3.1. Characterization of PLA Reinforcement Ligament
Resorbaid
® reinforcement ligaments are made of poly(
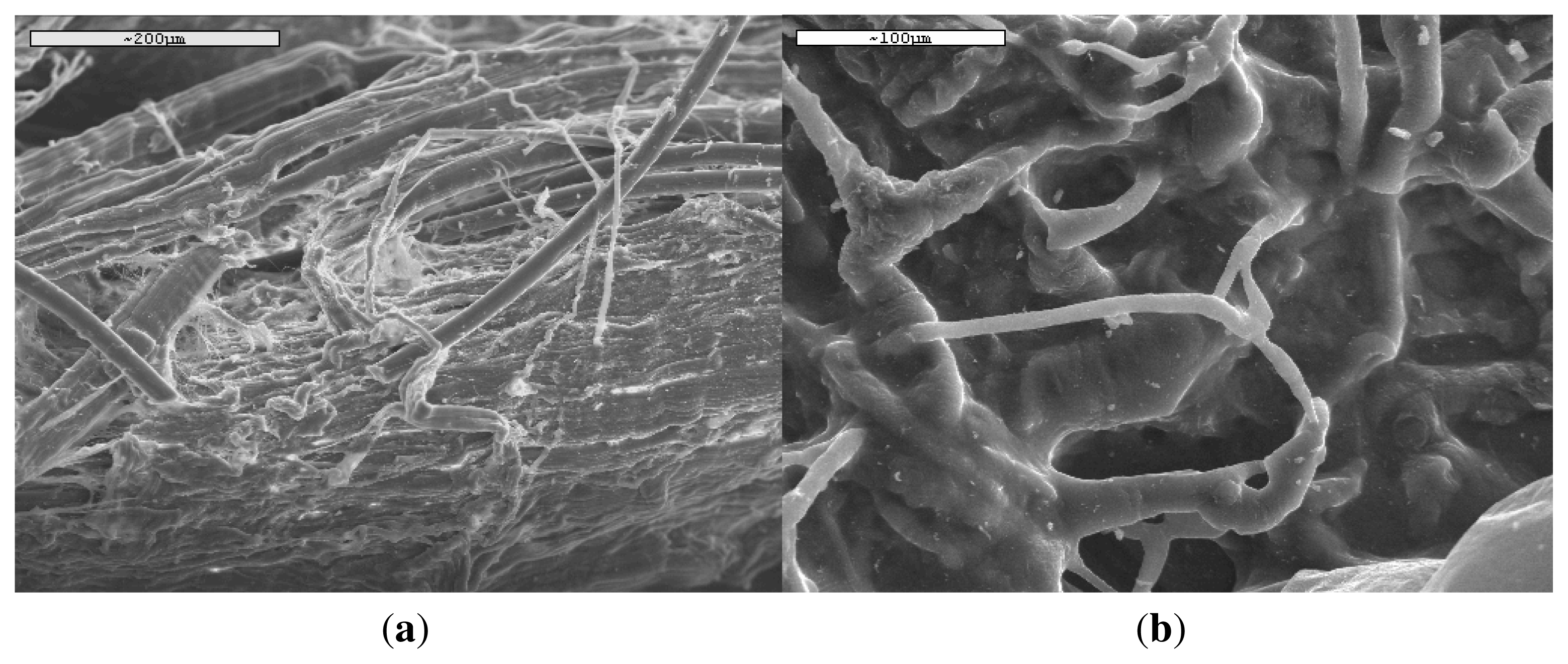
l-lactic acid) which is a bio-compatible material and fully bio-absorbable in the long term. The polyester has an intrinsic viscosity 1.41 dL/g. This corresponds to a high average number molecular weight (Mn) 120,000 g/mol which is necessary in order to prepare reinforcement ligaments with high mechanical properties. The ring opening polymerization of PLA is an important method to obtain such high molecular weight products, in which using high purity lactide is the most important factor in the whole process. The reinforcement ligament is designed for the repair and the reinforcement of articulation instabilities and can be fully desorbed in the human body within approximately 3–5 years. The ligament structure, which can be seen in
Figure 1(a), entails a quick in-growth of fibrosis and an excellent tissue re-colonization. Due to its design, consisting of fibers with an average diameter 15 μm (
Figure 1(b)), and its faultless mechanical properties, it enables improvement in the primary mechanical resistance of the articular ligament during the first 6 months.
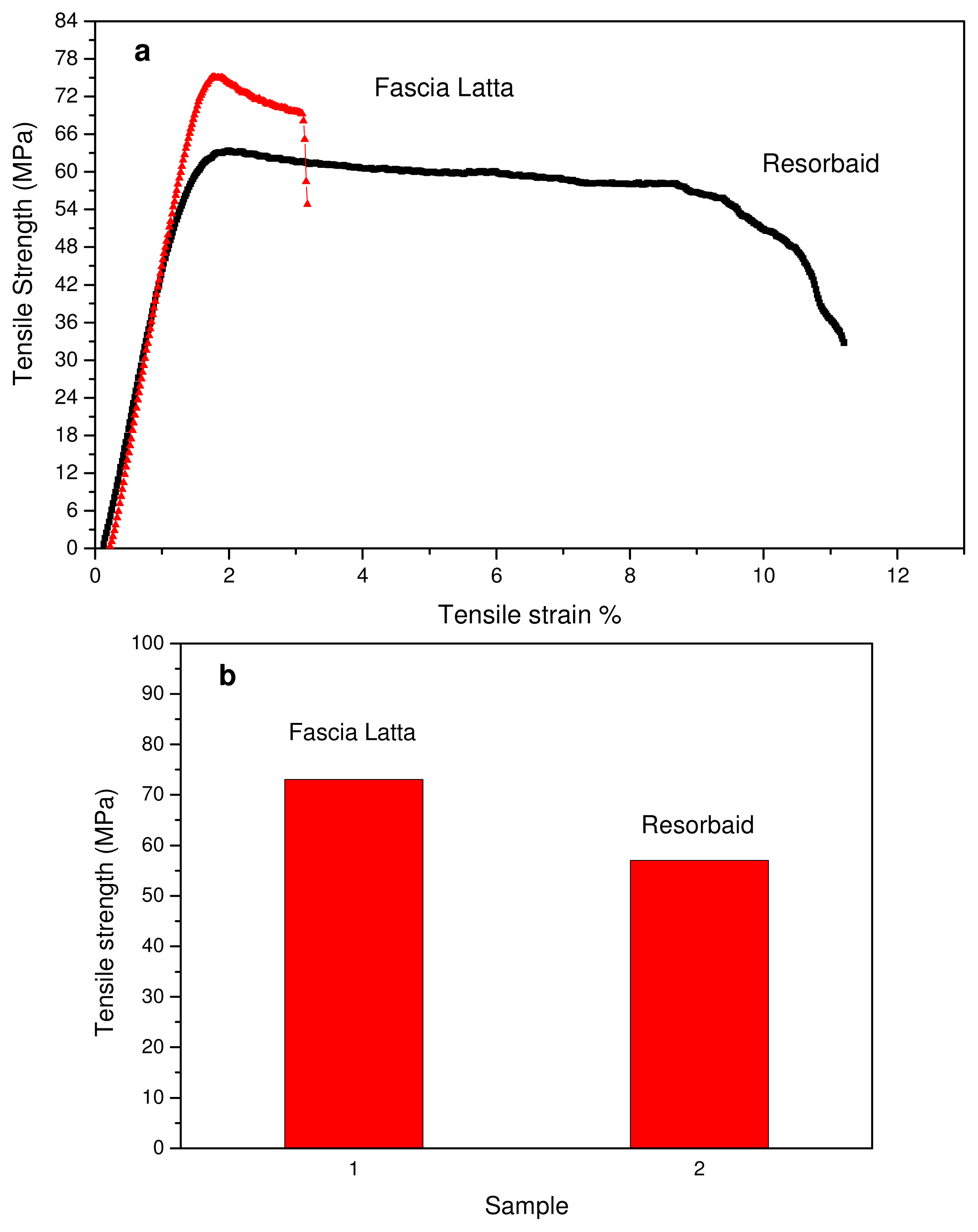
Tensile properties of the Resorbaid compared with Fascia Latta specimens were measured using an Instron tensile testing machine. Fascia Latta is a flexible natural collagen tissue scaffold that allows for new host tissue organization along its native fibers. From stress-strain curves (
Figure 2(a)) it is clear that Fascia Latta breaks almost immediately after yielding, while Resorbaid has slightly higher elongation at break (about 10%). However, both materials can be characterized as brittle and strong since they have high tensile strengths. In fact, Resorbaid shows a tensile strength of 57 MPa compared to 73 MPa of Fascia Latta. This is very important since, in orthopaedic applications such as reinforcement ligaments, the material may be subjected to significant loads. The polymer’s molecular weight affects its mechanical properties and degradation behavior and is therefore a critical property to evaluate. It was found that, in order to perform as a load-bearing orthopaedic implant, a L-PLA polymer with a molecular weight of at least 100 kDa should be used [
29,
30]. For this reason, the PLA used for the preparation of Resorbaid has a number average molecular weight about 120,000 g/mol. However, this high molecular weight can affect the hydrolysis rate of the reinforcement ligament.
