Bidirectionality and Compartmentation of Metabolic Fluxes Are Revealed in the Dynamics of Isotopomer Networks

Abstract
:1. Introduction
1.1. Motivation for exploiting the dynamic transient
2. Dynamic MFA in eukaryotic systems
2.1. Flux analysis with non-steady metabolism
2.2. Utilizing metabolic oscillations
3. Building predictive kinetic models
3.1. Measurement of in vivo kinetics
4. Simulation of isotopic transients
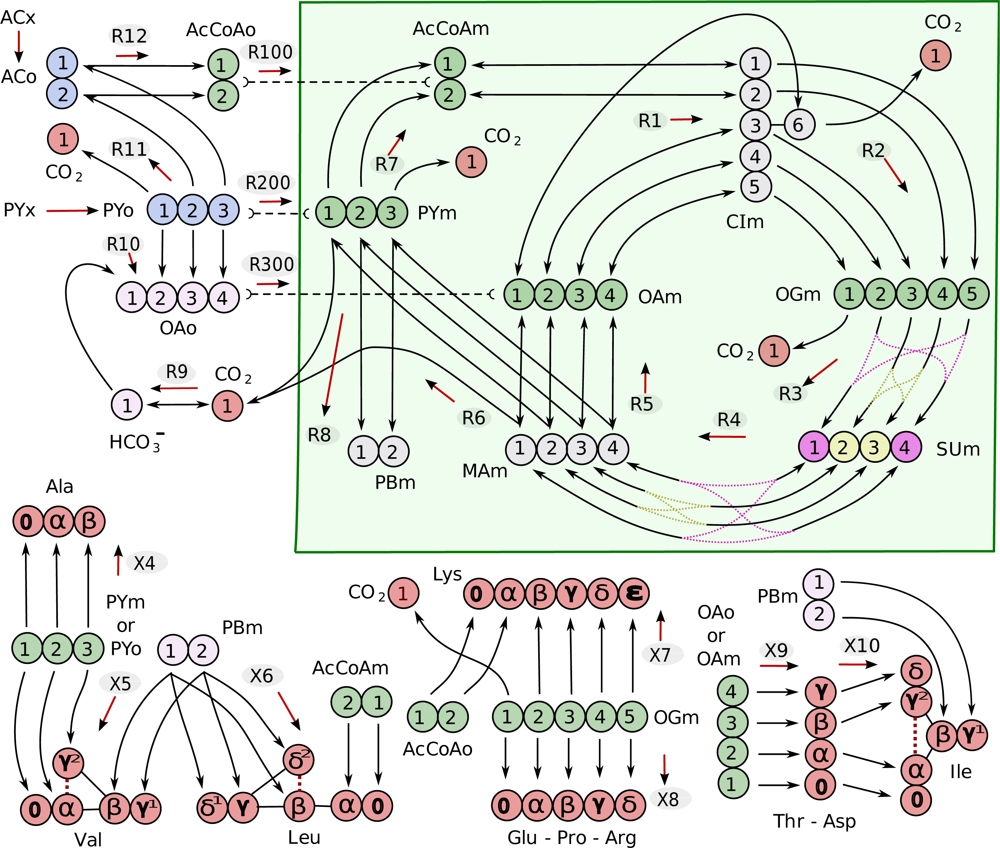
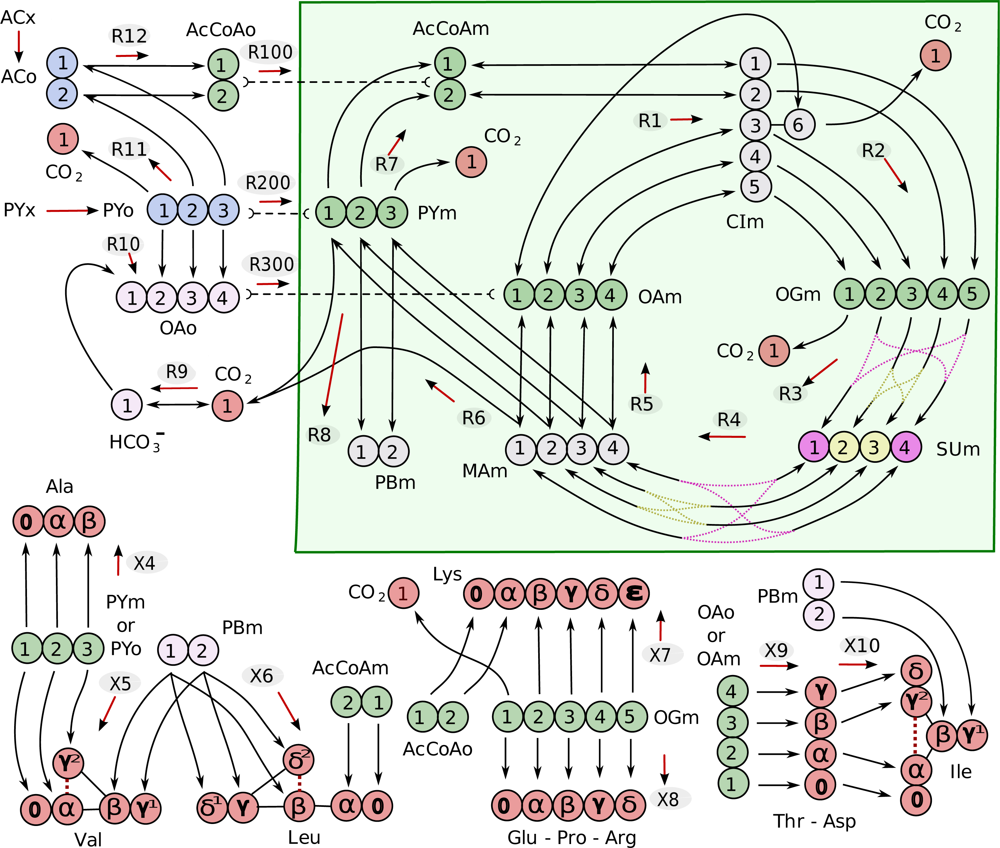
4.1. Composition of the metabolic network
4.2. Solving for the isotopic transient state
5. Extracting information from isotopomeric data
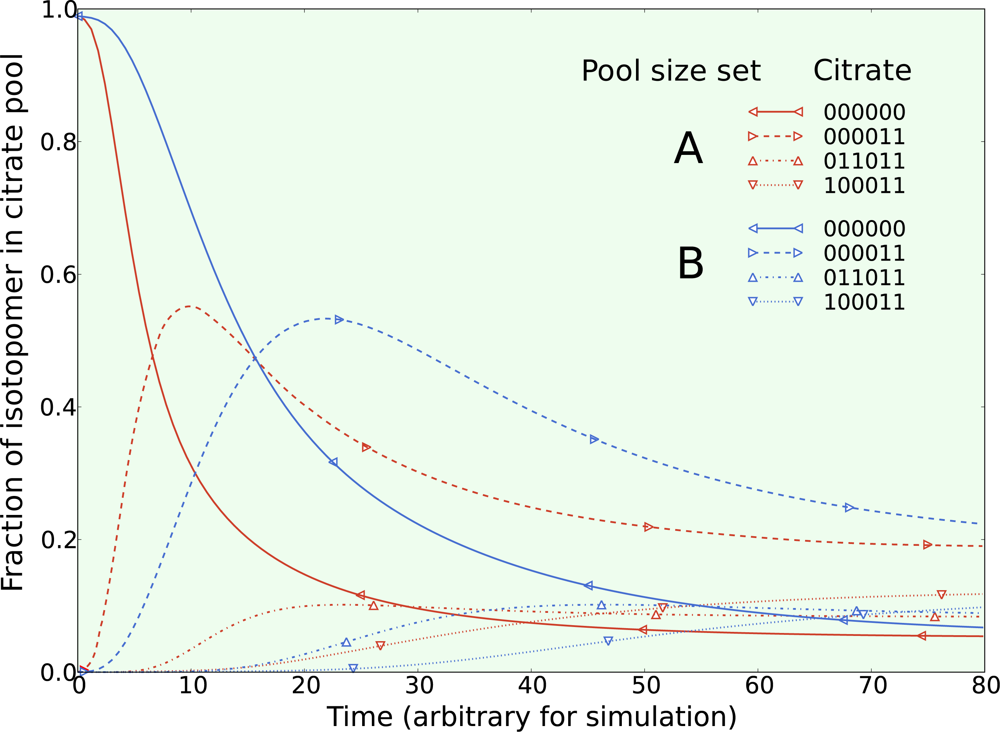
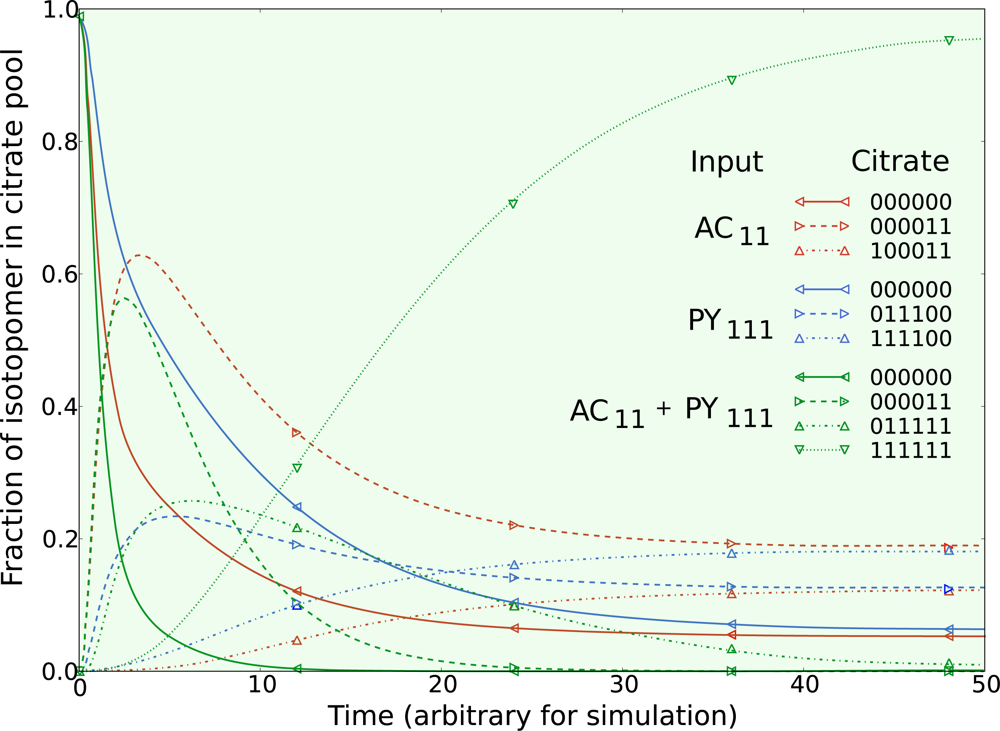
5.1. Inclusion of metabolic pool sizes
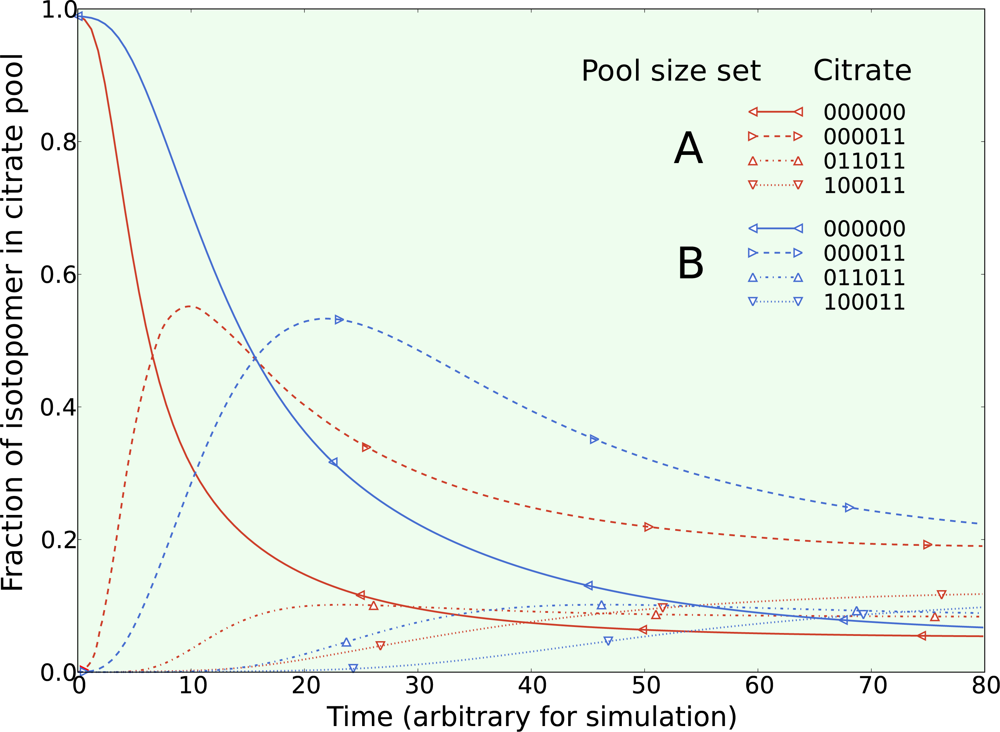
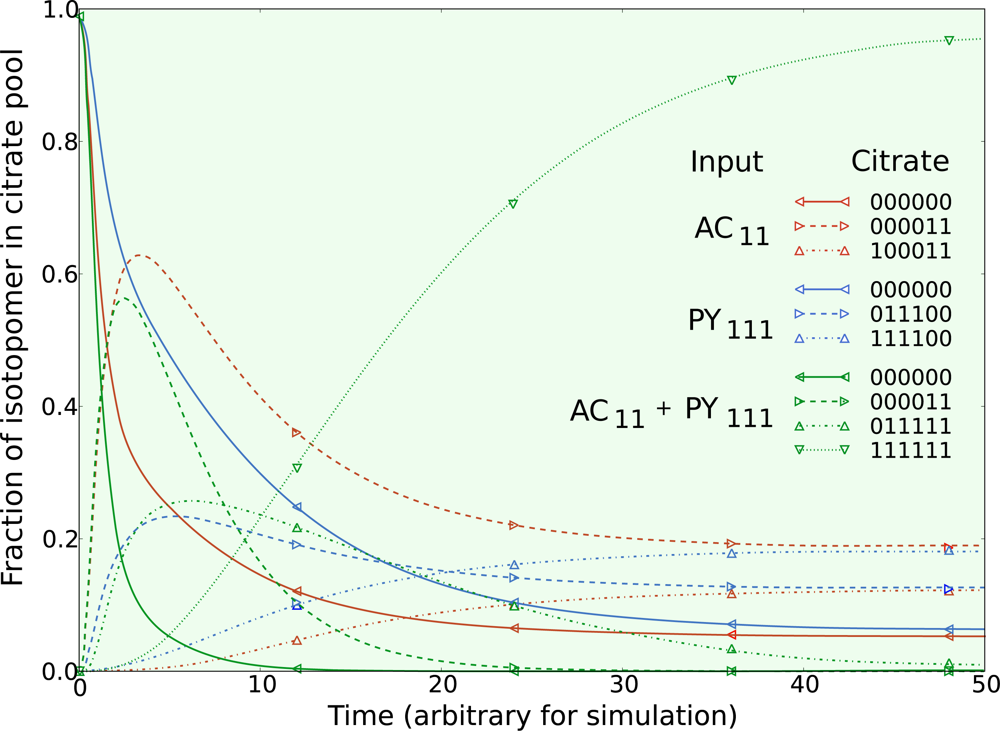
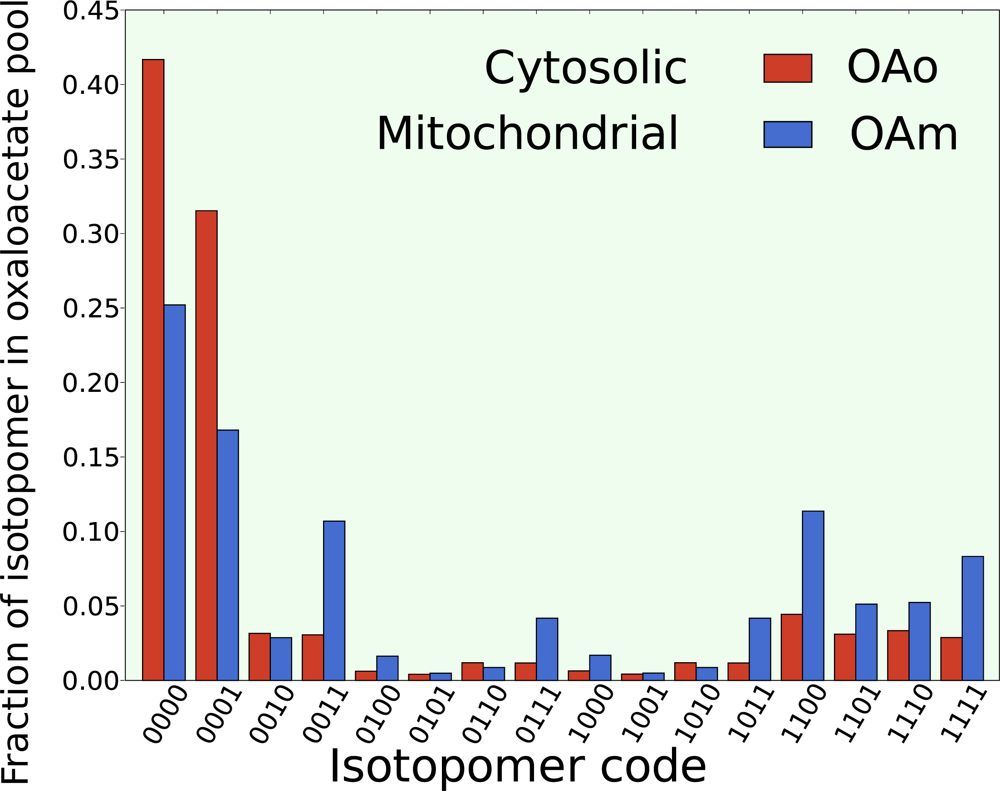
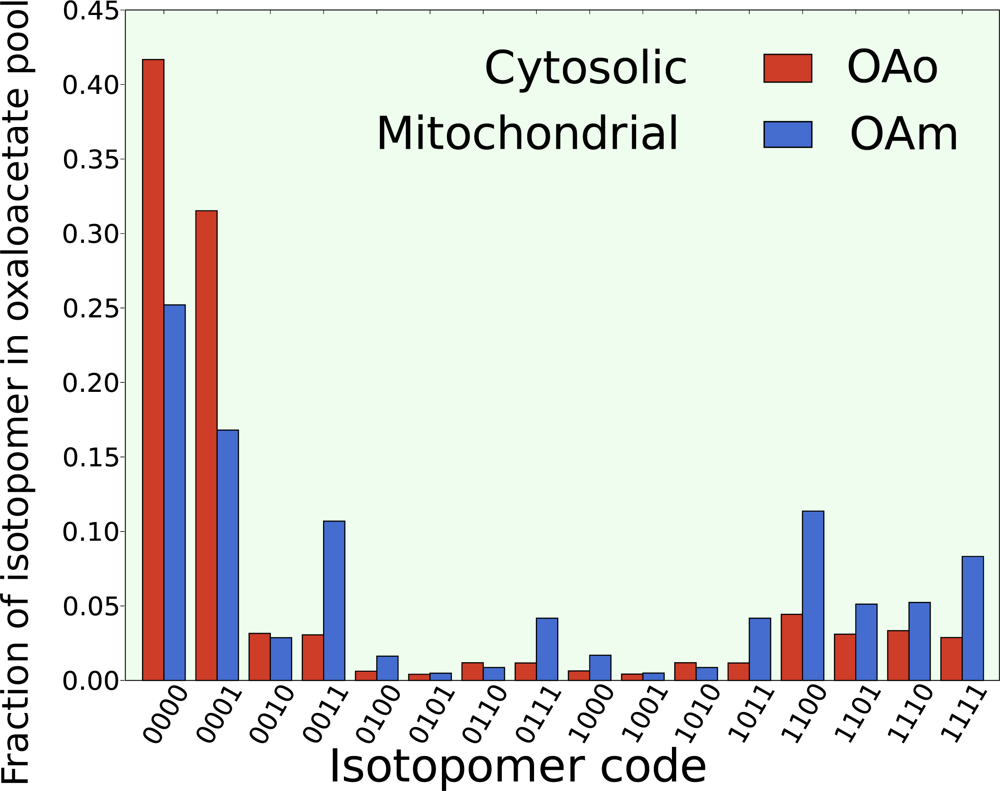
5.2. Compartmentalization is revealed in the dynamics
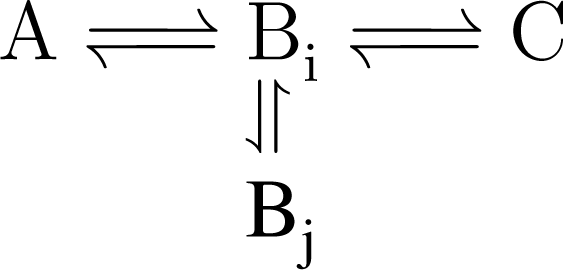
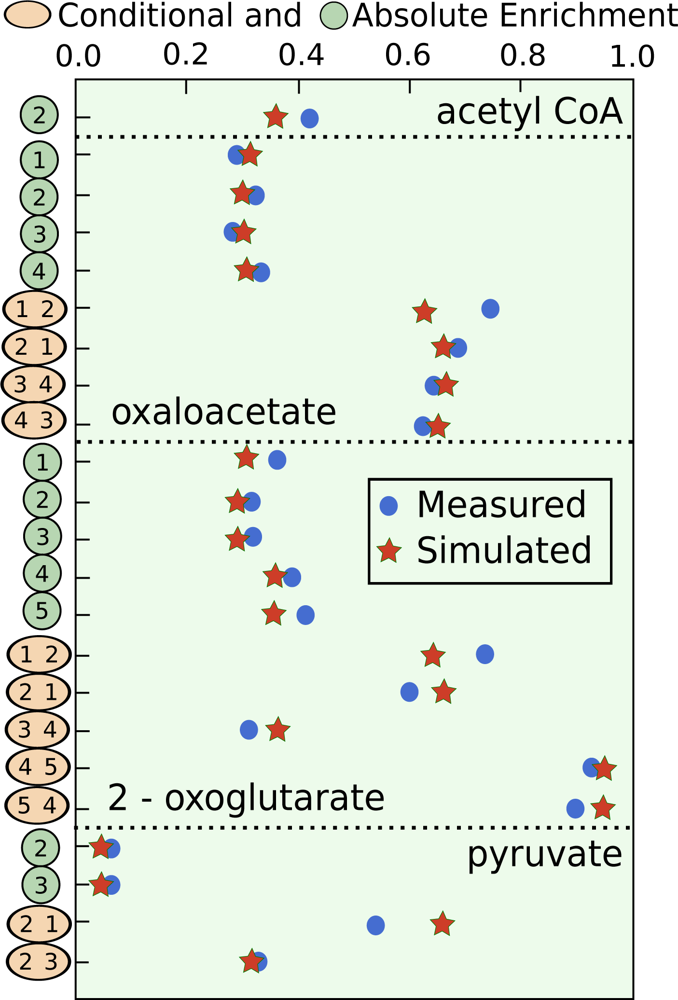
5.3. Example optimization of the TCA cycle in yeast
6. Conclusions
Acknowledgments
References and Notes
- Henry, P; Oz, G; Provencher, S; Gruetter, R. Toward dynamic isotopomer analysis in the rat brain in vivo: automatic quantitation of 13C NMR spectra using LCModel. NMR Biomed 2003, 16, 400–412. [Google Scholar]
- Rodrigues, TB; Cerdan, S. 13C MRS: an outstanding tool for metabolic studies. Concepts Magn. Reson. Part A 2005, 27A, 1–16. [Google Scholar]
- Hellerstein, M. In vivo measurement of fluxes through metabolic pathways: The missing link in functional genomics and pharmaceutical research. Ann. Rev. Nutr 2003, 23, 379–402. [Google Scholar]
- Iwatani, S; Yamada, Y; Usuda, Y. Metabolic flux analysis in biotechnology processes. Biotechnol. Lett 2008, 30, 791–799. [Google Scholar]
- Prather, KLJ; Martin, CH. De novo biosynthetic pathways: rational design of microbial chemical factories. Curr. Opin. Biotechnol 2008, 19, 468–474. [Google Scholar]
- Blank, LM; Lehmbeck, F; Sauer, U. Metabolic-flux and network analysis in fourteen hemiascomycetous yeasts. FEMS Yeast Res 2005, 5, 545–558. [Google Scholar]
