New Synthetic Routes to Furocoumarins and Their Analogs: A Review

Department of Organic Chemistry, D. Mendeleev University of Chemical Technology of Russia 125047, Moscow, Russia
Molecules 2004, 9(3), 50-66; https://doi.org/10.3390/90300050
Submission received: 27 January 2004
/
Accepted: 25 February 2004
/
Published: 28 February 2004
(This article belongs to the Special Issue Biologically Relevant Heterocyclic Compounds)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
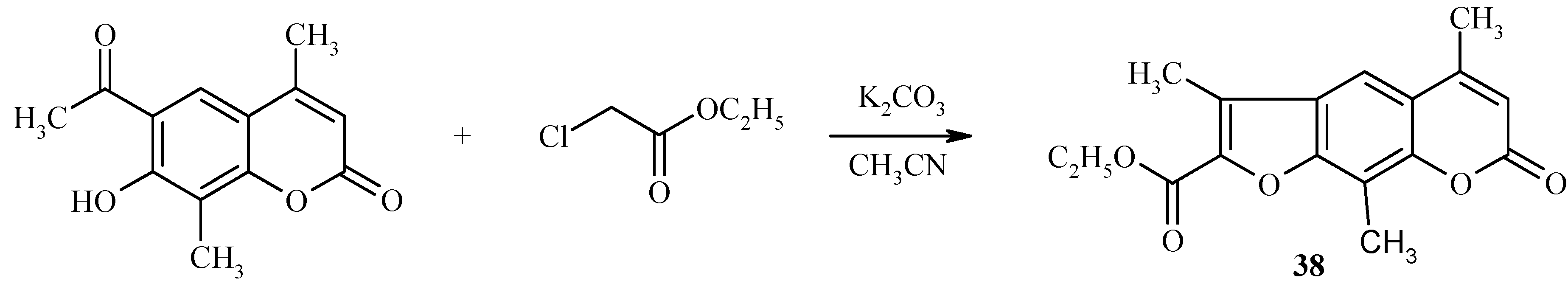{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
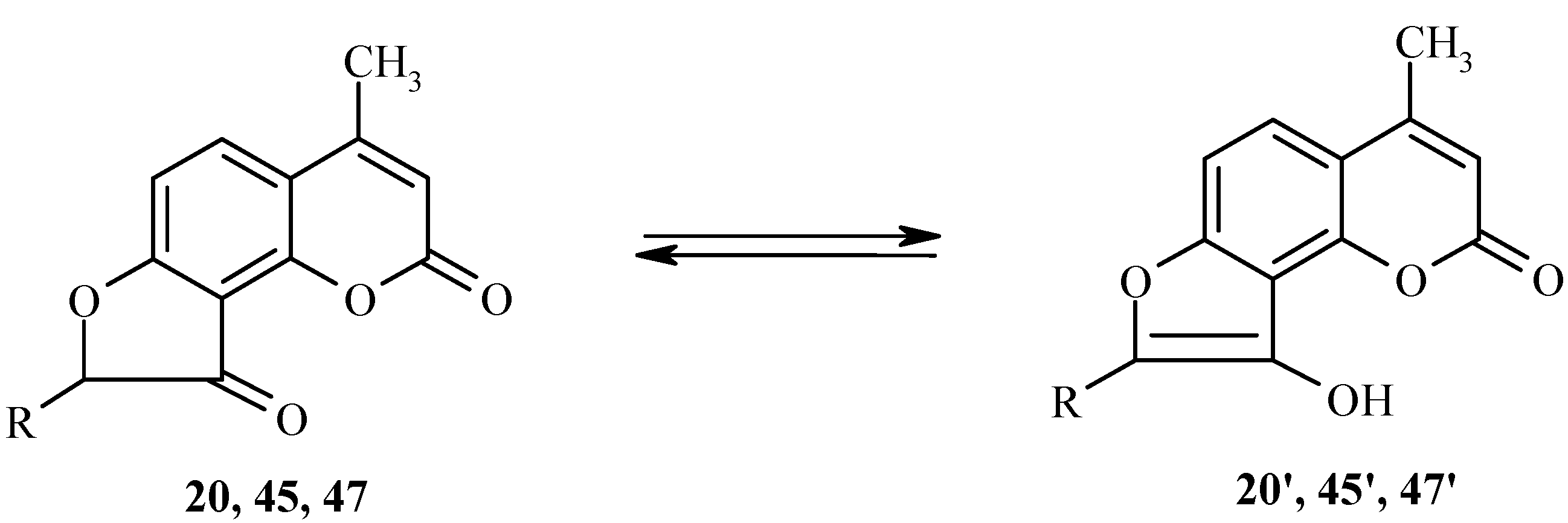{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
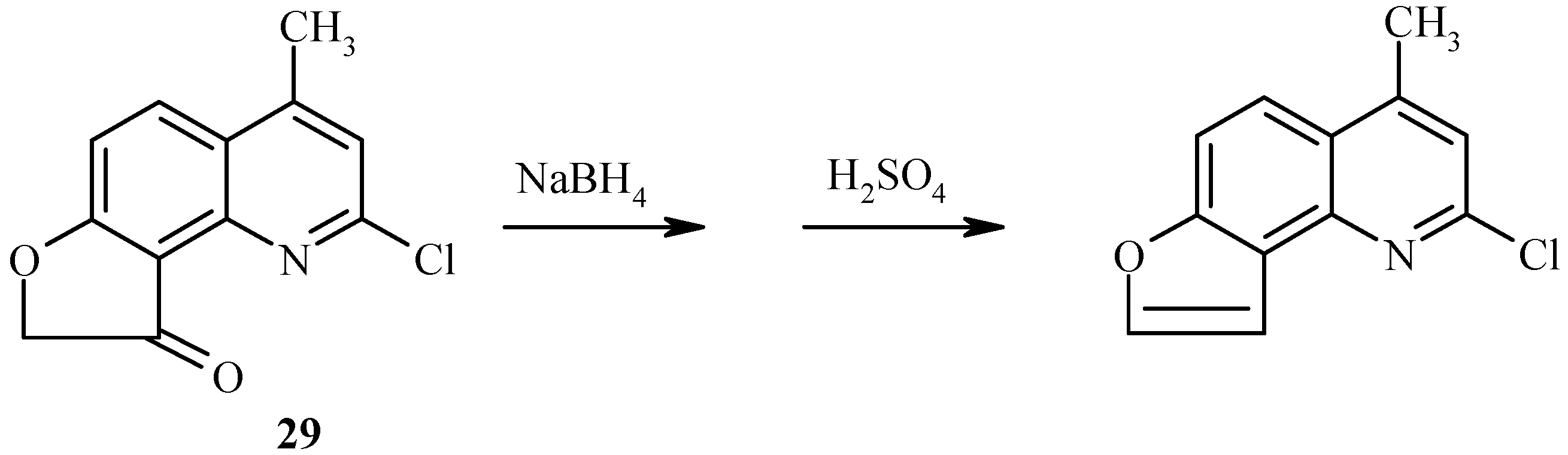{kind=link}
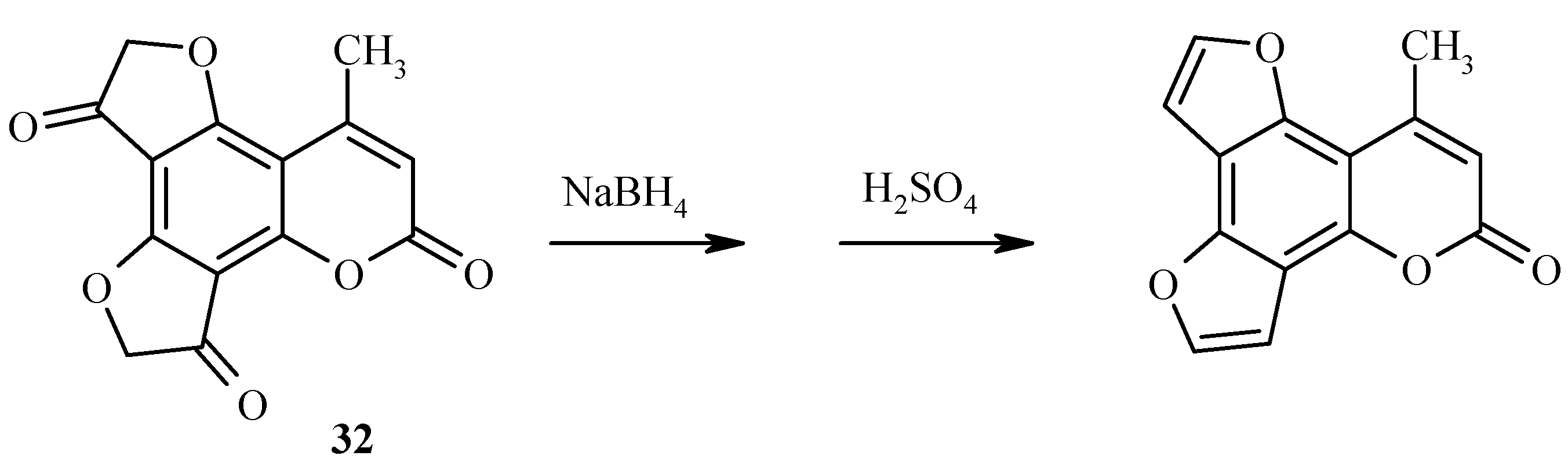{kind=link}
{kind=link}
Abstract
:Many furocoumarins and their analogs possess a prominent photobiological activity. Some of them are successfully used as drugs in phototherapy of skin diseases. Fries rearrangement of acyloxyheteroarenes, condensation of acylhydroxyheteroarenes with α-carbonyl compounds under base catalysis and transformations of dihydrofurocoumarinones are new trends in synthesis of furocoumarins and their analogs.
