3.3. Descriptor Selection and Model Building
The basic strategy of QSPR analysis is to find optimum quantitative relationships between the molecular descriptors and desired property, which can then be used for the prediction of the property from only molecular structures. One of the most important problems involved in QSPR studies is to select optimal subset of descriptors that have significant contribution to the desired property. The well-known genetic algorithm is just a well-accepted method for solving this kind of problems.
After correlation analysis of the descriptors, we used MLR analysis on the molecular descriptors that resulted in genetic algorithm (GA) variable selection procedure. The GA-algorithm applied in this paper uses a binary representation as the coding technique for the given problem; the presence or absence of a descriptor in a chromosome is coded by 1 or 0. The GA performs its optimization by variation and selection via the evaluation of the fitness function (RMSECV). The algorithm used in this paper is an evolution of the algorithm described in Ref. [
31], whose parameters are reported in
Table 6. In our study, a genetic algorithm procedure was used for selection of descriptors using the PLS Toolbox (version 2.0, Eigenvector Company, USA). The GA is implemented in MATLAB (version 7.1, MathWorks, Inc.). By performing GA, 22 descriptors were retained for next analysis step.
Finally, descriptor-screening methods were used to select the most relevant descriptor to establish the models for prediction of the molecular property. Here, the stepwise regression method was used to choose the subset of the molecular descriptors.
After the descriptor was selected, multiple linear regression (MLR)[
32] was used to develop the linear model of the property of interest, which takes the form below:
In this equation, y is the property, that is, the dependent variable, x1−xn represent the specific descriptor, while b1− bn represent the coefficients of those descriptors, and b0 is the intercept of the equation. The statistical evaluation of the data was obtained by the software SPSS. The SPSS software, (SPSS Ver. 11.5, SPSS Inc.), performed MLR analysis and variable selection by using stepwise method for the variable selection and modeling.
3.4. Theory of SVM
The foundation of support vector machines (SVM) has been developed by Vapnik, and they are gaining popularity due to many attractive features and promising empirical performance [
33]. The formulation embodies the structural risk minimization (SRM) principle [
32,
33], which has been shown to be superior to the traditional empirical risk minimization (ERM) principle, employed by conventional neural networks. SRM minimizes an upper bound on VC dimension (“generalization error”), as opposed to ERM that minimizes the error on the training data. It is the difference that equips SVM with good generalization performance, which is the goal in statistical learning. Originally, SVM were developed for classification problems [
34], and now, with the introduction of
ɛ-insensitive loss function, SVM have been extended to solve nonlinear regression estimation [
36].
Compared to other neural network regressors, there are three distinct characteristics when SVM are used to estimate the regression function. First of all, SVM estimate the regression using a set of linear functions that are defined in a high dimensional space. Second, SVM carry out the regression estimation by risk minimization where the risk is measured using Vapnik’s ɛ-insensitive loss function. Third, SVM use a risk function consisting of the empirical error and a regularization term which is derived from the SRM principle.
In support vector regression (SVR), the basic idea is to map the data x into a higher-dimensional feature space F via a nonlinear mapping Φ, and then to do linear regression in this space. Therefore, regression approximation addresses the problem of estimating a function based on a given data set G = {(xi,di)}in (xi is the input vector, di is the desired value, and n is the total number of data patterns). SVM approximate the function using the following
where Φ(x) denotes the element wise mapping from x into feature space. The coefficients w and b are estimated by minimizing
In
Equation 5,
RSVMs is the regularized risk function, and the first term
is the empirical error (risk). They are measured by the
ɛ-insensitiveloss function (
Lɛ) given by
Equation 6. This loss function provides the advantage of enabling one to use sparse data points to represent the decision function given by
Equation 4. The second term
, on the other hand, is the regularization term.
