, 1023–1025. [Google Scholar]
- Liang, Z.; Zhou, H.; He, Z.; Zheng, H.; Wu, J. mirAct: A web tool for evaluating microRNA activity based on gene expression data. Nucleic Acids Res 2011, 39. [Google Scholar] [CrossRef]
- Tiscornia, G.; Izpisua Belmonte, J.C. MicroRNAs in embryonic stem cell function and fate. Genes Dev 2010, 24, 2732–2741. [Google Scholar]
- MiRaGE Server Home Page. Available online: http://www.granular.com/MiRaGE/ accessed on 8 December 2011.
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Stat. Methodol. ) 1995, 57, 289–300. [Google Scholar]
- Aiba, K.; Nedorezov, T.; Piao, Y.; Nishiyama, A.; Matoba, R.; Sharova, L.V.; Sharov, A.A.; Yamanaka, S.; Niwa, H.; Ko, M.S. Defining developmental potency and cell lineage trajectories by expression profiling of differentiating mouse embryonic stem cells. DNA Res 2009, 16, 73–80. [Google Scholar]
- Yi, R.; Fuchs, E. MicroRNAs and their roles in mammalian stem cells. J. Cell Sci 2011, 124, 1775–1783. [Google Scholar]
- Li, Z.; Yang, C.S.; Nakashima, K.; Rana, T.M. Small RNA-mediated regulation of iPS cell generation. EMBO J 2011, 30, 823–834. [Google Scholar]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet 2008, 40, 43–50. [Google Scholar]
- Liu, C.; Teng, Z.Q.; Santistevan, N.J.; Szulwach, K.E.; Guo, W.; Jin, P.; Zhao, X. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell 2010, 6, 433–444. [Google Scholar]
- Siegel, G.; Obernosterer, G.; Fiore, R.; Oehmen, M.; Bicker, S.; Christensen, M.; Khudayberdiev, S.; Leuschner, P.F.; Busch, C.J.; Kane, C.; et al. A functional screen implicates microRNA-138- dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol 2009, 11, 705–716. [Google Scholar]
- Sharova, L.V.; Sharov, A.A.; Nedorezov, T.; Piao, Y.; Shaik, N.; Ko, M.S. Database for mRNA half-life of 19,977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res 2009, 16, 45–58. [Google Scholar]
- Tripathi, R.; Saini, H.K.; Rad, R.; Abreu-Goodger, C.; van Dongen, S.; Enright, A.J. Messenger RNA and microRNA profiling during early mouse EB formation. Gene Expr. Patterns 2011, 11, 334–344. [Google Scholar]
- Genome Browser Table. Available online: http://genome.ucsc.edu/cgi-bin/hgTables accessed 8 December 2011.
- miRBase Home Page. Available online: http://www.mirbase.org/ accessed 8 December 2011.
- Yeung, K.Y.; Ruzzo, W.L. Principal component analysis for clustering gene expression data. Bioinformatics 2001, 17, 763–774. [Google Scholar]

{kind=link}
{kind=link}
| miRNA underrepresented after neural differentiation | P-values | ||
|---|---|---|---|
| KS | t | Wilcoxon | |
| mmu-miR-200b | 4.14 × 10−16 | 9.17 × 10−17 | 5.39 × 10−17 |
| mmu-miR-200c | 4.14 × 10−16 | 9.17 × 10−17 | 5.39 × 10−17 |
| mmu-miR-429 | 4.14 × 10−16 | 9.17 × 10−17 | 5.39 × 10−17 |
| mmu-miR-291a-3p | 2.43 × 10−13 | 5.88 × 10−15 | 1.36 × 10−16 |
| mmu-miR-294 | 2.43 × 10−13 | 5.88 × 10−15 | 1.36 × 10−16 |
| mmu-miR-295 | 2.43 × 10−13 | 5.88 × 10−15 | 1.36 × 10−16 |
| mmu-miR-302a | 2.43 × 10−13 | 5.88 × 10−15 | 1.36 × 10−16 |
| mmu-miR-302b | 2.43 × 10−13 | 5.88 × 10−15 | 1.36 × 10−16 |
| mmu-miR-302d | 2.43 × 10−13 | 5.88 × 10−15 | 1.36 × 10−16 |
| mmu-miR-141 | 4.66 × 10−13 | 3.08 × 10−11 | 1.68 × 10−11 |
| mmu-miR-200a | 4.66 × 10−13 | 3.08 × 10−11 | 1.68 × 10−11 |
| mmu-miR-23b | 2.18 × 10−12 | 1.30 × 10−14 | 7.32 × 10−16 |
| mmu-miR-23a | 2.18 × 10−12 | 1.30 × 10−14 | 7.32 × 10−16 |
| mmu-miR-30a | 1.02 × 10−11 | 3.09 × 10−10 | 3.96 × 10−10 |
| mmu-miR-30b | 1.02 × 10−11 | 3.09 × 10−10 | 3.96 × 10−10 |
| mmu-miR-30e | 1.02 × 10−11 | 3.09 × 10−10 | 3.96 × 10−10 |