Introduction
Furocoumarins, such as psoralene and the angelicine derivatives are naturally occurring compounds. They are known to possess a high photobiological activity [1]. Psoralene derivatives have been used for many years in the treatment of skin diseases [2]. Furocoumarin/ultraviolet therapy, known as photopheresis, has recently become an effective treatment of cutaneous T cell lymphoma, Sezary syndrome and related diseases [3,4]. The photochemotherapeutic effects of furocoumarins are based on intercalation of the molecules between the pyrimidine bases of the microorganism’s DNA. The intercalation is then followed by the UV light activated cycloaddition reactions of furocoumarins with the pyrimidine bases. These [2+2] photocycloaddition reactions result in a cross-linking of DNA and prevent a microorganism’s reproduction.
Psoralens, which are linear furocoumarins, have the highest photosensitivity. Their molecules have two active sites in the [2+2] photocycloaddition reactions: the pyrone ring and furan ring double bonds. This kind of difunctionality of the psoralens (and of the angelicines in to a lesser extent) has been suggested to cause undesirable side effects in their medical use. Mutagenicity and carcinogenicity should be mentioned among these side effects of some psoralens [5,6,7,8,9,10,11].
Even though some methods of furocoumarin synthesis have been known for a long time, the successful use of furocoumarins as effective medicines requires access to as many new derivatives as possible. For example, it has been found that furocoumarin analogs that contain other heteroatoms besides the oxygen atom in the lactone ring are free of some side effects [4]. Better intercalation properties and higher hydrophilicity should be also specific for new furocoumarins recommended for pharmaceutical testing. Therefore, the search for new synthetic approaches to the fucoumarins and their analogous heterocyclic compounds is a promising trend in photochemotherapy [12,13,14,15,16,17,18,19].
Fries rearrangement of acetoxyheteroarenes
Acylhydroxyheteroarenes are convenient intermediates in furoheteroarene synthesis. The key reaction for preparation of these intermediates is the Fries rearrangement of acyloxyheteroarenes. Nevertheless, this reaction with heteroarene derivatives has not been studied much when compared with that of benzene derivatives.
