Functional Analysis of Familial Asp67Glu and Thr1051Ser BRCA1 Mutations in Breast/Ovarian Carcinogenesis

Abstract
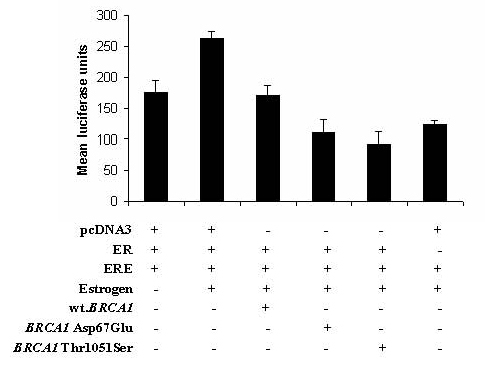
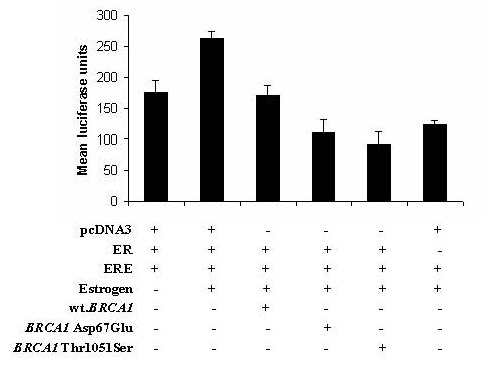
:
{kind=link}
{kind=link}
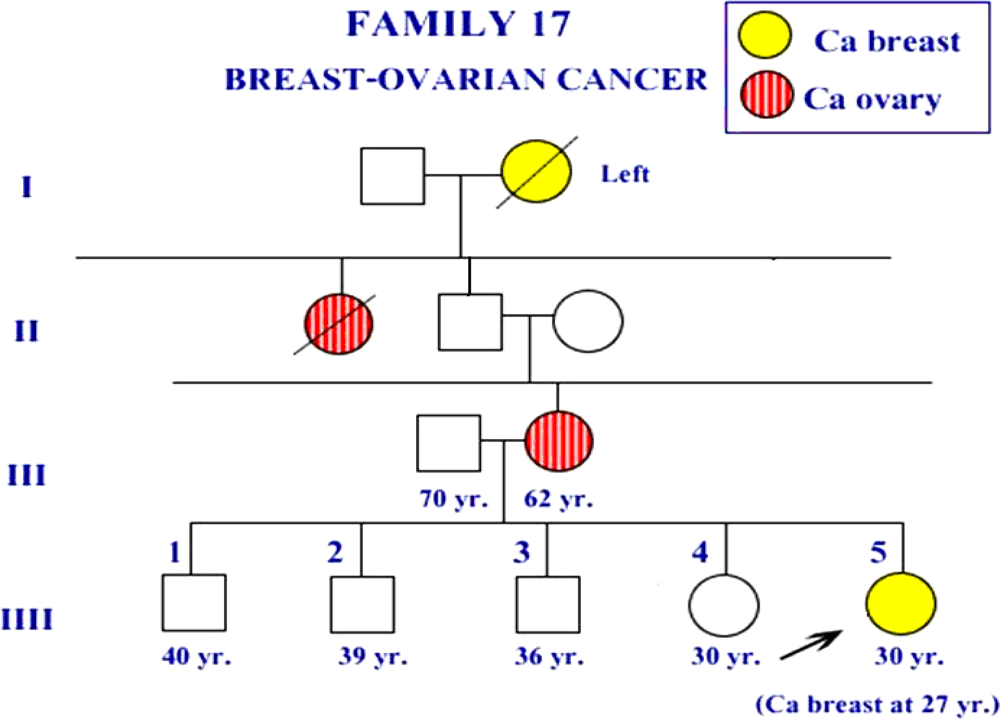{kind=link}
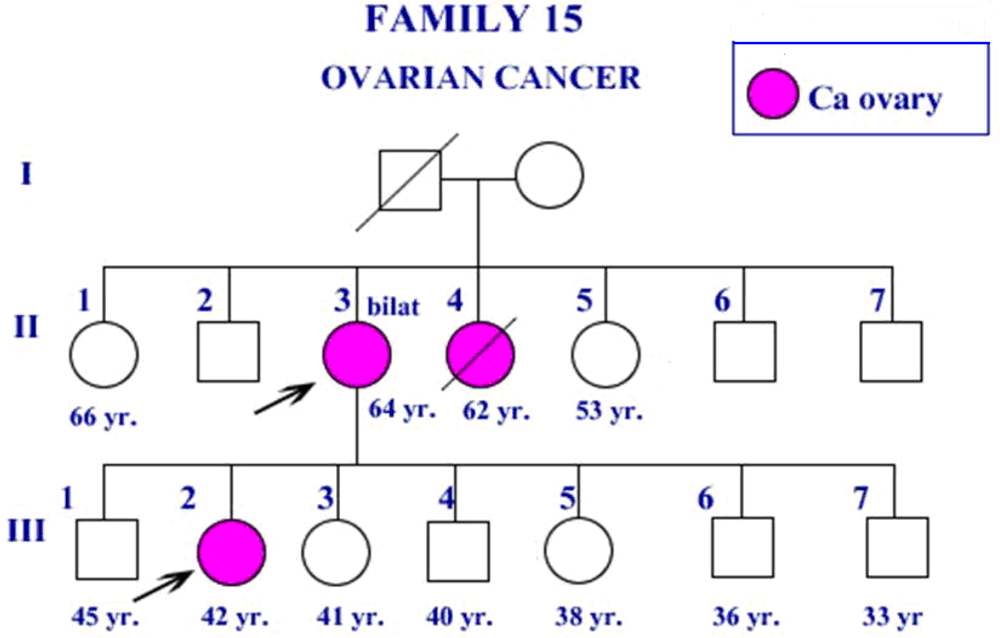{kind=link}
{kind=link}
1. Introduction
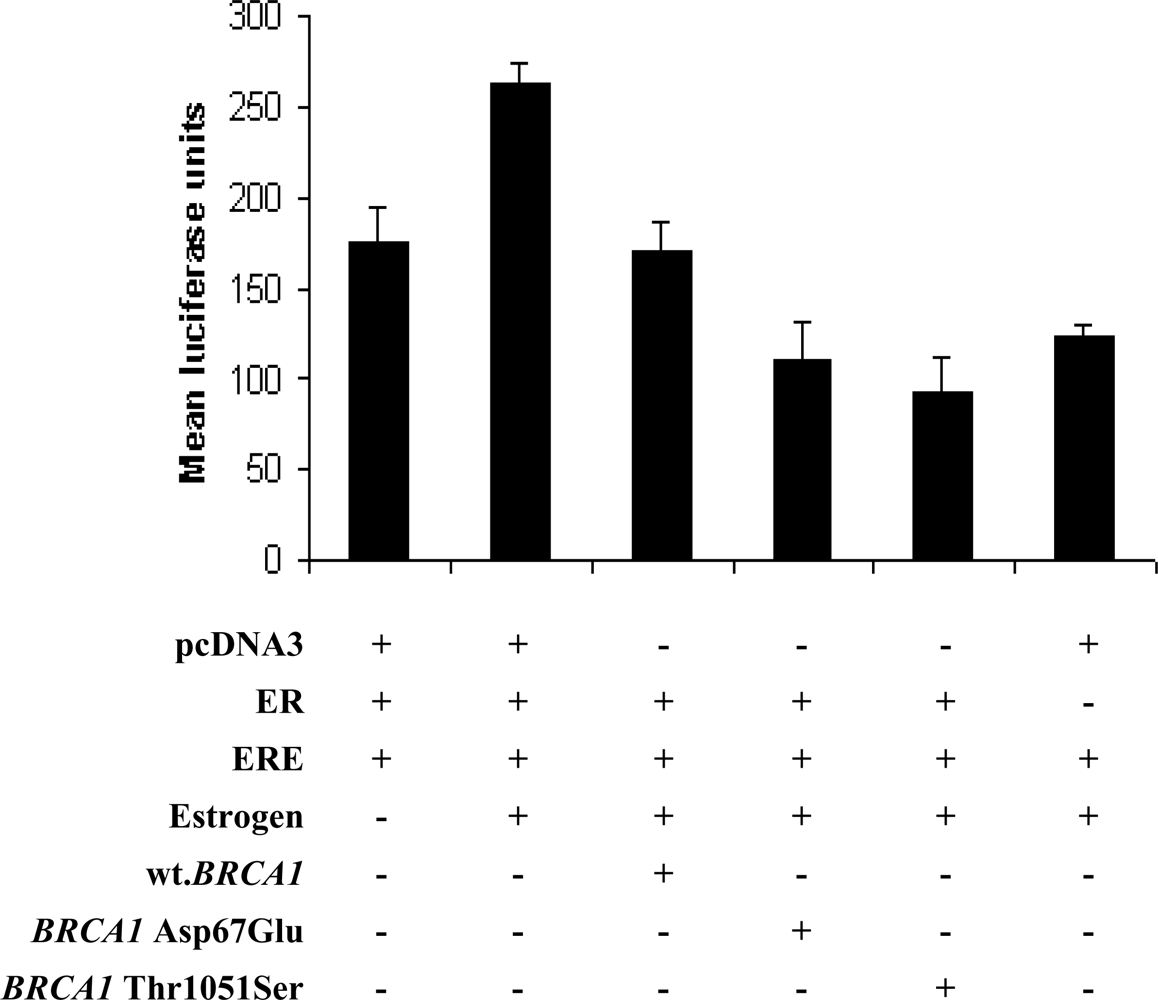
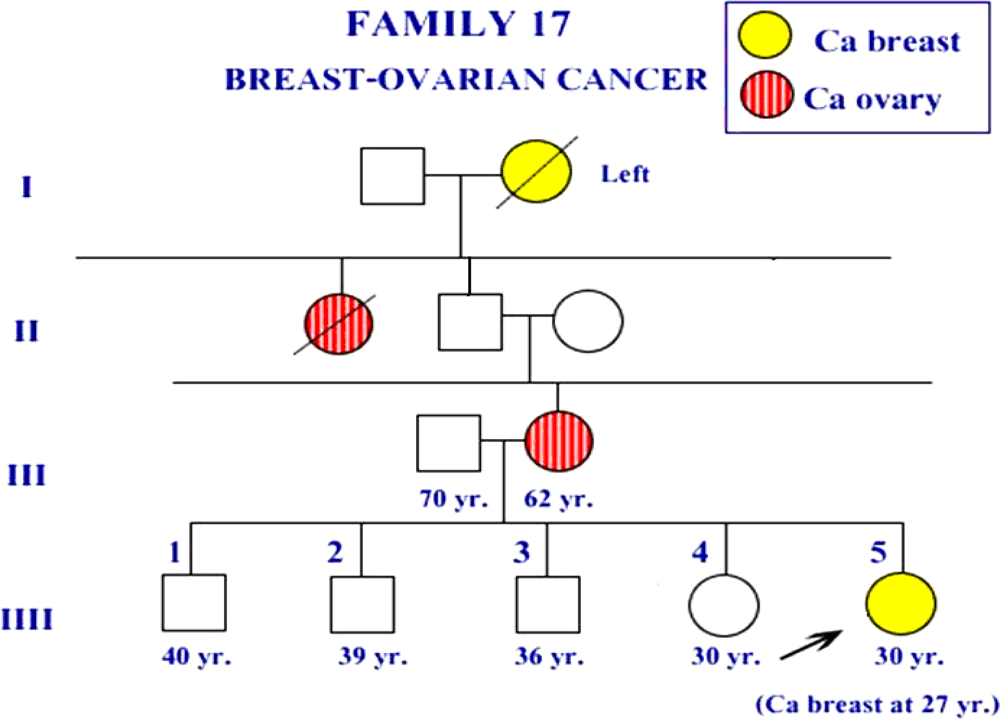
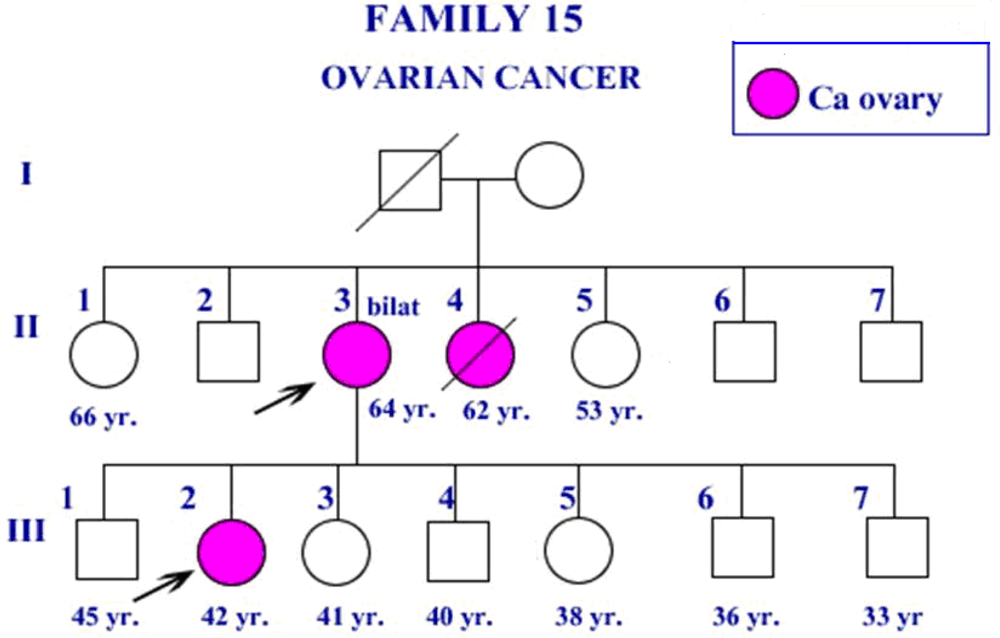
2. Results and Discussion
3. Experimental Section
3.1. Construction of vectors
3.2. Cell line and culture
3.3. Transient transfection
4. Conclusions
Acknowledgments
References
- Rahman, N; Stratton, MR. The genetics of breast cancer susceptibility. Annu. Rev. Genet 1998, 32, 95–121. [Google Scholar]
- Miki, Y; Swensen, J; Shattuck-Eidens, D; Futreal, PA; Harshman, K; Tavtigian, S; Liu, Q; Cochran, C; Bennett, M; Ding, W; Bell, R; Rosenthal, J; Hussey, C; Tran, T; McClure, M; Frye, C; Hattier, T; Phelps, R; Haugen-Strano, A; Katcher, H; Yakumo, K; Gholami, Z; Shaffer, D; Stone, S; Bayer, S; Wray, C; Bogden, R; Dayananth, P; Ward, J; Tonin, P; Narod, S; Bristow, PK; Norris, FH; Helvering, L; Morris, P; Rosteck, P; Lai, M; Barrett, JC; Lewis, C; Neuhausen, S; Cannon-Albright, L; Goldgar, D; Wiseman, R; Kamb, A; Skolnick, MH. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Jensen, DE; Proctor, M; Marquis, ST; Gardner, HP; Ha, SI; Chodosh, LA; Ishov, AM; Tommerup, N; Vissing, H; Sekido, Y; Minna, J; Borodovsky, A; Schultz, DC; Wilkinson, KD; Maul, GG; Barlev, N; Berger, SL; Prendergast, GC; Rauscher, FJ, 3rd. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1- mediated cell growth suppression. Oncogene 1998, 16, 1097–1112. [Google Scholar]
- Hashizume, R; Fukuda, M; Maeda, I; Nishikawa, H; Oyake, D; Yabuki, Y; Ogata, H; Ohata, T. The ring heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Bio. Chem 2001, 276, 14537–14540. [Google Scholar]
- Monterio, AN; August, A; Hanafusa, H. Evidence for a transcriptional activation of BRCA1 C-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar]
- Callebaut, I; Mormon, JP. From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett 1997, 400, 25–30. [Google Scholar]
- George, FW; Wilson, JD. The Physiology of Reproduction; Knobil, E, Ed.; New York Press Inc.: New York, NY, USA, 1988; pp. 2–27. [Google Scholar]
- Jensen, EV. Nuclear Hormone Receptors; Parker, MJ, Ed.; Academic Press: London, UK, 1992; pp. 1–13. [Google Scholar]
- Hilakivi-Clarke, L; Onojafe, I; Raygada, M; Cho, E; Lippman, ME. A method diet high in n-6 polyunsaturated fats alters mammary gland development, puberty onset, and breast cancer risk among female rat offspring. Proc. Natl. Acad. Sci. USA 1997, 94, 9372–9377. [Google Scholar]
- Gudas, JM; Nguyen, H; Li, T; Cowan, KH. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res 1995, 55, 4561–4565. [Google Scholar]
- Liehr, JG. Is estradiol a genotoxic mutagenic carcinogen? Endocr. Rev 2000, 21, 40–54. [Google Scholar]