3.2. Crystallization Studies
In addition to molecular weight, the degree of crystallinity and crystallization rates can have an effect on the hydrolysis rate of PLA. Thus, crystallization studies on PLA ligament are very important since, as previously reported, further crystallization can take place during
in vivo hydrolysis and this can affect the degradation rates of PLA [
4,
31]. During hydrolysis, PLA oligomers can be formed due to the ester bond cleavage as well as acidic end groups. This results in the formation of a skin composed of a polymer which degrades less rapidly than the polymer located away from the surface. Thus, it can be said that the surface of PLA fibers is less susceptible to hydrolysis rate than the interior part. The whole mechanism can result in hollow residual structures when the matrix remains amorphous for the whole degradation process. It is critical to note that if the matrix is initially crystalline or crystallizes during degradation, the inner part of large size devices degrades faster than the surface but does not lead to hollow residual structures. However, it leads to tiny crystalline residues which can be inflammatory when they remain at the surgery site, even if they are less inflammatory than the residues issued from a quenched device [
4].
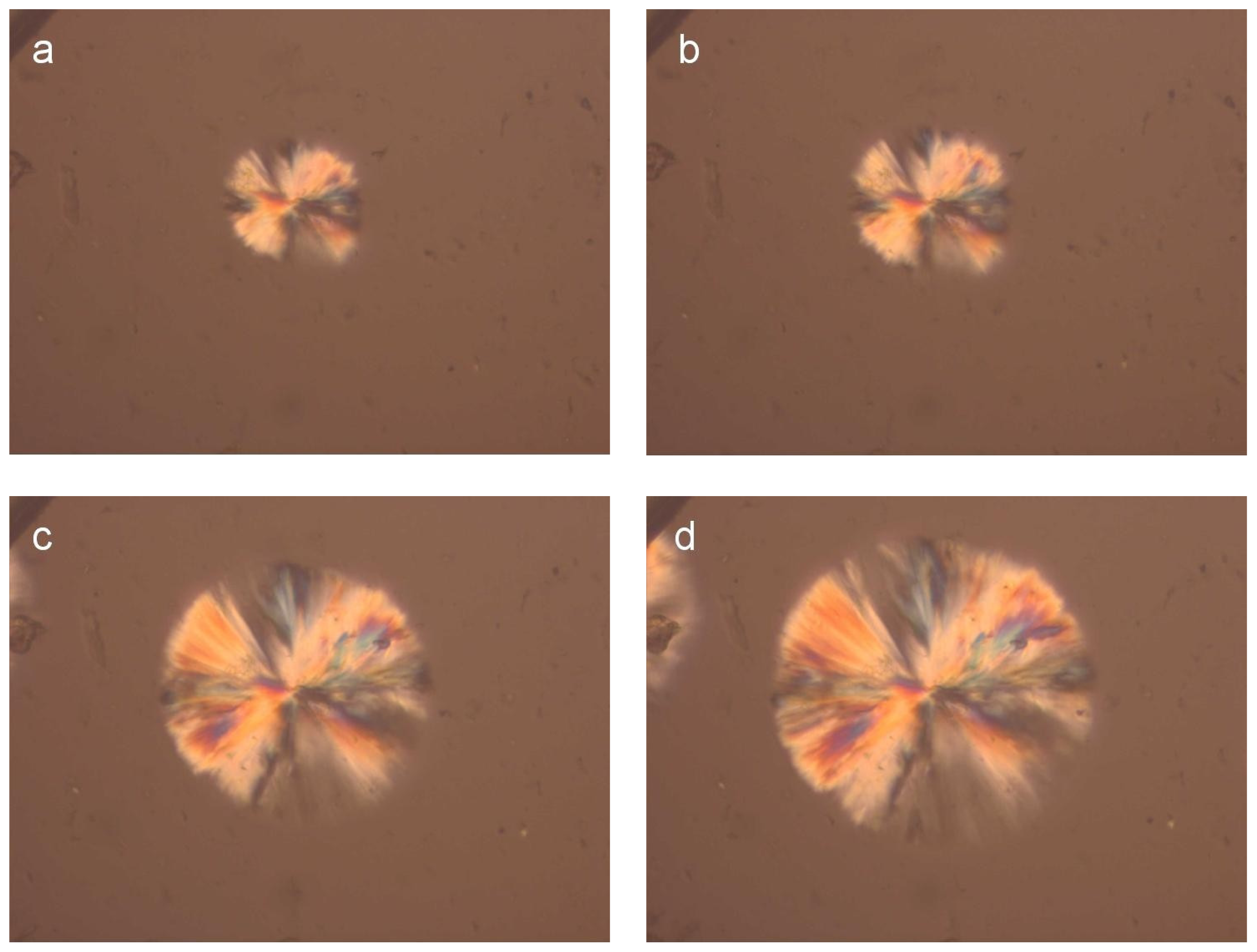
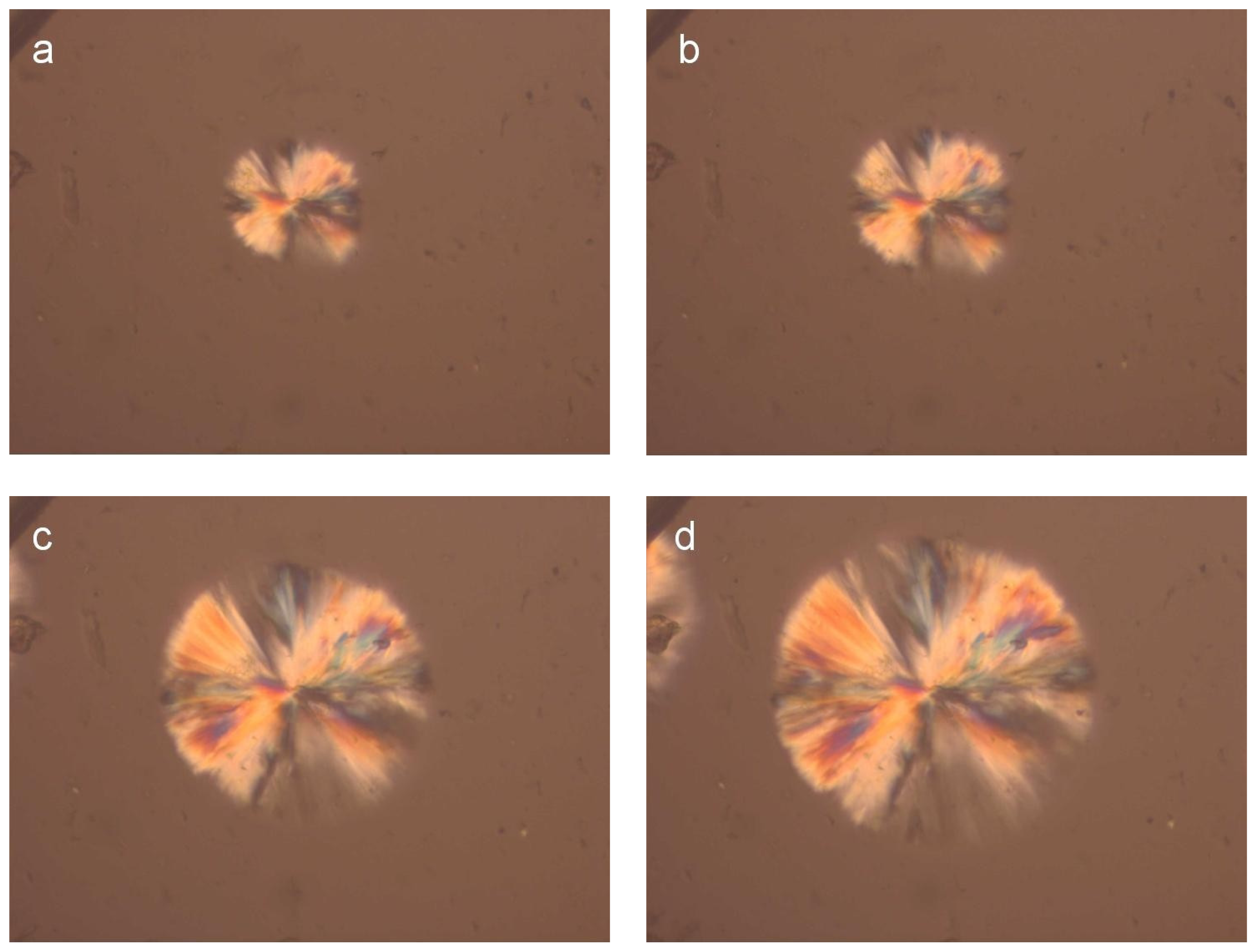
For a direct observation of the crystallization of the PLA reinforcement ligament and for a better understanding of related phenomena relating to aspects of nucleation and growth, polarized light microscopy was employed.
Figure 3 shows photographs taken during isothermal crystallization of PLA samples cut from the reinforcement ligaments (
Figure 3) at 155 °C after cooling from melt. The spherulites formed after 2 min at 155 °C and their size increased progressively with increasing isothermal crystallization time.
