A New Graphical Model for Proton NMR (De)shielding over a Carbon-Carbon Double Bond to Replace the Shielding Cone Model

Abstract
:Introduction
Methods and Calculations
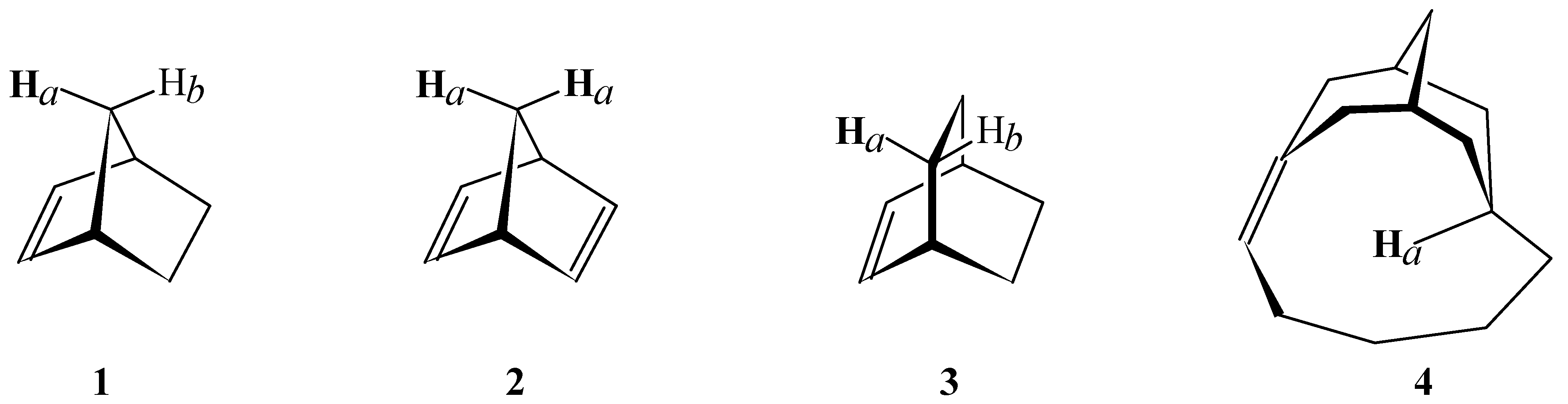


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | McConnell Predicted Δδ | Ab Initio GIAO-SCF Predicted Δδ | Experimentally Observed Δδ |