- Vo, TD; Lim, SK; Lee, WNP; Palsson, BO. Isotopomer analysis of cellular metabolism in tissue culture: A comparative study between the pathway and network-based methods. Metabolomics 2006, 2, 243–256. [Google Scholar]
- Allen, DK; Shachar-Hill, Y; Ohlrogge, JB. Compartment-specific labeling information in 13C metabolic flux analysis of plants. Phytochemistry 2007, 68, 2197–2210. [Google Scholar]
- Kruger, NJ; Lay, PL; Ratcliffe, RG. Vacuolar compartmentation complicates the steady-state analysis of glucose metabolism and forces reappraisal of sucrose cycling in plants. Phytochemistry 2007, 68, 2189–2196. [Google Scholar]
- Stelling, J; Sauer, U; Szallasi, Z; Doyle, FJ, III; Doyle, J. Robustness of cellular functions. Cell 2004, 118, 675–685. [Google Scholar]
- Schaub, J; Mauch, K; Reuss, M. Metabolic flux analysis in escherichia coli by integrating isotopic dynamic and isotopic stationary 13C labeling data. Biotechnol. Bioeng 2008, 99, 1170–1185. [Google Scholar]
- Grotkjaer, T; Akesson, M; Christensen, B; Gombert, AK; Nielsen, J. Impact of transamination reactions and protein turnover on labeling dynamics in 13C-labeling experiments. Biotechnol. Bioeng 2004, 86, 209–216. [Google Scholar]
- Den Hollander, JA; Behar, KL; Shulman, RG. 13C NMR study of transamination during acetate utilization by saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1981, 78, 2693–2697. [Google Scholar]
- Wiechert, W; Noh, K. From stationary to instationary metabolic flux analysis. Adv. Biochem. Eng. Biotechnol 2005, 92, 145–172. [Google Scholar]
- Matsuda, F; Wakasa, K; Miyagawa, H. Metabolic flux analysis in plants using dynamic labeling technique: Application to tryptophan biosynthesis in cultured rice cells. Phytochemistry 2007, 68, 2290–2301. [Google Scholar]
- Heinzle, E; Matsuda, F; Miyagawa, H; Wakasa, K; Nishioka, T. Estimation of metabolic fluxes, expression levels and metabolite dynamics of a secondary metabolic pathway in potato using label pulse-feeding experiments combined with kinetic network modelling and simulation. Plant J 2007, 50, 176–187. [Google Scholar]
- Shastri, A; Morgan, J. A transient isotopic labeling methodology for 13C metabolic flux analysis of photo auto trophic microorganisms. Phytochemistry 2007, 68, 2302–2312. [Google Scholar]