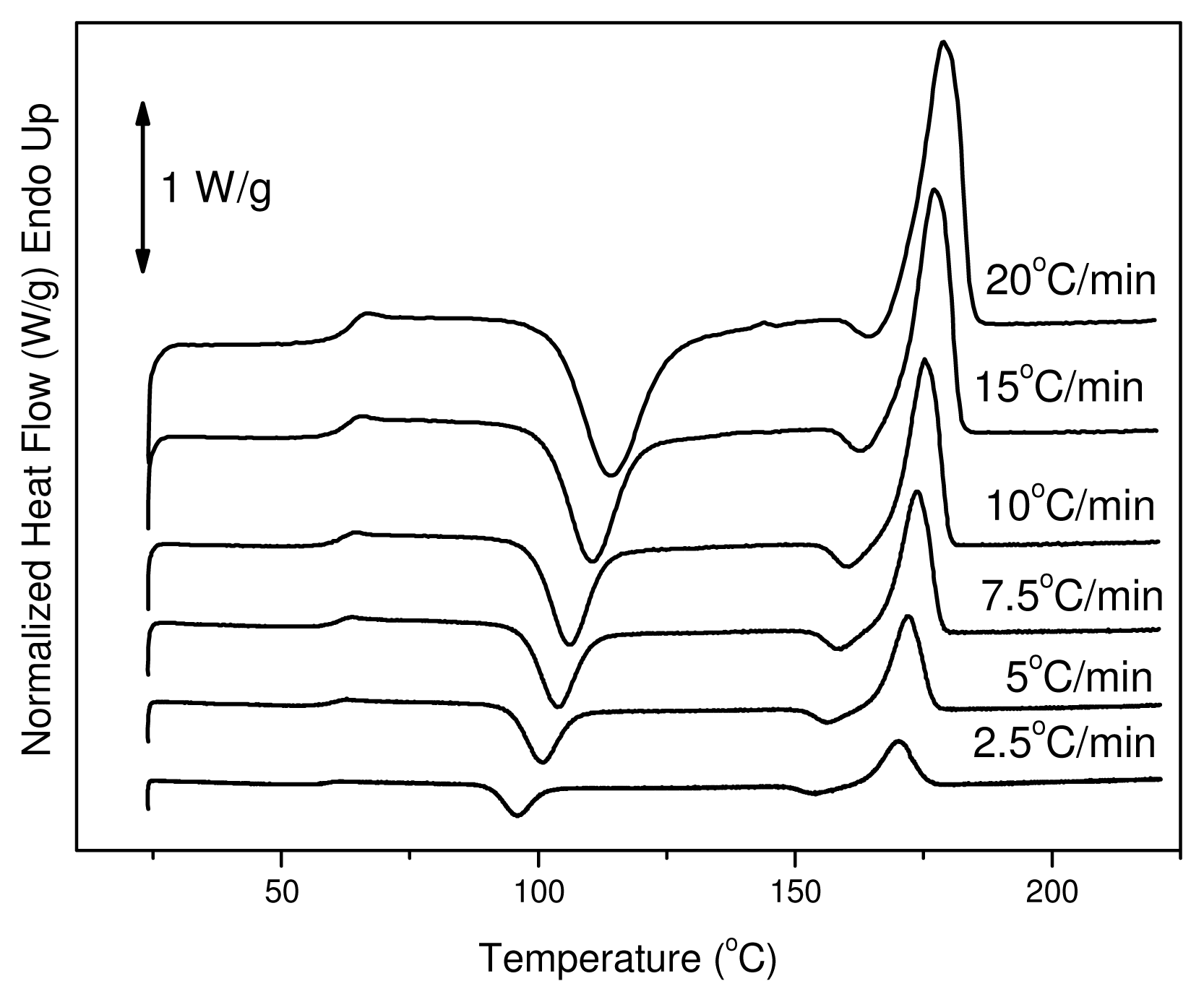
The reinforcement ligament consists of semicrystalline PLA, thus dynamic crystallization has also been studied after quenching from the melt. Cold crystallization of the polymers of this work was studied with DSC. Thus, heating traces of amorphous samples were recorded at various heating rates from 2.5 to 20 °C/min. In these traces it is evident that PLA has a glass transition temperature around 60 °C, which shifted upwards to higher temperatures with increasing heating rate. This is a very high value since glass transition below body temperature ensures that the material is flexible and adaptable in response to mechanical loading. The same trend can be observed for the cold-crystallization peak (
Tcc). At a heating rate of 2.5 °C/min the T
cc value is 96 °C and shifted to 115 °C at heating rate of 20 °C/min. Also, this was broadened with increasing heating rate (see
Figure 4). After cold crystallization the material melts at about 170 °C.
To quantitatively describe the evolution of the relative degree of crystallinity
X during nonisothermal crystallization, a number of models have been proposed in the literature [
32]. The most common approach is that based on a modified Avrami equation [
32,
33]. Thus, the Avrami model can be modified to describe the crystallization kinetics under non-isothermal conditions:
where Zt and n denote the growth rate constant and the Avrami exponent, respectively.
Taking the logarithms
Equation 2 can be written as:
According to the Ozawa theory the non-isothermal crystallization process is the result of an infinite number of small isothermal crystallization steps and the degree of conversion at temperature
T,
X(
T), can be calculated as [
34]:
where
m is the Ozawa exponent that depends on the dimension of crystal growth and
K* is the cooling or heating crystallization function.
K* is related to the overall crystallization rate and indicates how fast crystallization occurs.
a is the heating or cooling rate. Taking the double-logarithmic form of
Equation 4, it follows:
Mo and co-workers [
35] proposed a different kinetic model by combining the Ozawa and Avrami equations. As the degree of crystallinity was related to the cooling rate α and the crystallization time
t or temperature
T the relation between α and
t could be defined for a given degree of crystallinity. Consequently, combining
Equations 3 and
5 derived a new kinetic model for non-isothermal crystallization:
By rearrangement at a given degree of crystallinity and solving for the cooling or heating rate
a,
Equation 6 becomes:
where
