Investigating Ionic Effects Applied to Water Based Organocatalysed Aldol Reactions

Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Diprolinamide Organocatalyst
2.2. Evaluation of Ionic Solution Effects
3. Conclusion
Supplementary Material
ijms-12-09083-s001.pdfAcknowledgements
References and Notes
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc 2000, 122, 2395–2396. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar]
- List, B. Introduction: Organocatalysis. Chem. Rev 2007, 107, 5413–5415. [Google Scholar]
- Mase, N.; Barbas, C.F., III. In water, on water, and by water: mimicking nature’s aldolases with organocatalysis and water. Org. Biomol. Chem. 2010, 8, 4043–4050. [Google Scholar]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Meo, P.L.; Noto, R. Advances towards highly active and stereoselective simple and cheap proline-based organocatalysts. Eur. J. Org. Chem 2010, 29, 5696–5704. [Google Scholar]
- Ricci, A.; Bernardi, L.; Gioia, C.; Vierucci, S.; Robitzer, M.; Quignard, F. Chitosan aerogel: A recyclable, heterogeneous organocatalyst for the asymmetric direct aldol reaction in water. Chem. Commun 2010, 46, 6288–6290. [Google Scholar]
- Wu, C.; Fu, X.; Ma, X.; Li, S. One-step, efficient synthesis of combined threonine-surfactant organocatalysts for the highly enantioselective direct aldol reactions of cyclic ketones with aromatic aldehydes in the presence of water. Tetrahedron: Asymmetry 2010, 21, 2465–2470. [Google Scholar]
- Raja, M.; Singh, V.K. Organocatalytic reactions in water. Chem. Commun 2009, 6687–6703. [Google Scholar]
- Wu, C.; Fu, X.; Li, S. Highly efficient, large-scale, asymmetric direct aldol reaction employing simple threonine derivatives as recoverable organocatalysts in the presence of water. Eur. J. Org. Chem 2011, 7, 1291–1299. [Google Scholar]
- Ramasastry, S.S.V.; Albertshofer, K.; Utsumi, N.; Barbas, C.F., III. Water-compatible organocatalysts for direct asymmetric syn-aldol reactions of dihydroxyacetone and aldehydes. Org. Lett 2008, 10, 1621–1624. [Google Scholar]
- Zheng, Z.; Perkins, B.L.; Ni, B. Diarylprolinol silyl ether salts as new, efficient, water-soluble, and recyclable organocatalysts for the asymmetric michael addition on water. J. Am. Chem. Soc 2010, 132, 50–51. [Google Scholar]
- Huang, J.; Zhang, X.; Armstrong, D.W. Highly efficient asymmetric direct stoichiometric aldol reactions on/in water. Angew. Chem. Int. Ed 2007, 46, 9073–9077. [Google Scholar]
- Aratake, S.; Itoh, T.; Okano, T.; Usui, T.; Shoji, M.; Hayashi, Y. Small organic molecule in enantioselective, direct aldol reaction “in water”. Chem. Commun 2007, 24, 2524–2526. [Google Scholar]
- Tzeng, H.-Z.; Chen, H.-Y.; Huang, C.-T.; Chen, K. Camphor containing organocatalysts in asymmetric aldol reaction on water. Tetrahedron Lett 2008, 45, 4134–4137. [Google Scholar]
- Chimni, S.S.; Singh, S.; Mahajan, D. Protonated (S)-prolinamide derivatives—water compatible organocatalysts for direct asymmetric aldol reaction. Tetrahedron: Asymmetry 2008, 19, 2276–2284. [Google Scholar]
- Córdova, A.; Notz, W.; Barbas, C.F., III. Direct organocatalytic aldol reactions in buffered aqueous media. Chem. Commun. 2002, 24, 3024–3025. [Google Scholar]
- Brogan, A.P.; Dickerson, T.J.; Janda, K.D. Enamine-based aldol organocatalysis in water: Are they really “All Wet”? Angew. Chem. Int. Ed 2006, 45, 8100–8102. [Google Scholar]
