Drosophila melanogaster as a Model Organism of Brain Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Drosophila as a brain disease model
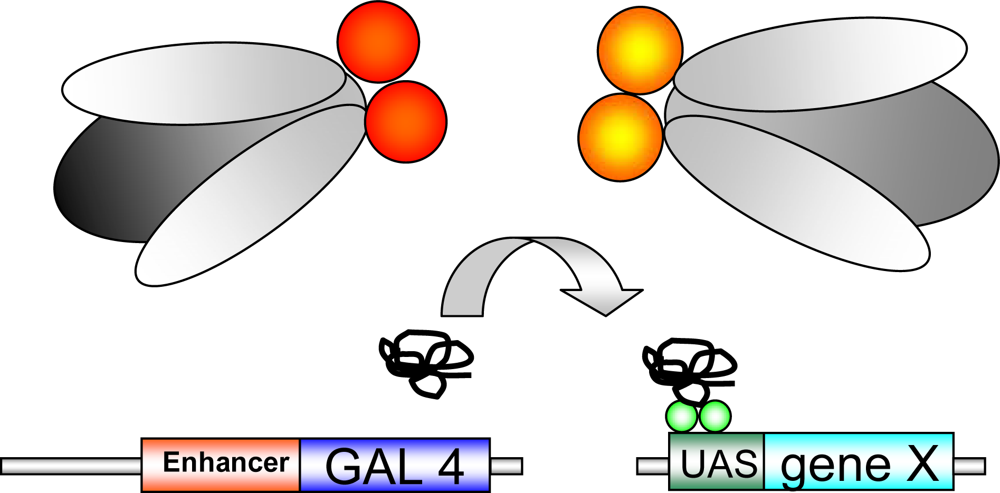
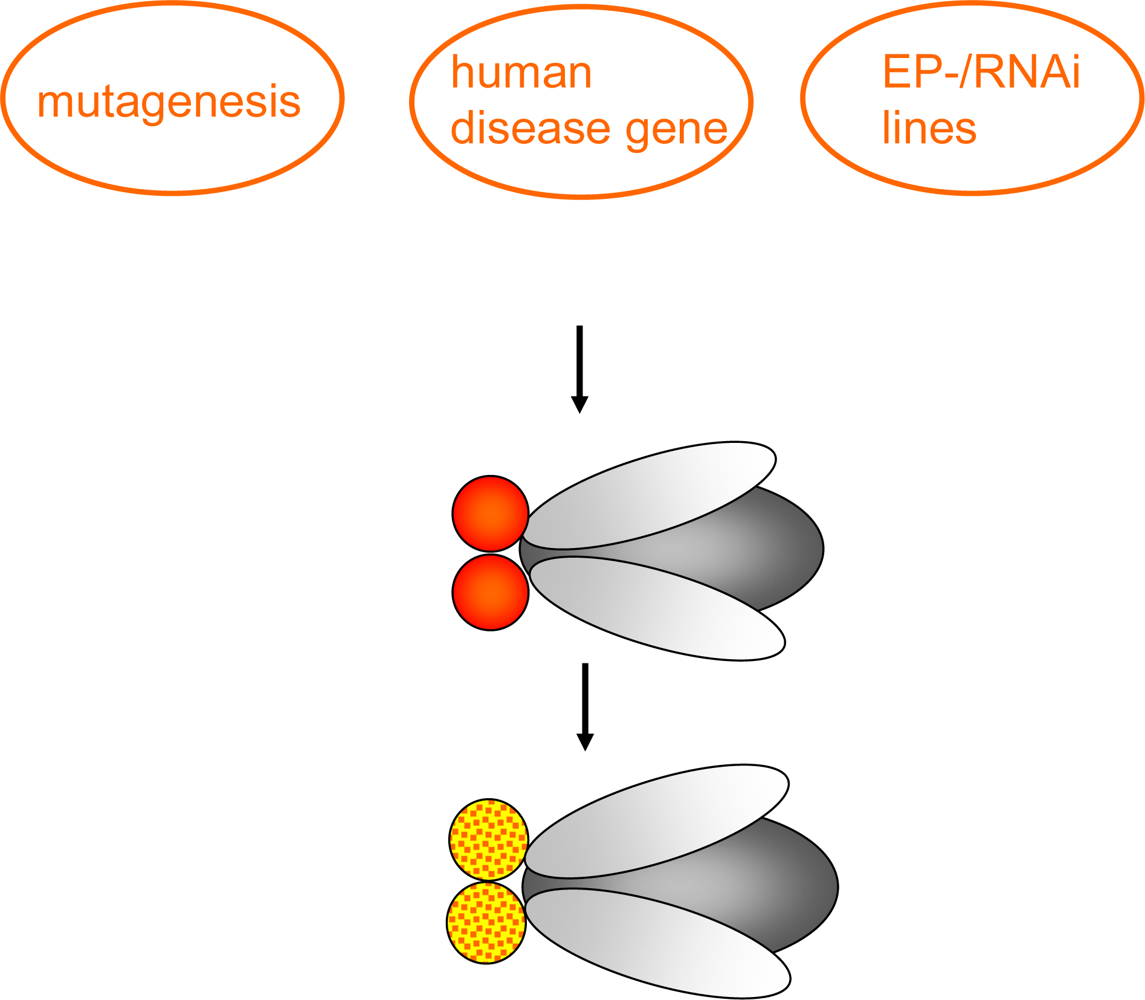
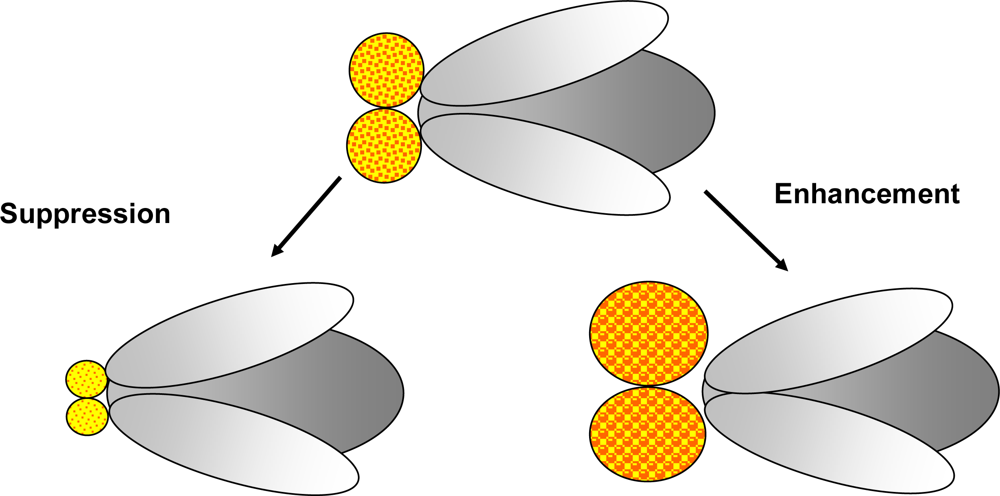
1.2. Genetic tools in Drosophila
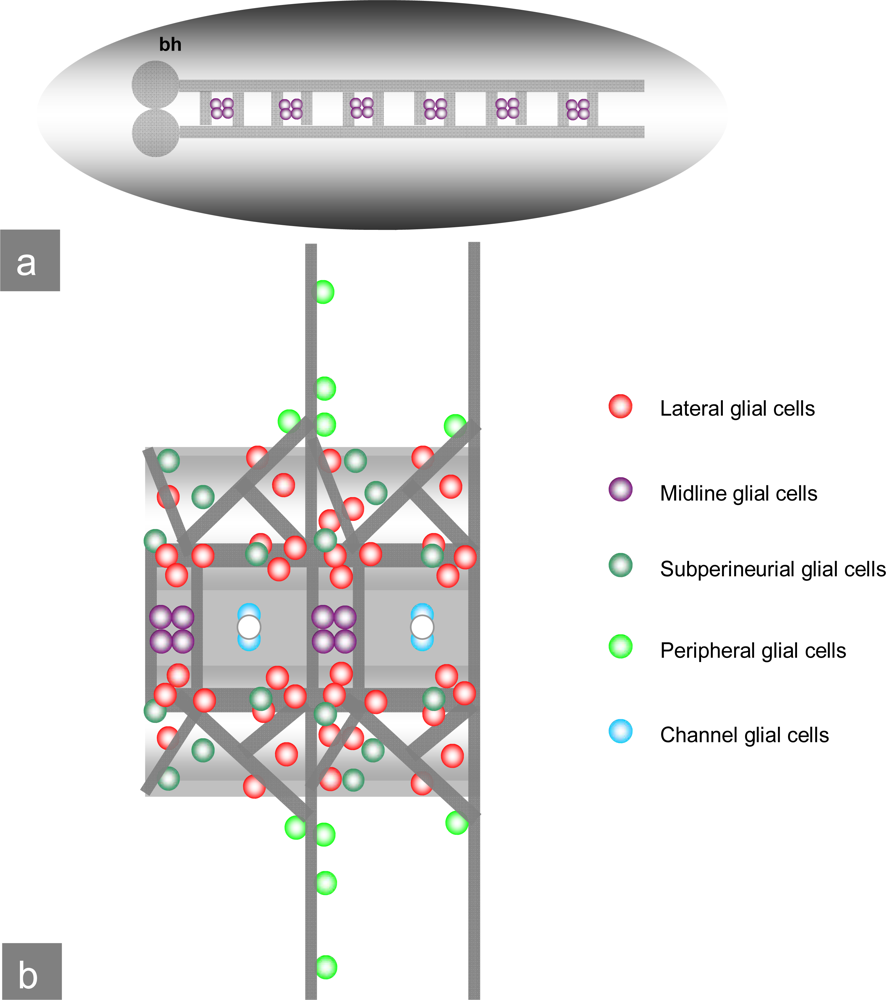
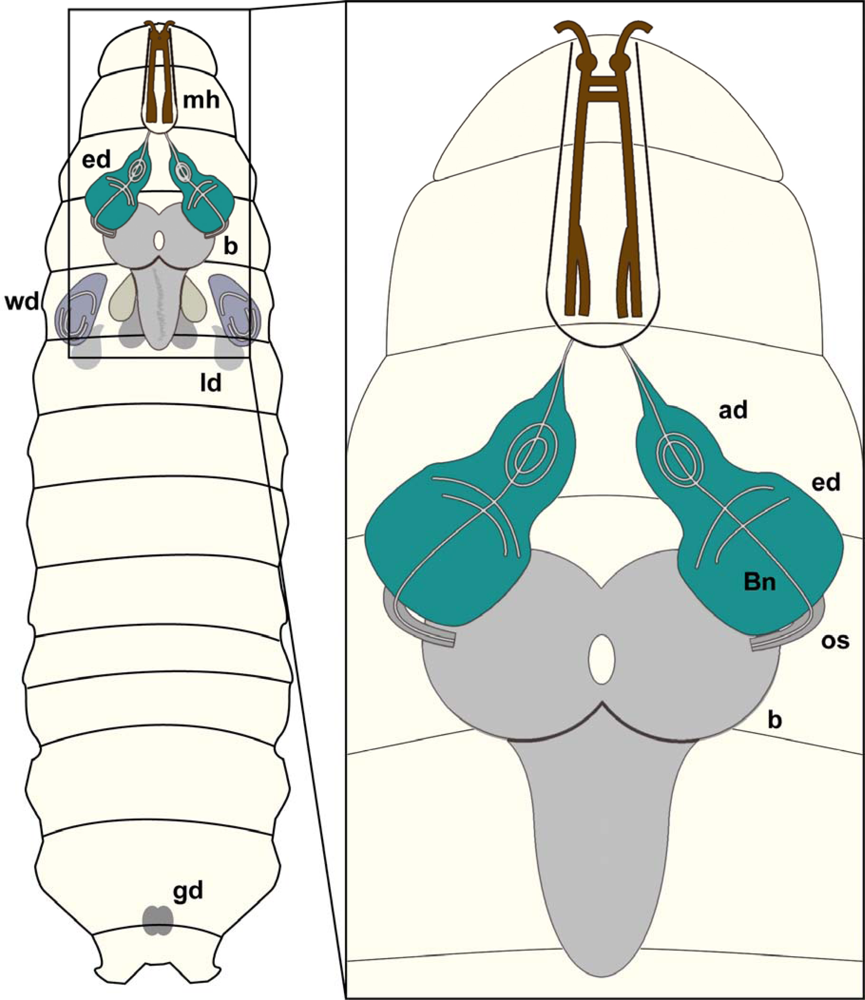
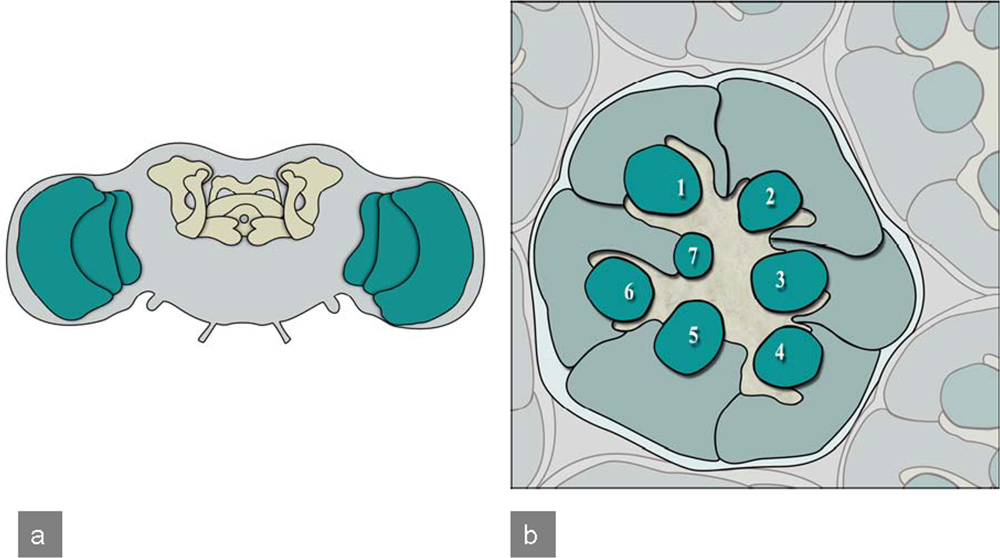
1.3. Drosophila CNS development
2. Neurodegeneration
2.1. Alzheimer’s Disease
2.2. Tauopathies
2.3. Parkinson’s Disease
2.4. Prion Diseases
2.5. Polyglutamine Disorders
2.5.1. Huntington’s disease
2.5.2. Spinocerebellar ataxia type 3 (Machado-Joseph disease; SCA3)
2.5.3. X-linked spinobulbar muscular atrophy (SBMA; Kennedy Disease)
2.5.4. Non coding trinucleotide repeat diseases
2.5.5. Spinocerebellar ataxia type 8 (SCA8)
2.5.6. Spinal muscular atrophy (SMA)
2.6. Amyotrophic lateral sclerosis
3. Metabolic disorders
3.1. Leigh Disease
3.2. Nieman-Pick-Disease
3.3. Ceroid lipofuscinoses
4. Tumors
4.1. Neurofibromatosis 1
4.2. Neurofibromatosis 2
4.3. Tuberous sclerosis
4.4. Neuronal/neuroblastic tumors
5. Epilepsy
6. Trauma
7. Conclusions
References
- Matthews, KA; Kaufman, TC; Gelbart, WM. Research resources for Drosophila: The expanding universe. Nat Rev Genet 2005, 6(3), 179–193. [Google Scholar]
- Venken, KJ; Bellen, HJ. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat Rev Genet 2005, 6(3), 167–178. [Google Scholar]
- Dietzl, G; Chen, D; Schnorrer, F; Su, KC; Barinova, Y; Fellner, M; Gasser, B; Kinsey, K; Oppel, S; Scheiblauer, S; Couto, A; Marra, V; Keleman, K; Dickson, BJ. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448(7150), 151–156. [Google Scholar]
- Rubin, GM; Lewis, EB. A brief history of Drosophila’s contributions to genome research. Science 2000, 287(5461), 2216–2218. [Google Scholar]
- Adams, MD; Celniker, SE; Holt, RA; Evans, CA; Gocayne, JD; Amanatides, PG; Scherer, SE; Li, PW; Hoskins, RA; Galle, RF; George, RA; Lewis, SE; Richards, S; Ashburner, M; Henderson, SN; Sutton, GG; Wortman, JR; Yandell, MD; Zhang, Q; Chen, LX; Brandon, RC; Rogers, YH; Blazej, RG; Champe, M; Pfeiffer, BD; Wan, KH; Doyle, C; Baxter, EG; Helt, G; Nelson, CR; Gabor, GL; Abril, JF; Agbayani, A; An, HJ; Andrews-Pfannkoch, C; Baldwin, D; Ballew, RM; Basu, A; Baxendale, J; Bayraktaroglu, L; Beasley, EM; Beeson, KY; Benos, PV; Berman, BP; Bhandari, D; Bolshakov, S; Borkova, D; Botchan, MR; Bouck, J; Brokstein, P; Brottier, P; Burtis, KC; Busam, DA; Butler, H; Cadieu, E; Center, A; Chandra, I; Cherry, JM; Cawley, S; Dahlke, C; Davenport, LB; Davies, P; de Pablos, B; Delcher, A; Deng, Z; Mays, AD; Dew, I; Dietz, SM; Dodson, K; Doup, LE; Downes, M; Dugan-Rocha, S; Dunkov, BC; Dunn, P; Durbin, KJ; Evangelista, CC; Ferraz, C; Ferriera, S; Fleischmann, W; Fosler, C; Gabrielian, AE; Garg, NS; Gelbart, WM; Glasser, K; Glodek, A; Gong, F; Gorrell, JH; Gu, Z; Guan, P; Harris, M; Harris, NL; Harvey, D; Heiman, TJ; Hernandez, JR; Houck, J; Hostin, D; Houston, KA; Howland, TJ; Wei, MH; Ibegwam, C; Jalali, M; Kalush, F; Karpen, GH; Ke, Z; Kennison, JA; Ketchum, KA; Kimmel, BE; Kodira, CD; Kraft, C; Kravitz, S; Kulp, D; Lai, Z; Lasko, P; Lei, Y; Levitsky, AA; Li, J; Li, Z; Liang, Y; Lin, X; Liu, X; Mattei, B; McIntosh, TC; McLeod, MP; McPherson, D; Merkulov, G; Milshina, NV; Mobarry, C; Morris, J; Moshrefi, A; Mount, SM; Moy, M; Murphy, B; Murphy, L; Muzny, DM; Nelson, DL; Nelson, DR; Nelson, KA; Nixon, K; Nusskern, DR; Pacleb, JM; Palazzolo, M; Pittman, GS; Pan, S; Pollard, J; Puri, V; Reese, MG; Reinert, K; Remington, K; Saunders, RD; Scheeler, F; Shen, H; Shue, BC; Siden-Kiamos, I; Simpson, M; Skupski, MP; Smith, T; Spier, E; Spradling, AC; Stapleton, M; Strong, R; Sun, E; Svirskas, R; Tector, C; Turner, R; Venter, E; Wang, AH; Wang, X; Wang, ZY; Wassarman, DA; Weinstock, GM; Weissenbach, J; Williams, SM; Woodage, T; Worley, KC; Wu, D; Yang, S; Yao, QA; Ye, J; Yeh, RF; Zaveri, JS; Zhan, M; Zhang, G; Zhao, Q; Zheng, L; Zheng, XH; Zhong, FN; Zhong, W; Zhou, X; Zhu, S; Zhu, X; Smith, HO; Gibbs, RA; Myers, EW; Rubin, GM; Venter, JC. The genome sequence of Drosophila melanogaster. Science 2000, 287(5461), 2185–2195. [Google Scholar]
- Lander, ES; Linton, LM; Birren, B; Nusbaum, C; Zody, MC; Baldwin, J; Devon, K; Dewar, K; Doyle, M; FitzHugh, W; Funke, R; Gage, D; Harris, K; Heaford, A; Howland, J; Kann, L; Lehoczky, J; LeVine, R; McEwan, P; McKernan, K; Meldrim, J; Mesirov, JP; Miranda, C; Morris, W; Naylor, J; Raymond, C; Rosetti, M; Santos, R; Sheridan, A; Sougnez, C; Stange-Thomann, N; Stojanovic, N; Subramanian, A; Wyman, D; Rogers, J; Sulston, J; Ainscough, R; Beck, S; Bentley, D; Burton, J; Clee, C; Carter, N; Coulson, A; Deadman, R; Deloukas, P; Dunham, A; Dunham, I; Durbin, R; French, L; Grafham, D; Gregory, S; Hubbard, T; Humphray, S; Hunt, A; Jones, M; Lloyd, C; McMurray, A; Matthews, L; Mercer, S; Milne, S; Mullikin, JC; Mungall, A; Plumb, R; Ross, M; Shownkeen, R; Sims, S; Waterston, RH; Wilson, RK; Hillier, LW; McPherson, JD; Marra, MA; Mardis, ER; Fulton, LA; Chinwalla, AT; Pepin, KH; Gish, WR; Chissoe, SL; Wendl, MC; Delehaunty, KD; Miner, TL; Delehaunty, A; Kramer, JB; Cook, LL; Fulton, RS; Johnson, DL; Minx, PJ; Clifton, SW; Hawkins, T; Branscomb, E; Predki, P; Richardson, P; Wenning, S; Slezak, T; Doggett, N; Cheng, JF; Olsen, A; Lucas, S; Elkin, C; Uberbacher, E; Frazier, M; Gibbs, RA; Muzny, DM; Scherer, SE; Bouck, JB; Sodergren, EJ; Worley, KC; Rives, CM; Gorrell, JH; Metzker, ML; Naylor, SL; Kucherlapati, RS; Nelson, DL; Weinstock, GM; Sakaki, Y; Fujiyama, A; Hattori, M; Yada, T; Toyoda, A; Itoh, T; Kawagoe, C; Watanabe, H; Totoki, Y; Taylor, T; Weissenbach, J; Heilig, R; Saurin, W; Artiguenave, F; Brottier, P; Bruls, T; Pelletier, E; Robert, C; Wincker, P; Smith, DR; Doucette-Stamm, L; Rubenfield, M; Weinstock, K; Lee, HM; Dubois, J; Rosenthal, A; Platzer, M; Nyakatura, G; Taudien, S; Rump, A; Yang, H; Yu, J; Wang, J; Huang, G; Gu, J; Hood, L; Rowen, L; Madan, A; Qin, S; Davis, RW; Federspiel, NA; Abola, AP; Proctor, MJ; Myers, RM; Schmutz, J; Dickson, M; Grimwood, J; Cox, DR; Olson, MV; Kaul, R; Shimizu, N; Kawasaki, K; Minoshima, S; Evans, GA; Athanasiou, M; Schultz, R; Roe, BA; Chen, F; Pan, H; Ramser, J; Lehrach, H; Reinhardt, R; McCombie, WR; de la Bastide, M; Dedhia, N; Blocker, H; Hornischer, K; Nordsiek, G; Agarwala, R; Aravind, L; Bailey, JA; Bateman, A; Batzoglou, S; Birney, E; Bork, P; Brown, DG; Burge, CB; Cerutti, L; Chen, HC; Church, D; Clamp, M; Copley, RR; Doerks, T; Eddy, SR; Eichler, EE; Furey, TS; Galagan, J; Gilbert, JG; Harmon, C; Hayashizaki, Y; Haussler, D; Hermjakob, H; Hokamp, K; Jang, W; Johnson, LS; Jones, TA; Kasif, S; Kaspryzk, A; Kennedy, S; Kent, WJ; Kitts, P; Koonin, EV; Korf, I; Kulp, D; Lancet, D; Lowe, TM; McLysaght, A; Mikkelsen, T; Moran, JV; Mulder, N; Pollara, VJ; Ponting, CP; Schuler, G; Schultz, J; Slater, G; Smit, AF; Stupka, E; Szustakowski, J; Thierry-Mieg, D; Thierry-Mieg, J; Wagner, L; Wallis, J; Wheeler, R; Williams, A; Wolf, YI; Wolfe, KH; Yang, SP; Yeh, RF; Collins, F; Guyer, MS; Peterson, J; Felsenfeld, A; Wetterstrand, KA; Patrinos, A; Morgan, MJ; de Jong, P; Catanese, JJ; Osoegawa, K; Shizuya, H; Choi, S; Chen, YJ. Initial sequencing and analysis of the human genome. Nature 2001, 409(6822), 860–921. [Google Scholar]
- Venter, JC; Adams, MD; Myers, EW; Li, PW; Mural, RJ; Sutton, GG; Smith, HO; Yandell, M; Evans, CA; Holt, RA; Gocayne, JD; Amanatides, P; Ballew, RM; Huson, DH; Wortman, JR; Zhang, Q; Kodira, CD; Zheng, XH; Chen, L; Skupski, M; Subramanian, G; Thomas, PD; Zhang, J; Gabor Miklos, GL; Nelson, C; Broder, S; Clark, AG; Nadeau, J; McKusick, VA; Zinder, N; Levine, AJ; Roberts, RJ; Simon, M; Slayman, C; Hunkapiller, M; Bolanos, R; Delcher, A; Dew, I; Fasulo, D; Flanigan, M; Florea, L; Halpern, A; Hannenhalli, S; Kravitz, S; Levy, S; Mobarry, C; Reinert, K; Remington, K; Abu-Threideh, J; Beasley, E; Biddick, K; Bonazzi, V; Brandon, R; Cargill, M; Chandramouliswaran, I; Charlab, R; Chaturvedi, K; Deng, Z; Di Francesco, V; Dunn, P; Eilbeck, K; Evangelista, C; Gabrielian, AE; Gan, W; Ge, W; Gong, F; Gu, Z; Guan, P; Heiman, TJ; Higgins, ME; Ji, RR; Ke, Z; Ketchum, KA; Lai, Z; Lei, Y; Li, Z; Li, J; Liang, Y; Lin, X; Lu, F; Merkulov, GV; Milshina, N; Moore, HM; Naik, AK; Narayan, VA; Neelam, B; Nusskern, D; Rusch, DB; Salzberg, S; Shao, W; Shue, B; Sun, J; Wang, Z; Wang, A; Wang, X; Wang, J; Wei, M; Wides, R; Xiao, C; Yan, C; Yao, A; Ye, J; Zhan, M; Zhang, W; Zhang, H; Zhao, Q; Zheng, L; Zhong, F; Zhong, W; Zhu, S; Zhao, S; Gilbert, D; Baumhueter, S; Spier, G; Carter, C; Cravchik, A; Woodage, T; Ali, F; An, H; Awe, A; Baldwin, D; Baden, H; Barnstead, M; Barrow, I; Beeson, K; Busam, D; Carver, A; Center, A; Cheng, ML; Curry, L; Danaher, S; Davenport, L; Desilets, R; Dietz, S; Dodson, K; Doup, L; Ferriera, S; Garg, N; Gluecksmann, A; Hart, B; Haynes, J; Haynes, C; Heiner, C; Hladun, S; Hostin, D; Houck, J; Howland, T; Ibegwam, C; Johnson, J; Kalush, F; Kline, L; Koduru, S; Love, A; Mann, F; May, D; McCawley, S; McIntosh, T; McMullen, I; Moy, M; Moy, L; Murphy, B; Nelson, K; Pfannkoch, C; Pratts, E; Puri, V; Qureshi, H; Reardon, M; Rodriguez, R; Rogers, YH; Romblad, D; Ruhfel, B; Scott, R; Sitter, C; Smallwood, M; Stewart, E; Strong, R; Suh, E; Thomas, R; Tint, NN; Tse, S; Vech, C; Wang, G; Wetter, J; Williams, S; Williams, M; Windsor, S; Winn-Deen, E; Wolfe, K; Zaveri, J; Zaveri, K; Abril, JF; Guigo, R; Campbell, MJ; Sjolander, KV; Karlak, B; Kejariwal, A; Mi, H; Lazareva, B; Hatton, T; Narechania, A; Diemer, K; Muruganujan, A; Guo, N; Sato, S; Bafna, V; Istrail, S; Lippert, R; Schwartz, R; Walenz, B; Yooseph, S; Allen, D; Basu, A; Baxendale, J; Blick, L; Caminha, M; Carnes-Stine, J; Caulk, P; Chiang, YH; Coyne, M; Dahlke, C; Mays, A; Dombroski, M; Donnelly, M; Ely, D; Esparham, S; Fosler, C; Gire, H; Glanowski, S; Glasser, K; Glodek, A; Gorokhov, M; Graham, K; Gropman, B; Harris, M; Heil, J; Henderson, S; Hoover, J; Jennings, D; Jordan, C; Jordan, J; Kasha, J; Kagan, L; Kraft, C; Levitsky, A; Lewis, M; Liu, X; Lopez, J; Ma, D; Majoros, W; McDaniel, J; Murphy, S; Newman, M; Nguyen, T; Nguyen, N; Nodell, M; Pan, S; Peck, J; Peterson, M; Rowe, W; Sanders, R; Scott, J; Simpson, M; Smith, T; Sprague, A; Stockwell, T; Turner, R; Venter, E; Wang, M; Wen, M; Wu, D; Wu, M; Xia, A; Zandieh, A; Zhu, X. The sequence of the human genome. Science 2001, 291(5507), 1304–1351. [Google Scholar]