F(
T) = [
K*(
T)
/Zt ]1
/m, refers to the value of cooling rate chosen at unit crystallization time, when the system has a certain degree of crystallinity,
b is the ratio of the Avrami exponent to Ozawa exponents,
i.e.,
b = n/
m. According to
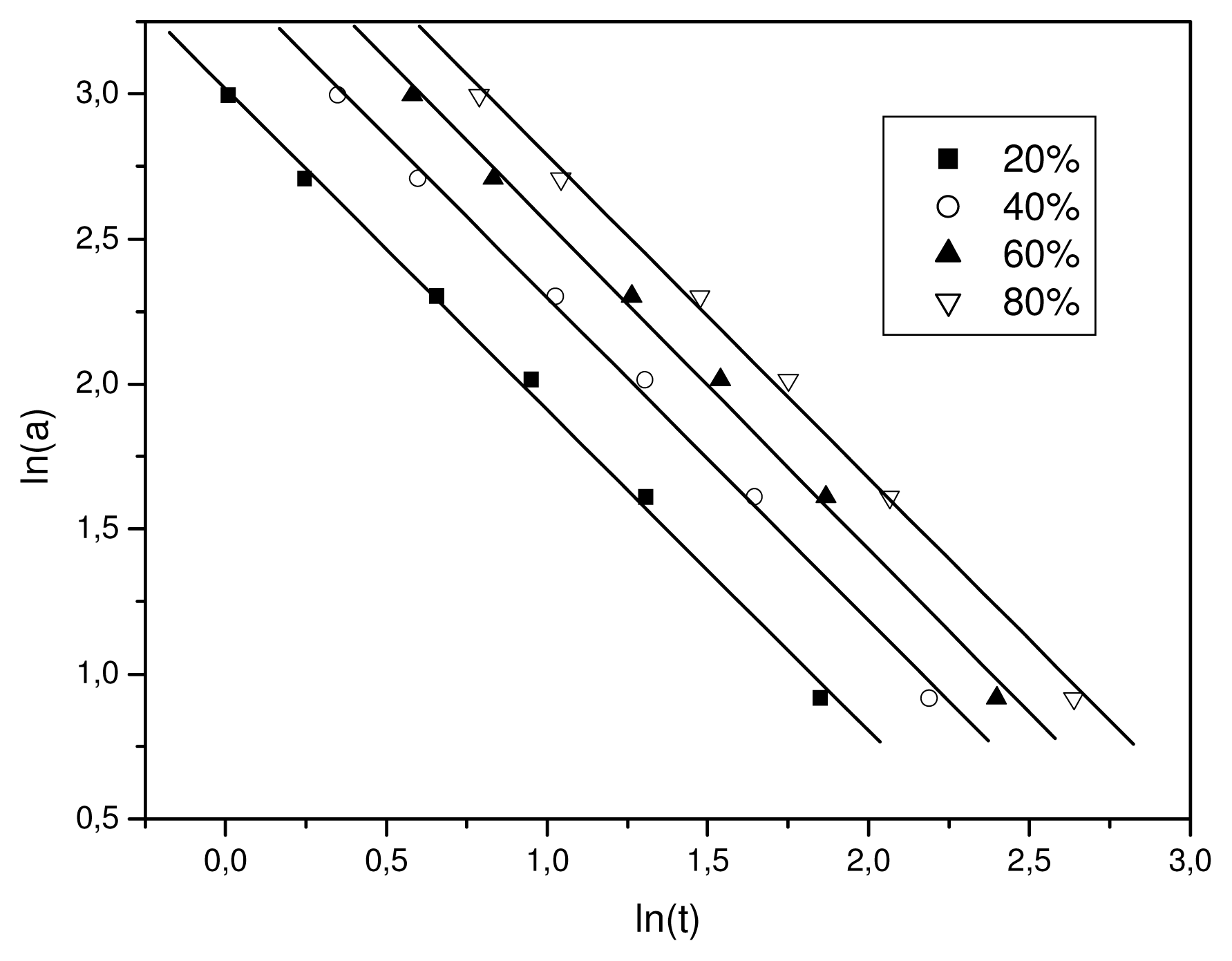
Equation 7 at a given degree of crystallinity the plot of ln α against ln
t will give a straight line with an intercept of ln
F(
T) and a slope of −
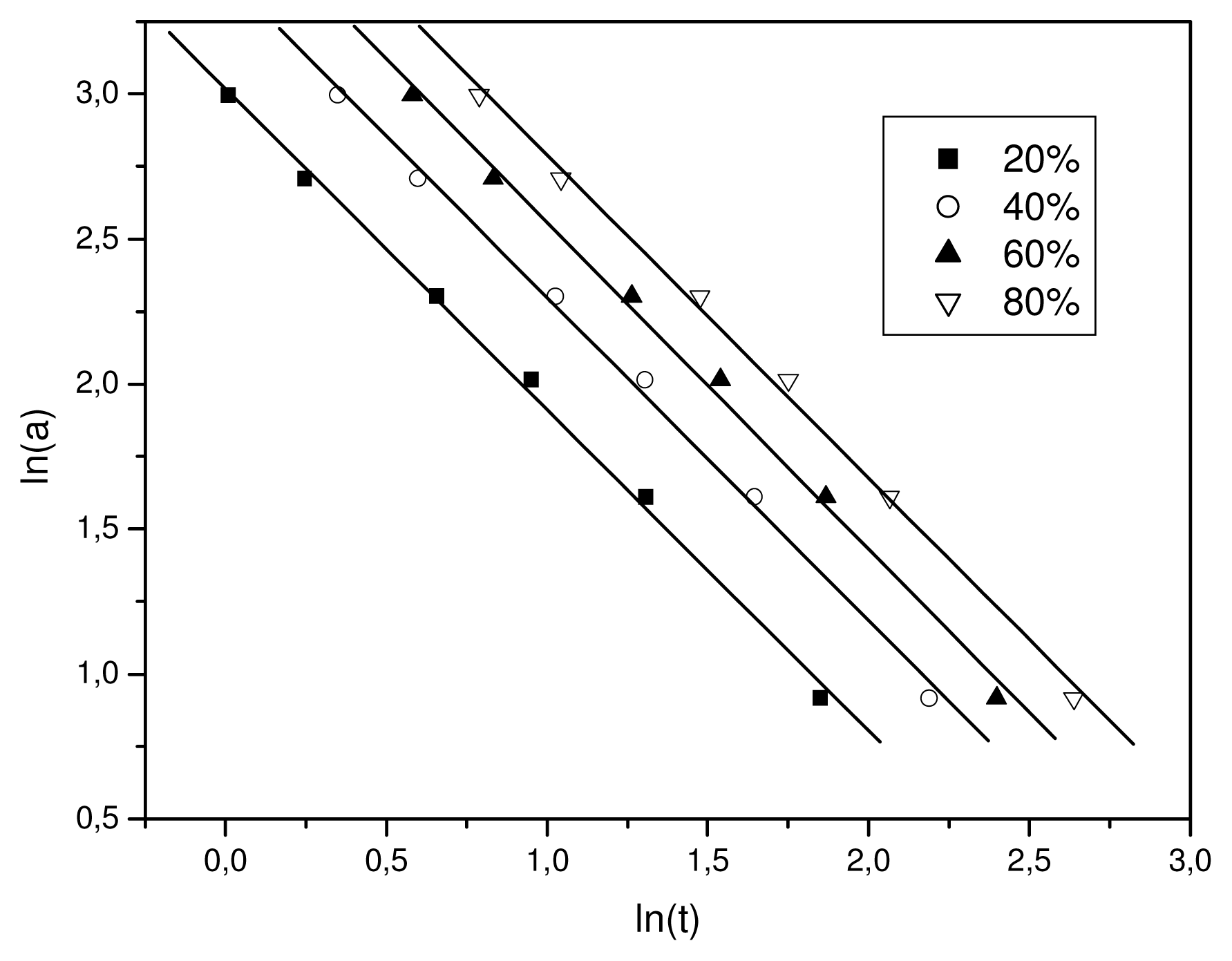
b. As it is shown in
Figure 5, plotting ln
a against ln
t, at a given degree of crystallinity, a linear relationship was observed (correlation coefficient
R > 0.997). The values of
F(
T) and the slope
b are listed in
Table 1. The
F(
T) values increased with the relative degree of crystallinity. However,
b was practically constant as it ranged from 1.47 to 1.51. In addition, these values mean that the Avrami exponent
n is always slightly greater than the Ozawa exponent,
m, as has also been reported in literature [
32]. Non-isothermal crystallization is difficult to describe with a single equation since there are a lot of parameters that have to be taken into account simultaneously. The importance of this method is that it correlates the cooling rate to temperature, time, and morphology.
The Avrami model is suitable for describing the early stages of crystallization. Complications arise from the effects of growth site impingement and secondary crystallization process, which were disregarded for the sake of simplicity in the original derivation of the model. Tobin proposed a theory for crystallization with growth site impingement [
36–
38]. According to this approach, the relative crystallinity function of time X(t) can be expressed in the following form:
where
KT and
nT are the Tobin crystallization rate constant and the Tobin exponent, respectively and t is the time of crystallization. The exponent
nT need not be an integer and is governed by different types of nucleation and growth mechanisms.
Equation 8 can be rewritten in its logarithmic form as follows:
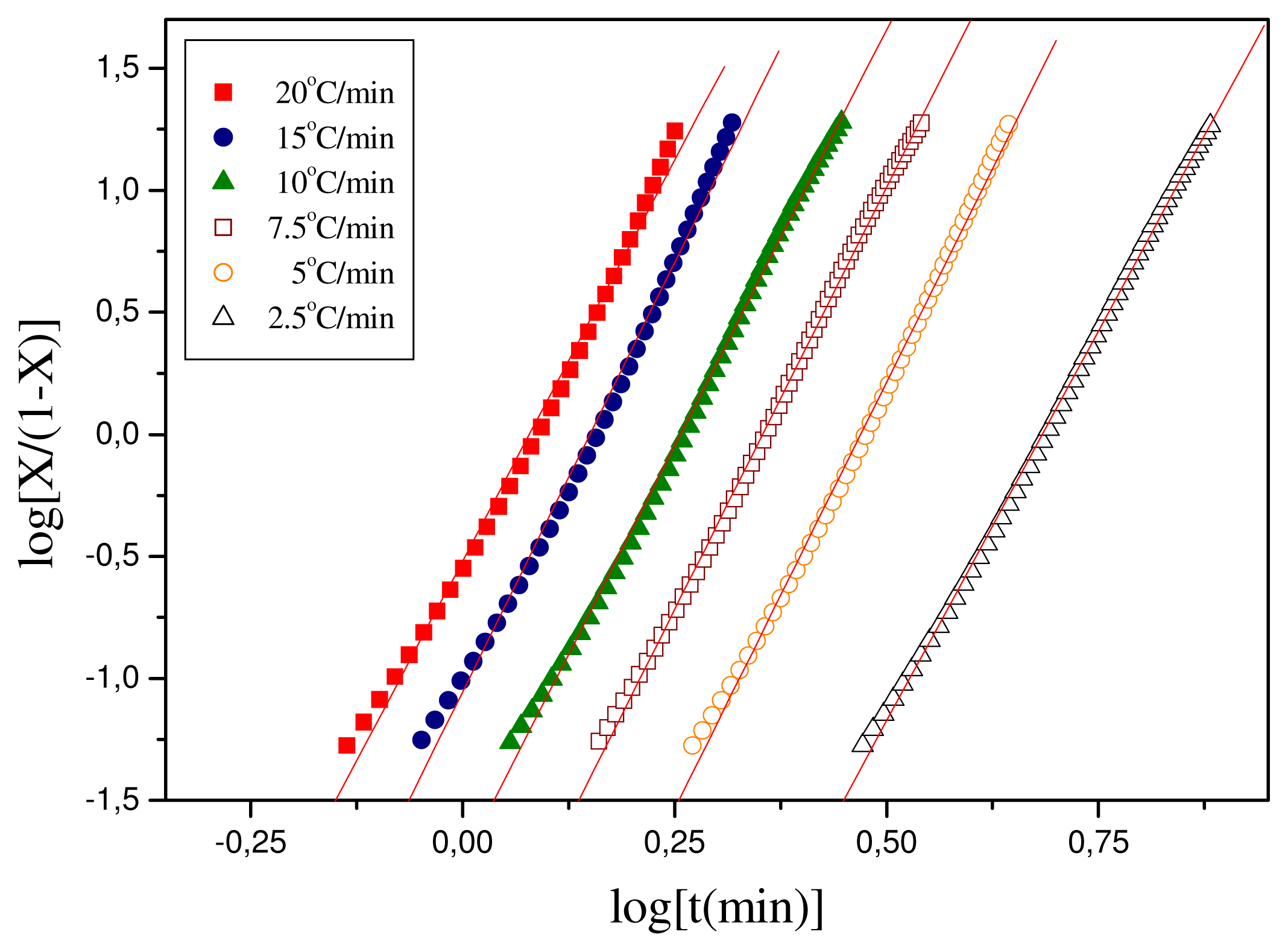
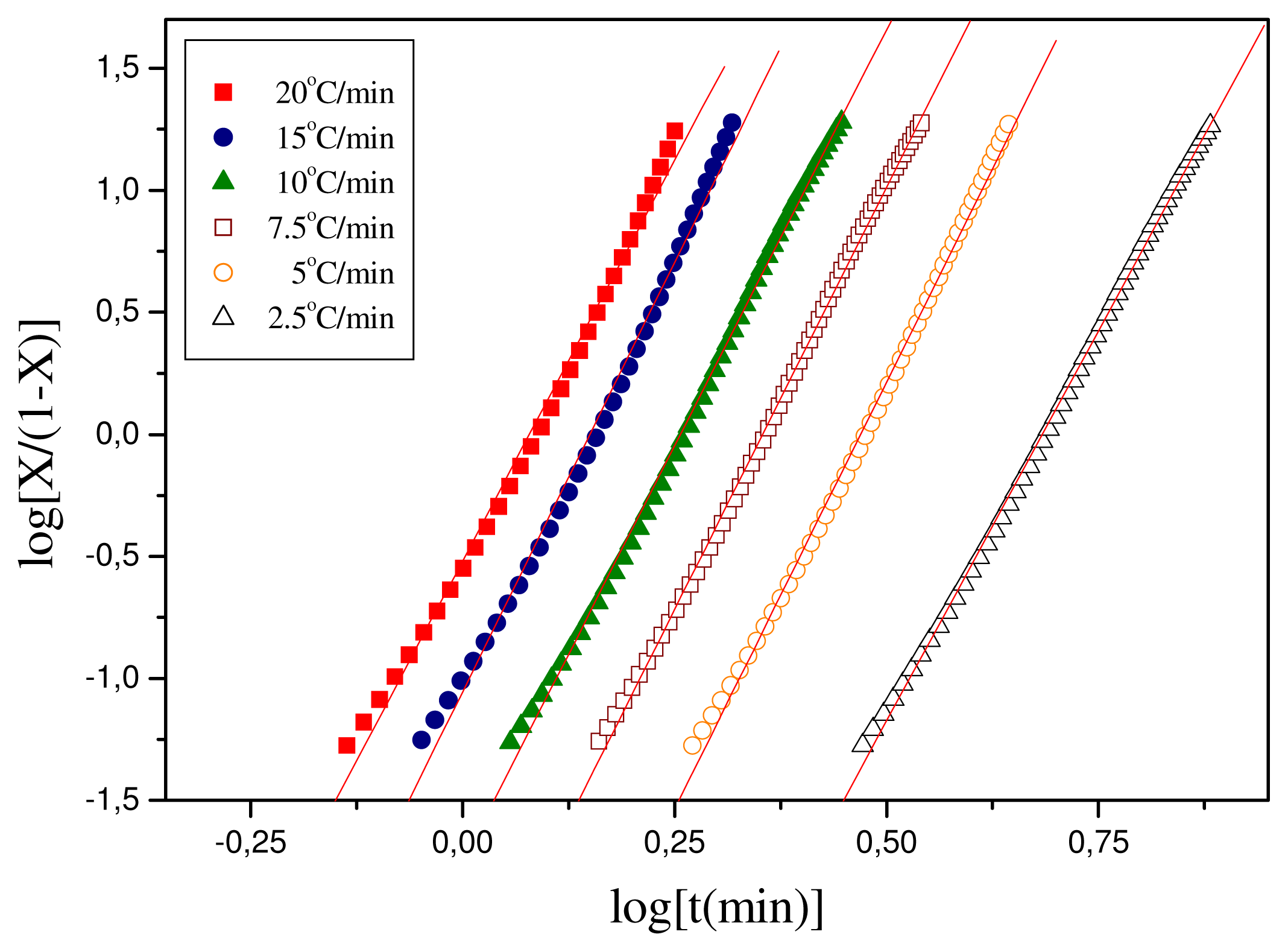
The parameters
nT and
KT can be obtained from the slope and intercept of the plots of log [
X(t)/(1 −
X(t))] against
log t. The respective plots for the polymeric materials of this study are shown in
Figure 6 and the values of the calculated parameters are shown in
Table 2. For Resorbaid, the Tobin plots seem to be linear for almost the full range of crystallization. Only for fast heating rates,
i.e., 15 or 20 °C/min there appears some limited curvature.