- Robak, M.A.T.; Herbage, M.A.; Ellman, J.A. Development of an N-sulfinyl prolinamide for the asymmetric aldol reaction. Tetrahedron 2011, 67, 4412–4416. [Google Scholar]
- Larionova, N.A.; Kucherenko, A.S.; Siyutkin, D.E.; Zlotin, S.G. (S)-Threonine/α, α-(S) diphenylvalinol-derived chiral ionic liquid: an immobilized organocatalyst for asymmetric syn-aldol reactions. Tetrahedron 2011, 67, 1948–1954. [Google Scholar]
- Notz, W.; Tanaka, F.; Barbas, C.F., III. Enamine-based organocatalysiswith proline and diamines: The development of direct catalytic asymmetric aldol, mannich, michael, and diels-alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar]
- Tian, H.; Gao, J.-L.; Xu, H.; Zheng, L.-Y.; Huang, W.-B.; Liu, Q.-W.; Zhang, S.-Q. Proline-based dipeptides as efficient organocatalysts for asymmetric aldolreactions in brine. Tetrahedron: Asymmetry 2011, 22, 1074–1080. [Google Scholar]
- Miura, T.; Ina, M.; Imai, K.; Nakashima, K.; Yasaku, Y.; Koyata, N.; Murakami, Y.; Imai, N.; Tada, N.; Itoh, A. Direct asymmetric aldol reactions in water with a β-aminosulfonamide organocatalyst. Tetrahedron: Asymmetry 2011, 22, 1028–1034. [Google Scholar]
- Huang, W.; Tian, H.; Xu, H.; Zheng, L.; Liu, Q.; Zhang, S. l-Valine dipeptide organocatalysts with two amide units for the direct asymmetric aldol reaction in brine. Catal. Lett 2011, 141, 872–876. [Google Scholar]
- Hayashi, Y.; Sumiya, T.; Takahashi, J.; Gotoh, H.; Urushima, T.; Shoji, M. Highly diastereo- and enantioselective direct aldol reactions in water. Angew. Chem. Int. Ed 2006, 118, 958–961. [Google Scholar]
- Mase, N.; Noshiro, N.; Mokuya, A.; Takabe, K. Effect of long chain fatty acids on organocatalytic aqueous direct aldol reactions. Adv. Synth. Catal 2009, 351, 2791–2796. [Google Scholar]
- Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F., III. Organocatalytic direct asymmetric aldol reactions in water. J. Am. Chem. Soc. 2006, 128, 734–735. [Google Scholar]
- Wang, B.; Liu, X.-W.; Liu, L.-Y.; Chang, W.-X.; Li, J. Highly efficient direct asymmetric aldol reactions catalyzed by a prolinethioamide derivative in aqueous media. Eur. J. Org. Chem 2010, 31, 5951–5954. [Google Scholar]
- Casiraghi, G.; Zanardi, F. The vinylogous aldol reaction: A valuable, yet understated carbon-carbonbond-forming manoeuvre. Chem. Rev 2000, 100, 1929–1972. [Google Scholar]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev 2010, 39, 1600–1632. [Google Scholar]
- Delaney, J.P.; Henderson, L.C. Direct asymmetric aldol reactions in water catalysed by a highly active C2-symmetric bisprolinamide organocatalyst. Adv. Synth. Catal 2011. [Google Scholar] [CrossRef]
- Representative experimental procedure for organocatalyzed aldol reactions (for 4- nitrobenzaldehyde): Water (1.6 mL) containing a saturated solution of the indicated salt was added to a round bottom flask charged with benzaldehyde (0.244 mmol, 37 mg, 1 equivalent), ketone (1.22 mmol, 0.127 mL, 5 equivalents) and catalyst 5(24 μmol, 2 mg, 1 mol%). The emulsion was then stirred vigorously (approx 1400 rpm) at room temperature for 24 h. The emulsion was then extracted with CH2Cl2 the solvent removed in vacuo to give the crude product as a yellow solid. Analysis by 1H NMR spectroscopy was used to determine the desired product, reaction conversion and the diastereomeric ratio. The crude product purified by silica gel column chromatography and the pure product analysed by chiral HPLC (Chiral Pak AD-H, hexane:isopropanol, 90:10, 1 mL/min) to determine enantiomeric excess.