- Aquadro, CF; Bauer DuMont, V; Reed, FA. Genome-wide variation in the human and fruitfly: A comparison. Curr Opin Genet Dev 2001, 11(6), 627–634. [Google Scholar]
- Brand, AH; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118(2), 401–415. [Google Scholar]
- Min, KT; Benzer, S. Spongecake and eggroll: Two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr Biol 1997, 7(11), 885–888. [Google Scholar]
- Kretzschmar, D; Hasan, G; Sharma, S; Heisenberg, M; Benzer, S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci 1997, 17(19), 7425–7432. [Google Scholar]
- Min, KT; Benzer, S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science 1999, 284(5422), 1985–1988. [Google Scholar]
- Cauchi, RJ; van den Heuvel, M. The fly as a model for neurodegenerative diseases: Is it worth the jump. Neurodegener Dis 2006, 3(6), 338–356. [Google Scholar]
- Mitchell, KJ; Doyle, JL; Serafini, T; Kennedy, TE; Tessier-Lavigne, M; Goodman, CS; Dickson, BJ. Genetic analysis of Netrin genes in Drosophila: Netrins guide CNS commissural axons and peripheral motor axons. Neuron 1996, 17(2), 203–215. [Google Scholar]
- Hummel, T; Schimmelpfeng, K; Klambt, C. Commissure formation in the embryonic CNS of Drosophila. Development 1999, 126(4), 771–779. [Google Scholar]
- Klambt, C; Hummel, T; Granderath, S; Schimmelpfeng, K. Glial cell development in Drosophila. Int J Dev Neurosci 2001, 19(4), 373–378. [Google Scholar]
- Hartenstein, V; Nassif, C; Lekven, A. Embryonic development of the Drosophila brain. II. Pattern of glial cells. J Comp Neurol 1998, 402(1), 32–47. [Google Scholar]
- Heisenberg, M. Mushroom body memoir: from maps to models. Nat Rev Neurosci 2003, 4(4), 266–275. [Google Scholar]
- Gu, H; O’Dowd, DK. Cholinergic synaptic transmission in adult Drosophila Kenyon cells in situ. J. Neurosci 2006, 26(1), 265–272. [Google Scholar]
- Marsh, JL; Thompson, LM. Drosophila in the study of neurodegenerative disease. Neuron 2006, 52(1), 169–178. [Google Scholar]
- Clandinin, TR; Lee, CH; Herman, T; Lee, RC; Yang, AY; Ovasapyan, S; Zipursky, SL. Drosophila LAR regulates R1–R6 and R7 target specificity in the visual system. Neuron 2001, 32(2), 237–248. [Google Scholar]
- Tomlinson, A. Cellular interactions in the developing Drosophila eye. Development 1988, 104(2), 183–193. [Google Scholar]
- Morante, J; Desplan, C; Celik, A. Generating patterned arrays of photoreceptors. Curr Opin Genet Dev 2007, 17(4), 314–319. [Google Scholar]
- Ball, MJ; Murdoch, GH. Neuropathological criteria for the diagnosis of Alzheimer’s disease: Are we really ready yet? Neurobiol Aging 1997, 18(4 Suppl), S3–12. [Google Scholar]
- Braak, H; Alafuzoff, I; Arzberger, T; Kretzschmar, H; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006, 112(4), 389–404. [Google Scholar]
- van der Zee, J; Sleegers, K; Van Broeckhoven, C. Invited article: The Alzheimer disease-frontotemporal lobar degeneration spectrum. Neurology 2008, 71(15), 1191–1197. [Google Scholar]
- Martin-Morris, LE; White, K. The Drosophila transcript encoded by the beta-amyloid protein precursor-like gene is restricted to the nervous system. Development 1990, 110(1), 185–195. [Google Scholar]
- Rosen, DR; Martin-Morris, L; Luo, LQ; White, K. A Drosophila gene encoding a protein resembling the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 1989, 86(7), 2478–2482. [Google Scholar]
- Fossgreen, A; Bruckner, B; Czech, C; Masters, CL; Beyreuther, K; Paro, R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci USA 1998, 95(23), 13703–13708. [Google Scholar]
- Bonini, NM; Fortini, ME. Human neurodegenerative disease modeling using Drosophila. Annu. Rev. Neurosci 2003, 26, 627–656. [Google Scholar]
- Driscoll, M; Gerstbrein, B. Dying for a cause: Invertebrate genetics takes on human neurodegeneration. Nat Rev Genet 2003, 4(3), 181–194. [Google Scholar]
- Kopan, R; Goate, A. Aph-2/Nicastrin: An essential component of gamma-secretase and regulator of Notch signaling and Presenilin localization. Neuron 2002, 33(3), 321–324. [Google Scholar]
- Iijima, K; Liu, HP; Chiang, AS; Hearn, SA; Konsolaki, M; Zhong, Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci USA 2004, 101(17), 6623–6628. [Google Scholar]
- Iijima, K; Iijima-Ando, K. Drosophila Models of Alzheimer’s Amyloidosis: The Challenge of Dissecting the Complex Mechanisms of Toxicity of Amyloid-beta 42. J Alzheimers Dis 2008, 15(4), 523–540. [Google Scholar]
- Finelli, A; Kelkar, A; Song, HJ; Yang, H; Konsolaki, M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci 2004, 26(3), 365–375. [Google Scholar]
- Greeve, I; Kretzschmar, D; Tschape, JA; Beyn, A; Brellinger, C; Schweizer, M; Nitsch, RM; Reifegerste, R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci 2004, 24(16), 3899–3906. [Google Scholar]