In general, it can be concluded that the Resorbaid material shows slow crystallization. However, the Resorbaid material crystallization rates were found to be faster than those in most cases of PLA resins [
31].
3.3. In Vitro Hydrolysis
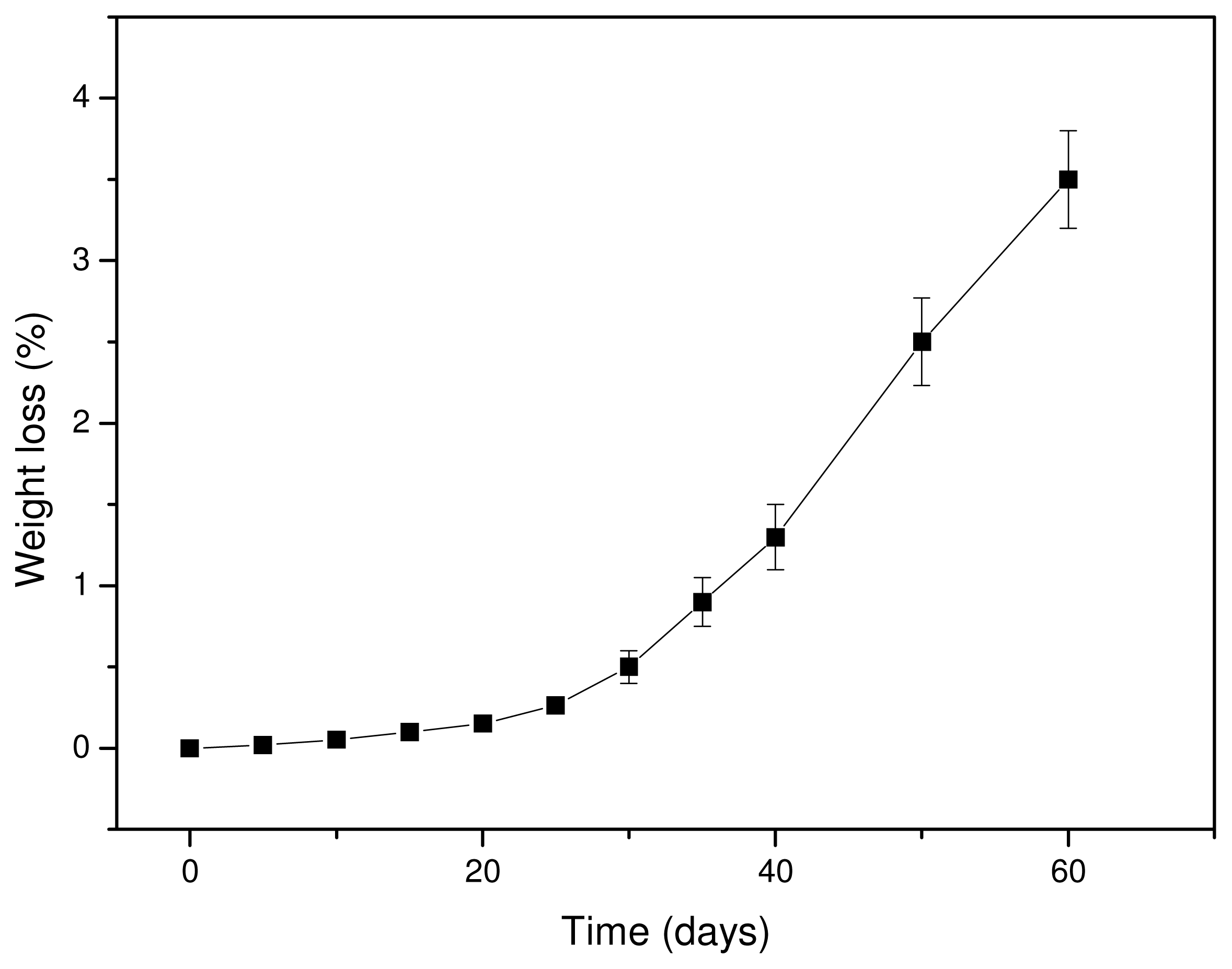
It is well known that hydrolytic scission of the macromolecular chains of aliphatic polyesters starts upon contact with water, which can hydrolyze the ester bonds. Since the hydrolysis rate of PLA is very slow, tests were performed at a high temperature (50 °C) to accelerate the phenomenon. Water can hydrolyze the macromolecular chains randomly, reducing the molecular weight and producing soluble oligomers. Thus, the hydrolysis rate can be deduced from weight loss measurements. As can be seen from
Figure 7, the weight loss of Resorbaid ligament is very small at initial stages and increases dramatically after 30–40 days of hydrolysis. It seems that, at the first days of hydrolysis, only small fragments are removed, probably because the hydrolysis rate is very small or takes place at the ends of macromolecular chains and thus fragments with low molecular weights are formed. However, it was reported that the low weight loss at the initial stages of hydrolysis could be attributed to the hydrolysis of polyesters that take place randomly along the macromolecular chains, reducing only the molecular weight of the polyesters [
39]. If this happens, a high molecular weight reduction should be recorded, followed by a small weight loss since the formed oligomers at initial degradation stages are too large, hence it has difficulty diffusing through the bulk material. Only after an extended period of time are they sufficiently reduced in size by hydrolysis to diffuse out as oligomers and result in a significant mass reduction [
40]. Furthermore, the formation of water-soluble lactic acid oligomers is hydrolysis time-dependent. Höglund and co-workers found that pure PLA can form water soluble oligomers after 28 days of hydrolysis at 60 °C and after 133 days of hydrolysis at 37 °C [
41]. The mass loss was considerably slower, and over 90% of the original mass remained in all materials after 28 days of degradation. This is due to the hydrophobic nature of PLA. The degradation products formed during hydrolysis are not water-soluble until they have a molar mass of >1,000 g/mol and therefore remain in the polymer bulk. It seems that the same also happens in our study. Thus, the mechanism of hydrolysis could be revealed in comparison with the changes in molecular weight of the PLA ligament during hydrolysis.