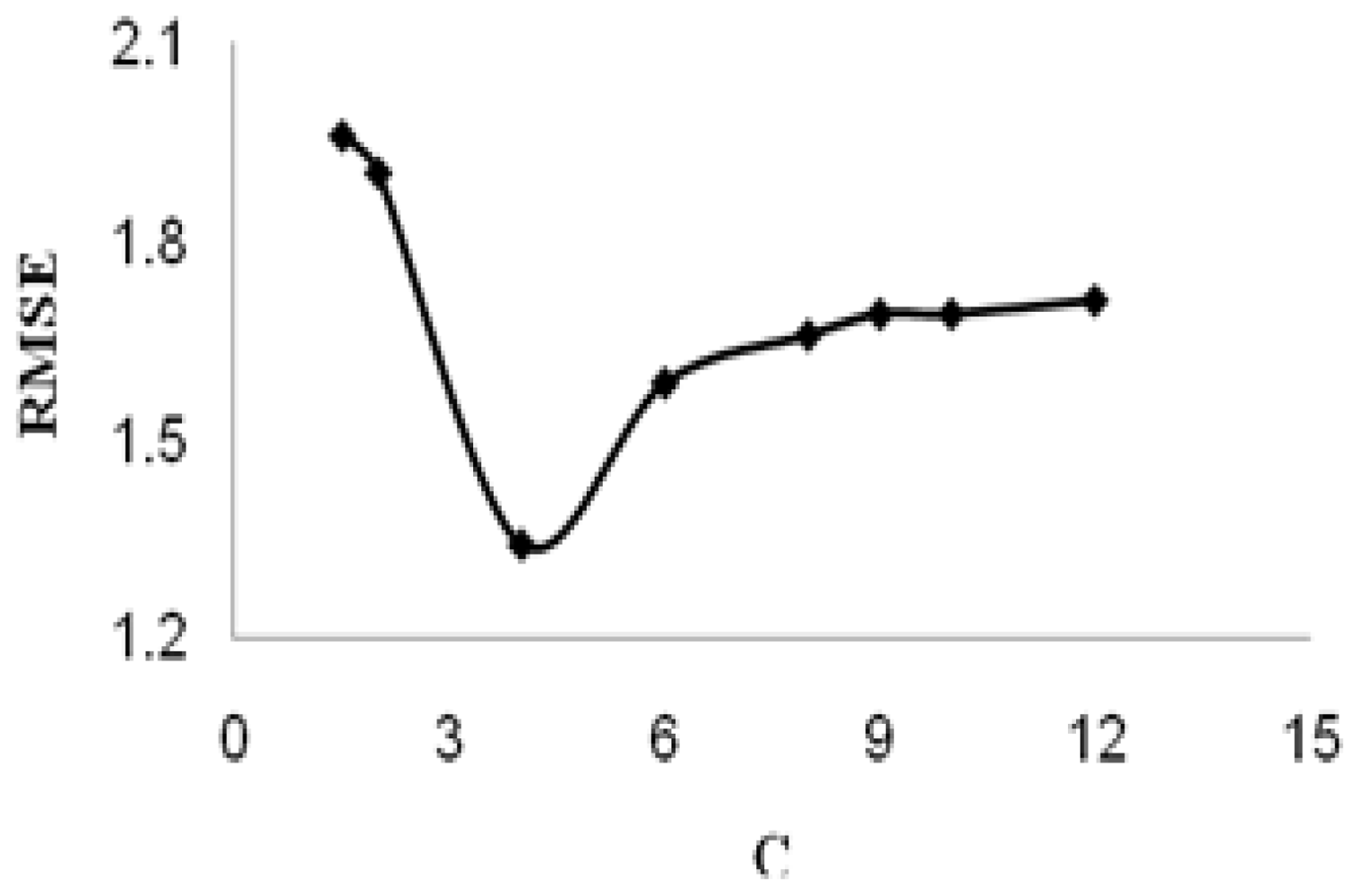
C is referred to as the regularized constant, and it determines the tradeoff between the empirical risk and the regularization term. Increasing the value of
C will result in the relative importance of the empirical risk with respect to the regularization term to grow.