- Ratcliffe, R; Shachar-Hill, Y. Measuring multiple fluxes through plant metabolic networks. Plant J 2006, 45, 490–511. [Google Scholar]
- Rios-Estepa, R; Lange, BM. Experimental and mathematical approaches to modeling plant metabolic networks. Phytochemistry 2007, 68, 2351–2374. [Google Scholar]
- Kresnowati, MTAP; van Winden, WA; Almering, MJH; ten Pierick, A; Ras, C; Knijnenburg, TA; Daran-Lapujade, P; Pronk, JT; Heijnen, JJ; Daran, JM. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol 2006, 2(49). [Google Scholar]
- Wahl, S; Noh, K; Wiechert, W. 13C labeling experiments at metabolic nonstationary conditions: An exploratory study. BMC Bioinf 2008, 9. [Google Scholar]
- Baxter, C; Liu, J; Fernie, A; Sweetlove, L. Determination of metabolic fluxes in a non-steady-state system. Phytochemistry 2007, 68, 2313–2319. [Google Scholar]
- Tu, BP; McKnight, SL. Metabolic cycles as an underlying basis of biological oscillations. Nat. Rev. Mol. Cell Biol 2006, 7, 696–701. [Google Scholar]
- Murray, DB; Beckmann, M; Kitano, H. Regulation of yeast oscillatory dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 2241–2246. [Google Scholar]
- Tu, BP; Kudlicki, A; Rowicka, M; McKnight, SL. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science 2005, 310, 1152–1158. [Google Scholar]
- Visser, D; van Zuylen, GA; van Dam, JC; Oudshoorn, A; Eman, MR; Ras, C; van Gulik, WM; Frank, J; van Dedem, GWK; Heijnen, JJ. Rapid sampling for analysis of in vivo kinetics using the BioScope: a system for continuous-pulse experiments. Biotechnol. Bioeng 2002, 79, 674–681. [Google Scholar]
- Kholodenko, BN. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol 2006, 7, 165–176. [Google Scholar]
- Vendelin, M; Kongas, O; Saks, V. Regulation of mitochondrial respiration in heart cells analyzed by reaction-diffusion model of energy transfer. Am. J. Physiol. Cell. Physiol 2000, 278, C747–C764. [Google Scholar]
- Saks, V; Kongas, O; Vendelin, M; Kay, L. Role of the creatine/phosphocreatine system in the regulation of mitochondrial respiration. Acta Physiol. Scand 2000, 168, 635–641. [Google Scholar]