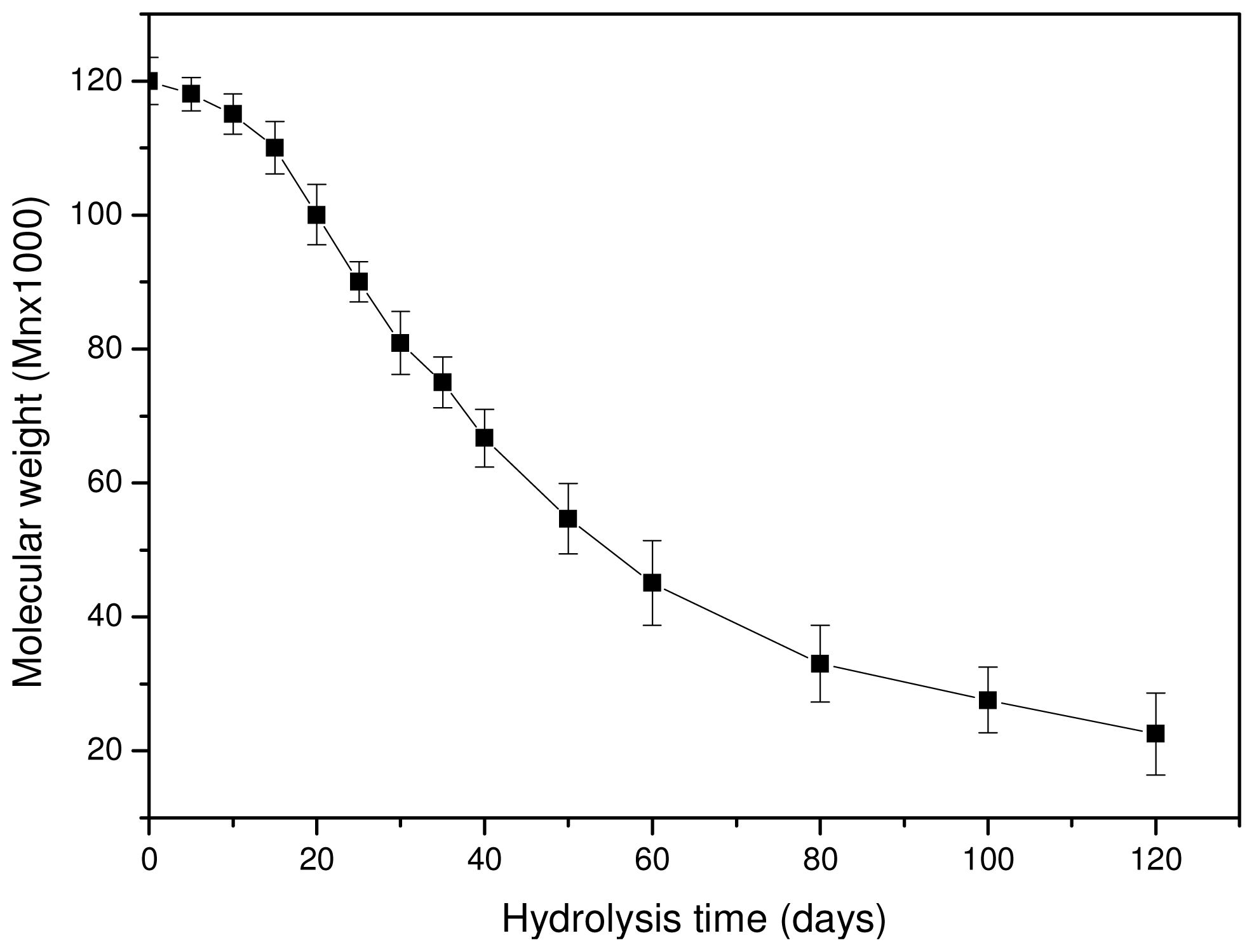
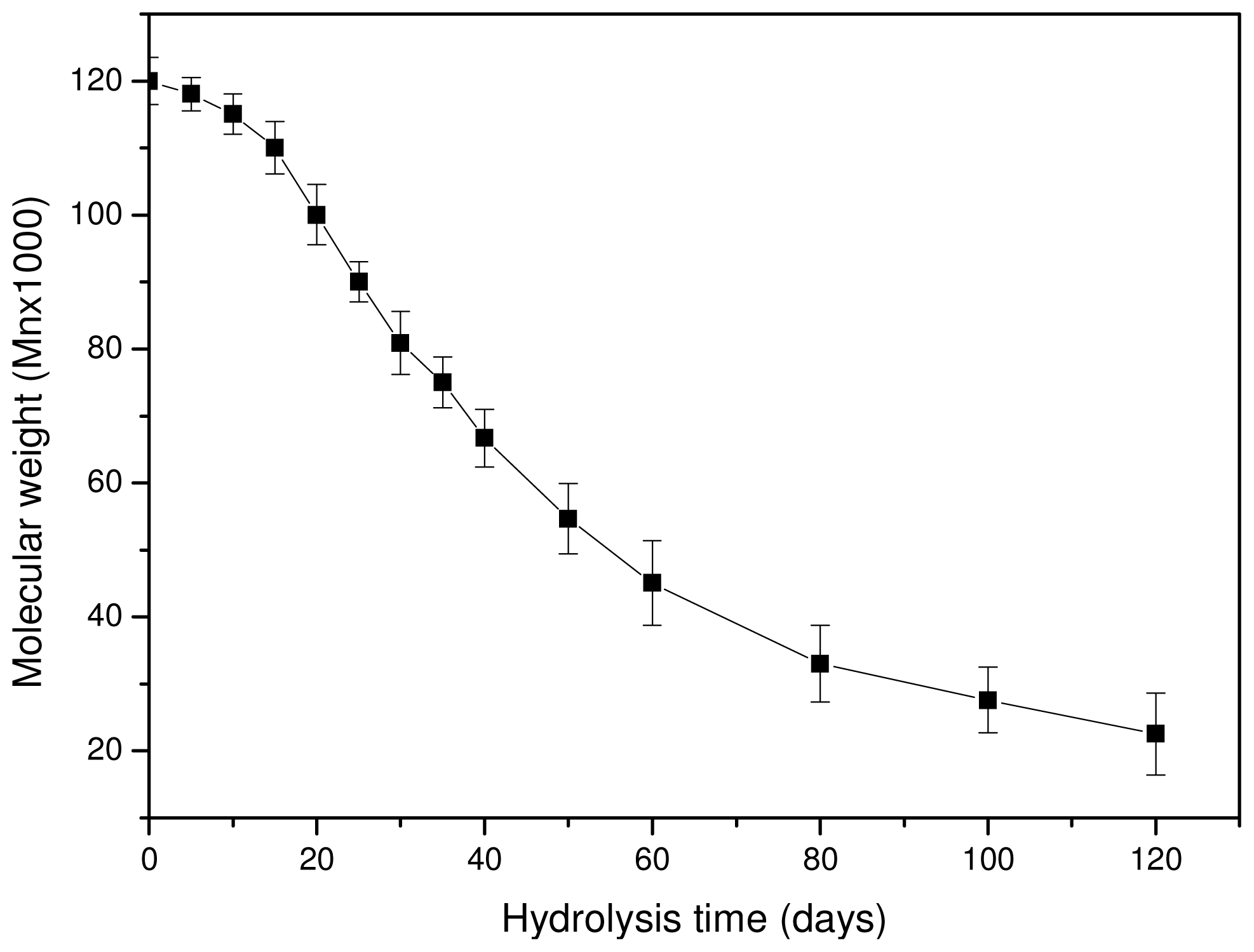
As can be seen in
Figure 8, the molecular weight is slightly reduced at the initial stages of hydrolysis and the rate becomes higher after 15–20 days of hydrolysis. However, after 80–100 days the rate seems to become progressively slower. The whole reduction seems to be S-type, which agrees with previously reported results [
23,
39]. The scission of ester bonds at initial hydrolysis times is slow, becoming progressively more rapid [
42]. This can also explain the low weight loss that is recorded at the initial 15–20 days of hydrolysis. The high molecular weight of used PLA for the preparation of reinforcement ligament or its surface hydrophobicity could be responsible for such behavior [
43]. Fukuzaki and coworkers found that the molecular weight reduction of high molecular weight L-lactide/glycolide copolymers becomes higher only after a certain time of hydrolysis [
39]. Thus, at this time, low molecular weight oligomers are formed and this explains the high weight loss that is observed after 30–40 days of hydrolysis. These results are in disagreement with a recent study by Dånmark
et al. [
44], where it was found that, during hydrolysis, the mass loss is very small but at the same time the molecular weight reduction is very high. As can be seen in the case of this work, the weight loss reduction at the initial stages is small since less than 0.2 wt % of the initial material is lost, followed by a small reduction in molecular weight. Soluble oligomers are formed after that and the rate is higher after 35–40 days of hydrolysis. However, as can be seen in
Figure 8, the high reduction in molecular weight appears at lower hydrolysis times (20 days). This is an indication that the macromolecular chains are first randomly broken during hydrolysis and, when this happens to a large extent, soluble oligomers are formed, resulting in a reduction to mass loss.