|---|---|---|---|
| 1 | -0.10 | -0.28 | -0.24 |
| 215 | -0.10 | -0.46 | -0.80 |
| 3 | -0.06 | 0.30 | 0.27 |
| 4 | 0.12 | -1.99 | -2.12 |
Results and Discussion
 or
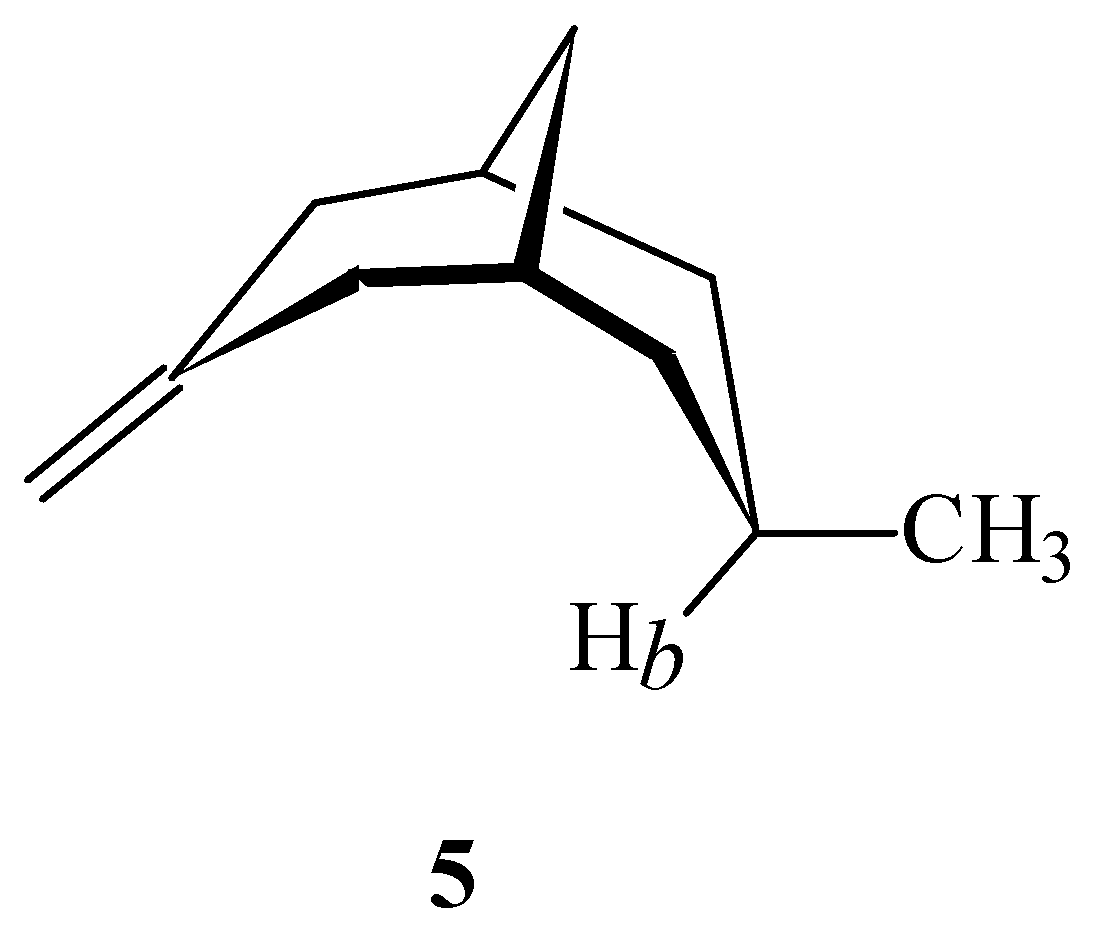
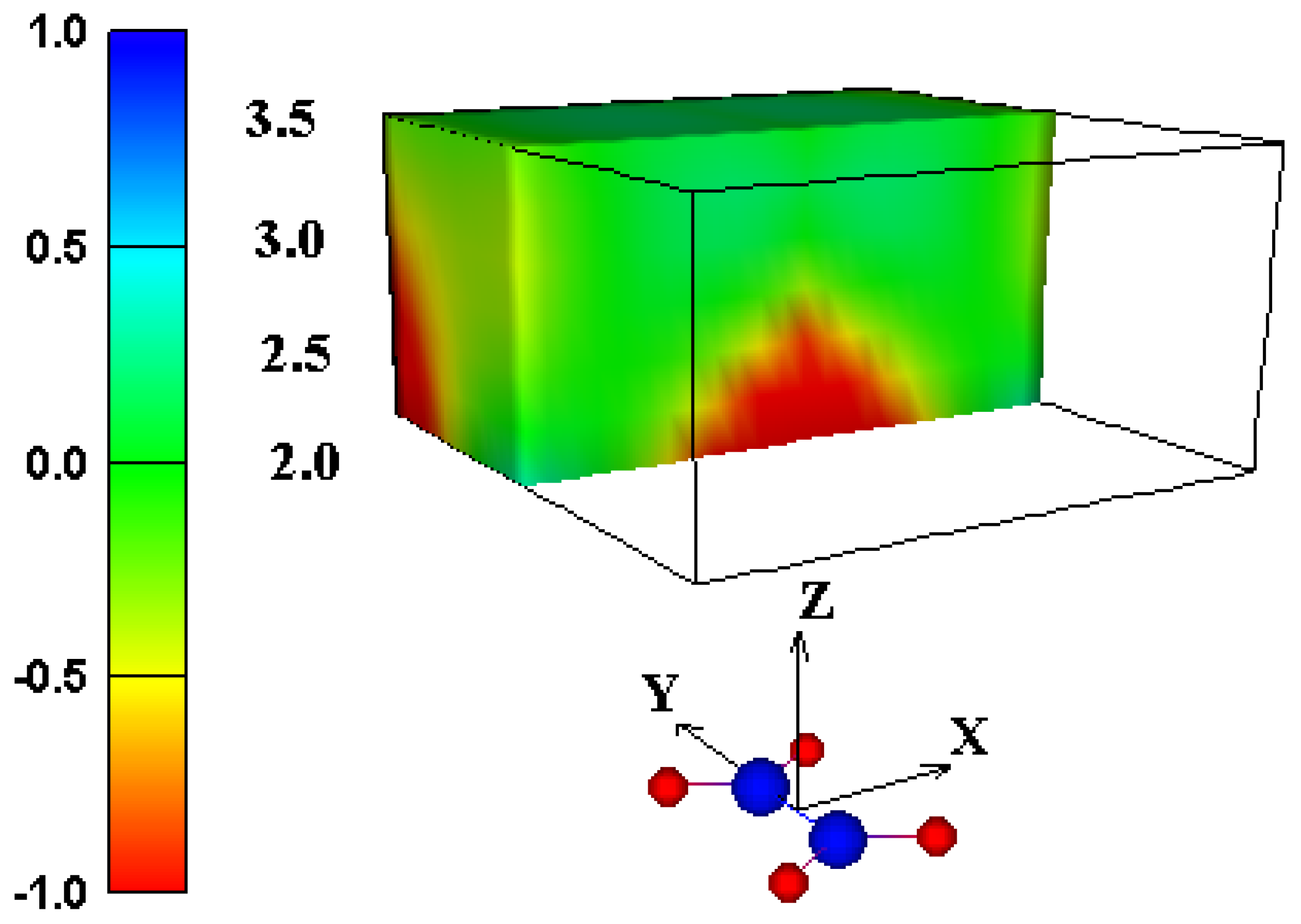
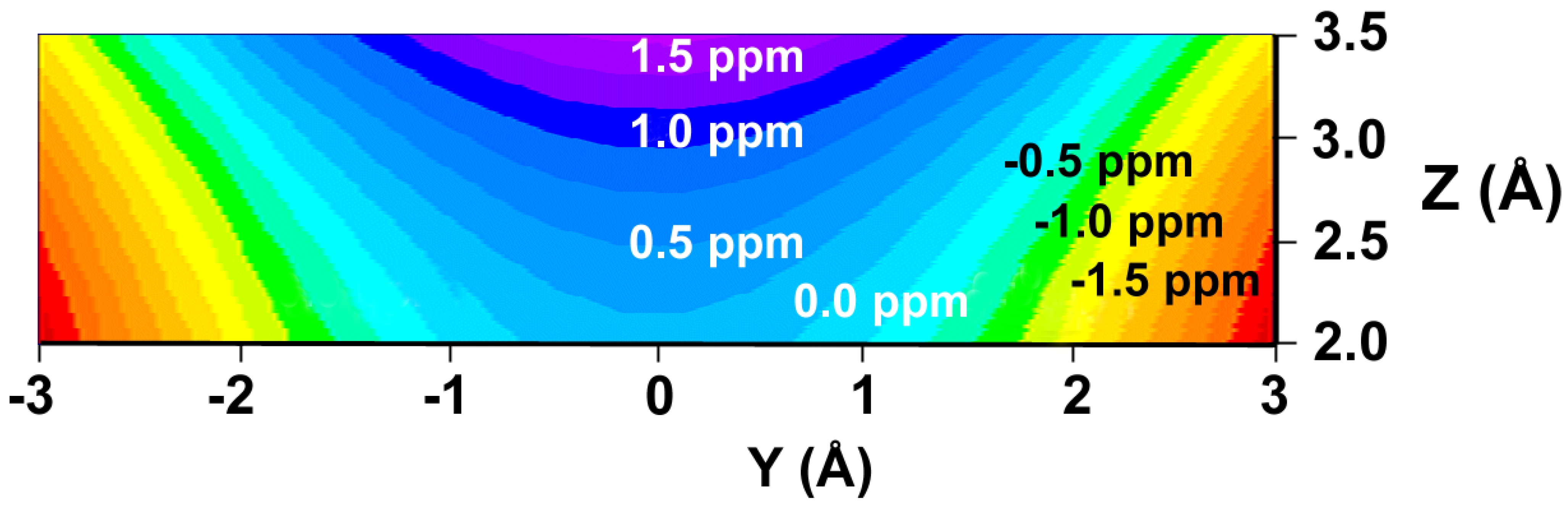
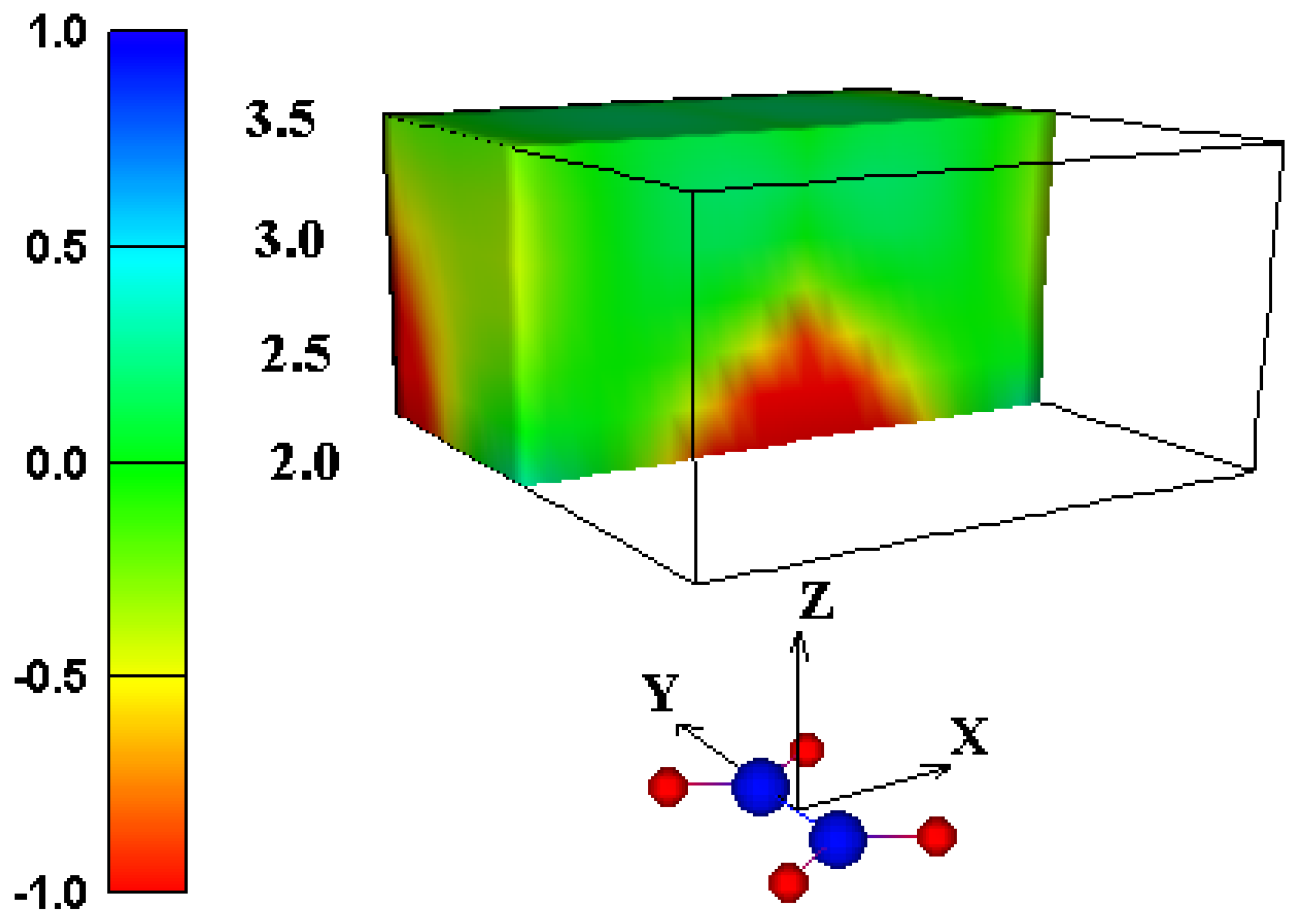
or  in a plane along the carbon-carbon double bond axis normal to the molecular plane of ethene from 2.0 Å to 3.5 Å above the molecular plane. The McConnell equation is the basis of the “shielding cone” model in common (but improper) use for predicting NMR shielding in the vicinity of a carbon-carbon double bond. The dramatic difference between the McConnell shielding model and the GIAO-SCF model is quite evident from a comparison of the two plots. When shielding increments calculated using the two methods are compared to experimental observations of chemical shift differences (Table 1) it is clear that the (mostly deshielding) values derived from the ab initio GIAO-SCF computations agree with experimental data much more closely.
