Structural Determinants of CX-4945 Derivatives as Protein Kinase CK2 Inhibitors: A Computational Study

Abstract
:1. Introduction
2. Materials and Methods
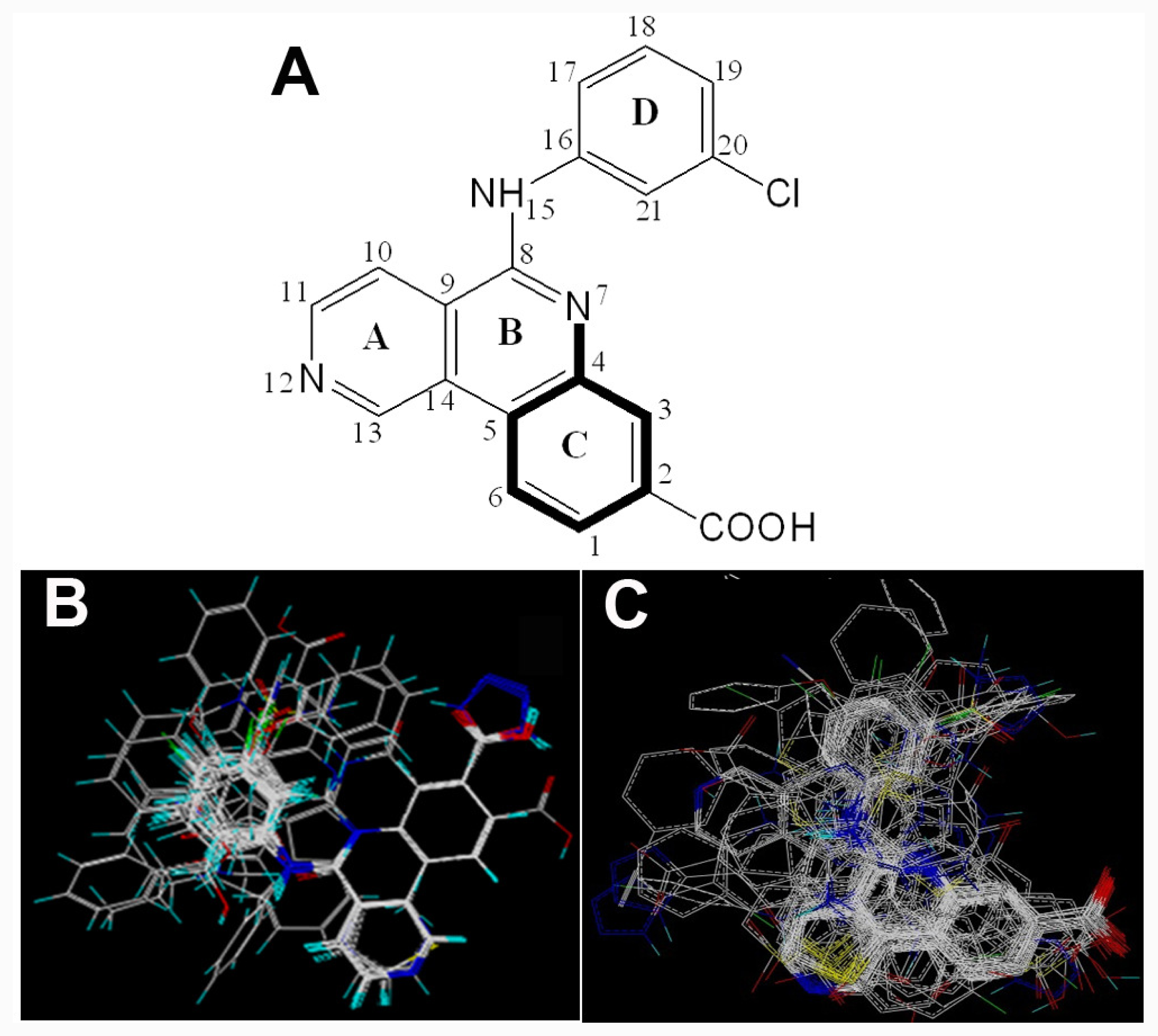
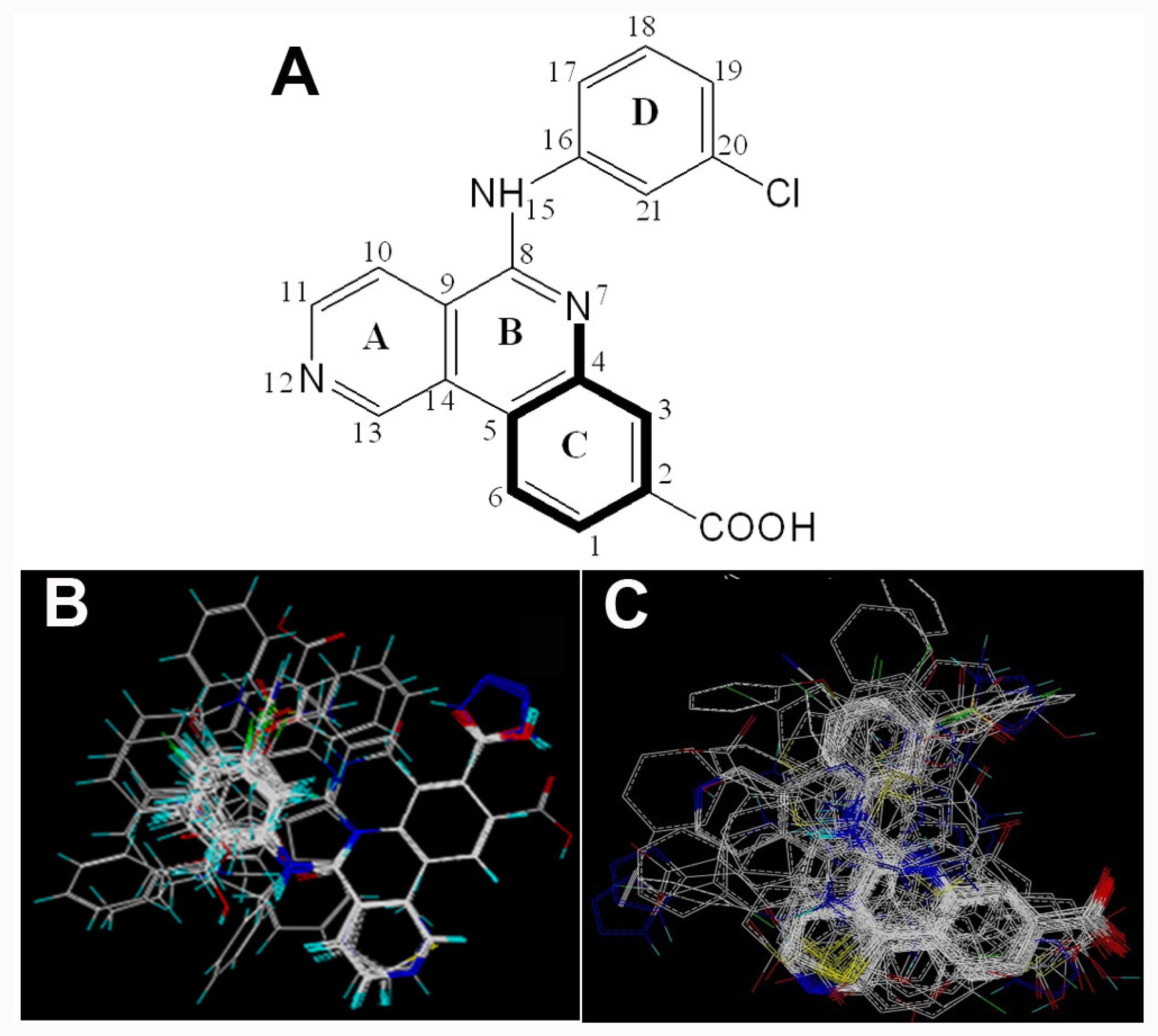
2.1. Data Sets
2.2. Conformational Sampling and Alignment
2.3. CoMFA and CoMSIA 3D-QSAR Models
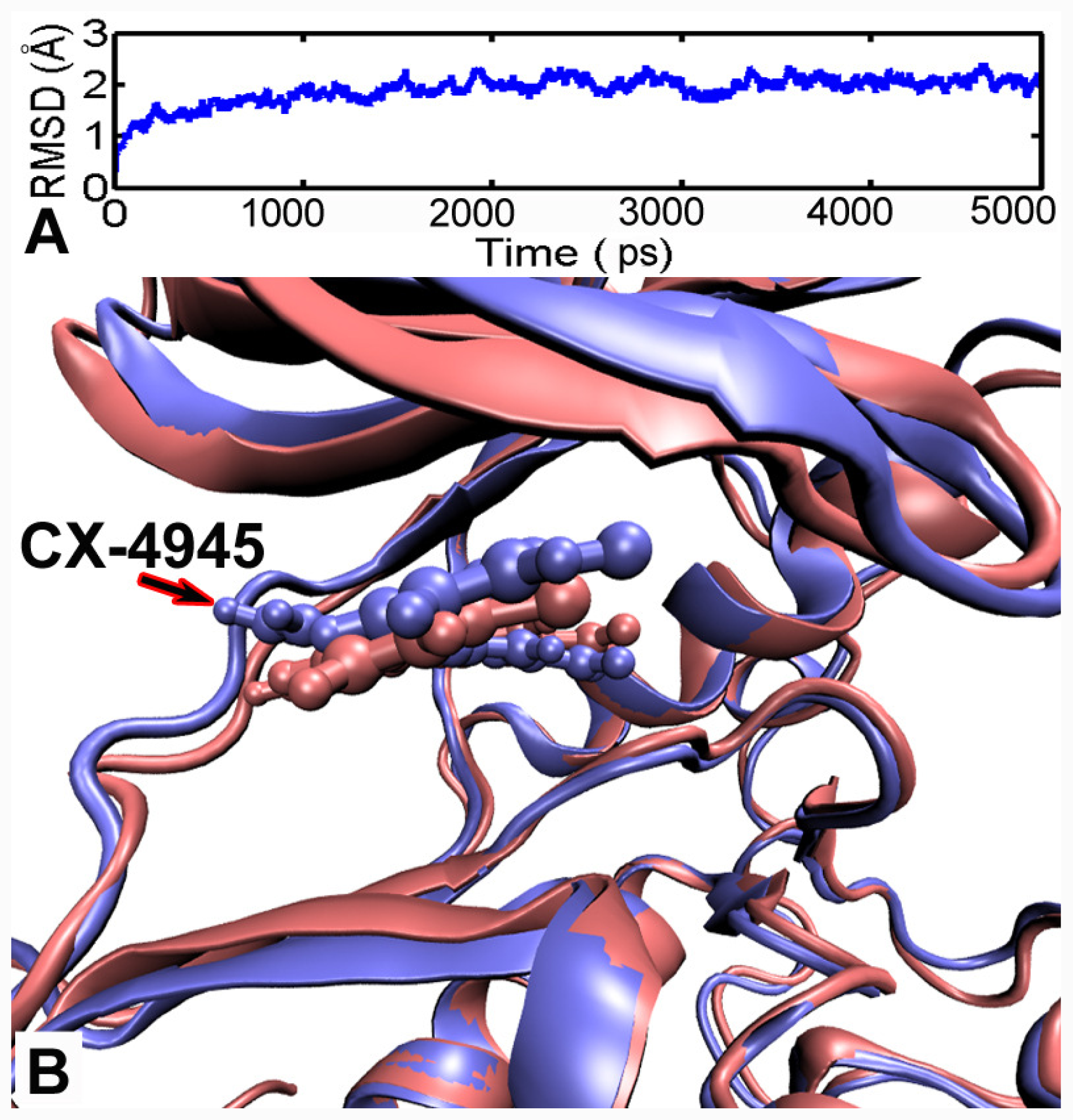
2.4. Molecular Dynamics
3. Results and Discussion
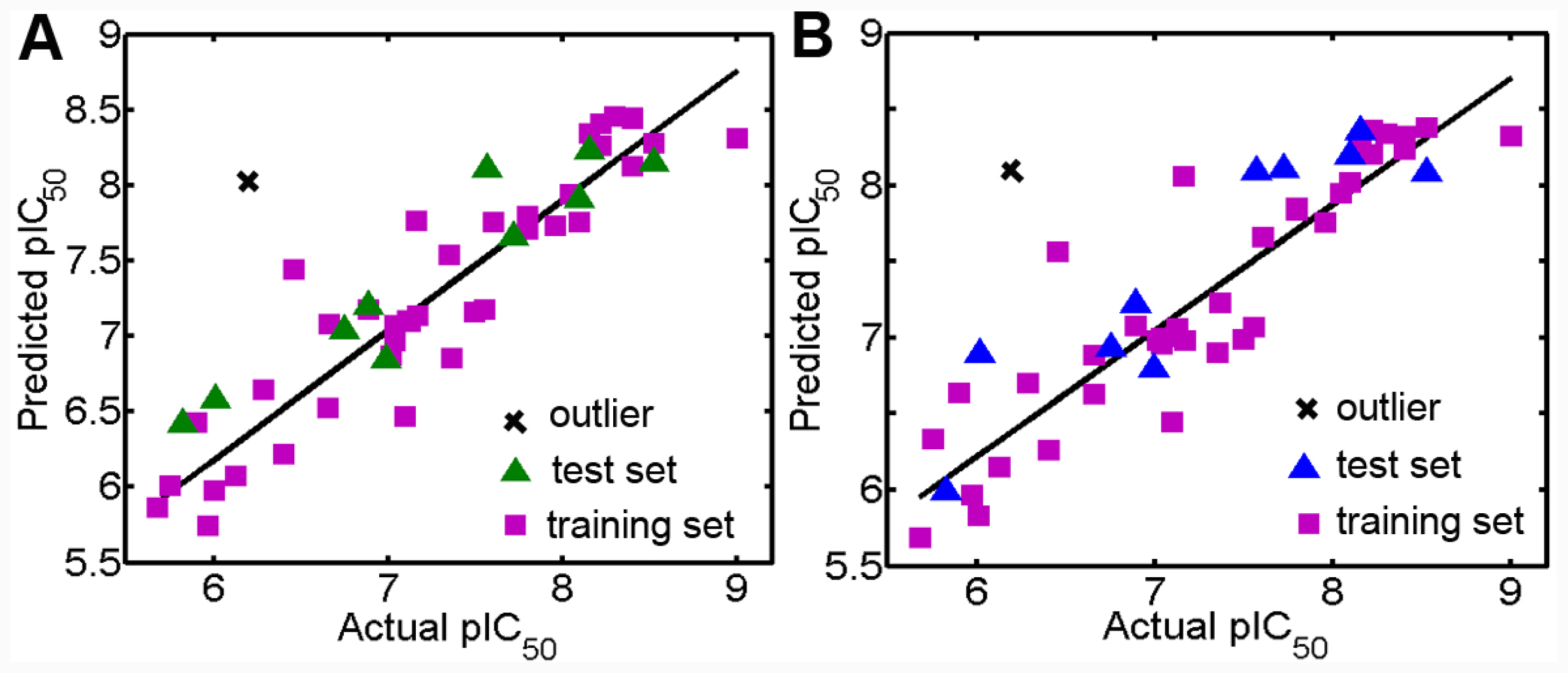
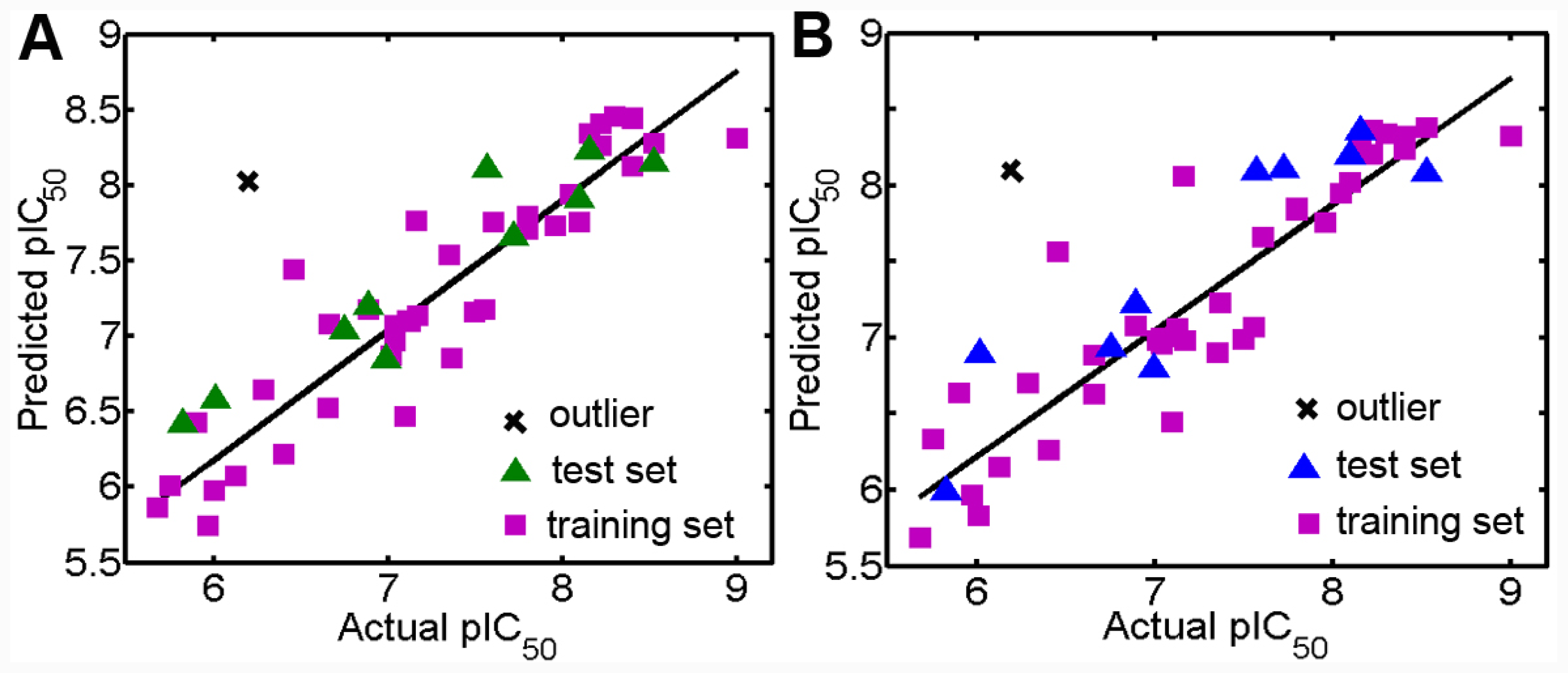
3.1. Validation of the 3D QSAR Models
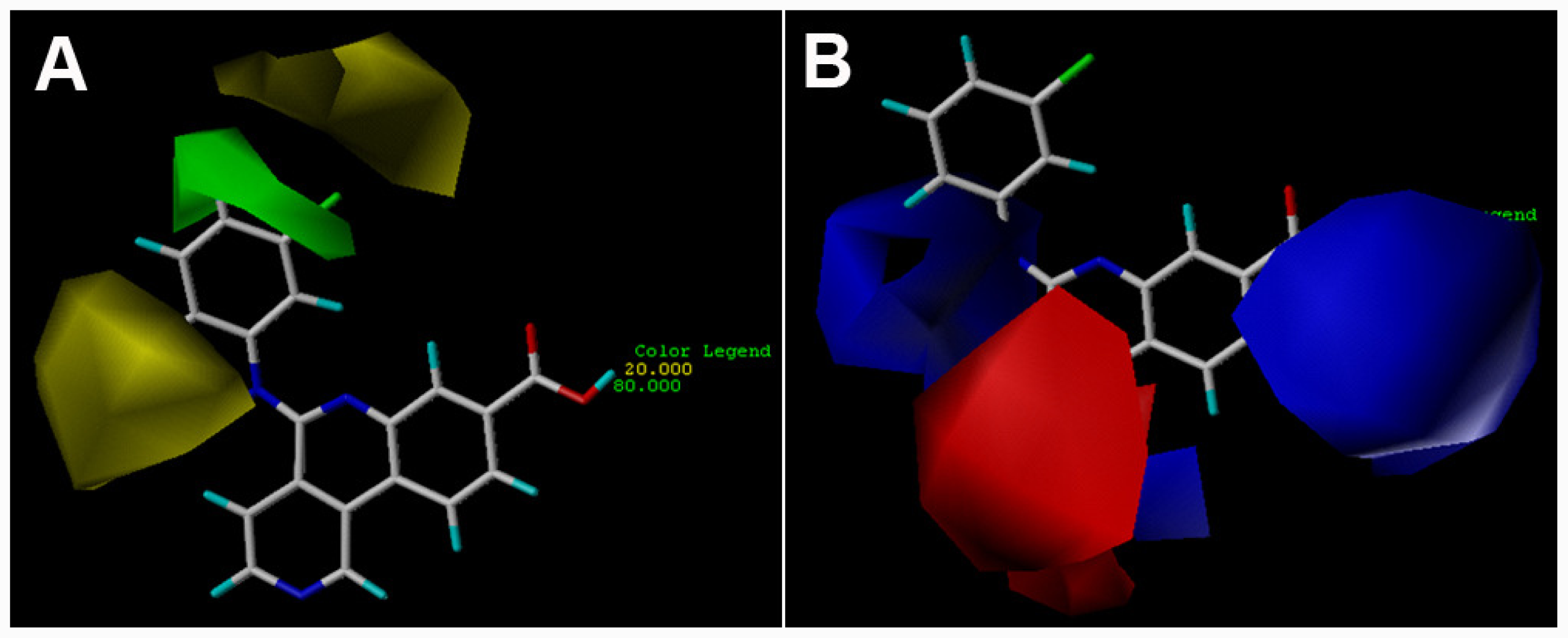
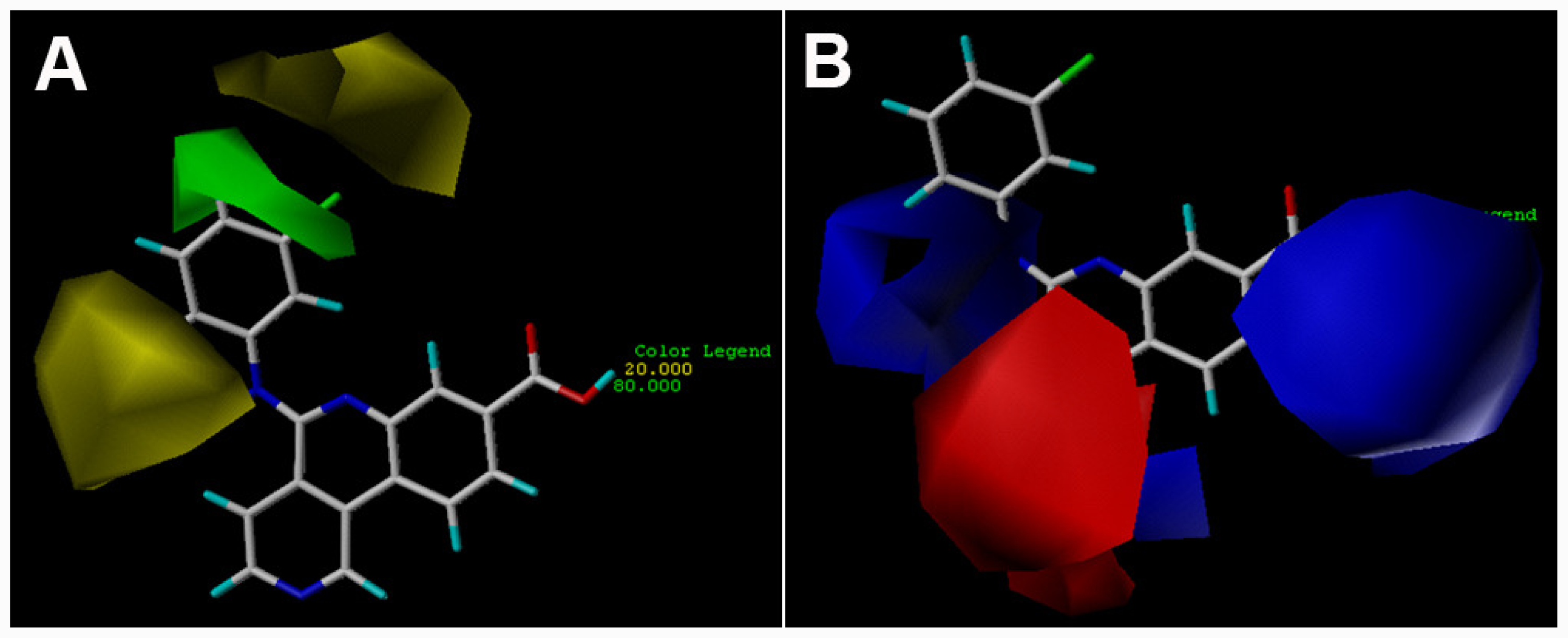
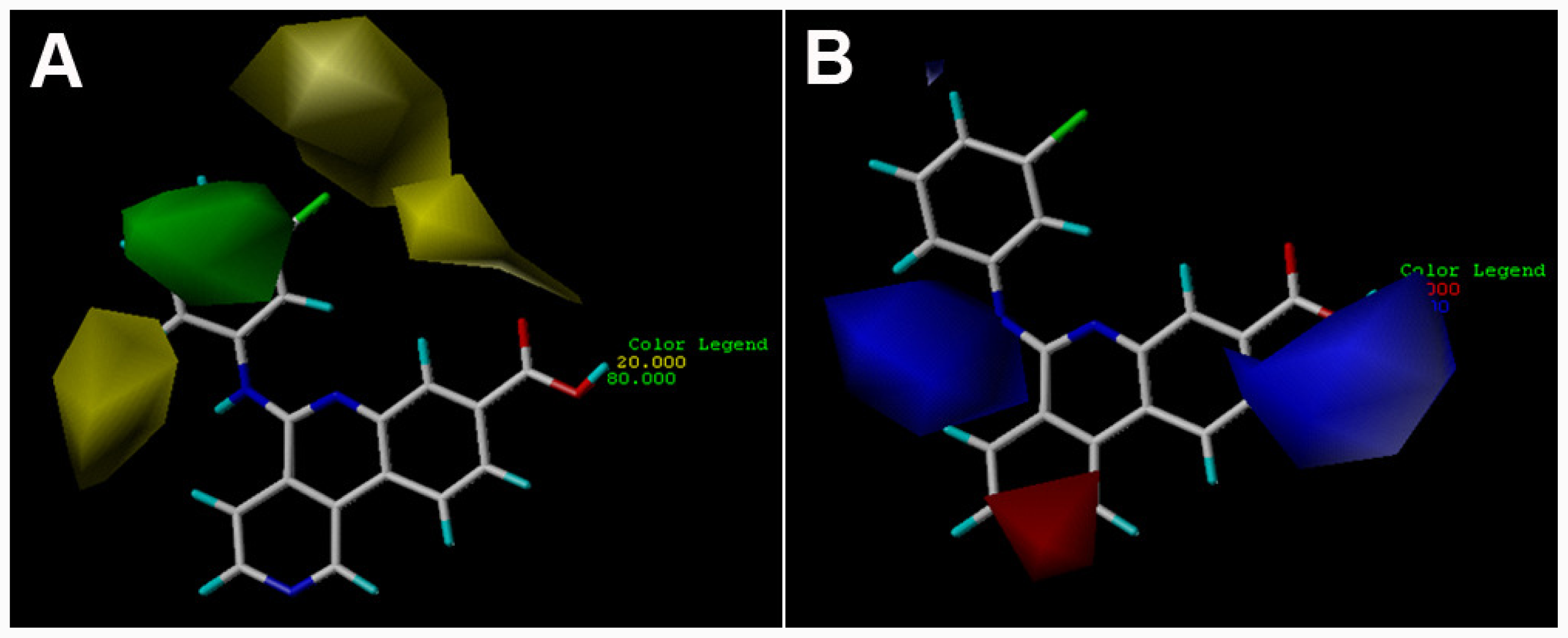
3.2. 3D-QSAR Contour Maps
 ) is more active than compound 24 (with
) is more active than compound 24 (with
 group at the same location).
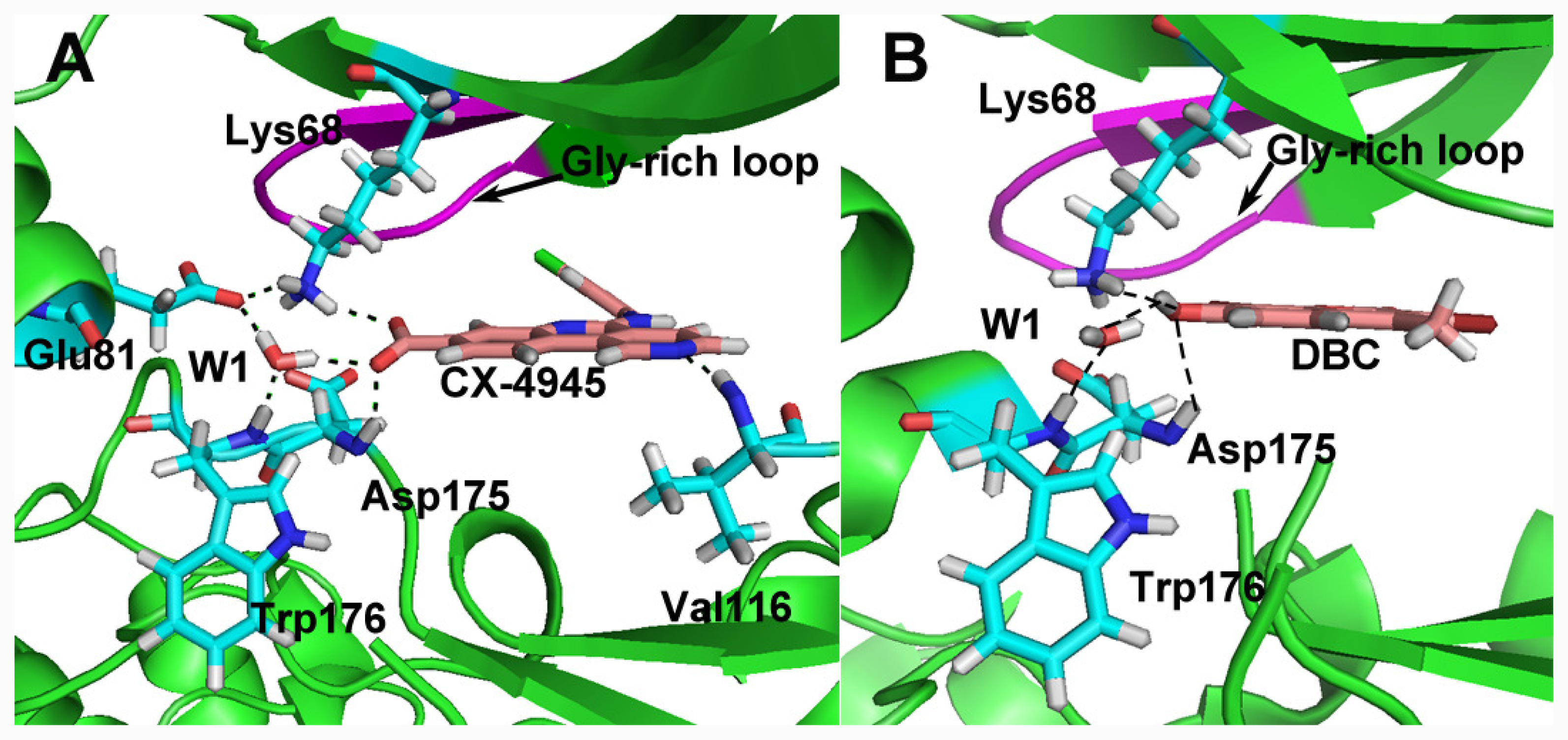
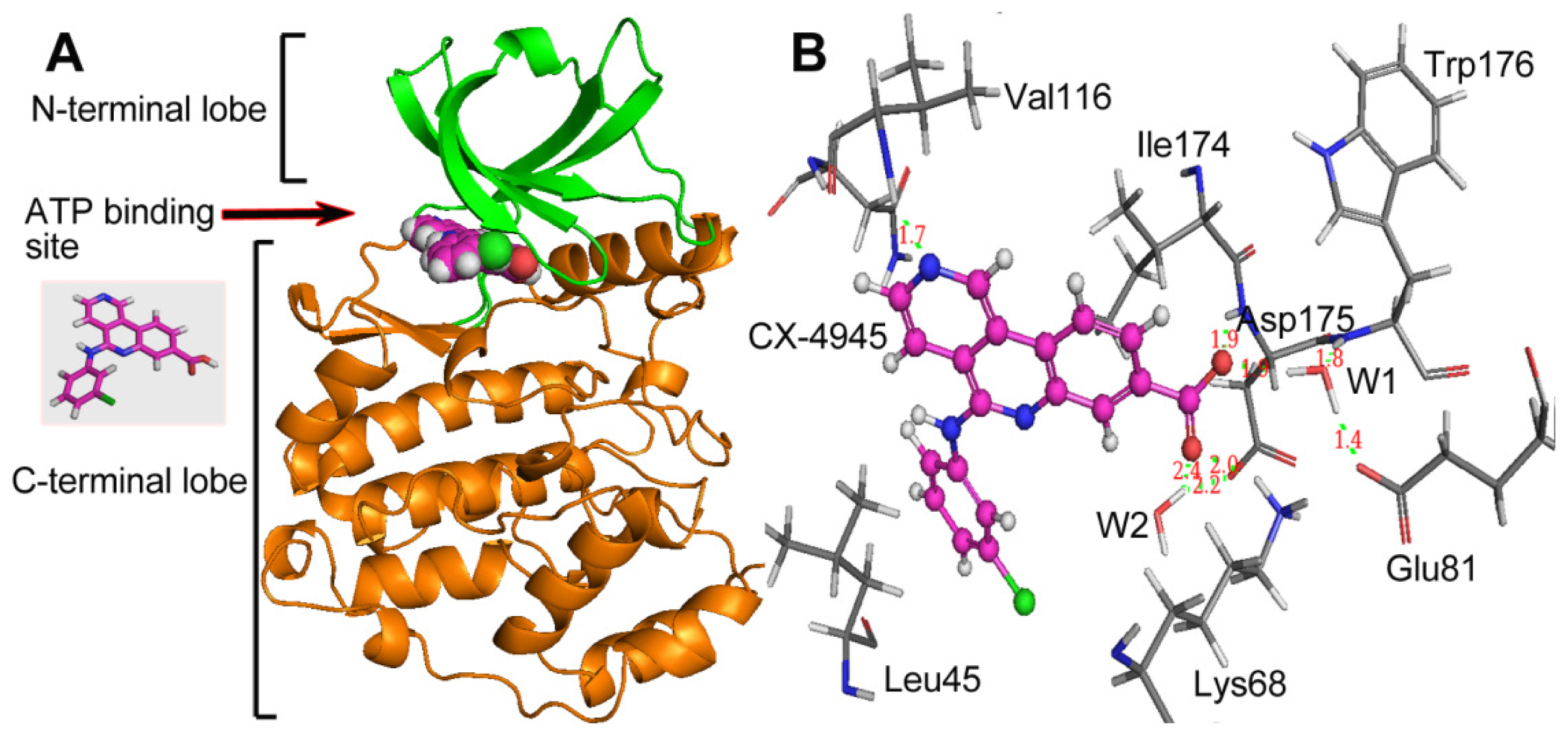
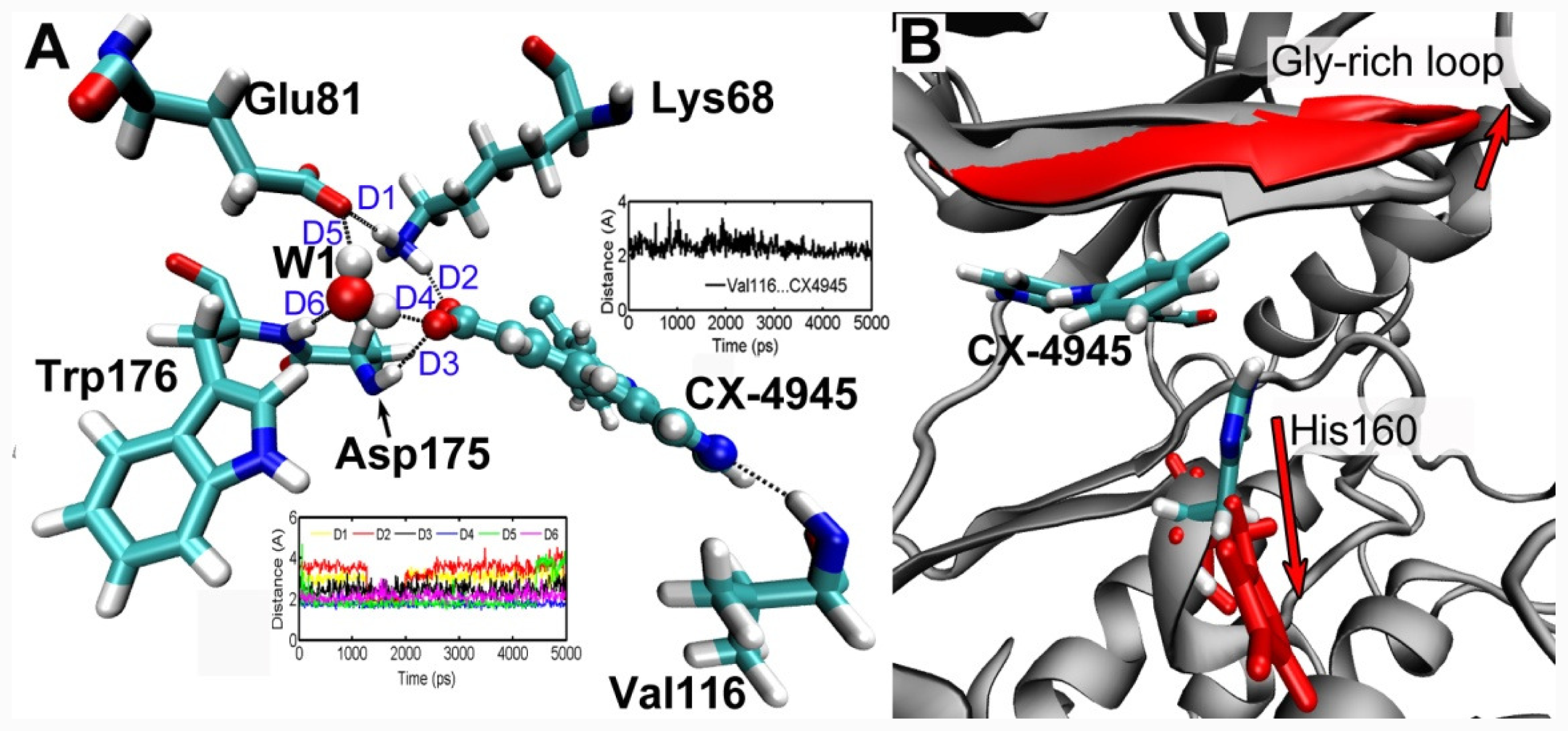
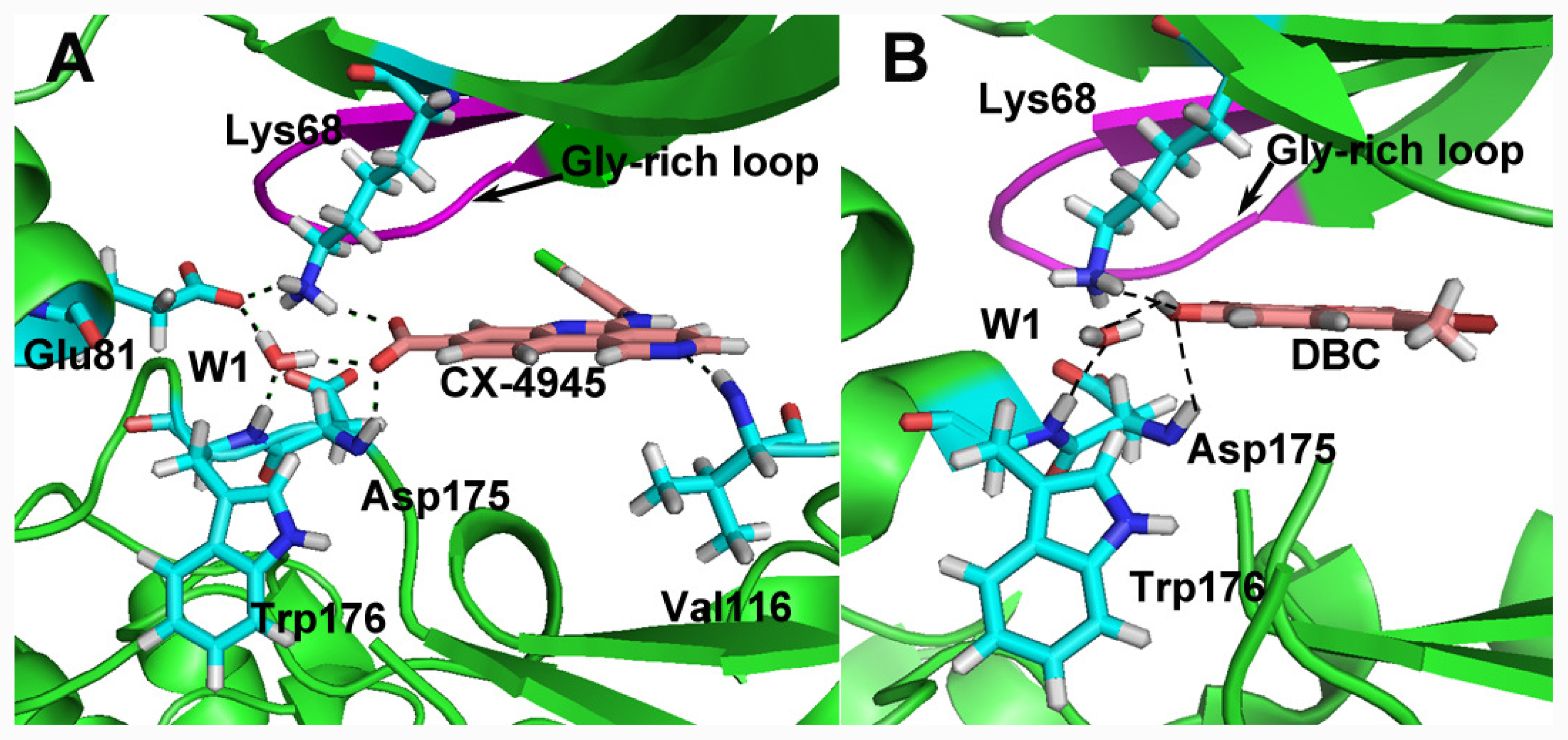
group at the same location).3.3. Docking Analysis and Comparison with 3D Contour Maps
3.4. Comparison with Binding Modes of 3,8-Dibromo-7-hydroxy-4-methylchromen-2-one (DBC)
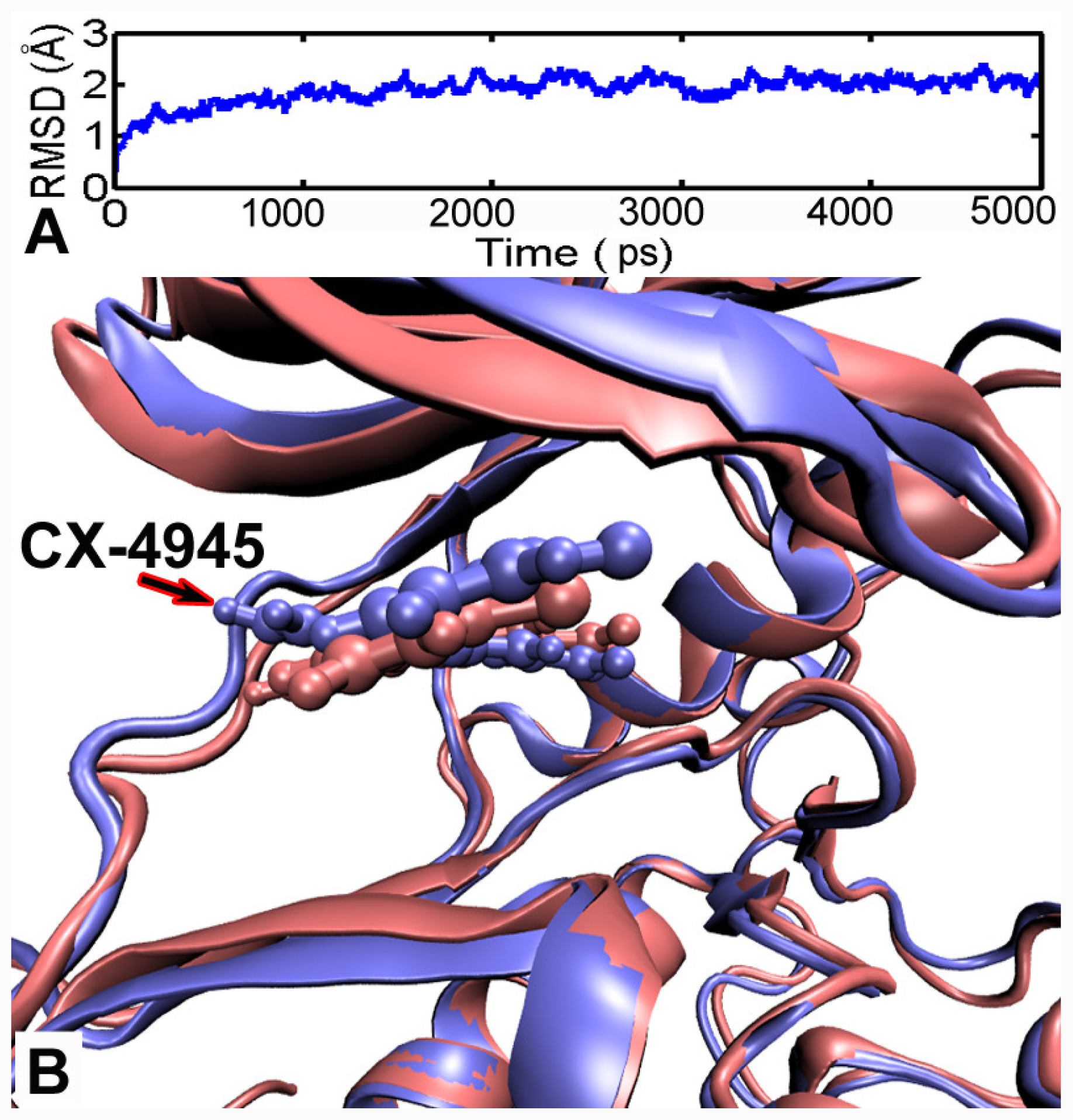
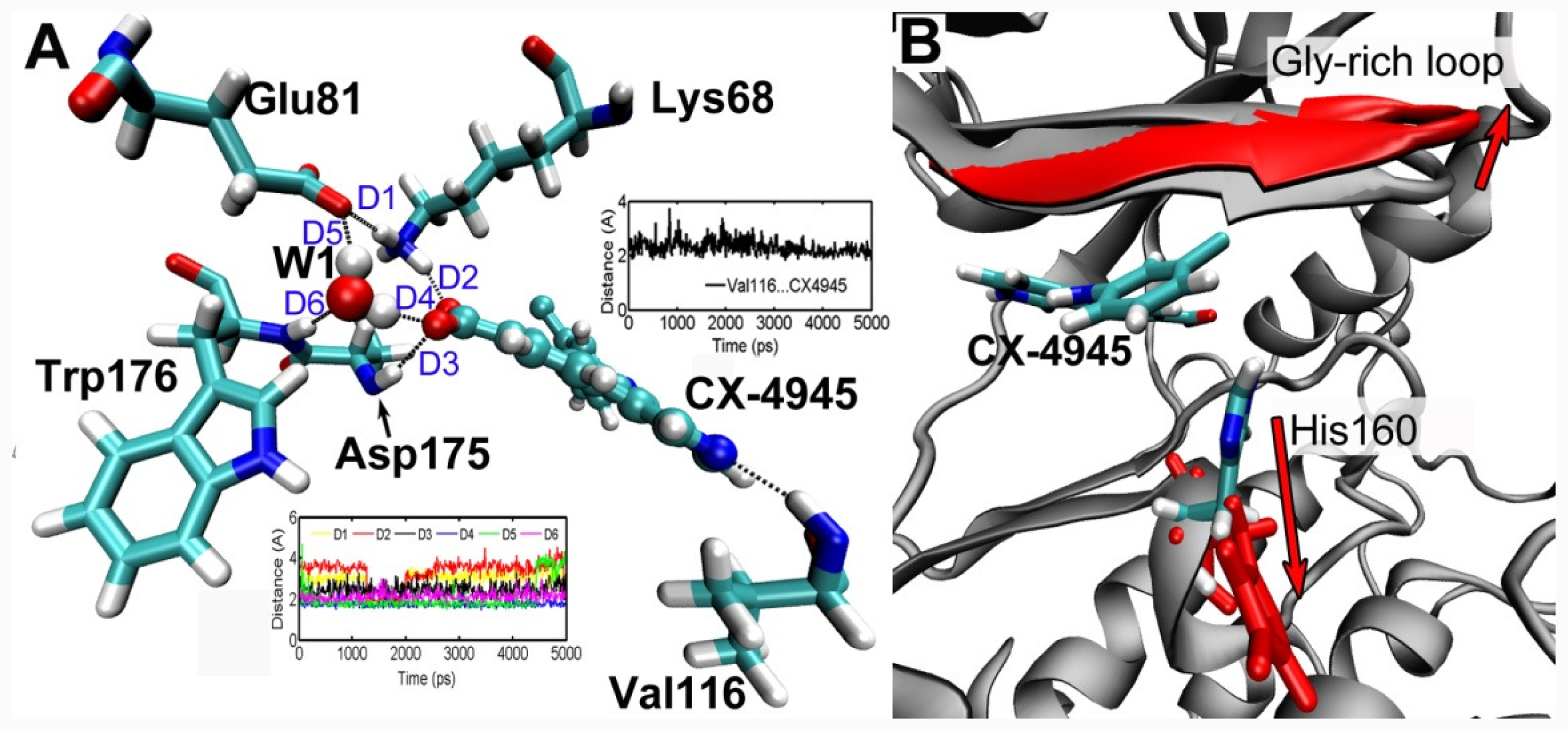
3.5. MD Simulations Analysis
4. Conclusion
Acknowledgments
References
- Duncan, JS; Litchfield, DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2007, 1784, 33–47. [Google Scholar]
