Biomarkers in Rare Disorders: The Experience with Spinal Muscular Atrophy

Abstract
:1. Introduction
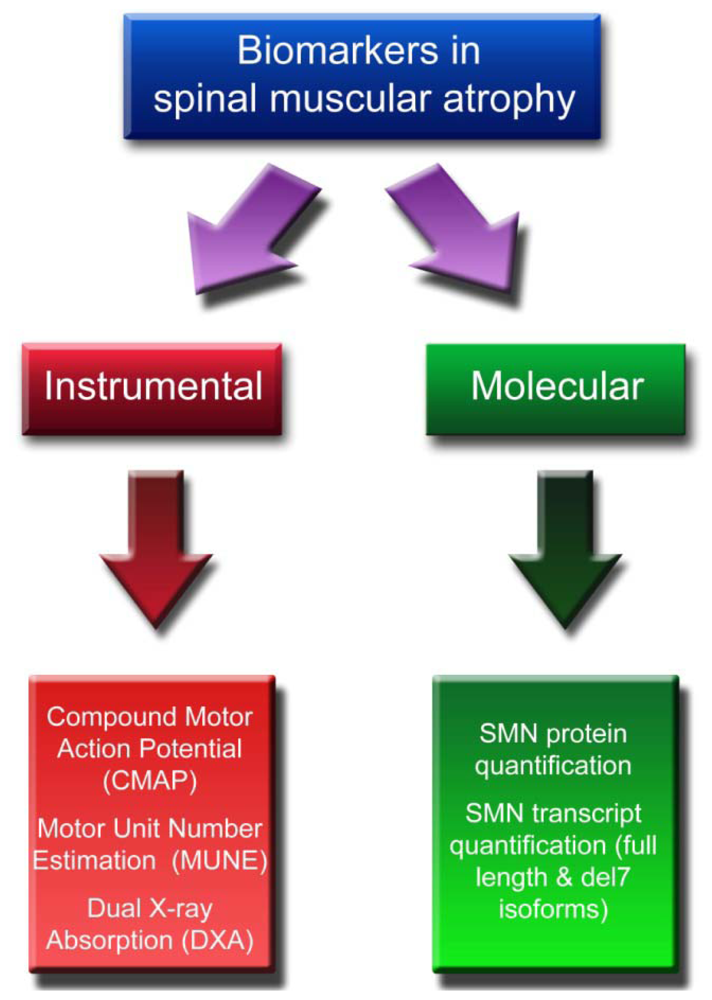
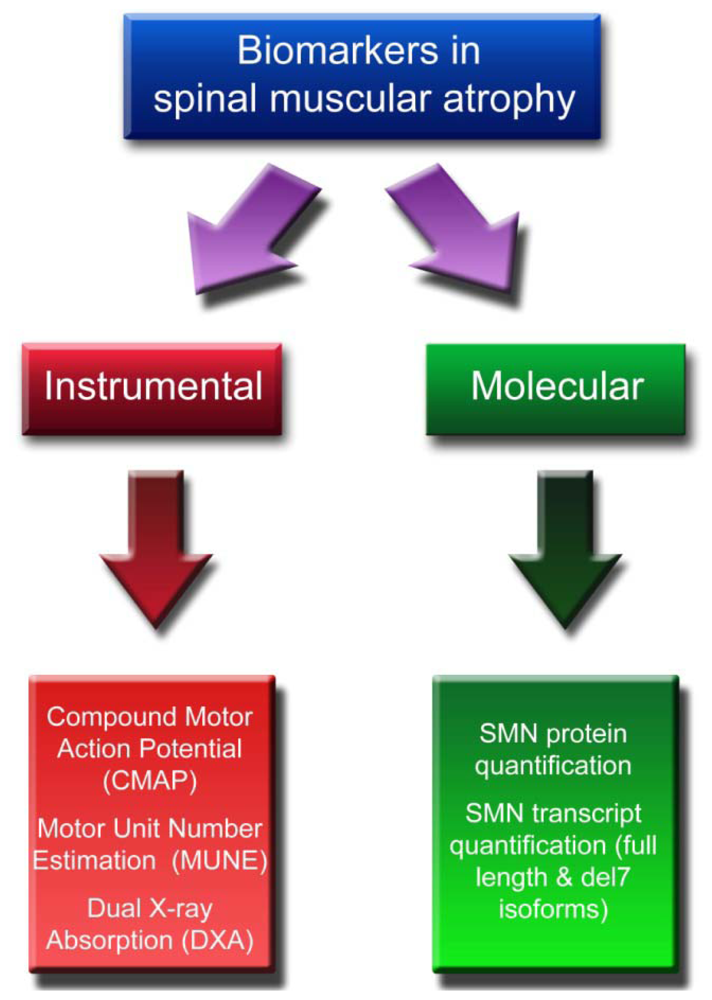
2. Instrumental Biomarkers
2.1. CMAP and MUNE
2.2. DXA
3. Molecular Biomarkers
3.1. SMN Protein Quantification
3.2. SMN Transcript Quantification
4. Conclusions
References
- Lunn, MR; Wang, CH. Spinal muscular atrophy. Lancet 2008, 371, 2120–2033. [Google Scholar]
- Dubowitz, V. Chaos in the classification of SMA: a possible resolution. Neuromuscul. Disord 1995, 5, 3–5. [Google Scholar]
- Lefebvre, S; Burglen, L; Reboullet, S; Clermont, O; Burlet, P; Viollet, L; Benichou, B; Cruaud, C; Millasseau, P; Zeviani, M; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mut 2000, 15, 228–237. [Google Scholar]
- Lefebvre, S; Burlet, P; Liu, Q; Bertrandy, S; Clermont, O; Munnich, A; Dreyfuss, G; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet 1997, 16, 265–269. [Google Scholar]
- Coovert, DD; Le, TT; McAndrew, PE; Strasswimmer, J; Crawford, TO; Mendell, JR; Coulson, SE; Androphy, EJ; Prior, TW; Burghes, AHM. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet 1997, 6, 1205–1214. [Google Scholar]
- Patrizi, AL; Tiziano, F; Zappata, S; Donati, A; Neri, G; Brahe, C. SMN protein analysis in fibroblast, amniocyte, and CVS cultures from spinal muscular atrophy patients and its relevance for diagnosis. Eur. J. Hum. Genet 1999, 7, 301–309. [Google Scholar]
- Rossoll, W; Bassell, GJ. Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. Results Probl. Cell Differ 2009, 48, 289–326. [Google Scholar]
- Feldkötter, M; Schwarzer, V; Wirth, R; Wienker, TI; Wirth, B. Quantitative analysis of SMN1 and SMN2 based on real-time LightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet 2002, 70, 358–368. [Google Scholar]
- Tiziano, FD; Bertini, E; Messina, S; Angelozzia, C; Panec, M; D’Amicob, A; Alfieric, P; Fioria, S; Battinie, R; Berardinellif, A; et al. The Hammersmith functional score correlates with the SMN2 copy number: a multicentric study. Neuromuscul. Disord 2007, 17, 400–403. [Google Scholar]
- Wirth, B; Brichta, L; Schrank, B; Lochmüller, H; Blick, S; Baasner, A; Heller, R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet 2006, 119, 422–428. [Google Scholar]
- Chang, JG; Hsieh-Li, HM; Jong, YJ; Wang, NM; Tsai, CH; Li, H. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci USA 2001, 98, 9808–9813. [Google Scholar]
- Brichta, L; Hofmann, Y; Hahnen, E; Siebzehnrubl, FA; Raschke, H; Blumcke, I; Eyupoglu, IY; Wirth, B. Valproic acid increases the SMN2 protein level: A well-known drug as a potential therapy for spinal muscular atrophy. Hum. Mol. Genet 2003, 12, 2481–2489. [Google Scholar]
- Sumner, CJ; Huynh, TN; Markowitz, JA; Perhac, JS; Hill, B; Coovert, DD; Schussler, K; Chen, XC; Jarecki, J; Burghes, AHM; et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol 2003, 54, 647–654. [Google Scholar]