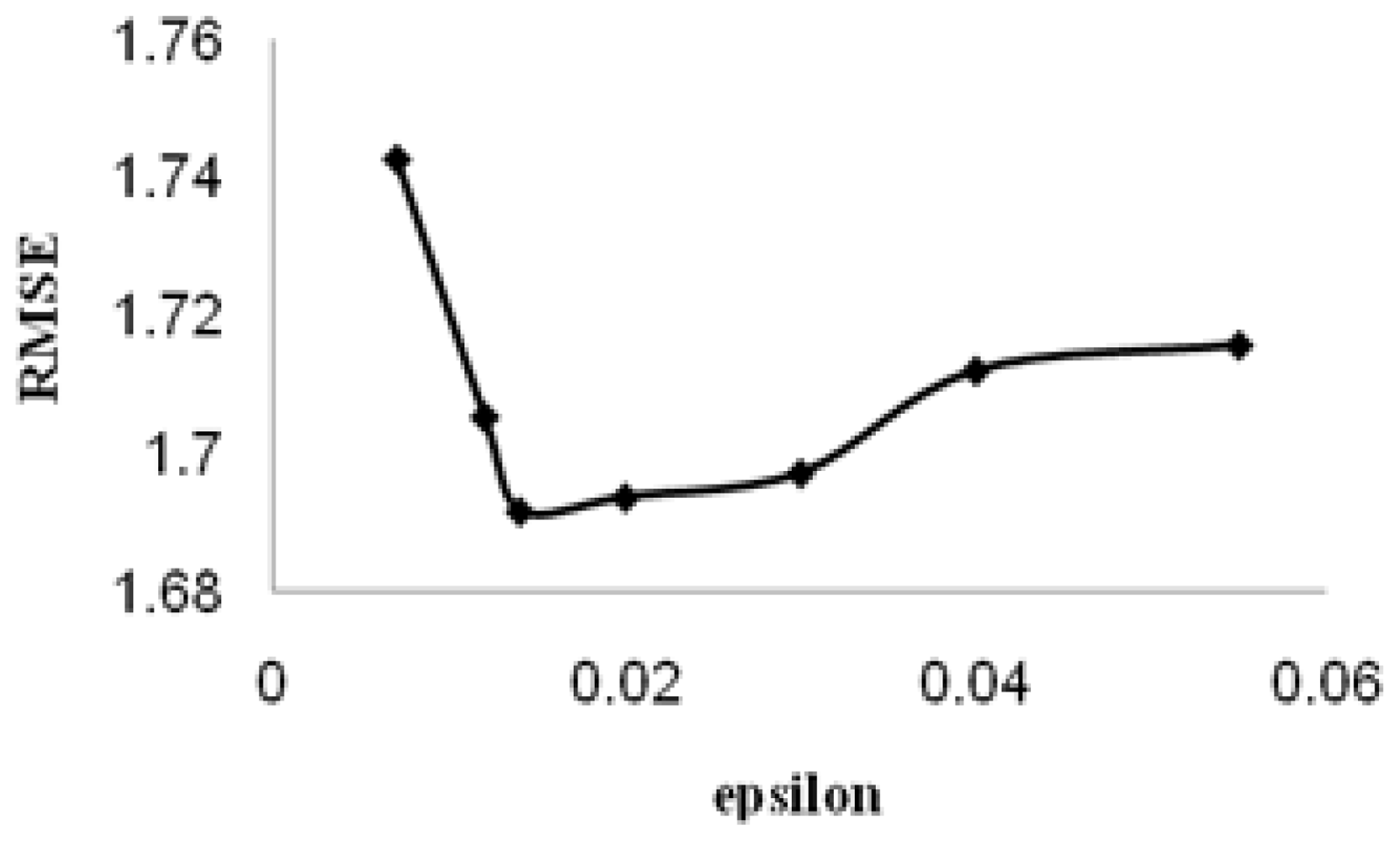
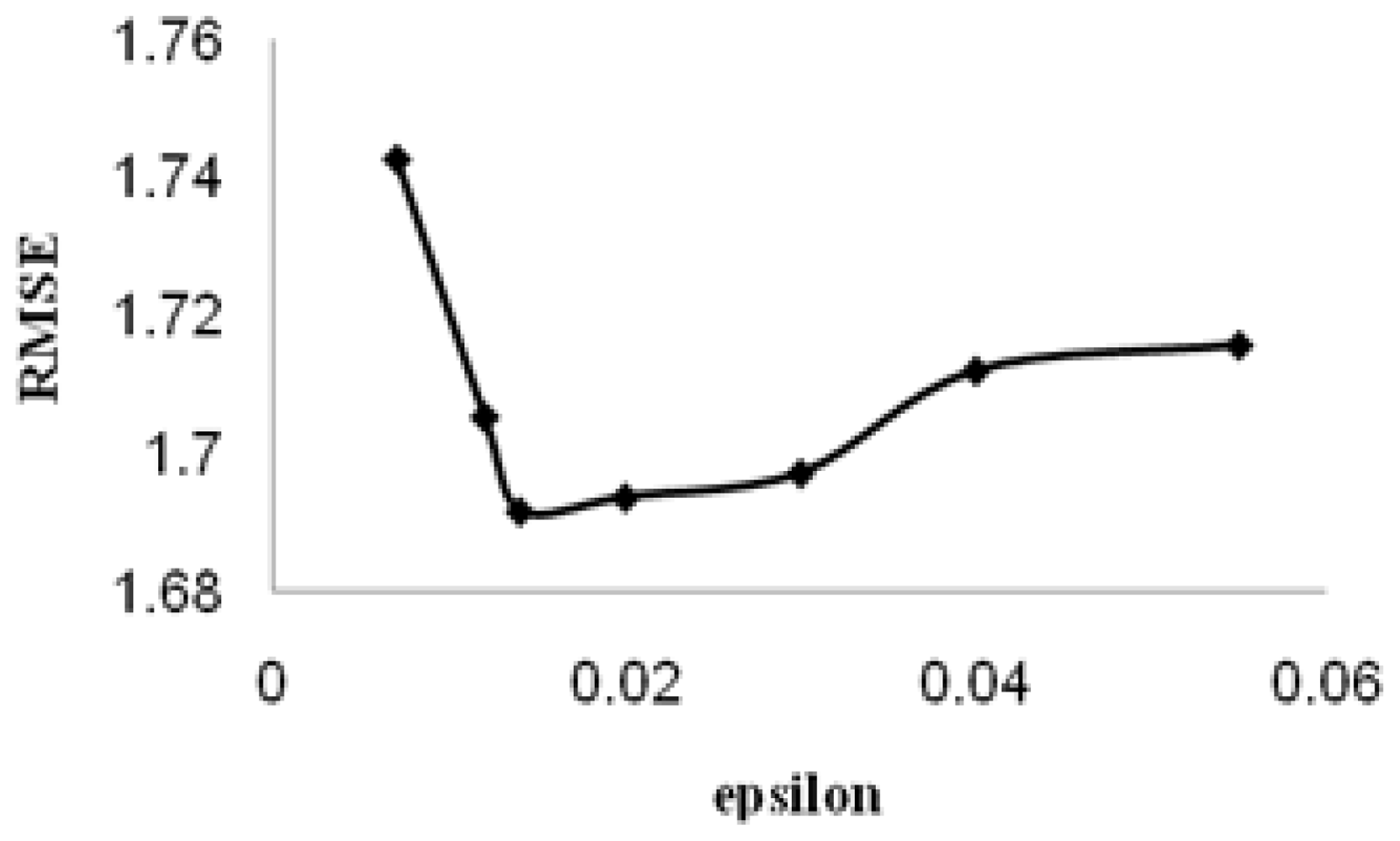
ɛ is called the tube size, and it is equivalent to the approximation accuracy placed on the training data points. Both C and ɛ are user-prescribed parameters.
Finally, by introducing Lagrange multipliers (
ai,
ai*) andexploiting the optimality constraints, the decision functiongiven by
Equation 4 has the following explicit form:
Based on the Karush-Kuhn-Tucker (KKT) conditions of quadratic programming, only a number of coefficients (
ai,
ai*) will assume nonzero values, and the data points associated with them could be referred to as support vectors. In
Equation 7, the kernel function
K corresponds to
K(
x,
xi) = Φ(
x).Φ(
xi). One has several possibilities for the choice of this kernel function, including linear, polynomial, splines, and radial basis function. The elegance of using the kernel function lies in the fact that one can deal with feature spaces of arbitrary dimensionality without having to compute the map Φ(
x) explicitly.
The overall performances of SVM models were evaluated in terms of root mean square error (RMSE), which was defined as below:
where yk is the desired output, ŷk is the predicted value and ns is the number of samples in the analyzed set.
The predictive power of the models developed on the calculated statistical parameters standard error of prediction (SEP) and relative error of prediction (REP %) as follows:
where ỹi, yi and ȳ are the predicted, experimental and mean activity property, respectively.