- Meggio, F; Pinna, LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J 2003, 17, 349–368. [Google Scholar]
- Pagano, MA; Cesaro, L; Meggio, F; Pinna, LA. Protein kinase CK2: a newcomer in the “druggable kinome”. Biochem. Soc. Trans 2006, 34, 1303–1306. [Google Scholar]
- Guerra, B; Issinger, OG. Protein kinase CK2 in human diseases. Curr. Med. Chem 2008, 15, 1870–1886. [Google Scholar]
- Unger, GM; Davis, AT; Slaton, JW; Ahmed, K. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr. Cancer Drug Targets 2004, 4, 77–84. [Google Scholar]
- Sarno, S; Salvi, M; Battistutta, R; Zanotti, G; Pinna, LA. Features and potentials of ATP-site directed CK2 inhibitors. Biochim. Biophys. Acta 2005, 1754, 263–270. [Google Scholar]
- Sarno, S; Pinna, LA. Protein kinase CK2 as a druggable target. Mol. Biosyst 2008, 4, 889–894. [Google Scholar]
- Pagano, MA; Bain, J; Kazimierczuk, Z; Sarno, S; Ruzzene, M; Di Maira, G; Elliott, M; Orzeszko, A; Cozza, G; Meggio, F; Pinna, LA. The selectivity of inhibitors of protein kinase CK2: an update. Biochem. J 2008, 415, 353–365. [Google Scholar]
- Sarno, S; deMoliner, E; Ruzzene, M; Pagano, MA; Battistutta, R; Bain, J; Fabbro, D; Schoepfer, J; Elliott, M; Furet, P; et al. Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl]-acetic acid (IQA). Biochem. J 2003, 374, 639–646. [Google Scholar]
- Meggio, F; Pagano, MA; Moro, S; Zagotto, G; Ruzzene, M; Sarno, S; Cozza, G; Bain, J; Elliott, M; Deana, AD; et al. Inhibition of protein kinase CK2 by condensed polyphenolic derivatives. An in vitro and in vivo study. Biochemistry 2004, 43, 12931–12936. [Google Scholar]