- Enmark, E; Gustafsson, JA. Oestrogen receptors–An overview. J. Intern. Med 1999, 246, 133–138. [Google Scholar]
- Halachmi, S; Marden, E; Martin, G; MacKay, H; Abbondanza, C; Brown, M. Estrogen receptor-associated protein: Possible mediators of hormone–induced transcription. Science 1994, 264, 1455–1458. [Google Scholar]
- Fan, S; Wang, JA; Yuan, R; Ma, Y; Meng, Q; Erdos, MR; Pestell, RG; Yuan, F; Auborn, KJ; Goldberg, ID; Rosen, EM. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 1999, 284, 1354–1356. [Google Scholar]
- Keen, JC; Davidson, NE. The biology of breast carcinoma. Cancer 2003, 97, 825–833. [Google Scholar]
- Patmasiriwat, P; Bhothisuwan, K; Sinilnikova, OM; Chopin, S; Methakijvaroon, S; Badzioch, M; Padungsutt, P; Vattanaviboon, P; Vattanasapt, V; Szabo, CI; Saunders, GF; Goldgar, D; Lenoir, GM. Analysis of breast cancer susceptibility genes BRCA1 and BRCA2 in Thai familial and isolated early-onset breast and ovarian cancer. Human Mutat 2002, 20, 230. [Google Scholar]
- Couse, J; Korach, K. Estrogen receptor null mice: What have we learned and where will they lead us? Endocrinol. Rev 1999, 20, 358–417. [Google Scholar]
- Colditz, G. Relationship between estrogen level, use of hormone replacement therapy, and breast cancer. J. Natl. Cancer Inst 1998, 90, 814–823. [Google Scholar]
- Migliaccio, A; Domenico, M; Castoria, G; de Falco, A; Bontempo, P; Nola, E; Auricchio, F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 1996, 15, 1292–1300. [Google Scholar]