- Jung, Y.; Marcus, R.A. On the theory of organic catalysis “on Water”. J. Am. Chem. Soc 2007, 129, 5492–5502. [Google Scholar]
- Lombardo, A.; Quintavalla, A.; Chiarucci, M.; Trombini, C. Multiphase homogeneous catalysis: Common procedures and recent applications. Synlett 2010, 12, 1746–1765. [Google Scholar]
- Butler, R.N.; Coyne, A.G. Water: Nature’s reaction enforcer-comparative effects for organic synthesis “in-water” and “on-water”. Chem. Rev 2010, 110, 6302–6337. [Google Scholar]
- Penhoat, M.; Barbry, D.; Rolando, C. Direct asymmetric aldol reaction co-catalyzed by l-proline and group 12 elements Lewis acids in the presence of water. Tetrahedron Lett 2011, 52, 159–162. [Google Scholar]
- Andreu, C.; Asensio, G. The role of Zn2+ in enhancing the rate and stereoselectivity of the aldol reactions catalyzed by the simple prolinamide model. Tetrahedron 2011, 67, 7050–7056. [Google Scholar]
- Akagawa, K.; Sakamoto, S.; Kudo, K. Direct asymmetric aldol reaction in aqueous mediausing polymer-supported peptide. Tetrahedron Lett 2005, 46, 8185–8187. [Google Scholar]