- Prudent, R; Cochet, C. New protein kinase CK2 inhibitors: jumping out of the catalytic box. Chem. Biol 2009, 16, 112–120. [Google Scholar]
- Morshed, MN; Muddassar, M; Pasha, FA; Cho, SJ. Pharmacophore identification and validation study of CK2 inhibitors using CoMFA/CoMSIA. Chem. Biol. Drug Des 2009, 74, 148–158. [Google Scholar]
- Wang, X; Yang, W; Xu, X; Zhang, H; Li, Y; Wang, Y. Studies of benzothiadiazine derivatives as hepatitis C virus NS5B polymerase inhibitors using 3D-QSAR, molecular docking and molecular dynamics. Curr. Med. Chem 2010, 17, 2788–2803. [Google Scholar]
- Cramer, RD; Patterson, DE; Bunce, JD. Comparative molecular field analysis (CoMFA). 1: effect of shape on binding of steroids to carrier proteins. J. Acoust. Soc. Am 1988, 110, 5959–5967. [Google Scholar]
- Klebe, G; Abraham, U; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem 1994, 37, 4130–4146. [Google Scholar]
- Siddiqui-Jain, A; Drygin, D; Streiner, N; Chua, P; Pierre, F; O’Brien, S; Bliesath, J; Omori, M; Huser, N; Ho, C; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits anti-tumor efficacy. Cancer Res 2010, 70, 10288–10298. [Google Scholar]
- Pierre, F; Chua, PC; O’Brien, SE; Siddiqui-Jain, A; Bourbon, P; Haddach, M; Michaux, J; Nagasawa, J; Schwaebe, MK; Stefan, E; et al. Discovery and SAR of 5-(3-Chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem 2011, 54, 635–654. [Google Scholar]