- Castoria, G; Barone, M; Domenico, M; Bilancio, A; Ametrano, D; Migliaccio, A; Auricchio, F. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J 1999, 18, 2500–2510. [Google Scholar]
- Sutherland, R; Reddel, R; Green, M. Effects of oestrogen on cell proliferation and cell cycle kinetics: A hypothesis on the cell cycle effects of antioestrogens. Eur. J. Cancer Clin. Oncol 1983, 19, 307–318. [Google Scholar]
- Leung, B; Potter, A. Mode of estrogen action on cell proliferation in CAMA-1 cells, sensitivity of G1 phase population. J. Cell Biochem 1987, 34, 213–225. [Google Scholar]
- Zuppan, P; Hall, JM; Lee, MK; Ponglikitmongkol, M; King, MC. Possible linkage of the estrogen receptor gene to breast cancer in a family with late-onset disease. Am. J. Hum. Genet 1991, 48, 1065. [Google Scholar]
- Zheng, L; Annab, LA; Afshari, CA; Lee, WH; Boyer, TG. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9587–9592. [Google Scholar]
- Pestell, RG; Albanese, C; Benlens, AT; Segall, JF; Lee, BJ; Arnold, A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr. Rev 1999, 20, 501–534. [Google Scholar]
- Prall, O; Carroll, J; Sutherland, R. A low abundance pool of nascent p21WAF1/Cip1 is targeted by estrogen to active cyclin E-CDK2. J. Bio. Chem 2001, 276, 45433–45442. [Google Scholar]
- Carroll, J; Swarbrick, A; Musgrove, E; Sutherland, R. Mechanisms of growth arrest by c-myc antisense oligonucleotides in MCF-7 breast cancer cells: Implications for the antiproliferative effects of antiestrogens. Cancer Res 2002, 62, 3126–3131. [Google Scholar]