- Selivanov, V; Sukhomlin, T; Centelles, J; Lee, P; Cascante, M. Integration of enzyme kinetic models and isotopomer distribution analysis for studies of in situ cell operation. BMC Neurosci 2006, 7, S7. [Google Scholar]
- Liebermeister, W; Klipp, E. Bringing metabolic networks to life: integration of kinetic, metabolic, and proteomic data. Theor. Biol. Med. Model 2006, 3, 42. [Google Scholar]
- McConnell, HM. Reaction rates by nuclear magnetic resonance. J. Chem. Phys 1958, 28, 430–431. [Google Scholar]
- Forsen, S; Hoffman, RA. Study of moderately rapid chemical exchange reactions by means of nuclear magnetic double resonance. J. Chem. Phys 1963, 39, 2892–2901. [Google Scholar]
- Led, J; Gesmar, H. The applicability of the magnetization transfer NMR technique to determine chemical-exchange rates in extreme cases - the importance of complementary experiments. J. Magn. Reson 1982, 49, 444–463. [Google Scholar]
- Ugurbil, K. Magnetization-transfer measurements of individual rate constants in the presence of multiple reactions. J. Magn. Reson 1985, 64, 207–219. [Google Scholar]
- Brindle, KM. NMR methods for measuring enzyme kinetics in vivo. Prog. Nucl. Magn. Reson. Spectrosc 1988, 20, 257–293. [Google Scholar]
- Ugurbil, K; Petein, M; Maidan, R; Michurski, S; From, A. Measurement of an individual rateconstant in the presence of multiple exchanges - application to myocardial creatine-kinase reaction. Biochemistry 1986, 25, 100–107. [Google Scholar]
- Spencer, RG; Balschi, JA; Leigh, JS; Ingwall, JS. ATP synthesis and degradation rates in the perfused rat heart. 31P-nuclear magnetic resonance double saturation transfer measurements. Biophys. J 1988, 54, 921–929. [Google Scholar]
- Koretsky, A; Weiner, M. 31Phosphorus nuclear magnetic resonance magnetization transfer measurements of exchange reactions in vivo; Radiology Research and Education Foundation: San Francisco, CA, USA, 1984; pp. 209–230. [Google Scholar]
- Joubert, F; Hoerter, JA; Mazet, J. Discrimination of cardiac subcellular creatine kinase fluxes by NMR spectroscopy: A new method of analysis. Biophys. J 2001, 81, 2995–3004. [Google Scholar]
- Joubert, F; Mazet, J; Mateo, P; Hoerter, JA. 31P NMR detection of subcellular creatine kinase fluxes in the perfused rat heart. contractility modifies energy transfer pathways. J. Biol. Chem 2002, 277, 18469–18476. [Google Scholar]