- Yamaguchi, H; Yamazaki, T; Ishiguro, K; Shoji, M; Nakazato, Y; Hirai, S. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol 1992, 85(1), 15–22. [Google Scholar]
- Gunawardena, S; Goldstein, LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 2001, 32(3), 389–401. [Google Scholar]
- Fiala, JC. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol 2007, 114(6), 551–571. [Google Scholar]
- Duyckaerts, C; Potier, MC; Delatour, B. Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathol 2008, 115(1), 5–38. [Google Scholar]
- Cairns, NJ; Bigio, EH; Mackenzie, IR; Neumann, M; Lee, VM; Hatanpaa, KJ; White, CL, 3rd; Schneider, JA; Grinberg, LT; Halliday, G; Duyckaerts, C; Lowe, JS; Holm, IE; Tolnay, M; Okamoto, K; Yokoo, H; Murayama, S; Woulfe, J; Munoz, DG; Dickson, DW; Ince, PG; Trojanowski, JQ; Mann, DM. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007, 114(1), 5–22. [Google Scholar]
- Frank, S; Clavaguera, F; Tolnay, M. Tauopathy models and human neuropathology: similarities and differences. Acta Neuropathol 2008, 115(1), 39–53. [Google Scholar]
- Heidary, G; Fortini, ME. Identification and characterization of the Drosophila tau homolog. Mech Dev 2001, 108. [Google Scholar]
- Wittmann, CW; Wszolek, MF; Shulman, JM; Salvaterra, PM; Lewis, J; Hutton, M; Feany, MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 2001, 293(5530), 711–714. [Google Scholar]
- Jackson, GR; Wiedau-Pazos, M; Sang, TK; Wagle, N; Brown, CA; Massachi, S; Geschwind, DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34(4), 509–519. [Google Scholar]
- Augustinack, JC; Schneider, A; Mandelkow, EM; Hyman, BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 2002, 103(1), 26–35. [Google Scholar]
- Iwatsubo, T; Hasegawa, M; Ihara, Y. Neuronal and glial tau-positive inclusions in diverse neurologic diseases share common phosphorylation characteristics. Acta Neuropathol 1994, 88(2), 129–136. [Google Scholar]
- Chen, X; Li, Y; Huang, J; Cao, D; Yang, G; Liu, W; Lu, H; Guo, A. Study of tauopathies by comparing Drosophila and human tau in Drosophila. Cell Tissue Res 2007, 329(1), 169–178. [Google Scholar]
- Mudher, A; Shepherd, D; Newman, TA; Mildren, P; Jukes, JP; Squire, A; Mears, A; Drummond, JA; Berg, S; MacKay, D; Asuni, AA; Bhat, R; Lovestone, S. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry 2004, 9(5), 522–530. [Google Scholar]
- Shulman, JM; Shulman, LM; Weiner, WJ; Feany, MB. From fruit fly to bedside: Translating lessons from Drosophila models of neurodegenerative disease. Curr Opin Neurol 2003, 16(4), 443–449. [Google Scholar]
- Nishimura, I; Yang, Y; Lu, B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 2004, 116(5), 671–682. [Google Scholar]
- Meredith, GE; Sonsalla, PK; Chesselet, MF. Animal models of Parkinson’s disease progression. Acta Neuropathol 2008, 115(4), 385–398. [Google Scholar]
- Kuzuhara, S; Mori, H; Izumiyama, N; Yoshimura, M; Ihara, Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol 1988, 75(4), 345–353. [Google Scholar]
- Feany, MB; Bender, WW. A Drosophila model of Parkinson’s disease. Nature 2000, 404(6776), 394–8. [Google Scholar]
- Pendleton, RG; Parvez, F; Sayed, M; Hillman, R. Effects of pharmacological agents upon a transgenic model of Parkinson’s disease in Drosophila melanogaster. J Pharmacol Exp Ther 2002, 300(1), 91–96. [Google Scholar]
- Greene, JC; Whitworth, AJ; Kuo, I; Andrews, LA; Feany, MB; Pallanck, LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 2003, 100(7), 4078–4083. [Google Scholar]
- Pesah, Y; Pham, T; Burgess, H; Middlebrooks, B; Verstreken, P; Zhou, Y; Harding, M; Bellen, H; Mardon, G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 2004, 131(9), 2183–2194. [Google Scholar]
- Yang, Y; Nishimura, I; Imai, Y; Takahashi, R; Lu, B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 2003, 37(6), 911–924. [Google Scholar]
- Sang, TK; Chang, HY; Lawless, GM; Ratnaparkhi, A; Mee, L; Ackerson, LC; Maidment, NT; Krantz, DE; Jackson, GR. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci 2007, 27(5), 981–992. [Google Scholar]
- Park, J; Lee, SB; Lee, S; Kim, Y; Song, S; Kim, S; Bae, E; Kim, J; Shong, M; Kim, JM; Chung, J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441(7097), 1157–1161. [Google Scholar]
- Meulener, M; Whitworth, AJ; Armstrong-Gold, CE; Rizzu, P; Heutink, P; Wes, PD; Pallanck, LJ; Bonini, NM. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol 2005, 15(17), 1572–1577. [Google Scholar]
- Park, J; Kim, SY; Cha, GH; Lee, SB; Kim, S; Chung, J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar]
- Zimprich, A; Biskup, S; Leitner, P; Lichtner, P; Farrer, M; Lincoln, S; Kachergus, J; Hulihan, M; Uitti, RJ; Calne, DB; Stoessl, AJ; Pfeiffer, RF; Patenge, N; Carbajal, IC; Vieregge, P; Asmus, F; Muller-Myhsok, B; Dickson, DW; Meitinger, T; Strom, TM; Wszolek, ZK; Gasser, T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44(4), 601–607. [Google Scholar]
- Lee, SB; Kim, W; Lee, S; Chung, J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun 2007, 358(2), 534–539. [Google Scholar]