in a plane along the carbon-carbon double bond axis normal to the molecular plane of ethene from 2.0 Å to 3.5 Å above the molecular plane. The McConnell equation is the basis of the “shielding cone” model in common (but improper) use for predicting NMR shielding in the vicinity of a carbon-carbon double bond. The dramatic difference between the McConnell shielding model and the GIAO-SCF model is quite evident from a comparison of the two plots. When shielding increments calculated using the two methods are compared to experimental observations of chemical shift differences (Table 1) it is clear that the (mostly deshielding) values derived from the ab initio GIAO-SCF computations agree with experimental data much more closely.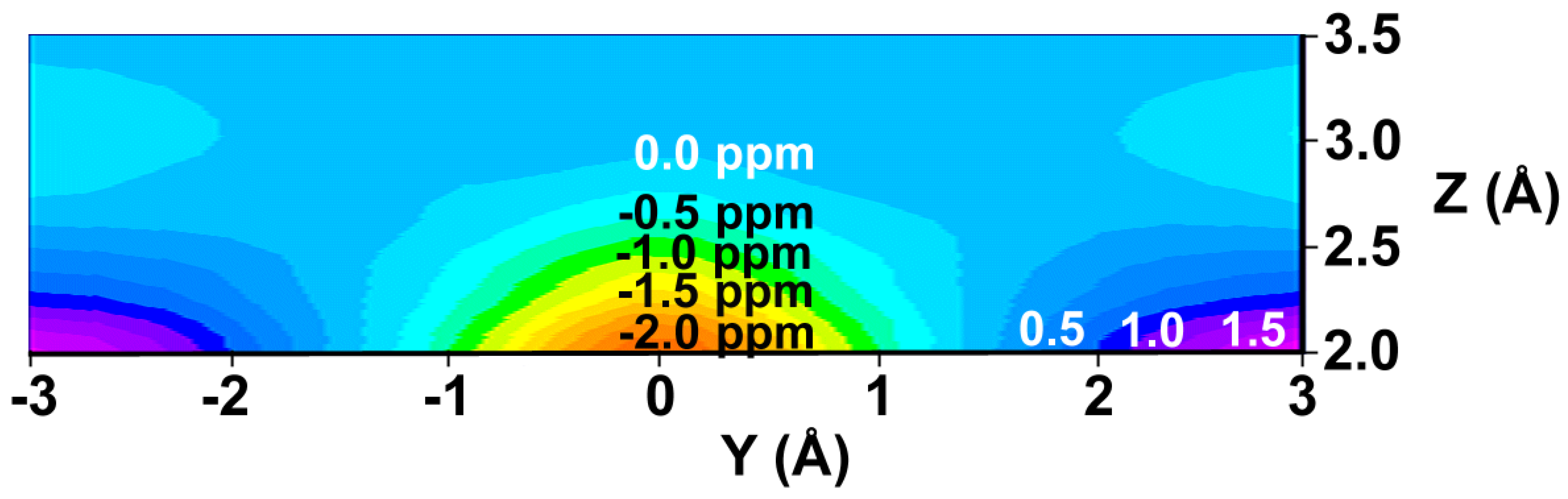
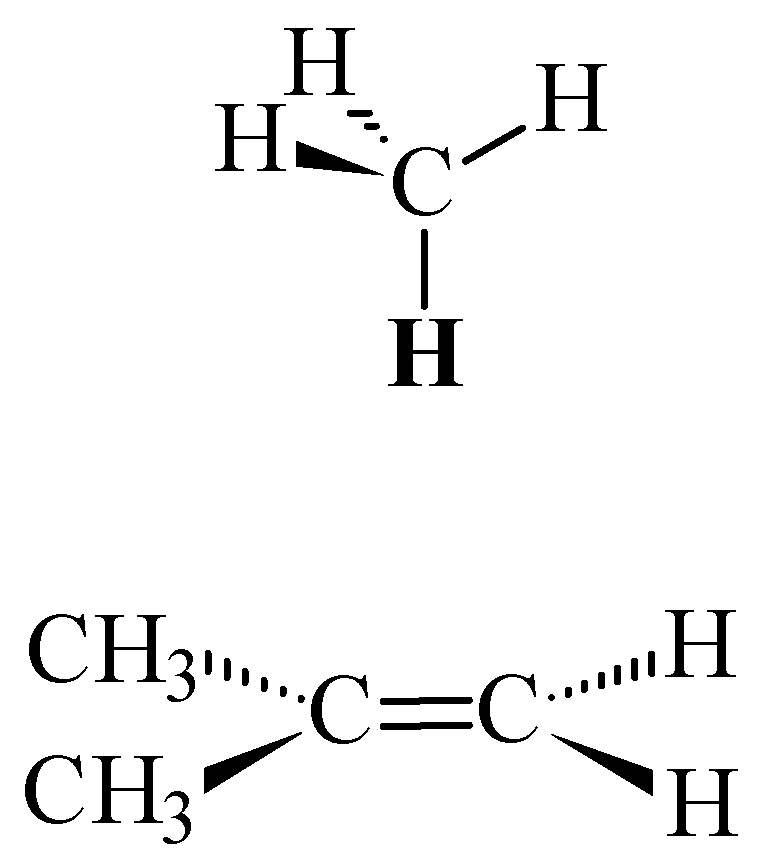
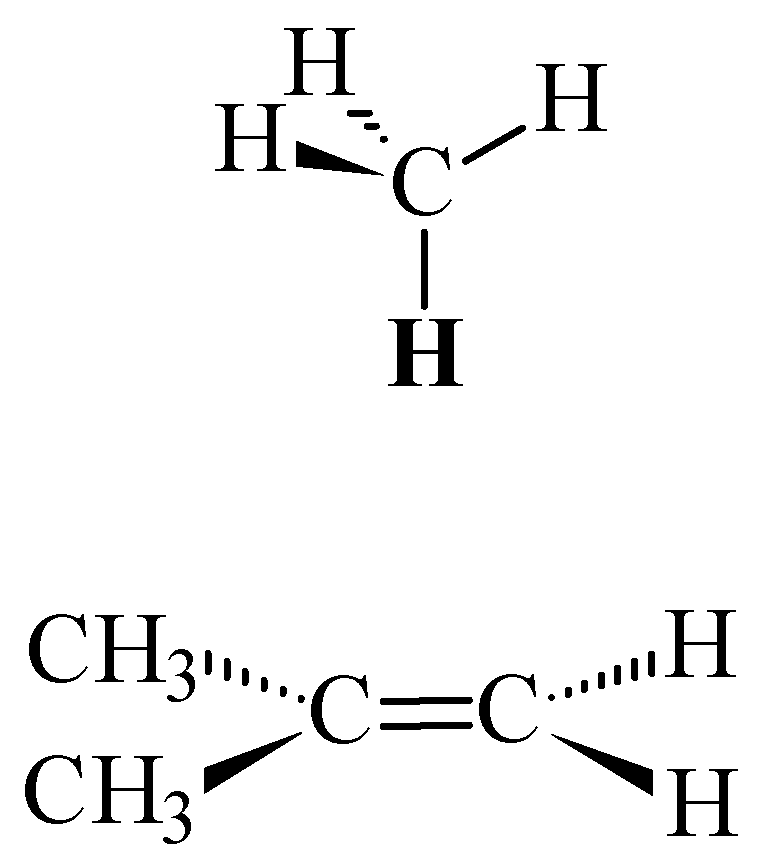
 and moved along the double bond axis and methane similarly moved over ethene