- Andreassi, C; Angelozzi, C; Tiziano, FD; Vitali, T; de Vincenzi, E; Boninsegna, A; Villanova, M; Bertini, E; Pini, A; Neri, G; et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur. J. Hum. Genet 2004, 12, 59–65. [Google Scholar]
- Brahe, C; Vitali, T; Tiziano, FD; Angelozzi, C; Pinto, AM; Borgo, F; Moscato, U; Bertini, E; Mercuri, E; Neri, G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur. J. Hum. Genet 2005, 13, 256–259. [Google Scholar]
- Brichta, L; Holker, I; Haug, K; Klockgether, T; Wirth, B. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann. Neurol 2006, 59, 970–975. [Google Scholar]
- Grzeschik, SM; Ganta, M; Prior, TW; Heavlin, WD; Wang, CH. Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann. Neurol 2005, 58, 194–202. [Google Scholar]
- Jarecki, J; Chen, X; Bernardino, A; Coovert, DD; Whitney, M; Burghes, A; Stack, J; Pollok, BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet 2005, 14, 2003–2018. [Google Scholar]
- Angelozzi, C; Borgo, F; Tiziano, FD; Martella, A; Neri, G; Brahe, C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J. Med. Genet 2008, 45, 29–31. [Google Scholar]
- Pane, M; Staccioli, S; Messina, S; D’Amicoc, A; Pelliccioniad, M; Mazzonea, ES; Cuttinie, M; Alfieria, P; Battinif, R; Maing, M; et al. Daily salbutamol in young patients with SMA type II. Neuromuscul. Disord 2008, 18, 536–540. [Google Scholar]
- Mercuri, E; Bertini, E; Messina, S; Solari, A; D’Amico, A; Angelozzi, C; Battini, R; Berardinelli, A; Boffi, P; Bruno, C; et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007, 68, 51–55. [Google Scholar]
- Weihl, CC; Connolly, AM; Pestronk, A. Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology 2006, 67, 500–501. [Google Scholar]
- Liang, WC; Yuo, CY; Chang, JG; Chen, YC; Chang, YF; Wang, HY; Ju, YH; Chiou, SS; Jong, YJ. The effect of hydroxyurea in spinal muscular atrophy cells and patients. J. Neurol. Sci 2008, 268, 87–94. [Google Scholar]
- Tiziano, FD; Lomastro, R; Pinto, AM; Messina, S; D’Amico, A; Fiori, S; Angelozzi, C; Pane, M; Mercuri, E; Bertini, E; et al. Salbutamol increases SMN transcript levels in leukocytes of spinal muscular atrophy patients: relevance for clinical trial design. J. Med. Genet 2010, 47, 856–858. [Google Scholar]
- Hua, Y; Sahashi, K; Hung, G; Rigo, F; Passini, MA; Bennett, CF; Krainer, AR. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010, 24, 1634–1644. [Google Scholar]
- Williams, JH; Schray, RC; Patterson, CA; Ayitey, SO; Tallent, MK; Lutz, GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci 2009, 29, 7633–7638. [Google Scholar]
- Singh, NN; Shishimorova, M; Cao, LC; Gangwani, L; Singh, RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol 2009, 6, 341–350. [Google Scholar]
- Russman, BS; Iannaccone, ST; Samaha, FJ. A phase 1 trial of riluzole in spinal muscular atrophy. Arch. Neurol 2003, 60, 1601–1603. [Google Scholar]