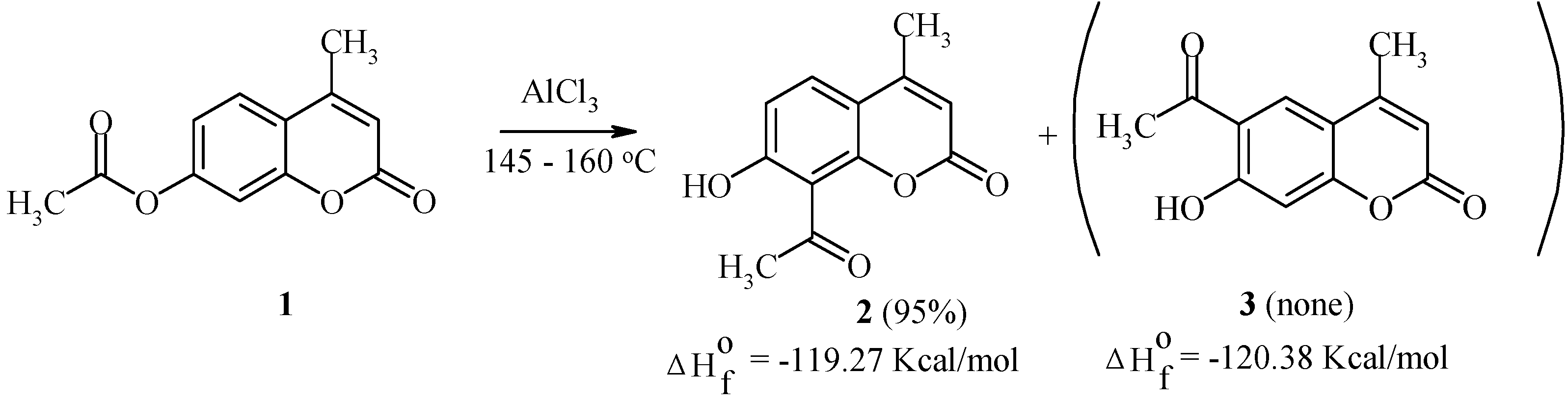
We have studied the Fries rearrangement of 7-acyloxycoumarins in detail [20,21,22]. Some of the results are given below. For example, 7-acetoxy-4-methylcoumarin (1) undergoes a smooth transformation into 8-acetyl-7-hydroxy-4-methylcoumarin (2) as the exclusive product of the rearrangement (Scheme 1). This rearrangement is largely temperature independent.
Scheme 1.
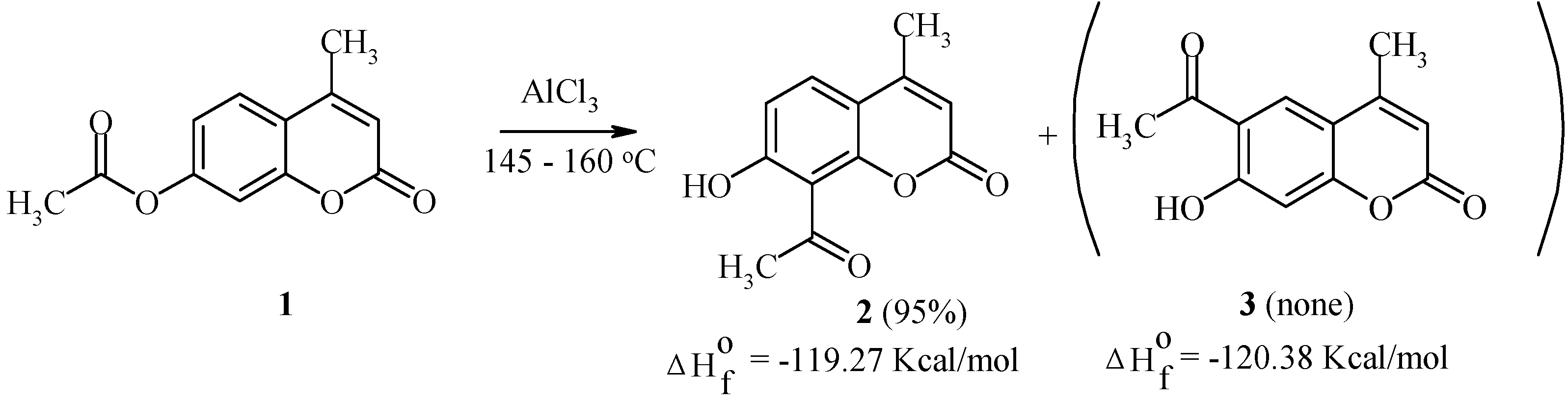
Kinetic factors seem to explain this result, since both 8-acetyl- and 6-acetyl-isomers (compounds 2 and 3, respectively) possess similar thermodynamic stability.