- Nichols, WC; Pankratz, N; Hernandez, D; Paisan-Ruiz, C; Jain, S; Halter, CA; Michaels, VE; Reed, T; Rudolph, A; Shults, CW; Singleton, A; Foroud, T. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005, 365(9457), 410–412. [Google Scholar]
- Gilks, WP; Abou-Sleiman, PM; Gandhi, S; Jain, S; Singleton, A; Lees, AJ; Shaw, K; Bhatia, KP; Bonifati, V; Quinn, NP; Lynch, J; Healy, DG; Holton, JL; Revesz, T; Wood, NW. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365(9457), 415–416. [Google Scholar]
- Liu, Z; Wang, X; Yu, Y; Li, X; Wang, T; Jiang, H; Ren, Q; Jiao, Y; Sawa, A; Moran, T; Ross, CA; Montell, C; Smith, WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci USA 2008, 105(7), 2693–2698. [Google Scholar]
- Coulom, H; Birman, S. Chronic exposure to rotenone models sporadic Parkinson’s disease in Drosophila melanogaster. J Neurosci 2004, 24(48), 10993–10998. [Google Scholar]
- Jimenez-Del-Rio, M; Daza-Restrepo, A; Velez-Pardo, C. The cannabinoid CP55,940 prolongs survival and improves locomotor activity in Drosophila melanogaster against paraquat: Implications in Parkinson’s disease. Neurosci Res 2008, 61(4), 404–411. [Google Scholar]
- Unterberger, U; Voigtlander, T; Budka, H. Pathogenesis of prion diseases. Acta Neuropathol 2005, 109(1), 32–48. [Google Scholar]
- Prusiner, SB. Prions. Proc Natl Acad Sci USA 1998, 95(23), 13363–13383. [Google Scholar]
- Ghetti, B; Tagliavini, F; Takao, M; Bugiani, O; Piccardo, P. Hereditary prion protein amyloidoses. Clin Lab Med 2003, 23(1). [Google Scholar]
- Collins, S; McLean, CA; Masters, CL. Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, and kuru: A review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci 2001, 8(5), 387–397. [Google Scholar]
- Raeber, AJ; Muramoto, T; Kornberg, TB; Prusiner, SB. Expression and targeting of Syrian hamster prion protein induced by heat shock in transgenic Drosophila melanogaster. Mech Dev 1995, 51. [Google Scholar]
- Gavin, BA; Dolph, MJ; Deleault, NR; Geoghegan, JC; Khurana, V; Feany, MB; Dolph, PJ; Supattapone, S. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann-Straussler-Scheinker syndrome. J Neurosci 2006, 26(48), 12408–12414. [Google Scholar]
- Vonsattel, JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 2008, 115(1), 55–69. [Google Scholar]
- Jackson, GR; Salecker, I; Dong, X; Yao, X; Arnheim, N; Faber, PW; MacDonald, ME; Zipursky, SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998, 21(3), 633–642. [Google Scholar]
- Gunawardena, S; Her, LS; Brusch, RG; Laymon, RA; Niesman, IR; Gordesky-Gold, B; Sintasath, L; Bonini, NM; Goldstein, LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40(1), 25–40. [Google Scholar]
- Jung, J; Bonini, N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science 2007, 315(5820), 1857–1859. [Google Scholar]
- Branco, J; Al-Ramahi, I; Ukani, L; Perez, AM; Fernandez-Funez, P; Rincon-Limas, D; Botas, J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet 2008, 17(3), 376–390. [Google Scholar]
- Lim, J; Crespo-Barreto, J; Jafar-Nejad, P; Bowman, AB; Richman, R; Hill, DE; Orr, HT; Zoghbi, HY. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008, 452(7188), 713–718. [Google Scholar]
- Yamada, M; Sato, T; Tsuji, S; Takahashi, H. CAG repeat disorder models and human neuropathology: Similarities and differences. Acta Neuropathol 2008, 115(1), 71–86. [Google Scholar]
- Warrick, JM; Paulson, HL; Gray-Board, GL; Bui, QT; Fischbeck, KH; Pittman, RN; Bonini, NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 1998, 93(6), 939–949. [Google Scholar]
- Warrick, JM; Chan, HY; Gray-Board, GL; Chai, Y; Paulson, HL; Bonini, NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 1999, 23(4), 425–428. [Google Scholar]
- Chan, HY; Warrick, JM; Gray-Board, GL; Paulson, HL; Bonini, NM. Mechanisms of chaperone suppression of polyglutamine disease: Selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 2000, 9(19), 2811–2820. [Google Scholar]
- Warrick, JM; Morabito, LM; Bilen, J; Gordesky-Gold, B; Faust, LZ; Paulson, HL; Bonini, NM. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell 2005, 18(1), 37–48. [Google Scholar]
- Bilen, J; Bonini, NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet 2007, 3(10), 1950–1964. [Google Scholar]
- Lessing, D; Bonini, NM. Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol 2008, 6(2), e29. [Google Scholar]
- Takeyama, K; Ito, S; Yamamoto, A; Tanimoto, H; Furutani, T; Kanuka, H; Miura, M; Tabata, T; Kato, S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 2002, 35(5), 855–864. [Google Scholar]
- Murata, T; Suzuki, E; Ito, S; Sawatsubashi, S; Zhao, Y; Yamagata, K; Tanabe, M; Fujiyama, S; Kimura, S; Ueda, T; Matsukawa, H; Kouzmenko, A; Furutani, T; Kuranaga, E; Miura, M; Takeyama, K; Kato, S. RNA-binding protein hoip accelerates polyQ-induced neurodegeneration in Drosophila. Biosci Biotechnol Biochem 2008, 72(9), 2255–2261. [Google Scholar]
- Ranum, LP; Day, JW. Pathogenic RNA repeats: an expanding role in genetic disease. Trends Genet 2004, 20(10), 506–512. [Google Scholar]