- Haddad, H; Cifuentes-Diaz, C; Miroglio, A; Roblot, N; Joshi, V; Melki, J. Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve 2003, 28, 432–437. [Google Scholar]
- Merlini, L; Solari, A; Vita, G; Bertini, E; Minetti, C; Mongini, T; Mazzoni, E; Angelini, C; Morandi, L. Role of gabapentin in spinal muscular atrophy: results of a multicenter, randomized Italian study. J. Child Neurol 2003, 18, 537–541. [Google Scholar]
- Swboda, KJ; Scott, CB; Reyna, SP; Prior, TW; LaSalle, B; Sorenson, SL; Wood, J; Acsadi, G; Crawford, TO; Kissel, JT; et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One 2009, 4, e5268. [Google Scholar]
- Swoboda, KJ; Scott, CB; Crawford, TO; Simard, LR; Reyna, SP; Krosschell, KJ; Acsadi, G; Elsheik, B; Schroth, MK; D’Anjou, G; et al. Project Cure Spinal Muscular Atrophy Investigators Network. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 2010, 5, e12140. [Google Scholar]
- Kaufmann, P; Muntoni, F. Issues in SMA clinical trial design. The International Coordinating Committee (ICC) for SMA Subcommittee on SMA Clinical Trial Design. Neuromuscul. Disord 2007, 17, 499–505. [Google Scholar]
- Lomen-Hoerth, C; Slawnych, MP. Statistical motor unit number estimation: from theory to practice. Muscle Nerve 2003, 28, 263–272. [Google Scholar]
- Slawnych, MP; Laszlo, CA; Hershler, C. A review of techniques employed to estimate the number of motor units in a muscle. Muscle Nerve 1990, 13, 1050–1064. [Google Scholar]
- Galea, V; Fehlings, D; Kirsch, S; McComas, A. Depletion and sizes of motor units in spinal muscular atrophy. Muscle Nerve 2001, 24, 1168–1172. [Google Scholar]
- Bromberg, MB; Swoboda, KJ; Lawson, VH. Counting motor units in chronic motor neuropathies. Exp. Neurol 2003, 184, 53–57. [Google Scholar]
- Swoboda, KJ; Prior, TW; Scott, CB; McNaught, TP; Wride, MC; Reyna, SP; Bromberg, MB. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol 2005, 57, 704–712. [Google Scholar]
- Oskoui, M; Levy, G; Garland, CJ; Gray, JM; O’Hagen, J; de Vivo, DC; Kaufmann, P. The changing natural history of spinal muscular atrophy type I. Neurology 2007, 69, 1931–1936. [Google Scholar]
- Zerres, K; Rudnik-Schoneborn, S; Forrest, E; Lusakowska, A; Borkowska, J; Hausmanowa-Petrusewicz, I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J. Neurol. Sci 1997, 146, 67–72. [Google Scholar]
- Bono, R; Inverno, M; Botteon, G; Iotti, E; Estienne, M; Berardinelli, A; Lanzi, G; Fedrizzi, E. Prospective study of gross motor development in children with SMA type II. Ital. J. Neurol. Sci 1995, 16, 223–230. [Google Scholar]
- Deymeer, F; Serdaroglu, P; Parman, Y; Poda, M. Natural history of SMA IIIb: Muscle strength decreases in a predictable sequence and magnitude. Neurology 2008, 71, 644–649. [Google Scholar]
- Monani, UR; Coovert, DD; Burghes, AH. Animal models of spinal muscular atrophy. Hum. Mol. Genet 2000, 9, 2451–2457. [Google Scholar]