and moved along the double bond axis and methane similarly moved over ethene  . Dotted lines are HF results; solid lines are MP2 results.
and moved along the double bond axis and methane similarly moved over ethene . Dotted lines are HF results; solid lines are MP2 results.
. Dotted lines are HF results; solid lines are MP2 results.
and moved along the double bond axis and methane similarly moved over ethene . Dotted lines are HF results; solid lines are MP2 results.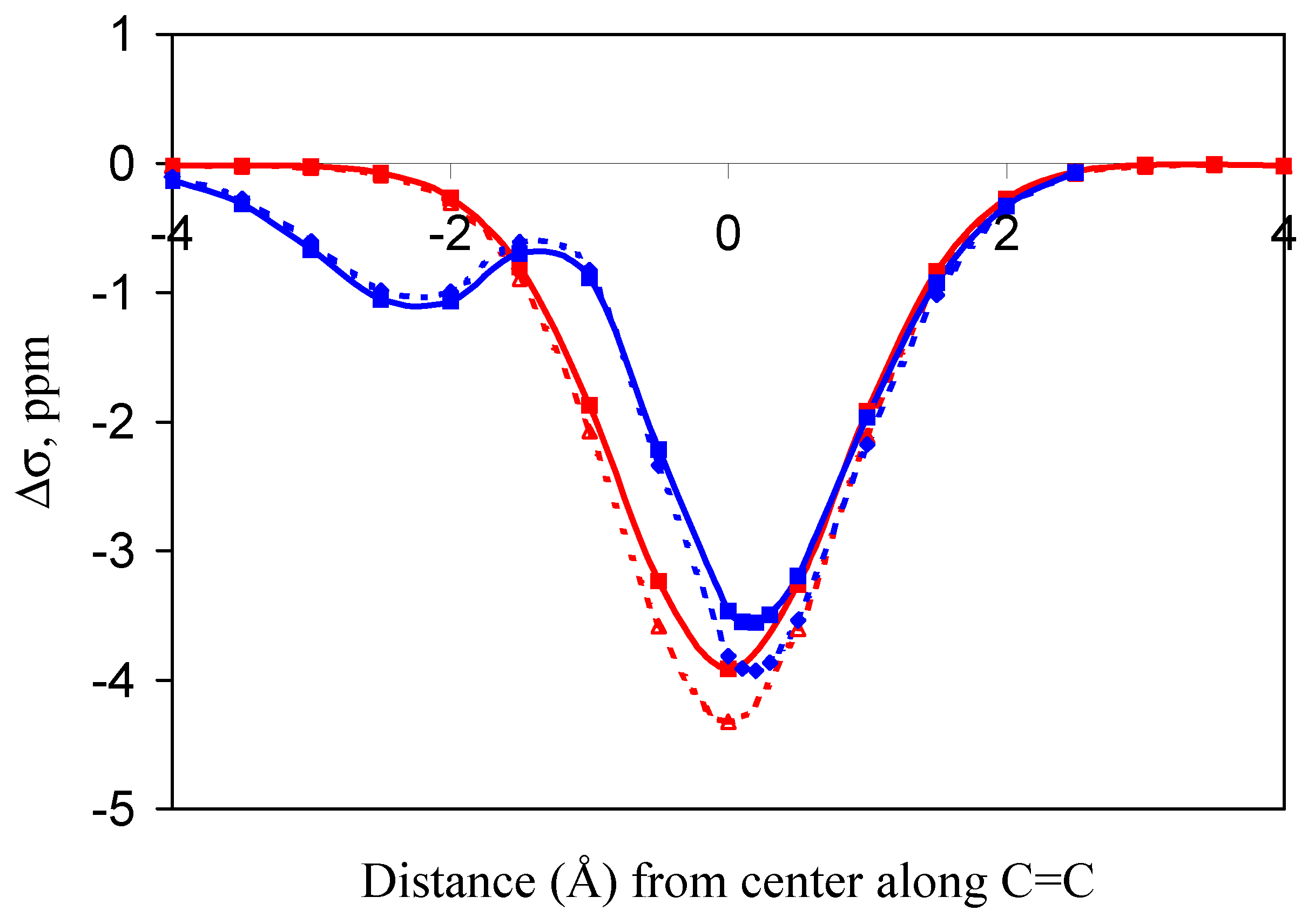 and the 2-methylpropene series . This is within the uncertainty of the predictions based on the SCF (HF) shielding function previously described [8].
and the 2-methylpropene series . This is within the uncertainty of the predictions based on the SCF (HF) shielding function previously described [8].Conclusions
Acknowledgements
References and Notes
- For example, Gunther, H. NMR Spectroscopy: Basic Principles, Concepts, and Applications in Chemistry, 2nd ed.; Wiley: Chichester, 1995; pp. 78–84. [Google Scholar]
- McConnell, H. M. Theory of Nuclear Magnetic Shielding in Molecules. I. Long-Range Dipolar Shielding of Protons. J. Chem. Phys. 1957, 27, 226–229. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. Computational Evidence of NMR Deshielding of Protons over a Carbon-Carbon Double Bond. J. Am. Chem. Soc. 1998, 120, 11510–11511. [Google Scholar]
- Elguero, J.; Alkorta, I. Ab Initio hybrid DFT-GIAO calculations of the shielding produced by carbon-carbon bonds and aromatic rings in 1H NMR. New J. Chem. 1998, 22, 381–385. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. An Empirical Proton NMR Shielding Equation for Alkenes Based on Ab Initio Calculations. Struct. Chem. 1998, 9(6), 403–410. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. Proceedings of ACS Symposium "Modeling NMR Chemical Shifts: Gaining Insights into Structure and Environment," American Chemical Society; Washington, D.C., 1999; pp. 207–219.