SEM was also used to study the microstructure of PLA and its variation during hydrolysis. It can been seen from
Figure 9 that significant etching character appeared on the surfaces of PLA films, while the surface of blank specimen was very smooth. Although the etching effect was not uniform, it primarily demonstrated that PLA ligaments can be hydrolyzed with time. However, as has already been reported, hydrolysis of PLA ester bonds starts homogeneously until soluble oligomers are formed, which can be removed from the matrix. At this time, those soluble oligomers which are close to the surface can leach out before total degradation, whereas those located well inside the matrix remain entrapped. As can be seen, small holes were created in the PLA ligament surface after 30 days of hydrolysis. These initially have small diameter, less than 1–2 μm, which increases progressively. Thus, after 60 days of hydrolysis, the size of the formed holes is in the range of 2–5 μm. This is in agreement with the recorded weight loss and molecular weight reduction during this time.
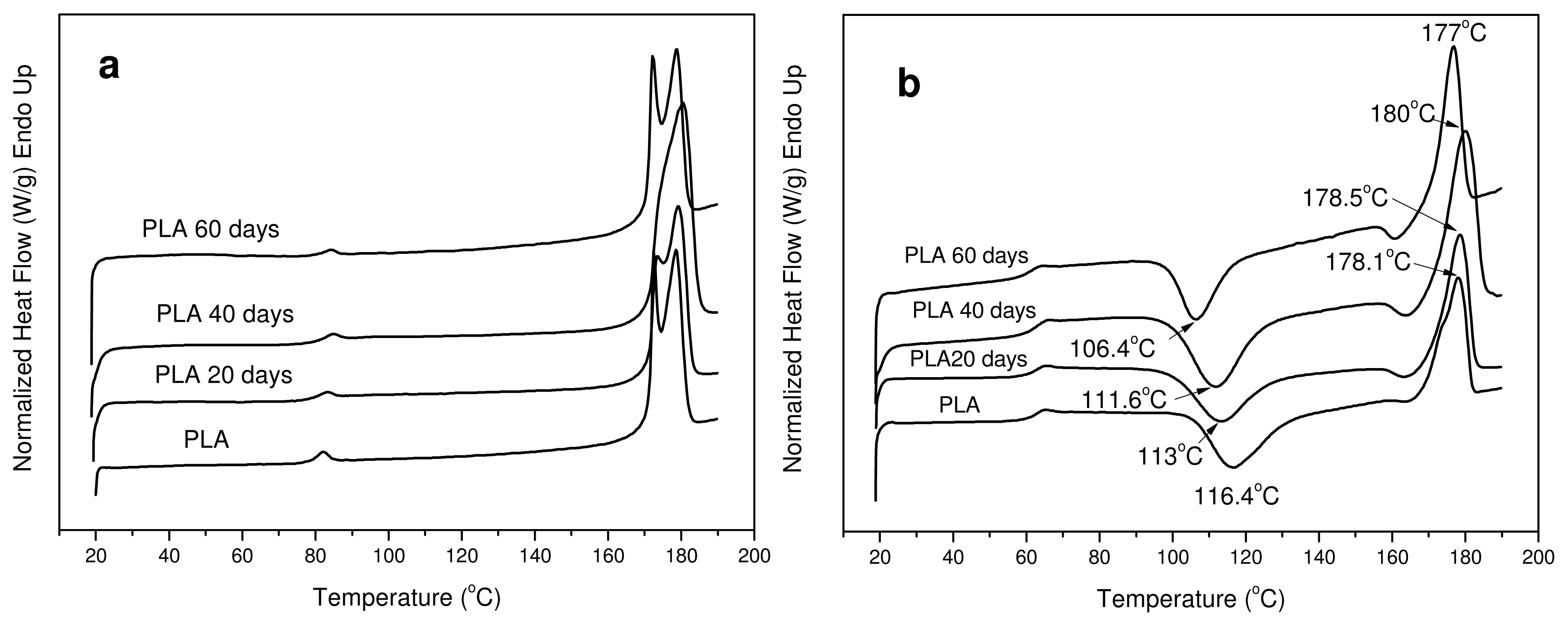
All the samples after hydrolysis were also tested with DSC since the degree of crystallinity can change during this treatment. As can be seen, PLA ligament is a semicrystalline material with two melting peaks (
Figure 10(a)). In the samples taken after hydrolysis at different times, two peaks are also visible but with some small differences. The melting points of these peaks at the first 40 days of hydrolysis have shifted to higher temperatures, but after 60 days they are shifted to lower values. However, most differences are in the values of heat of fusion. As can be seen in
Table 3, the heat of fusion was gradually increased to higher values during hydrolysis. This was expected because the amorphous phase of aliphatic polyesters degraded more rapidly than the crystalline phase during hydrolysis. Thus, the degree of crystallinity (X
c), as was calculated based on the enthalpy of fusion of 100% crystalline PLA, which is 93 J/g [
45], increases during hydrolysis. However, after 60 days there is a stabilization, indicating that, after this time, the crystalline parts of PLA might also start to hydrolyze. This is because the molecular weight of the samples was drastically reduced at the same time.
The reduction in molecular weight during hydrolysis also has an effect on cold crystallization temperature (
Tcc) (
Figure 10(b)). As can be seen, T
cc progressively shifts to lower temperatures by increasing the hydrolysis time. This is an indication that the samples crystallize faster than the initial sample; which was expected as it is well known that polymers with lower molecular weights have higher crystallization rates.
3.4. In Vivo Hydrolysis
In addition to
in vitro hydrolysis, the
in vivo behavior of the PLA ligaments is also interesting. A time of 6 months after the addition of ligament in a human body was selected for tests, which is very short for a complete hydrolysis of PLA ligament. It was reported that PLA in the form of screws needs almost 60 months (5 years) during
in vivo tests for complete disappearance of the screw [
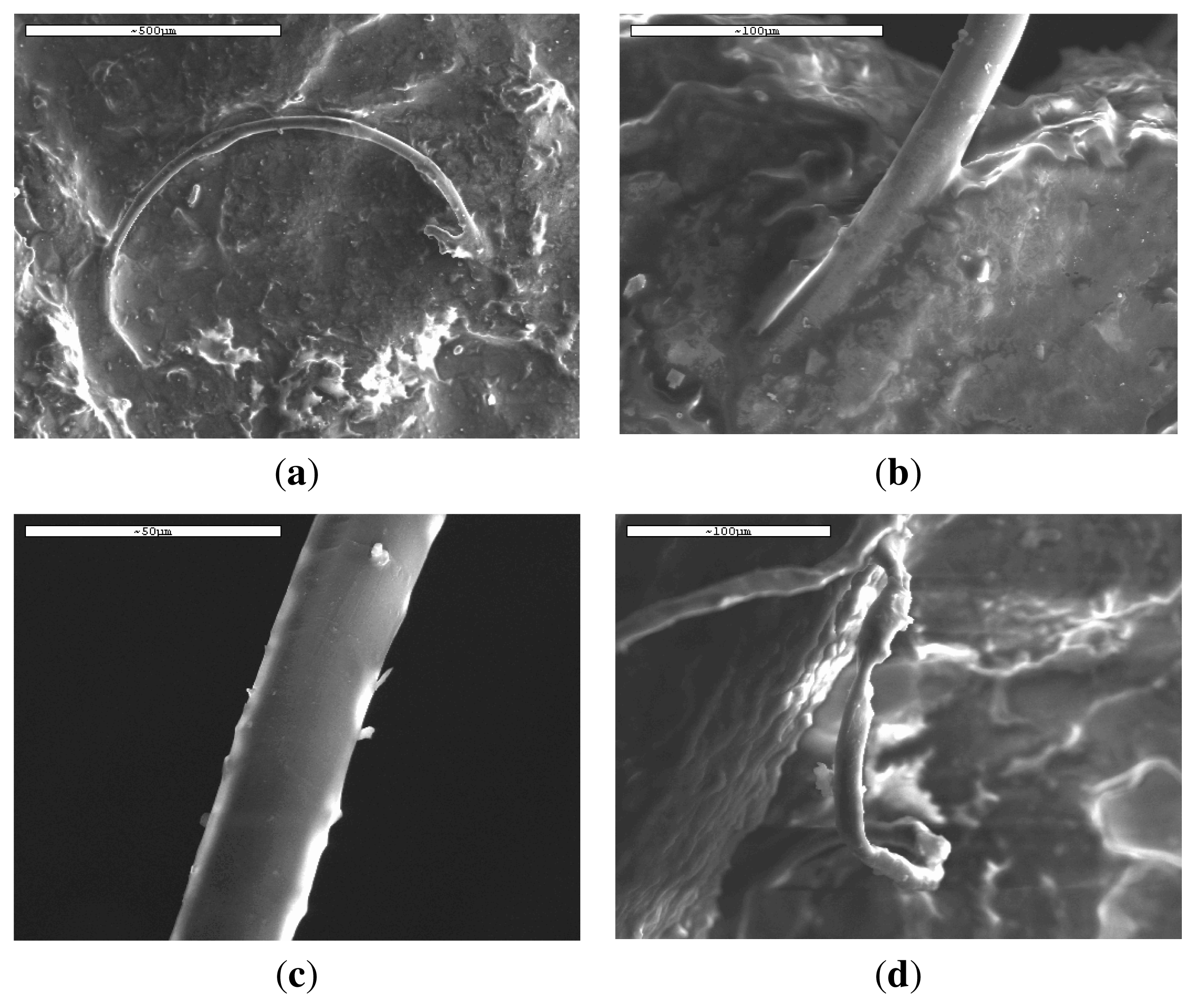
4]. In our case, a small sample was removed after 6 months from a human body and studied with SEM. The samples have been tasted without any cleaning. As can be seen from
Figure 11, there are a lot of differences in the PLA ligament behavior. At some positions the ligament is almost intact (
Figure 11(a)) and the fibers are well recognized due to their shape and mean diameter about 15 μm, exactly the same as the neat fibers (
Figure 1), and in some other points the fibers have been covered with flesh (
Figure 11(b)). This is a proof of high biocompatibility between the PLA ligament and the human body.