- Mawson, A; Lai, A; Carroll, J; Sergio, M; Mitchell, C; Sarcevic, B. Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer cells through differential regulation of c-Myc and cyclin D1. Mol. Cell Endocrinol 2005, 229, 161–173. [Google Scholar]
- Prall, OW; Rogan, EM; Musgrove, EA; Watts, CK; Sutherland, RL. Estrogen-induced activation of Cdk4 and Cdk2 during G I–S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem 1997, 272, 10882–10894. [Google Scholar]
- Lai, A; Sarcevic, B; Prall, O; Sutherland, R. Insulin/insulin–like growth–I and estrogen cooperate to stimulate cyclin E-CDK2 activation and cell cycle progression in MCF-7 breast cancer cells through differential regulation of cyclin E and p21 WAF1/Cip1. J. Biol. Chem 2001, 276, 25823–25833. [Google Scholar]
- Starita, LM; Parvin, JD. The multiple nuclear functions of BRCA1: Transcription, ubiquitination and DNA repair. Curr. Opin. Cell Biol 2003, 15, 345–350. [Google Scholar]
- Lorick, KL; Jensen, JPM; Fang, S; Ong, AM; Hatakeyama, S; Weissman, AM. Ring finger mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 11364–11369. [Google Scholar]
- Xia, Y; Pao, GM; Chen, HW; Vema, IM; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem 2003, 278, 5255–5263. [Google Scholar]
- Mallery, DL; Vandenberg, CJ; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J 2002, 21, 6755–6762. [Google Scholar]