- Crabtree, NJ; Kibirige, MS; Fordham, JN; Banks, LM; Muntoni, F; Chinn, D; Boivin, CM; Shaw, NJ. The relationship between lean body mass and bone mineral content in paediatric health and disease. Bone 2004, 35, 965–972. [Google Scholar]
- Khatri, IA; Chaudhry, US; Seikaly, MG; Browne, RH; Iannaccone, ST. Low bone mineral density in spinal muscular atrophy. Neuromuscul. Disord 2008, 10, 11–17. [Google Scholar]
- Kinali, M; Banks, LM; Mercuri, E; Manzur, AY; Muntoni, F. Bone mineral density in a paediatric spinal muscular atrophy population. Neuropediatrics 2004, 35, 325–328. [Google Scholar]
- Shanmugarajan, S; Tsuruga, E; Swoboda, KJ; Maria, BL; Ries, WL; Reddy, SV. Bone loss in survival motor neuron (Smn(−/−) SMN2) genetic mouse model of spinal muscular atrophy. J. Pathol 2009, 219, 52–60. [Google Scholar]
- Kolb, SJ; Gubitz, AK; Olszewski, RF, Jr; Ottinger, E; Sumner, CJ; Fischbeck, KH; Dreyfuss, G. A novel cell immunoassay to measure survival of motor neurons protein in blood cells. BMC Neurol 2006, 6, 6. [Google Scholar]
- Thi Man, N; Humphrey, E; Lam, LT; Fuller, HR; Lynch, TA; Sewry, CA; Goodwin, PR; MacKenzie, AE; Morris, GE. A two-site ELISA can quantify uperegulation of SMN protein by drugs for spinal muscular atrophy. Neurology 2008, 22, 1757–1763. [Google Scholar]
- Piepers, S; Cobben, JM; Sodaar, P; Jansen, MD; Wadman, RI; Meester-Delver, A; Poll-The, BT; Lemmink, HH; Wokke, JH; van der Pol, WL; van den Berg, LH. Quantification of SMN protein in leucocytes from spinal muscular atrophy patients: Effects of treatment with valproic acid. In J Neurol Neurosurg Psychiatry; 2010 2009; p. 200253. [CrossRef]
- Sumner, CJ; Kolb, SJ; Harmison, GG; Jeffries, NO; Schadt, K; Finkel, RS; Dreyfuss, G; Fischbeck, KH. SMN mRNA and protein levels in peripheral blood. Neurology 2006, 66, 1067–1073. [Google Scholar]
- Simard, LR; Bélanger, MC; Morissette, S; Wride, M; Prior, TW; Swoboda, KJ. Preclinical validation of a multiplex real-time assay to quantify SMN mRNA in patients with SMA. Neurology 2007, 68, 451–456. [Google Scholar]
- Vezain, M; Saugier-Veber, P; Melki, J; Toutain, A; Bieth, E; Husson, M; Pedespan, JM; Viollet, L; Pénisson-Besnier, I; Fehrenbach, S; et al. A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur. J. Hum. Genet 2007, 15, 1054–1062. [Google Scholar]
- Tiziano, FD; Pinto, AM; Fiori, S; Lomastro, R; Messina, S; Bruno, C; Pini, A; Pane, M; D’Amico, A; Ghezzo, A; Bertini, E; Mercuri, E; Neri, G; Brahe, C. SMN transcript levels in leukocytes of SMA patients determined by absolute real time PCR. Eur. J. Hum. Genet 2010, 18, 52–58. [Google Scholar]
- Tricarico, C; Pinzani, P; Bianchi, S; Paglieranib, M; Distantec, V; Pazzaglia, M; Bustin, SA; Orlandoa, C. Quantitative real-time reverse transcription polymerase chain reaction: normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal. Biochem 2002, 309, 293–300. [Google Scholar]