- Ventura-Clapier, R; Garnier, A; Veksler, V. Energy metabolism in heart failure. J Physiol 2004, 555, 1–13. [Google Scholar]
- Dawis, S; Walseth, T; Deeg, M; Heyman, R; Graeff, R; Goldberg, N. Adenosine triphosphate utilization rates and metabolic pool sizes in intact cells measured by transfer of 18O from water. Biophys. J 1989, 55, 79–99. [Google Scholar]
- Zeleznikar, R; Heyman, R; Graeff, R; Walseth, T; Dawis, S; Butz, E; Goldberg, N. Evidence for compartmentalized adenylate kinase catalysis serving a high energy phosphoryl transfer function in rat skeletal muscle. J. Biol. Chem 1990, 265, 300–311. [Google Scholar]
- Zeleznikar, R; Goldberg, N. Kinetics and compartmentation of energy-metabolism in intact skeletalmuscle determined from 18O labeling of metabolite phosphoryls. J. Biol. Chem 1991, 266, 15110–15119. [Google Scholar]
- Zeleznikar, RJ; Goldberg, ND. Adenylate kinase-catalyzed phosphoryl transfer couples ATP utilization with its generation by glycolysis in intact muscle. J. Biol. Chem 1995, 270, 7311–7319. [Google Scholar]
- Dzeja, PP; Zeleznikar, RJ; Goldberg, ND. Suppression of creatine kinase-catalyzed phosphotransfer results in increased phosphoryl transfer by adenylate kinase in intact skeletal muscle. J. Biol. Chem 1996, 271, 12847–12851. [Google Scholar]
- Dzeja, PP; Zeleznikar, RJ; Goldberg, ND. Adenylate kinase: Kinetic behavior in intact cells indicates it is integral to multiple cellular processes. Mol. Cell. Biochem 1998, 184, 169–182. [Google Scholar]
- Pucar, D; Janssen, E; Dzeja, PP; Juranic, N; Macura, S; Wieringa, B; Terzic, A. Compromised energetics in the adenylate kinase AK1 gene knockout heart under metabolic stress. J. Biol. Chem 2000, 275, 41424–41429. [Google Scholar]
- Pucar, D; Dzeja, PP; Bast, P; Juranic, N; Macura, S; Terzic, A. Cellular energetics in the preconditioned state. protective role for phosphotransfer reactions captured by 18O-assisted 31P NMR. J. Biol. Chem 2001, 276, 44812–44819. [Google Scholar]
- Pucar, D; Bast, P; Gumina, RJ; Lim, L; Drahl, C; Juranic, N; Macura, S; Janssen, E; Wieringa, B; Terzic, A; Dzeja, PP. Adenylate kinase AK1 knockout heart: energetics and functional performance under ischemia-reperfusion. Am. J. Physiol. Heart. Circ. Physiol 2002, 283, H776–H782. [Google Scholar]