All calculations in this work were carried out by using Matlab (V 7.1, The Mathworks, Inc.) and the SVM toolbox developed by Gunn [
37].
3.5. Validation Test
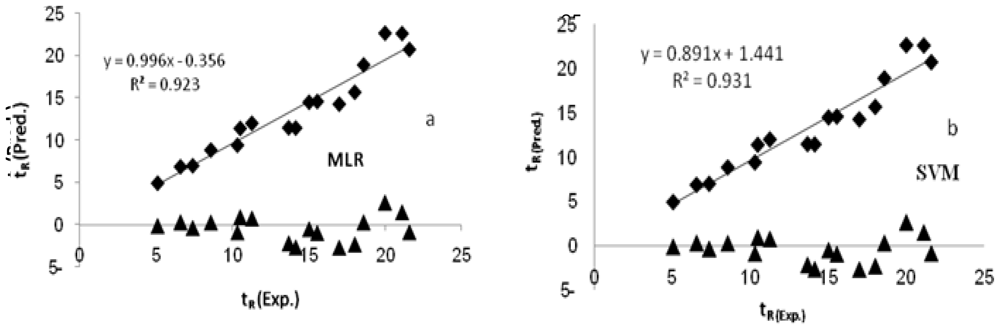
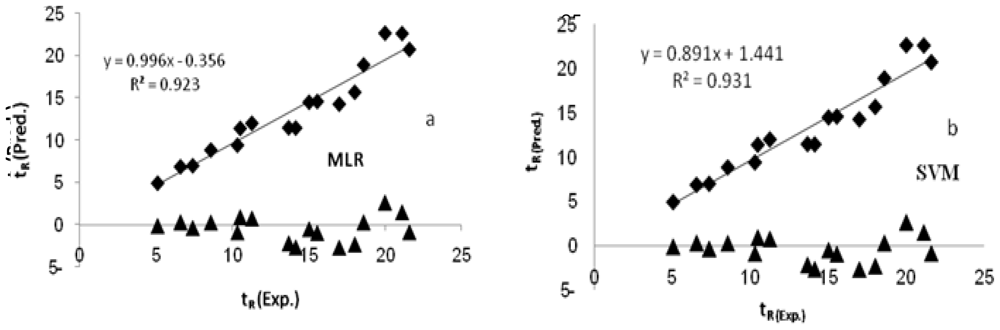
The main goal in QSPR studies is to obtain a model with the highest predictive ability. In order to evaluate the predictive ability of our QSPR model, we used the method described by Golbraikh and Tropsha [
38] and Roy and Roy [
39]. The determination coefficient in prediction (
q2test) was calculated using the following equation [
39]:
where
ypredtest and
yTest are the predicted values based on the QSPR equation (model response) and experimental activity values, respectively, of the external test set compounds.
ȳ is the mean activity value of the training set compounds. Further evaluation of the predictive ability of the QSAR model for the external test set compounds was done by determining the value of
rm2 using the following equation [
39]:
where r2 is the squared Pearson correlation coefficient for regression calculated using Y = a + bx; “a” is referred to as the y-intercept, “b” is the slope value of regression line, and r02 is the squared correlation coefficient for regression without using y-intercept and the regression equation was y = bx. Both r2 and r02 between experimental and predicted values for the external test set compounds were calculated using the regression of analysis Toolpak option of Excel. If rm2 value for a give model is >0.5, it indicates the good external predictability of the developed model.
The values of
k and
k′, slopes of the regression line of the predicted property
versus actual property and
vice versa, were calculated using the following equations [
38]:
where
ỹi and
yi are the predicted and experimental property, respectively. The values of
k and
k′ are within the specified range of 0.85 and 1.15 [
36]. The value of [r
2 − r
02/r
2] and [r
2 − r
02′/r
2] are less than 0.1 (stipulated value)[
38].
r02 and
r02′ are correlation coefficient of regression between the predicted and experimental property of compounds in the test set and
vice versa without using y-intercept.