- Cramer, RD, III; Patterson, DE; Bunce, JD. Recent advances in comparative molecular field analysis (CoMFA). Prog. Clin. Biol. Res 1989, 291, 161–165. [Google Scholar]
- Vriend, G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph 1990, 8, 52–56. [Google Scholar]
- Hilal, SH; Karickhoff, SW; Carreira, LA. A rigorous test for SPARC’s chemical reactivity models: estimation of more than 4300 ionization pKas. Quant Struct Act Relatsh 1995, 14, 348–355. [Google Scholar]
- Case, DA; Darden, T; Cheatham, TE, III; Simmerling, C; Wang, J; Duke, RE; Luo, R; Crowley, M; Walker, RC; Zhang, W; et al. AMBER 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Wang, J; Wolf, RM; Caldwell, JW; Kollman, PA; Case, DA. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Jakalian, A; Jack, DB; Bayly, CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem 2002, 23, 1623–1641. [Google Scholar]
- Duan, Y; Wu, C; Chowdhury, S; Lee, MC; Xiong, G; Zhang, W; Yang, R; Cieplak, P; Luo, R; Lee, T; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003, 24, 1999–2012. [Google Scholar]
- Jorgensen, WL; Chandrasekhar, J; Madura, JD; Klein, ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Essmann, U; Perera, L; Berkowitz, ML; Darden, T. A smooth particle mesh ewald method. J. Chem. Phys 1995, 103, 8577–9593. [Google Scholar]