- Pucar, D; Dzeja, P; Bast, P; Gumina, R; Drahl, C; Lim, L; Juranic, N; Macura, S; Terzic, A. Mapping hypoxia-induced bioenergetic rearrangements and metabolic signaling by 18O-assisted 31P NMR and 1H NMR spectroscopy. Mol Cell Biochem 2004, 256–257. [Google Scholar]
- Abstracts from the workshop: Non invasive investigation of muscle function. Marseille France, October 4–6, 2001, MAGMA. 2002, May; 14, 59–212.
- Bollard, ME; Murray, AJ; Clarke, K; Nicholson, JK; Griffin, JL. A study of metabolic compartmentation in the rat heart and cardiac mitochondria using high-resolution magic angle spinning 1H NMR spectroscopy. FEBS Lett 2003, 553, 73–78. [Google Scholar]
- Thelwall, PE. Detection of 17O-tagged phosphate by 31P MRS: a method with potential for in vivo studies of phosphorus metabolism. Magn. Reson. Med 2007, 57, 1168–1172. [Google Scholar]
- Maaheimo, H; Fiaux, J; Cakar, Z; Bailey, J; Sauer, U; Szyperski, T. Central carbon metabolism of saccharomyces cerevisiae explored by biosynthetic fractional 13C labeling of common amino acids. Eur. J. Biochem 2001, 268, 2464–2479. [Google Scholar]
- Sriram, G; Fulton, DB; Shanks, JV. Flux quantification in central carbon metabolism of catharanthus roseus hairy roots by 13C labeling and comprehensive bondomer balancing. Phytochemistry 2007, 68, 2243–2257. [Google Scholar]
- Young, J; Walther, J; Antoniewicz, M; Yon, H; Stephanopoulos, G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng 2008, 99, 686–699. [Google Scholar]
- Wiechert, W; Wurzel, M. Metabolic isotopomer labeling systems - Part I: global dynamic behavior. Math. Biosci 2001, 169, 173–205. [Google Scholar]
- Joubert, F; Gillet, B; Mazet, J; Mateo, P; Beloeil, J; Hoerter, J. Evidence for myocardial ATP compartmentation from NMR inversion transfer analysis of creatine kinase fluxes. Biophys. J 2000, 79, 1–13. [Google Scholar]
- Sonnewald, U; Schousboe, A; Qu, H; Waagepetersen, HS. Intracellular metabolic compartmentation assessed by 13C magnetic resonance spectroscopy. Neurochem. Int 2004, 45, 305–310. [Google Scholar]