- Martin, N. H.; Allen, N. W., III; Minga, E. K.; Ingrassia, S. T.; Brown, J. D. An Improved Model for Predicting Proton NMR Shielding by Alkenes Based on Ab Initio GIAO Calculations. Struct. Chem. 1999, 10(5), 375–380. [Google Scholar]
- Martin, N. H.; Allen, N. W., III; Brown, J. D.; Ingrassia, S. T.; Minga, E. K. An Algorithm for Predicting Proton NMR Deshielding over a Carbon-Carbon Double Bond. J. Mol. Graphics Mod. 2000, 18(1), 1–6. [Google Scholar]
- Sorensen, T. S.; Whitworth, S. M. □-Hydrido Bridging in Tricycloalkyl Carbocations. J. Am. Chem. Soc. 1990, 112, 8135–8144. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-Consistent Perturbation Theory of Diamagnetism. I. A Gauge-Invariant LCAO Method for N.M.R. Chemical Shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Chesnut, D. B.; Foley, C. K. Some Simple Basis Sets for Accurate 13C Chemical Shift Calculations. Chem. Phys. Lett. 1985, 118, 316–321. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J. F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98; Revision A.7; Gaussian, Inc.: Pittsburgh PA, 1998. [Google Scholar]
- Gauss, J. Effects of Electron Correlation on the Calculation of Nuclear Magnetic Resonance Chemical Shifts. J. Phys. Chem. 1993, 99(5), 3629–3643. [Google Scholar] [CrossRef]
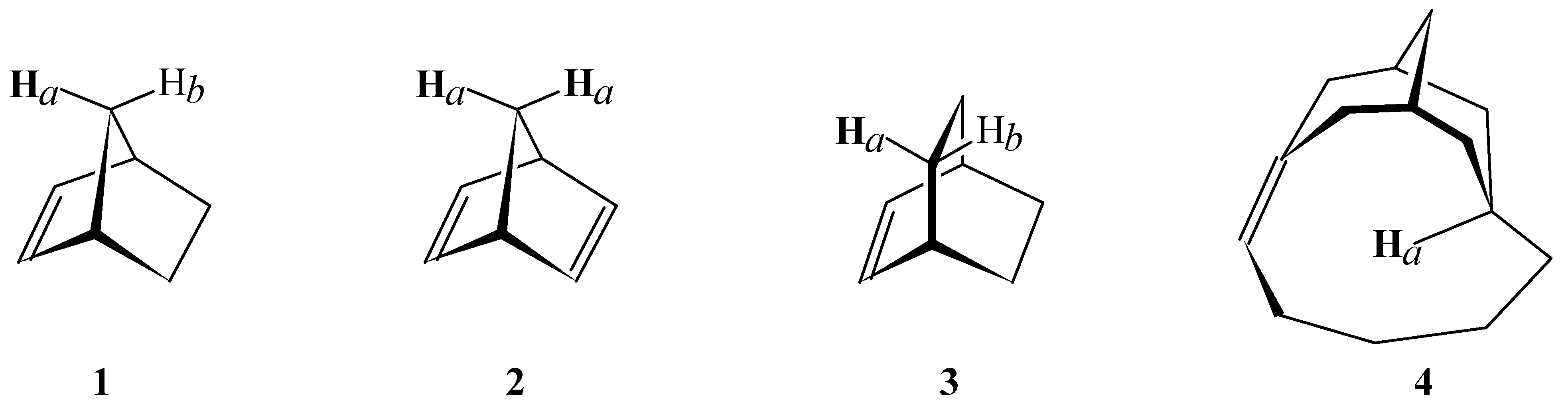
- For structure 2, the shielding and chemical shift values for Hb were taken from structure 1. For structure 4, the value for Hb in structure 5 (Figure 2) was used as reference.
- Boys, S. F.; Bernardi, F. The Calculations of Small Molecular Interaction by the Difference of Separate Total Energies. Some Procedures with Reduced Error. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Reed, A. E.; Weinstock, R. B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83(2), 735–746. [Google Scholar] [CrossRef]
© 2000 by MDPI (http://www.mdpi.org).
Share and Cite
Martin, N.H.; Brown, J.D. A New Graphical Model for Proton NMR (De)shielding over a Carbon-Carbon Double Bond to Replace the Shielding Cone Model. Int. J. Mol. Sci. 2000, 1, 84-91. https://doi.org/10.3390/ijms1040084
Martin NH, Brown JD. A New Graphical Model for Proton NMR (De)shielding over a Carbon-Carbon Double Bond to Replace the Shielding Cone Model. International Journal of Molecular Sciences. 2000; 1(4):84-91. https://doi.org/10.3390/ijms1040084
Chicago/Turabian StyleMartin, Ned H., and Justin D. Brown. 2000. "A New Graphical Model for Proton NMR (De)shielding over a Carbon-Carbon Double Bond to Replace the Shielding Cone Model" International Journal of Molecular Sciences 1, no. 4: 84-91. https://doi.org/10.3390/ijms1040084