- Buolamwini, JK; Assefa, H. CoMFA and CoMSIA 3D QSAR and docking studies on conformationally-restrained cinnamoyl HIV-1 integrase inhibitors: Exploration of a binding mode at the active site. J. Med. Chem 2002, 45, 841–852. [Google Scholar]
- Boeckler, F; Ohnmacht, U; Lehmann, T; Utz, W; Hubner, H; Gmeiner, P. CoMFA and CoMSIA investigations revealing novel insights into the binding modes of dopamine D3 receptor agonists. J. Med. Chem 2005, 48, 2493–2508. [Google Scholar]
- Gohlke, H; Schwarz, S; Gundisch, D; Tilotta, MC; Weber, A; Wegge, T; Seitz, G. 3D QSAR analyses-guided rational design of novel ligands for the (α4)2(β2)3 nicotinic acetylcholine receptor. J. Med. Chem 2003, 46, 2031–2048. [Google Scholar]
- Borges, F; Roleira, F; Milhazes, N; Santana, L; Uriarte, E. Simple coumarins and analogues in medicinal chemistry: occurrence, synthesis and biological activity. Curr. Med. Chem 2005, 12, 887–916. [Google Scholar]
- Lacy, A; O’Kennedy, R. Studies on coumarins and coumarin-related compounds to determine their therapeutic role in the treatment of cancer. Curr. Pharm. Des 2004, 10, 3797–3811. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Structure | IC50 (μM) | pIC50 | |||||
|---|---|---|---|---|---|---|---|---|
| 1 |  | 0.08 | 7.097 | |||||
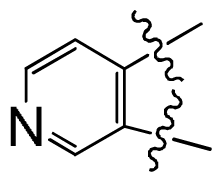
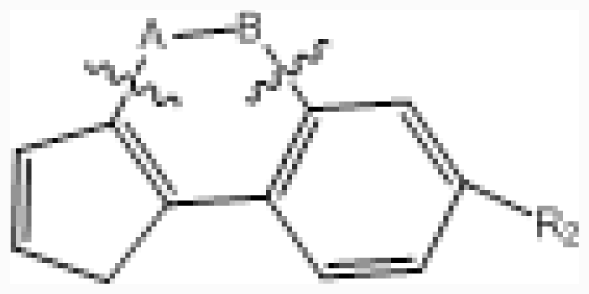 | ||||||||
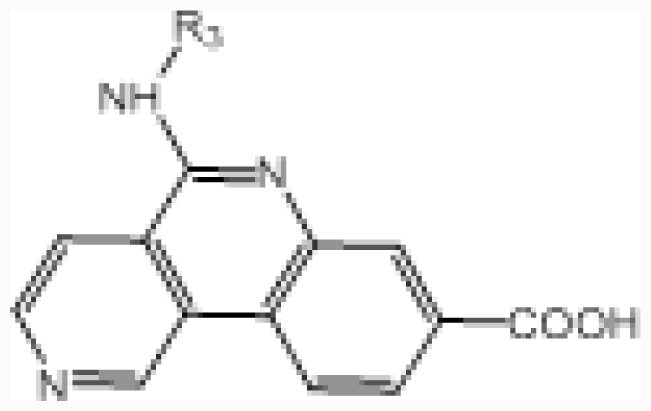
| No. |  | R2 | IC50 (μM) | pIC50 | ||||
| 2 | –CO–NH– | –COOH | 2.1 | 5.678 | ||||