| mmu-miR-30c | 1.02 × 10−11 | 3.09 × 10−10 | 3.96 × 10−10 |
| mmu-miR-30d | 1.02 × 10−11 | 3.09 × 10−10 | 3.96 × 10−10 |
| mmu-miR-384-5p | 1.02 × 10−11 | 3.09 × 10−10 | 3.96 × 10−10 |
| mmu-miR-129-5p | 4.31 × 10−11 | 8.12 × 10−11 | 7.63 × 10−11 |
| mmu-miR-218 | 1.06 × 10−10 | 3.11 × 10−11 | 1.33 × 10−11 |
| mmu-miR-144 | 1.75 × 10−10 | 8.24 × 10−11 | 1.34 × 10−10 |
| mmu-miR-130a | 3.36 × 10−10 | 1.20 × 10−8 | 3.35 × 10−8 |
| mmu-miR-301a | 3.36 × 10−10 | 1.20 × 10−8 | 3.35 × 10−8 |
| mmu-miR-130b | 3.36 × 10−10 | 1.20 × 10−8 | 3.35 × 10−8 |
| mmu-miR-301b | 3.36 × 10−10 | 1.20 × 10−8 | 3.35 × 10−8 |
| mmu-miR-721 | 3.36 × 10−10 | 1.20 × 10−8 | 3.35 × 10−8 |
| mmu-miR-135a | 4.83 × 10−10 | 1.91 × 10−10 | 6.04 × 10−10 |
| mmu-miR-135b | 4.83 × 10−10 | 1.91 × 10−10 | 6.04 × 10−10 |
| mmu-miR-106a | 5.35 × 10−10 | 4.36 × 10−9 | 1.98 × 10−8 |
| miRNA underrepresented after differentiation | P-values * | ||
|---|---|---|---|
| KS | t | Wilcoxon | |
| mmu-miR-106a | 1.07 × 10−15 | 6.83 × 10−13 | 9.39 × 10−15 |
| mmu-miR-106b | 1.07 × 10−15 | 6.83 × 10−13 | 9.39 × 10−15 |
| mmu-miR-20a | 1.07 × 10−15 | 6.83 × 10−13 | 9.39 × 10−15 |
| mmu-miR-93 | 1.07 × 10−15 | 6.83 × 10−13 | 9.39 × 10−15 |
| mmu-miR-17 | 1.07 × 10−15 | 6.83 × 10−13 | 9.39 × 10−15 |
| mmu-miR-20b | 1.07 × 10−15 | 6.83 × 10−13 | 9.39 × 10−15 |
| mmu-miR-291a-3p | 1.81 × 10−12 | 2.05 × 10−13 | 1.72 × 10−14 |
| mmu-miR-294 | 1.81 × 10−12 | 2.05 × 10−13 | 1.72 × 10−14 |
| mmu-miR-295 | 1.81 × 10−12 | 2.05 × 10−13 | 1.72 × 10−14 |
| mmu-miR-302a | 1.81 × 10−12 | 2.05 × 10−13 | 1.72 × 10−14 |
| mmu-miR-302b | 1.81 × 10−12 | 2.05 × 10−13 | 1.72 × 10−14 |
| mmu-miR-302d | 1.81 × 10−12 | 2.05 × 10−13 | 1.72 × 10−14 |
| mmu-miR-128 | 9.36 × 10−11 | 5.70 × 10−8 | 4.81 × 10−9 |
| mmu-miR-214 | 2.64 × 10−10 | 9.77 × 10−8 | 4.92 × 10−6 |
| mmu-miR-761 | 2.64 × 10−10 | 9.77 × 10−5 | 4.92 × 10−6 |
| mmu-miR-141 | 4.13 × 10−10 | 3.10 × 10−8 | 1.68 × 10−8 |
| mmu-miR-200a | 4.13 × 10−10 | 3.10 × 10−8 | 1.68 × 10−8 |
| mmu-miR-203 | 1.33 × 10−10 | 3.22 × 10−9 | 4.17 × 10−9 |
| mmu-miR-199a-5p | 4.65 × 10−7 | 4.72 × 10−5 | 1.91 × 10−5 |
| mmu-miR-200b | 1.16 × 10−6 | 2.69 × 10−6 | 1.50 × 10−6 |
| mmu-miR-200c | 1.16 × 10−6 | 2.69 × 10−6 | 1.50 × 10−6 |
| mmu-miR-429 | 1.16 × 10−6 | 2.69 × 10−6 | 1.50 × 10−6 |
| mmu-miR-96 | 4.50 × 10−6 | 3.64 × 10−6 | 4.93 × 10−7 |
| mmu-miR-130a | 6.00 × 10−6 | 3.46 × 10−5 | 1.53 × 10−5 |
| mmu-miR-301a | 6.00 × 10−6 | 3.46 × 10−5 | 1.53 × 10−5 |
| mmu-miR-130b | 6.00 × 10−6 | 3.46 × 10−5 | 1.53 × 10−5 |
| miRNA overrepresented after differentiation | P-values | ||
|---|---|---|---|
| KS | t | Wilcoxon | |
| mmu-miR-184 | 0.809 | 0.006 | 0.985 |
| mmu-miR-10b | 0.876 | 0.039 | 0.996 |
| mmu-miR-10a | 0.876 | 0.039 | 0.996 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yoshizawa, M.; Taguchi, Y.-h.; Yasuda, J. Inference of Gene Regulation via miRNAs During ES Cell Differentiation Using MiRaGE Method. Int. J. Mol. Sci. 2011, 12, 9265-9276. https://doi.org/10.3390/ijms12129265
Yoshizawa M, Taguchi Y-h, Yasuda J. Inference of Gene Regulation via miRNAs During ES Cell Differentiation Using MiRaGE Method. International Journal of Molecular Sciences. 2011; 12(12):9265-9276. https://doi.org/10.3390/ijms12129265
Chicago/Turabian StyleYoshizawa, Masato, Y-h. Taguchi, and Jun Yasuda. 2011. "Inference of Gene Regulation via miRNAs During ES Cell Differentiation Using MiRaGE Method" International Journal of Molecular Sciences 12, no. 12: 9265-9276. https://doi.org/10.3390/ijms12129265