- Zhu, X.; Xu, X.-P.; Sun, C.; Chen, T.; Shen, Z.-L.; Ji, S.-J. I-MCR-Ullmann cascade toward furo[2,3-b]indole scaffold. Tetrahedron 2011, 67, 6375–6381. [Google Scholar]
- Surry, D.S.; Buchwald, S.L. Diamine ligands in copper-catalyzed reactions. Chem. Sci 2010, 1, 13–31. [Google Scholar]
- Hare, P.E.; Gil-Av, E. Separation of D and L amino acids by liquid chromatography: Use of chiral eluants. Science 1979, 204, 1226–1228. [Google Scholar]
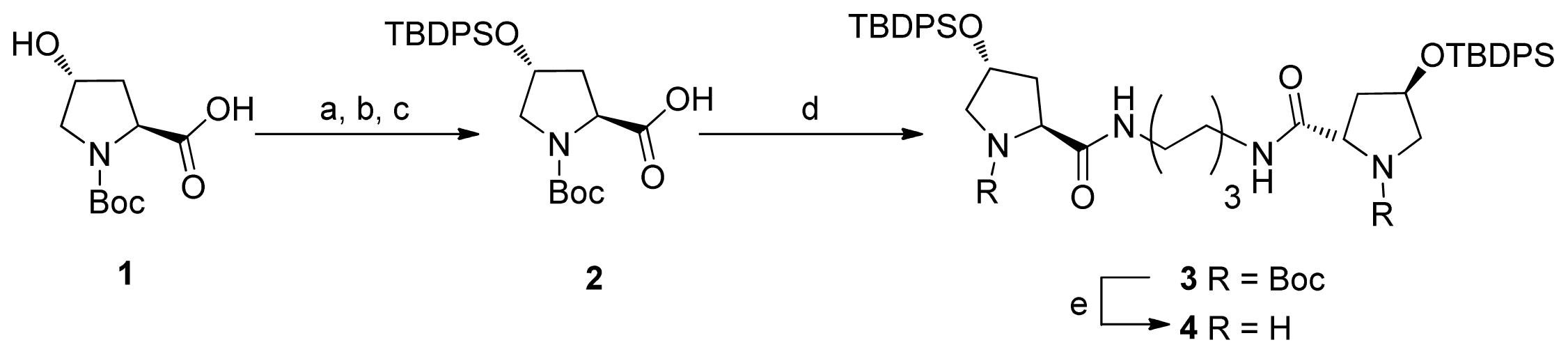
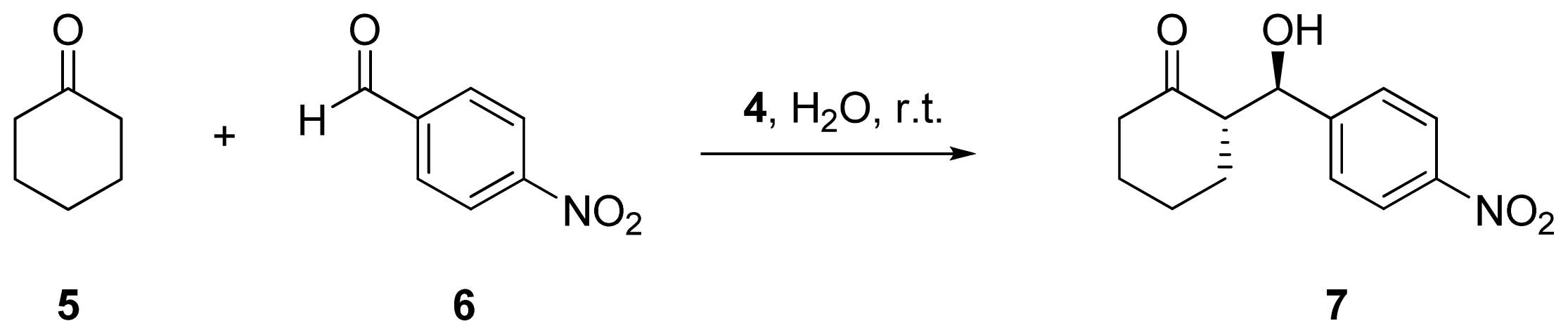

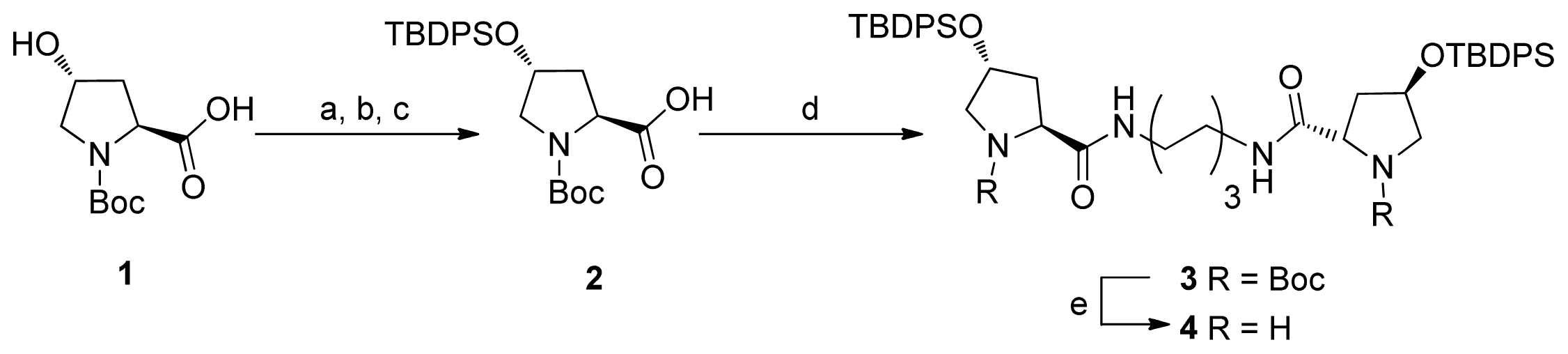{kind=link}
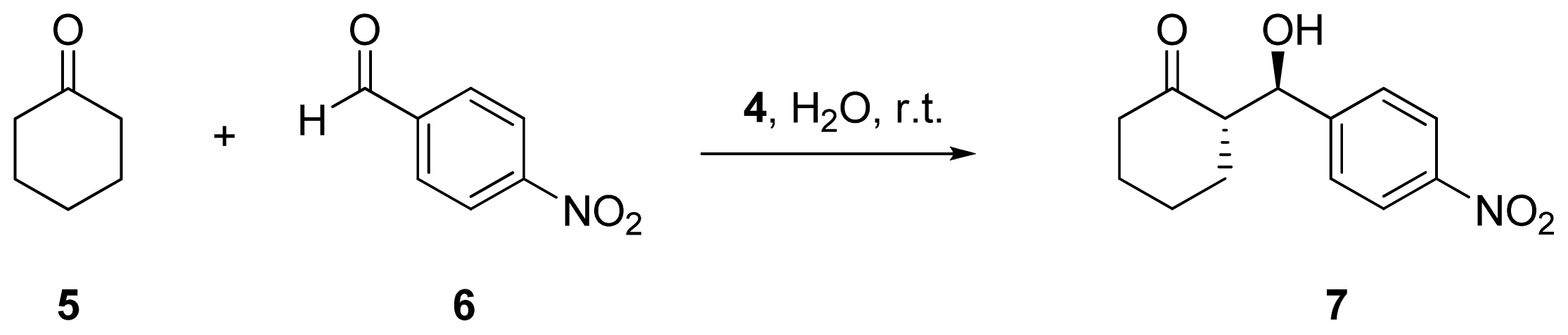{kind=link}
{kind=link}
| Entry | Cation | Anion | Yield (%) a | dr (%) a anti/syn | ee (%) b |
|---|---|---|---|---|---|
| 1 c | - | - | 91 | >99/1 | >99 |
| 2 d | - | - | 7 | 81/19 | 22 |
| 3 | Na | F | 49 | 83/17 | 67 |
| 4 | Na | Cl | 54 | 82/18 | 87 |
| 5 | Na | Br | 30 | 78/22 | 43 |
| 6 | Na | I | 17 | 79/21 | 34 |