- Hashizume, RM; Fukuda, M; Maeda, I; Nishikawa, H; Oyake, D; Yabuki, Y; Otaga, H; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer–derived mutation. J. Bio. Chem 2001, 276, 14537–14540. [Google Scholar]
- Brzovic, PS; Keeffe, JR; Nishikawa, H; Miyamoto, K; Fox, D; Fukuda, M; Ohta, T; Klevit, R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651. [Google Scholar]
- Morris, JR; Pangon, LM; Boutell, C; Katagiri, T; Keep, NH; Solomon, E. Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum. Mol. Genet 2006, 15, 599–606. [Google Scholar]
- Morris, JR; Solomon, E. BRCA1:BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol. Genet 2004, 13, 807–817. [Google Scholar]

© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pongsavee, M.; Patmasiriwat, P.; Saunders, G.F. Functional Analysis of Familial Asp67Glu and Thr1051Ser BRCA1 Mutations in Breast/Ovarian Carcinogenesis. Int. J. Mol. Sci. 2009, 10, 4187-4197. https://doi.org/10.3390/ijms10094187
Pongsavee M, Patmasiriwat P, Saunders GF. Functional Analysis of Familial Asp67Glu and Thr1051Ser BRCA1 Mutations in Breast/Ovarian Carcinogenesis. International Journal of Molecular Sciences. 2009; 10(9):4187-4197. https://doi.org/10.3390/ijms10094187
Chicago/Turabian StylePongsavee, Malinee, Pimpicha Patmasiriwat, and Grady F. Saunders. 2009. "Functional Analysis of Familial Asp67Glu and Thr1051Ser BRCA1 Mutations in Breast/Ovarian Carcinogenesis" International Journal of Molecular Sciences 10, no. 9: 4187-4197. https://doi.org/10.3390/ijms10094187