| 3 † | –CO–N((CH2)3OH)– | –COOH | 1.5 | 5.824 | ||||
| 4 | –(C–O(CH2)3OH)=N– | –COOH | 0.99 | 6.004 | ||||
| 5 | –(C–NH(CH2)3OH)=N– | –COOH | 0.75 | 6.125 | ||||
 | ||||||||
| No. | –NR3R4 | R2 | IC50 (μM) | pIC50 | ||||
| 6 | –NH–(CH2)2OH | –COOH | 1.26 | 5.900 | ||||
| 7 † | –NH–(CH2)2NMe2 | –COOH | 0.102 | 6.991 | ||||
| 8 | –Pyrrolidino | –COOH | 1.780 | 5.750 | ||||
| 9 | –NH–phenyl | –COOH | 0.092 | 7.036 | ||||
| 10 | –NMe–phenyl | –COOH | 1.070 | 5.971 | ||||
| 11 † | –NH–(2–Me–phenyl) | –COOH | 0.970 | 6.013 | ||||
| 12 | –NH–phenyl | –C-(1H-tetrazol-5-yl) | 0.096 | 7.018 | ||||
| 13 | –NH–(CH2)2Ph | –COOH | 0.516 | 6.287 | ||||
| 14 | –NH–(4–F–phenyl) | –COOH | 0.219 | 6.660 | ||||
| 15 | –NH–(3–F–phenyl) | –COOH | 0.068 | 7.168 | ||||
| 16 † | –NH–(4–Cl–phenyl) | –COOH | 0.178 | 6.750 | ||||
| 17 | –NH–(3–Cl–phenyl) | –COOH | 0.032 | 7.495 | ||||
| 18 | –NH–(3–MeO–phenyl) | –COOH | 0.077 | 7.114 | ||||
| 19 | –NH–(3–acetylenyl–phenyl) | –COOH | 0.028 | 7.553 | ||||
| 20 | –NH–(3–(PhO)–phenyl) | –COOH | 0.395 | 6.403 | ||||
| 21 † | –NH–(3–(CONHMe)–phenyl) | –COOH | 0.129 | 6.889 | ||||
| 22 | –NH–(3–Cl–phenyl) | –C-(1H-tetrazol-5-yl) | 0.129 | 6.889 | ||||
| 23 | –NH–(3–F–phenyl) | –C-(1H-tetrazol-5-yl) | 0.075 | 7.125 | ||||
 | ||||||||
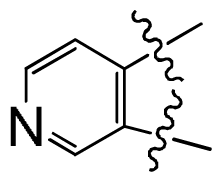
| No. | A ring | IC50 (μM) | pIC50 | |||||
| 24 |  | 0.092 | 7.036 | |||||
| 25 | 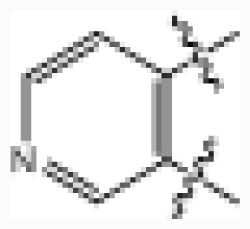 | 0.006 | 8.222 | |||||
 | ||||||||