| 7 | Na | OAc | 75 | 81/19 | 38 |
| 8 | K | F | 23 | 79/22 | 67 |
| 9 | K | Cl | 27 | 83/17 | 45 |
| 10 | K | Br | 44 | 80/20 | 35 |
| 11 | K | I | 32 | 75/25 | 21 |
| 12 | Mg | Cl2 | 55 | 80/20 | 51 |
| 13 | Ca | Cl2 | 99 | 45/55 | 2 |
| Entry | Cation | Anion | Yield (%) a | de (%) b anti/syn | ee (%) c |
|---|---|---|---|---|---|
| 1 | Cu | Cl | trace d | - | - |
| 2 | Cu | Br2 | trace d | - | - |
| 3 | Fe(III) | Cl3 | >99 | 42/58 | 0 |
| 4 | Ni | Cl2 | 3 | 82/18 | 83 |
| 5 | Zn | OAc | >99 | 80/20 | 92 |
| Entry | EDTA (% w/w) | Yield (%) a | de (%) a | ee (%) b |
|---|---|---|---|---|
| 1 | 0 | 7 | 81 | 22 |
| 2 c | 5 | 0 | - | - |
| 3 | 5 | 98 | 84 | 51 |
 | ||||||
|---|---|---|---|---|---|---|
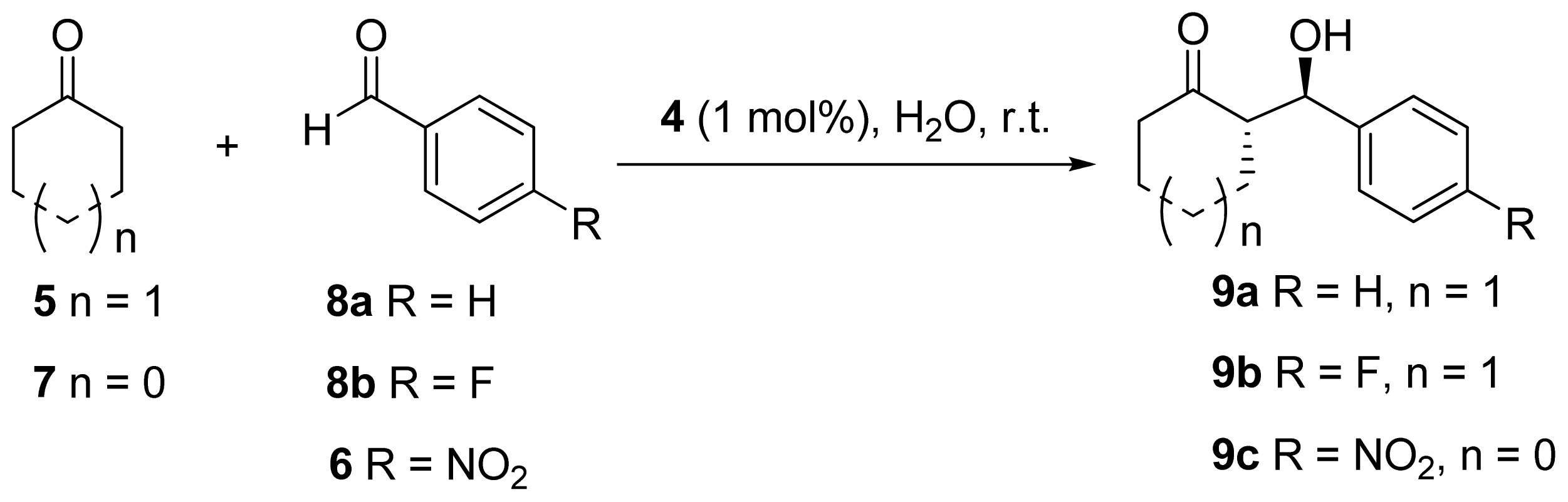
| Entry | Aldehyde | n | Salt | Conversion a | dr a (anti/syn) | ee b |
| 1 | 8a | 1 | N/A c | 45 | 92/8 | 68 |
| 2 | 8a | 1 | N/A d | 82 | 84/16 | 28 |
| 3 | 8a | 1 | EDTA e | 98 | 83/17 | 25 |
| 4 | 8a | 1 | Zn(OAc)2 | 44 | 76/24 | 4 |
| 5 | 8b | 1 | N/A c | 21 | 91/9 | 92 |