- Nemes, JP; Benzow, KA; Moseley, ML; Ranum, LP; Koob, MD. The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1). Hum Mol Genet 2000, 9(10), 1543–1551. [Google Scholar]
- Mutsuddi, M; Marshall, CM; Benzow, KA; Koob, MD; Rebay, I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol 2004, 14(4), 302–308. [Google Scholar]
- Simic, G; Mladinov, M; Seso Simic, D; Jovanov Milosevic, N; Islam, A; Pajtak, A; Barisic, N; Sertic, J; Lucassen, PJ; Hof, PR; Kruslin, B. Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol 2008, 115(3), 313–326. [Google Scholar]
- Lefebvre, S; Burglen, L; Reboullet, S; Clermont, O; Burlet, P; Viollet, L; Benichou, B; Cruaud, C; Millasseau, P; Zeviani, M; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80(1), 155–165. [Google Scholar]
- Miguel-Aliaga, I; Chan, YB; Davies, KE; van den Heuvel, M. Disruption of SMN function by ectopic expression of the human SMN gene in Drosophila. FEBS Lett 2000, 486(2), 99–102. [Google Scholar]
- Chan, YB; Miguel-Aliaga, I; Franks, C; Thomas, N; Trulzsch, B; Sattelle, DB; Davies, KE; van den Heuvel, M. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet 2003, 12(12), 1367–1376. [Google Scholar]
- Chang, HC; Dimlich, DN; Yokokura, T; Mukherjee, A; Kankel, MW; Sen, A; Sridhar, V; Fulga, TA; Hart, AC; Van Vactor, D; Artavanis-Tsakonas, S. Modeling spinal muscular atrophy in Drosophila. PLoS ONE 2008, 3(9), e3209. [Google Scholar]
- Lomen-Hoerth, C. Amyotrophic lateral sclerosis from bench to bedside. Semin Neurol 2008, 28(2), 205–211. [Google Scholar]
- Rosen, DR; Siddique, T; Patterson, D; Figlewicz, DA; Sapp, P; Hentati, A; Donaldson, D; Goto, J; O’Regan, JP; Deng, HX; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362(6415), 59–62. [Google Scholar]
- Kato, S. Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences. Acta Neuropathol 2008, 115(1), 97–114. [Google Scholar]
- Lin, WL; Dickson, DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol 2008, 116(2), 205–213. [Google Scholar]
- Watson, MR; Lagow, RD; Xu, K; Zhang, B; Bonini, NM. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem 2008, 283(36), 24972–24981. [Google Scholar]
- Ratnaparkhi, A; Lawless, GM; Schweizer, FE; Golshani, P; Jackson, GR. A Drosophila model of ALS: Human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE 2008, 3(6), e2334. [Google Scholar]
- Parkes, TL; Elia, AJ; Dickinson, D; Hilliker, AJ; Phillips, JP; Boulianne, GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet 1998, 19(2), 171–4. [Google Scholar]
- Elia, AJ; Parkes, TL; Kirby, K; St George-Hyslop, P; Boulianne, GL; Phillips, JP; Hilliker, AJ. Expression of human FALS SOD in motorneurons of Drosophila. Free Radic Biol Med 1999, 26. [Google Scholar]
- Birch-Machin, MA; Taylor, RW; Cochran, B; Ackrell, BA; Turnbull, DM. Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann Neurol 2000, 48(3), 330–335. [Google Scholar]
- Horvath, R; Abicht, A; Holinski-Feder, E; Laner, A; Gempel, K; Prokisch, H; Lochmuller, H; Klopstock, T; Jaksch, M. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry 2006, 77(1), 74–76. [Google Scholar]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 1951, 14(3), 216–221. [Google Scholar]
- Paulus, W; Peiffer, J. Intracerebral distribution of mitochondrial abnormalities in 21 cases of infantile spongy dystrophy. J Neurol Sci 1990, 95(1), 49–62. [Google Scholar]
- Mast, JD; Tomalty, KM; Vogel, H; Clandinin, TR. Reactive oxygen species act remotely to cause synapse loss in a Drosophila model of developmental mitochondrial encephalopathy. Development 2008, 135(15), 2669–2679. [Google Scholar]
- Zordan, MA; Cisotto, P; Benna, C; Agostino, A; Rizzo, G; Piccin, A; Pegoraro, M; Sandrelli, F; Perini, G; Tognon, G; De Caro, R; Peron, S; Kronnie, TT; Megighian, A; Reggiani, C; Zeviani, M; Costa, R. Post-transcriptional silencing and functional characterization of the Drosophila melanogaster homolog of human Surf1. Genetics 2006, 172(1), 229–241. [Google Scholar]
- Vanier, MT; Millat, G. Niemann-Pick disease type C. Clin Genet 2003, 64(4), 269–281. [Google Scholar]
- Liscum, L; Sturley, SL. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: Obligate components of subcellular lipid transport. Biochim Biophys Acta 2004, 1685. [Google Scholar]
- Carstea, ED; Morris, JA; Coleman, KG; Loftus, SK; Zhang, D; Cummings, C; Gu, J; Rosenfeld, MA; Pavan, WJ; Krizman, DB; Nagle, J; Polymeropoulos, MH; Sturley, SL; Ioannou, YA; Higgins, ME; Comly, M; Cooney, A; Brown, A; Kaneski, CR; Blanchette-Mackie, EJ; Dwyer, NK; Neufeld, EB; Chang, TY; Liscum, L; Strauss, JF, 3rd; Ohno, K; Zeigler, M; Carmi, R; Sokol, J; Markie, D; O’Neill, RR; van Diggelen, OP; Elleder, M; Patterson, MC; Brady, RO; Vanier, MT; Pentchev, PG; Tagle, DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997, 277(5323), 228–231. [Google Scholar]
- Naureckiene, S; Sleat, DE; Lackland, H; Fensom, A; Vanier, MT; Wattiaux, R; Jadot, M; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290(5500), 2298–2301. [Google Scholar]