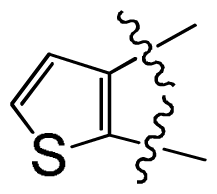
| No. | R3 | IC50 (μM) | pIC50 | |||||
| 26 | –(CH2)2NMe | 0.025 | 7.602 | |||||
| 27 † | –cyclopentyl | 0.027 | 7.569 | |||||
| 28 | –OMe | 0.008 | 8.097 | |||||
| 29 | –cyclopropyl | 0.016 | 7.796 | |||||
| 30 | –(CH2)2O-i-Pr | 0.011 | 7.959 | |||||
| 31 | –(CH2)phenyl | 0.009 | 8.046 | |||||
| 32 † | –(CH2)2phenyl | 0.003 | 8.523 | |||||
| 33 | –(CH2)3phenyl | 0.016 | 7.796 | |||||
| 34 | –(3-MeO-phenyl) | 0.004 | 8.398 | |||||
| 35 | –(3-Cl,4-F-phenyl) | 0.004 | 8.398 | |||||
| 36 | –(3-F-phenyl) | 0.005 | 8.301 | |||||
| 37 † | –(2-Cl-phenyl) | 0.008 | 8.097 | |||||
| 38 | –(3-Cl-phenyl) | 0.001 | 9.000 | |||||
| 39 | –(4-Cl-phenyl) | 0.007 | 8.155 | |||||
| 40 | –(3-acetylenyl-phenyl) | 0.003 | 8.523 | |||||
| 41 | –(3-CN-phenyl) | 0.004 | 8.398 | |||||
| 42 | –(4-(PhO)-phenyl) | 0.069 | 7.161 | |||||
| 43 † | –(3-(PhO)-phenyl) | 0.019 | 7.721 | |||||
| 44 | –(3-(SO2NH2)-phenyl) | 0.043 | 7.367 | |||||
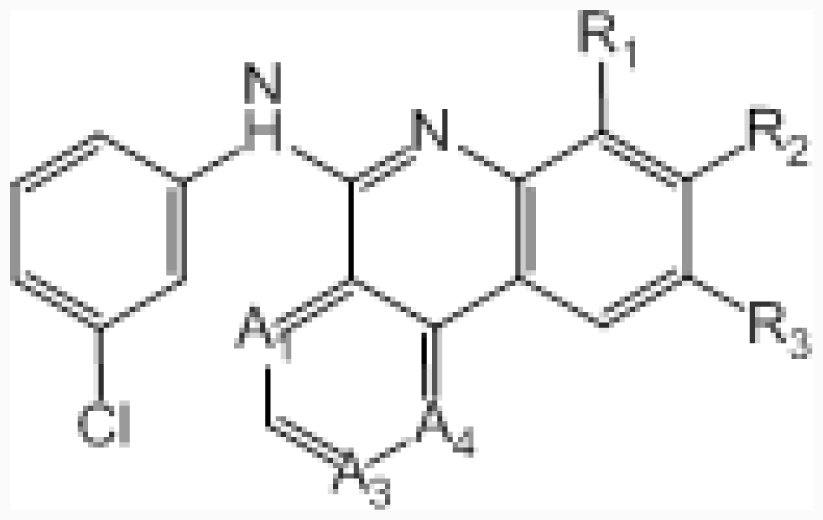 | ||||||||
| No. | A1 | A3 | A4 | R1 | R2 | R3 | IC50 (μM) | pIC50 |
| 45 | –CH= | –N= | –CH= | –H | –C-(1H-tetrazol-3-yl) | –H | 0.045 | 7.347 |
| 46 ‡ | –CH= | –N= | –CH= | –H | –CONH2 | –H | 0.417 | 6.380 |
| 47 | –CH= | –N= | –CH= | –Me | –COOH | –H | 0.006 | 8.222 |
| 48 | –CH= | –N= | –CH= | –H | –H | –COOH | 0.350 | 6.456 |
| 49 | –N= | –CH= | –N= | –H | –COOH | –H | 0.220 | 6.658 |
| 50 † | –N= | –N= | –CH= | –H | –COOH | –H | 0.007 | 8.155 |