| 6 | 8b | 1 | N/A d | 80 | 86/14 | 68 |
| 7 | 8b | 1 | EDTA e | 70 | 85/15 | 88 |
| 8 | 8b | 1 | Zn(OAc)2 | 70 | 76/24 | 64 |
| 9 | 6 | 0 | N/A c | 61 | 76/24 | 14 |
| 10 | 6 | 0 | N/A d | 56 | 39/61 | 22 |
| 11 | 6 | 0 | EDTA e | 74 | 34/66 | 13 |
| 12 | 6 | 0 | Zn(OAc)2 | 97 | 29/71 | 14 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delaney, J.P.; Henderson, L.C. Investigating Ionic Effects Applied to Water Based Organocatalysed Aldol Reactions. Int. J. Mol. Sci. 2011, 12, 9083-9094. https://doi.org/10.3390/ijms12129083
Delaney JP, Henderson LC. Investigating Ionic Effects Applied to Water Based Organocatalysed Aldol Reactions. International Journal of Molecular Sciences. 2011; 12(12):9083-9094. https://doi.org/10.3390/ijms12129083
Chicago/Turabian StyleDelaney, Joshua P., and Luke C. Henderson. 2011. "Investigating Ionic Effects Applied to Water Based Organocatalysed Aldol Reactions" International Journal of Molecular Sciences 12, no. 12: 9083-9094. https://doi.org/10.3390/ijms12129083