- Vance, JE. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett 2006, 580(23), 5518–5524. [Google Scholar]
- Fluegel, ML; Parker, TJ; Pallanck, LJ. Mutations of a Drosophila NPC1 gene confer sterol and ecdysone metabolic defects. Genetics 2006, 172(1), 185–196. [Google Scholar]
- Huang, X; Suyama, K; Buchanan, J; Zhu, AJ; Scott, MP. A Drosophila model of the Niemann-Pick type C lysosome storage disease: dnpc1a is required for molting and sterol homeostasis. Development 2005, 132(22), 5115–5124. [Google Scholar]
- Phillips, SE; Woodruff, EA, 3rd; Liang, P; Patten, M; Broadie, K. Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J Neurosci 2008, 28(26), 6569–6582. [Google Scholar]
- Huang, X; Warren, JT; Buchanan, J; Gilbert, LI; Scott, MP. Drosophila Niemann-Pick type C-2 genes control sterol homeostasis and steroid biosynthesis: A model of human neurodegenerative disease. Development 2007, 134(20), 3733–3742. [Google Scholar]
- Wisniewski, KE; Kida, E; Golabek, AA; Kaczmarski, W; Connell, F; Zhong, N. Neuronal ceroid lipofuscinoses: Classification and diagnosis. Adv. Genet 2001, 45, 1–34. [Google Scholar]
- Siintola, E; Lehesjoki, AE; Mole, SE. Molecular genetics of the NCLs — status and perspectives. Biochim Biophys Acta 2006, 1762(10), 857–864. [Google Scholar]
- Stogmann, E; El Tawil, S; Wagenstaller, J; Gaber, A; Edris, S; Abdelhady, A; Assem-Hilger, E; Leutmezer, F; Bonelli, S; Baumgartner, C; Zimprich, F; Strom, TM; Zimprich, A. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics 2008. [Google Scholar]
- Hickey, AJ; Chotkowski, HL; Singh, N; Ault, JG; Korey, CA; MacDonald, ME; Glaser, RL. Palmitoyl-protein thioesterase 1 deficiency in Drosophila melanogaster causes accumulation of abnormal storage material and reduced life span. Genetics 2006, 172(4), 2379–2390. [Google Scholar]
- Korey, CA; MacDonald, ME. An over-expression system for characterizing Ppt1 function in Drosophila. BMC Neurosci 2003, 4, 30. [Google Scholar]
- Buff, H; Smith, AC; Korey, CA. Genetic modifiers of Drosophila palmitoyl-protein thioesterase 1-induced degeneration. Genetics 2007, 176(1), 209–220. [Google Scholar]
- Ferner, RE. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol 2007, 6(4), 340–351. [Google Scholar]
- Louis, DN; Ohgaki, H; Wiestler, OD; Cavenee, WK; Burger, PC; Jouvet, A; Scheithauer, BW; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007, 114(2), 97–109. [Google Scholar]
- The, I; Hannigan, GE; Cowley, GS; Reginald, S; Zhong, Y; Gusella, JF; Hariharan, IK; Bernards, A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 1997, 276(5313), 791–794. [Google Scholar]
- Guo, HF; Tong, J; Hannan, F; Luo, L; Zhong, Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 2000, 403(6772), 895–898. [Google Scholar]
- Williams, JA; Su, HS; Bernards, A; Field, J; Sehgal, A. A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science 2001, 293(5538), 2251–2256. [Google Scholar]
- Tong, JJ; Schriner, SE; McCleary, D; Day, BJ; Wallace, DC. Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nat Genet 2007, 39(4), 476–485. [Google Scholar]
- Walker, JA; Bernards, A. Drosophila melanogaster neurofibromatosis-1: ROS, not Ras? Nat Genet 2007, 39(4), 443–445. [Google Scholar]
- Pellock, BJ; Buff, E; White, K; Hariharan, IK. The Drosophila tumor suppressors Expanded and Merlin differentially regulate cell cycle exit, apoptosis, and Wingless signaling. Dev Biol 2007, 304(1), 102–115. [Google Scholar]
- Hamaratoglu, F; Willecke, M; Kango-Singh, M; Nolo, R; Hyun, E; Tao, C; Jafar-Nejad, H; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 2006, 8(1), 27–36. [Google Scholar]
- Povey, S; Burley, MW; Attwood, J; Benham, F; Hunt, D; Jeremiah, SJ; Franklin, D; Gillett, G; Malas, S; Robson, EB; et al. Two loci for tuberous sclerosis: one on 9q34 and one on 16p13. Ann Hum Genet 1994, 58, 107–127. [Google Scholar]
- Crino, PB; Nathanson, KL; Henske, EP. The tuberous sclerosis complex. N Engl J Med 2006, 355(13), 1345–1356. [Google Scholar]
- Huttenlocher, PR; Heydemann, PT. Fine structure of cortical tubers in tuberous sclerosis: A Golgi study. Ann Neurol 1984, 16(5), 595–602. [Google Scholar]
- Tapon, N; Ito, N; Dickson, BJ; Treisman, JE; Hariharan, IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 2001, 105(3), 345–355. [Google Scholar]
- Brumby, AM; Richardson, HE. Using Drosophila melanogaster to map human cancer pathways. Nat Rev Cancer 2005, 5(8), 626–639. [Google Scholar]
- Betschinger, J; Mechtler, K; Knoblich, JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell 2006, 124(6), 1241–1253. [Google Scholar]
- Beaucher, M; Goodliffe, J; Hersperger, E; Trunova, S; Frydman, H; Shearn, A. Drosophila brain tumor metastases express both neuronal and glial cell type markers. Dev Biol 2007, 301(1), 287–297. [Google Scholar]
- Woodhouse, EC; Fisher, A; Bandle, RW; Bryant-Greenwood, B; Charboneau, L; Petricoin, EF, 3rd; Liotta, LA. Drosophila screening model for metastasis: Semaphorin 5c is required for l(2)gl cancer phenotype. Proc Natl Acad Sci USA 2003, 100(20), 11463–11468. [Google Scholar]