Examining more carefully these fibers it can be seen that there are a lot of differences in their surfaces (
Figure 12(a–c)) as well as in their shapes (
Figure 12(d)). The surfaces are not as smooth as in the initial fibers indicating their alteration during
in vivo hydrolysis. According to Pitt
et al., the
in vivo degradation of similar polyesters like poly(
dl-lactide) and poly(ɛ-caprolactone), proceeded in two stages: first there was only a decrease in molecular weight; subsequently the polymer experienced weight loss with an increase in the rate of chain scission [
46]. Furthermore, from
in vivo studies of PLA samples in the form of disks added in adult New Zealand white rabbits, small weight loss was found after 1-month implantation, corresponding to approximately 5% of its original weight and almost 30% after 6 months [
27]. The number average molecular weight M
n of PLA was also reduced from 47,200 to 43,300 after 1 month and 3,600 g/mol after 6 months. In our case, it seems that the
in vivo hydrolysis is slower than that reported previously, since only small differences are observed in the fibers’ appearance (
Figure 12(a–c)). This may be due to the high molecular weight of the PLA used and short time of hydrolysis. If the rate was higher and molecular weight was dramatically reduced, some holes could be created on fibers surface due to elimination of soluble oligomers. This seems to be in agreement with the
in vitro hydrolysis. However, from
Figure 12(d) it is clear that some fibers are not cylindrical anymore and it seems that they have collapsed. From a previous work concerning the
in vivo degradation of poly(a-hydroxy acids) derived from LA and/or GA monomers, it was found that all the intrinsically amorphous members of the family degrade hydrolytically and that degradation is faster in the centre than on the surface, at least for large devices [
26]. Taking this study into account it can be concluded that the
in vivo hydrolysis of the PLA ligament is also higher in the inner part of the fibers and lower on their surface. This may be a result of the higher crystallization rates of the PLA fibers’ surface during
in vivo hydrolysis, as previously reported [
4].
Further evidence for the small
in vivo hydrolysis rate was provided from the molecular weight measurement that was performed on the removed sample. The Mn was reduced from approximately 120,000 g/mol to about 100,000–110,000 g/mol. Taking into account that the
in vivo hydrolysis could be different between the fibers’ surface and their inner part, the molecular weight should be checked separately from these different areas. However, due to the small surface of the fibers, this was not possible and the molecular weight was measured from the whole sample. The small reduction in Mn after 12 months of
in vivo hydrolysis can be attributed to the fact that the hydrolysis rate of PLA is taking place from the end of macromolecular chains and thus only small fragments could be removed. Thus, the molecular weight was almost unaffected. However, this is not in agreement with previous studies on
in vivo hydrolysis in New Zealand white rabbits, where the rate was higher [
27], or with the results from the
in vitro hydrolysis of this particular sample discussed previously. This disagreement could be attributed to the higher temperatures during
in vitro hydrolysis (50 °C). Also, as can be seen from the
in vitro hydrolysis, the molecular weight reduction is very small for the initial 10–20 days, indicating that there may be an induction period after which the molecular weight decreases faster. Thus, it may be possible that 12 months is not time long enough for
in vivo hydrolysis to cause a drastic reduction in the molecular weight of the sample.
The removed sample was also examined with FTIR spectroscopy in order to find any detectable differences in its chemical structure. As can be seen from
Figure 13, the FTIR spectra of the initial sample and after
in vivo hydrolysis are almost identical. This was expected since only small differences in molecular weight were recorded. The only detectable difference is the higher absorbance of the
in vivo sample in the area of hydroxyl groups. However, this could not be attributed to the higher amount of hydroxyl groups that this sample contains but to the humidity that was absorbed by the sample.
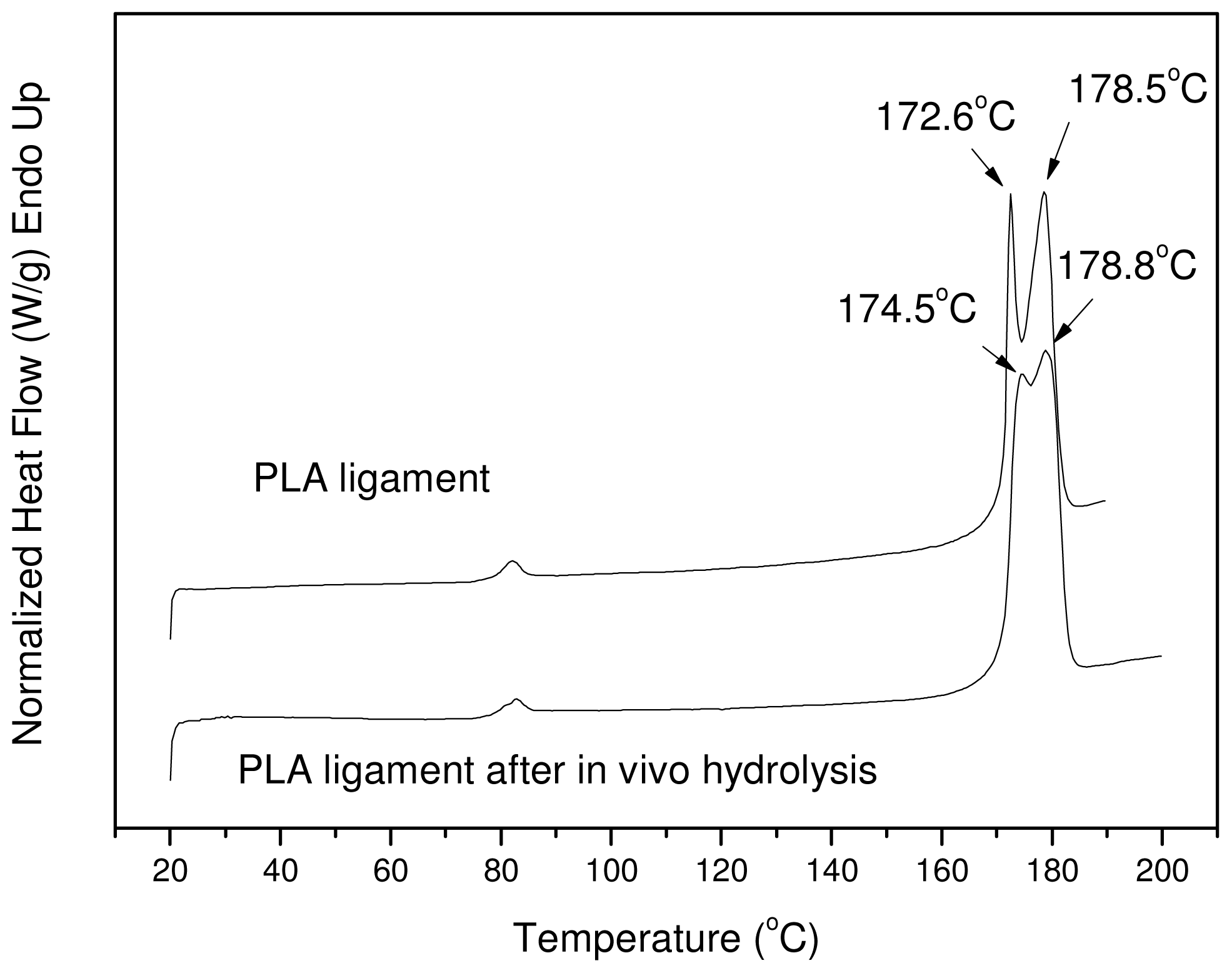
The
in vivo sample was also studied with DSC to see differences in its physical state and mainly in the degree of crystallinity; as found from the
in vitro hydrolysis, the latter is one of the most notable changes. The DSC thermograms of the PLA Resorbaid sample initially and also after 12 months of hydrolysis are presented in
Figure 14. As can be seen, the initial sample has two distinct peaks with maximum temperatures at 172.6 °C and at 178.5 °C. These two peaks are also recorded in the
in vivo sample but at slightly different temperatures: 174.5 and 178.8 °C, respectively. Furthermore, in addition to the first peak shifting to higher temperatures, these two peaks tend to merge together into one peak. A similar behavior was also found in the sample after 40 days of
in vitro hydrolysis (
Figure 10(a)), where the sample showed a peak at higher temperatures than the initial sample. The heat of fusion in the case of the
in vivo sample was also changed from 52.5 J/g to 57.8 J/g, which provides clear evidence that the degree of crystallinity increased from 56.45% to 62.15%, which is in accordance with the already detected changes during
in vitro hydrolysis (
Table 3). All these differences may be due to the
in vivo hydrolysis of the amorphous part of the used reinforcement ligament or due to the annealing procedure of the polyester taking place during its insertion into the human body. The increased degree of crystallinity in comparison with the molecular weight of the initial sample may be the most probable reason for the low
in vivo hydrolysis rate of the reinforcement ligament.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}