- Monge, C; Beraud, N; Kuznetsov, A; Rostovtseva, T; Sackett, D; Schlattner, U; Vendelin, M; Saks, V. Regulation of respiration in brain mitochondria and synaptosomes: restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol. Cell. Biochem 2008, 318, 147–165. [Google Scholar]
- Seppet, EK; Kaambre, T; Sikk, P; Tiivel, T; Vija, H; Tonkonogi, M; Sahlin, K; Kay, L; Appaix, F; Braun, U; Eimre, M; Saks, VA. Functional complexes of mitochondria with Ca,MgATPases of myofibrils and sarcoplasmic reticulum in muscle cells. Biochim. Biophys. Acta 2001, 1504, 379–395. [Google Scholar]
- Saks, VA; Kaambre, T; Sikk, P; Eimre, M; Orlova, E; Paju, K; Piirsoo, A; Appaix, F; Kay, L; Regitz-Zagrosek, V; Fleck, E; Seppet, E. Intracellular energetic units in red muscle cells. Biochem. J 2001, 356, 643657. [Google Scholar]
- Kaasik, A; Veksler, V; Boehm, E; Novotova, M; Minajeva, A; Ventura-Clapier, R. Energetic crosstalk between organelles: Architectural integration of energy production and utilization. Circ. Res 2001, 89, 153–159. [Google Scholar]
- Saks, V; Kuznetsov, A; Andrienko, T; Usson, Y; Appaix, F; Guerrero, K; Kaambre, T; Sikk, P; Lemba, M; Vendelin, M. Heterogeneity of ADP diffusion and regulation of respiration in cardiac cells. Biophys. J 2003, 84, 3436–3456. [Google Scholar]
- Vendelin, M; Eimre, M; Seppet, E; Peet, N; Andrienko, T; Lemba, M; Engelbrecht, J; Seppet, E; Saks, V. Intracellular diffusion of adenosine phosphates is locally restricted in cardiac muscle. Mol Cell Biochem 2004. [Google Scholar]
- Vendelin, M; Birkedal, R. Anisotropic diffusion of fluorescently labeled ATP in rat cardiomyocytes determined by raster image correlation spectroscopy. Am. J. Physiol. Cell. Physiol 2008, 295, C1302–C1315. [Google Scholar]
- Vendelin, M; Beraud, N; Guerrero, K; Andrienko, T; Kuznetsov, AV; Olivares, J; Kay, L; Saks, VA. Mitochondrial regular arrangement in muscle cells: a ”crystal-like” pattern. Am. J. Physiol. Cell. Physiol 2005, 288, C757–C767. [Google Scholar]
- Birkedal, R; Shiels, HA; Vendelin, M. Three-dimensional mitochondrial arrangement in ventricular myocytes: from chaos to order. Am. J. Physiol. Cell Physiol 2006, 291, C1148–C1158. [Google Scholar]