- Uhlirova, M; Bohmann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J 2006, 25(22), 5294–5304. [Google Scholar]
- Scheffer, IE; Berkovic, SF. The genetics of human epilepsy. Trends Pharmacol Sci 2003, 24(8), 428–433. [Google Scholar]
- Kalachikov, S; Evgrafov, O; Ross, B; Winawer, M; Barker-Cummings, C; Martinelli Boneschi, F; Choi, C; Morozov, P; Das, K; Teplitskaya, E; Yu, A; Cayanis, E; Penchaszadeh, G; Kottmann, AH; Pedley, TA; Hauser, WA; Ottman, R; Gilliam, TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002, 30(3), 335–341. [Google Scholar]
- Stromme, P; Mangelsdorf, ME; Shaw, MA; Lower, KM; Lewis, SM; Bruyere, H; Lutcherath, V; Gedeon, AK; Wallace, RH; Scheffer, IE; Turner, G; Partington, M; Frints, SG; Fryns, JP; Sutherland, GR; Mulley, JC; Gecz, J. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet 2002, 30(4), 441–445. [Google Scholar]
- Blumcke, I; Pauli, E; Clusmann, H; Schramm, J; Becker, A; Elger, C; Merschhemke, M; Meencke, HJ; Lehmann, T; von Deimling, A; Scheiwe, C; Zentner, J; Volk, B; Romstock, J; Stefan, H; Hildebrandt, M. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropathol 2007, 113(3), 235–244. [Google Scholar]
- Pinel, JP; Chorover, SL. Inhibition by arousal of epilepsy induced by chlorambucil in rats. Nature 1972, 236(5344), 232–234. [Google Scholar]
- Racine, R. Kindling: The first decade. Neurosurgery 1978, 3(2), 234–252. [Google Scholar]
- Pavlidis, P; Tanouye, MA. Seizures and failures in the giant fiber pathway of Drosophila bang-sensitive paralytic mutants. J Neurosci 1995, 15(8), 5810–5819. [Google Scholar]
- Ganetzky, B. Genetic analysis of ion channel dysfunction in Drosophila. Kidney Int 2000, 57(3), 766–771. [Google Scholar]
- Titus, SA; Warmke, JW; Ganetzky, B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci 1997, 17(3), 875–881. [Google Scholar]
- Reynolds, ER; Stauffer, EA; Feeney, L; Rojahn, E; Jacobs, B; McKeever, C. Treatment with the antiepileptic drugs phenytoin and gabapentin ameliorates seizure and paralysis of Drosophila bang-sensitive mutants. J Neurobiol 2004, 58(4), 503–513. [Google Scholar]
- Stilwell, GE; Saraswati, S; Littleton, JT; Chouinard, SW. Development of a Drosophila seizure model for in vivo high-throughput drug screening. Eur J Neurosci 2006, 24(8), 2211–2222. [Google Scholar]
- Song, J; Tanouye, MA. From bench to drug: Human seizure modeling using Drosophila. Prog Neurobiol 2008, 84(2), 182–191. [Google Scholar]
- Kuebler, D; Tanouye, MA. Modifications of seizure susceptibility in Drosophila. J Neurophysiol 2000, 83(2), 998–1009. [Google Scholar]
- Kuebler, D; Zhang, H; Ren, X; Tanouye, MA. Genetic suppression of seizure susceptibility in Drosophila. J Neurophysiol 2001, 86(3), 1211–1225. [Google Scholar]
- Song, J; Hu, J; Tanouye, M. Seizure suppression by top1 mutations in Drosophila. J Neurosci 2007, 27(11), 2927–2937. [Google Scholar]
- Song, J; Parker, L; Hormozi, L; Tanouye, MA. DNA topoisomerase I inhibitors ameliorate seizure-like behaviors and paralysis in a Drosophila model of epilepsy. Neuroscience 2008, 156(3), 722–728. [Google Scholar]
- Fergestad, T; Olson, L; Patel, KP; Miller, R; Palladino, MJ; Ganetzky, B. Neuropathology in Drosophila mutants with increased seizure susceptibility. Genetics 2008, 178(2), 947–956. [Google Scholar]
- Profyris, C; Cheema, SS; Zang, D; Azari, MF; Boyle, K; Petratos, S. Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiol Dis 2004, 15(3), 415–436. [Google Scholar]
- Niess, C; Grauel, U; Toennes, SW; Bratzke, H. Incidence of axonal injury in human brain tissue. Acta Neuropathol 2002, 104(1), 79–84. [Google Scholar]
- Batchelor, PE; Wills, TE; Hewa, AP; Porritt, MJ; Howells, DW. Stimulation of axonal sprouting by trophic factors immobilized within the wound core. Brain Res 2008, 1209, 49–56. [Google Scholar]
- Ayaz, D; Leyssen, M; Koch, M; Yan, J; Srahna, M; Sheeba, V; Fogle, KJ; Holmes, TC; Hassan, BA. Axonal injury and regeneration in the adult brain of Drosophila. J Neurosci 2008, 28(23), 6010–6021. [Google Scholar]
- Kaneko, M; Hall, JC. Neuroanatomy of cells expressing clock genes in Drosophila: Transgenic manipulation of the period and timeless genes to mark the perikarya of circadian pacemaker neurons and their projections. J Comp Neurol 2000, 422(1), 66–94. [Google Scholar]
- Silver, J; Miller, JH. Regeneration beyond the glial scar. Nat Rev Neurosci 2004, 5(2), 146–156. [Google Scholar]


© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jeibmann, A.; Paulus, W. Drosophila melanogaster as a Model Organism of Brain Diseases. Int. J. Mol. Sci. 2009, 10, 407-440. https://doi.org/10.3390/ijms10020407
Jeibmann A, Paulus W. Drosophila melanogaster as a Model Organism of Brain Diseases. International Journal of Molecular Sciences. 2009; 10(2):407-440. https://doi.org/10.3390/ijms10020407
Chicago/Turabian StyleJeibmann, Astrid, and Werner Paulus. 2009. "Drosophila melanogaster as a Model Organism of Brain Diseases" International Journal of Molecular Sciences 10, no. 2: 407-440. https://doi.org/10.3390/ijms10020407