- Tsai, LK; Yang, CC; Ting, CH; Su, YN; Hwu, WL; Li, H. Correlation of survival motor neuron expression in leukocytes and spinal cord in spinal muscular atrophy. J. Pediatr 2009, 154, 303–305. [Google Scholar]

{kind=link}
| Potential Biomarker | Pros | Cons |
|---|---|---|
| Instrumental | ||
| CMAP and MUNE | ||
| • Both measures are related to phenotypic severity | • MUNE does not appear related to motor function in a group of type II patients | |
| • Progressively decrease over time (MUNE is more stable in type III) | • There is no evidence yet of correlations between motor function and CMAP variations | |
| • Are related to SMN2 copy number | ||
| • Have been evaluated in an open phase II trial of valproic acid | ||
| • CMAP, but not MUNE, increases with VPA | ||
| DXA | ||
| • Bone density increased after VPA treatment | • The biological significance of BMD reduction in SMA patients is not established | |
| • It is not known whether BMD variations are related to the clinical outcome of treatment | ||
| Molecular | ||
| SMN protein quantification | ||
| • SMN protein levels, as determined by cell immunoassay, are related to SMN2 copy number | • SMN protein levels are not related to clinical severity | |
| • For cell immunoassay, small amount of PBMC are sufficient for SMN quantification | • No stabilization buffers are commercially available for total proteins | |
| • ELISA assay is sensitive down to magnitude of pg/mL of SMN protein | • PBMC should be manipulated within 2 hours from sampling | |
| • The minimum amount of peripheral blood necessary for SMN quantification is not known | ||
| • It is not indicated for evaluation of candidate compounds which do not modify SMN levels | ||
| SMN transcript quantification | ||
| • Small amounts of blood (2.5 mL or less) are sufficient for mRNA quantification | • It is not known if protein and transcript levels are related | |
| • Several stabilization buffers are available for multicenter clinical trials | • It is unknown if transcript level variations are related to the clinical outcome of treatment | |
| • SMN transcripts are stable over time | • It is unknown if transcript levels in blood and target tissues are related | |
| • It is not indicated for the evaluation of candidate compounds which do not modify SMN levels |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tiziano, F.D.; Neri, G.; Brahe, C. Biomarkers in Rare Disorders: The Experience with Spinal Muscular Atrophy. Int. J. Mol. Sci. 2011, 12, 24-38. https://doi.org/10.3390/ijms12010024
Tiziano FD, Neri G, Brahe C. Biomarkers in Rare Disorders: The Experience with Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2011; 12(1):24-38. https://doi.org/10.3390/ijms12010024
Chicago/Turabian StyleTiziano, Francesco D., Giovanni Neri, and Christina Brahe. 2011. "Biomarkers in Rare Disorders: The Experience with Spinal Muscular Atrophy" International Journal of Molecular Sciences 12, no. 1: 24-38. https://doi.org/10.3390/ijms12010024