- Cruz, F; Villalba, M; Garcia-Espinosa, MA; Ballesteros, P; Bogonez, E; Satrustegui, J; Cerdin, S. Intracellular compartmentation of pyruvate in primary cultures of cortical neurons as detected by 13C NMR spectroscopy with multiple 13C labels. J. Neurosci. Res 2001, 66, 771–781. [Google Scholar]
- Wächter, A; Biegler, L. On the implementation of an interior-point filter line-search algorithm for large-scale nonlinear programming. Math. Program 2006, 106, 25–57. [Google Scholar]
- Paalme, T; Nisamedtinov, I; Abner, K; Laht, T; Drews, M; Pehk, T. Application of 13C -[2] - and 13C -[1,2] acetate in metabolic labelling studies of yeast and insect cells. Antonie van Leeuwenhoek 2006, 89, 443–457. [Google Scholar]
- Cline, GW; LePine, RL; Papas, KK; Kibbey, RG; Shulman, GI. 13C NMR isotopomer analysis of anaplerotic pathways in INS-1 cells. J. Biol. Chem 2004, 279, 44370–44375. [Google Scholar]
- Noh, K; Gronke, K; Luo, B; Takors, R; Oldiges, M; Wiechert, W. Metabolic flux analysis at ultra short time scale: Isotopically non-stationary 13C labeling experiments. J. Biotechnol 2007, 129, 249–267. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Abbreviation | |
|---|---|---|
| Cytosolic | Mitochondrial | |
| acetate | ACo | |
| acetyl-CoA | AcCoAo | AcCoAm |
| pyruvate | PYo | PYm |
| PY biomass precursor | PBm | |
| citrate/isocitrate | CIm | |
| oxaloacetate | OAo | OAm |
| succinate | SUm | |
| malate | MAm | |
| 2-oxoglutarate | OGm | |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schryer, D.W.; Peterson, P.; Paalme, T.; Vendelin, M. Bidirectionality and Compartmentation of Metabolic Fluxes Are Revealed in the Dynamics of Isotopomer Networks. Int. J. Mol. Sci. 2009, 10, 1697-1718. https://doi.org/10.3390/ijms10041697
Schryer DW, Peterson P, Paalme T, Vendelin M. Bidirectionality and Compartmentation of Metabolic Fluxes Are Revealed in the Dynamics of Isotopomer Networks. International Journal of Molecular Sciences. 2009; 10(4):1697-1718. https://doi.org/10.3390/ijms10041697
Chicago/Turabian StyleSchryer, David W., Pearu Peterson, Toomas Paalme, and Marko Vendelin. 2009. "Bidirectionality and Compartmentation of Metabolic Fluxes Are Revealed in the Dynamics of Isotopomer Networks" International Journal of Molecular Sciences 10, no. 4: 1697-1718. https://doi.org/10.3390/ijms10041697