| Parameters | Ligand-based alignment | Docking-based alignment | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Rcv2 | 0.618 | 0.681 | 0.562 | 0.545 |
| ONC | 3 | 3 | 4 | 5 |
| SEP | 0.567 | 0.518 | 0.616 | 0.637 |
| Rncv2 | 0.860 | 0.828 | 0.877 | 0.791 |
| SEE | 0.343 | 0.380 | 0.326 | 0.187 |
| F value | 71.892 | 56.197 | 62.523 | 173.161 |
| Rpred2 | 0.892 | 0.843 | 0.556 | 0.580 |
| Field contribution | ||||
| Steric | 0.423 | 0.180 | 0.668 | 0.151 |
| Electrostatic | 0.567 | 0.820 | 0.332 | 0.246 |
| Hydrophobic | - | - | - | 0.321 |
| Acceptor | - | - | - | 0.282 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, H.; Wang, X.; Wang, J.; Wang, J.; Li, Y.; Yang, L.; Li, G. Structural Determinants of CX-4945 Derivatives as Protein Kinase CK2 Inhibitors: A Computational Study. Int. J. Mol. Sci. 2011, 12, 7004-7021. https://doi.org/10.3390/ijms12107004
Liu H, Wang X, Wang J, Wang J, Li Y, Yang L, Li G. Structural Determinants of CX-4945 Derivatives as Protein Kinase CK2 Inhibitors: A Computational Study. International Journal of Molecular Sciences. 2011; 12(10):7004-7021. https://doi.org/10.3390/ijms12107004
Chicago/Turabian StyleLiu, Hongbo, Xia Wang, Jian Wang, Jinghui Wang, Yan Li, Ling Yang, and Guohui Li. 2011. "Structural Determinants of CX-4945 Derivatives as Protein Kinase CK2 Inhibitors: A Computational Study" International Journal of Molecular Sciences 12, no. 10: 7004-7021. https://doi.org/10.3390/ijms12107004