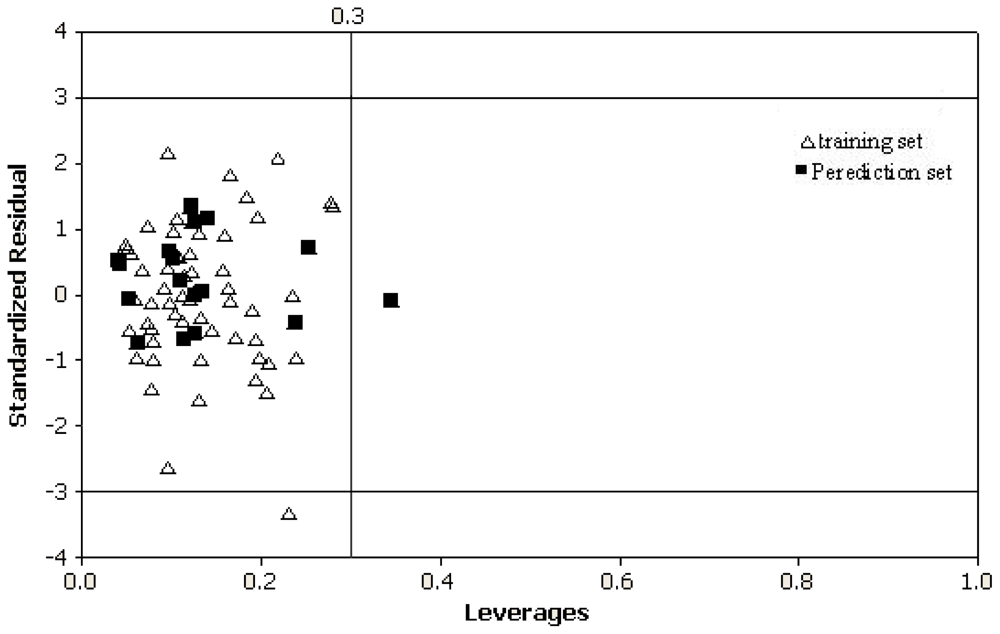
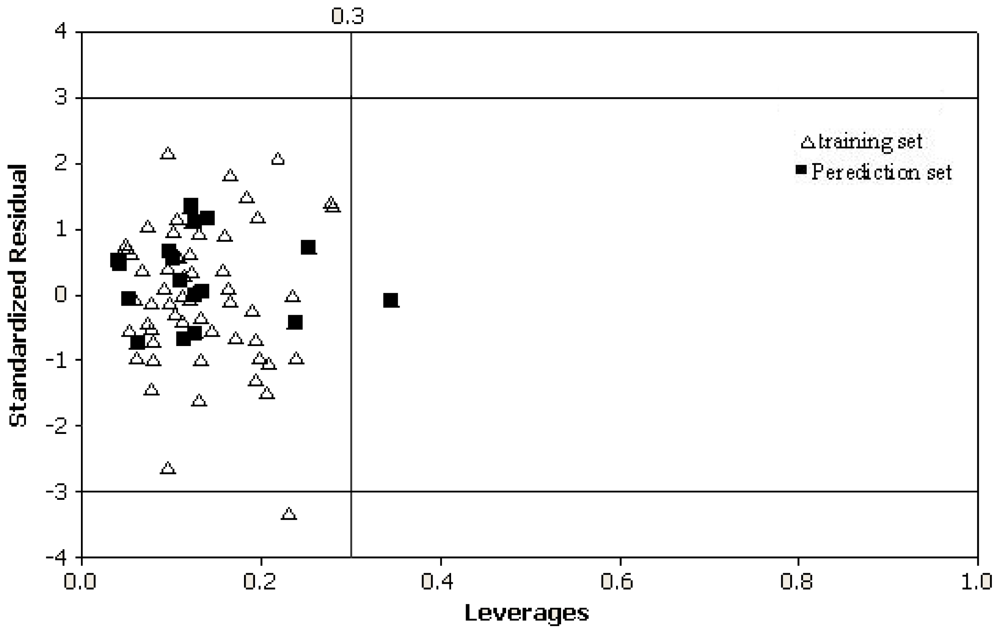
To further check the inter-correlation of descriptors variance inflation factor (VIF) analysis was performed. The VIF value is calculated from 1/1 − r
2, where
r2 is the multiplecorrelation coefficient of one descriptor’s effect regressed on the remaining molecular descriptors. If the VIF value is larger than 10, information of the descriptor could be hidden by correlation of descriptors [
40].



{kind=link}
{kind=link}
{kind=link}
{kind=link}