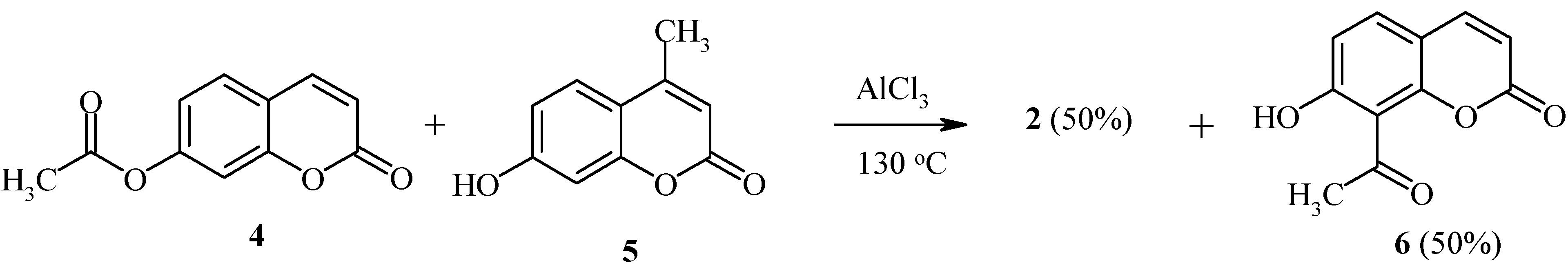
We have found that the Fries rearrangement of 7-acyloxycoumarins takes place by an intermolecular mechanism [20]. Thus, rearrangement of 7-acetoxycoumarin (4) in the presence of 7-hydroxy-4-methylcoumarin (5) results in the formation of equimolar amounts of 8-acetyl-7-hydroxycoumarin (6) and 8-acetyl-7-hydroxy-4-methylcoumarin (2) (Scheme 2).
Scheme 2.

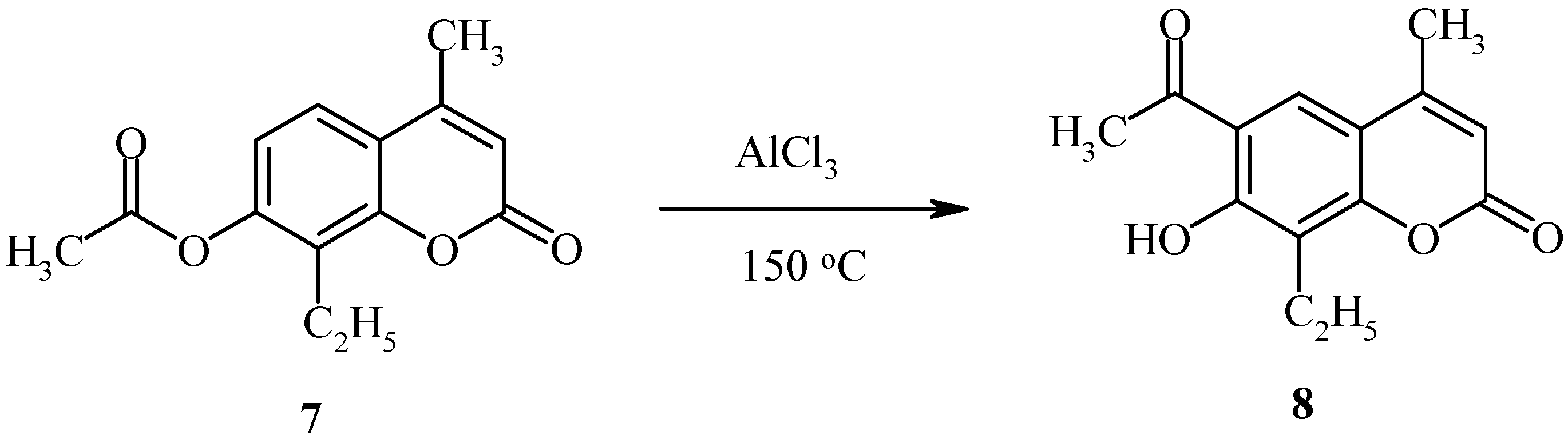
The deacylated product, 7-hydroxycoumarin, has also been found in the final reaction mixture. When the position 8 of the starting coumarin derivative is occupied (as in compound 7, Scheme 3), the acyl group rearrangement involves the 6 position.
Scheme 3.
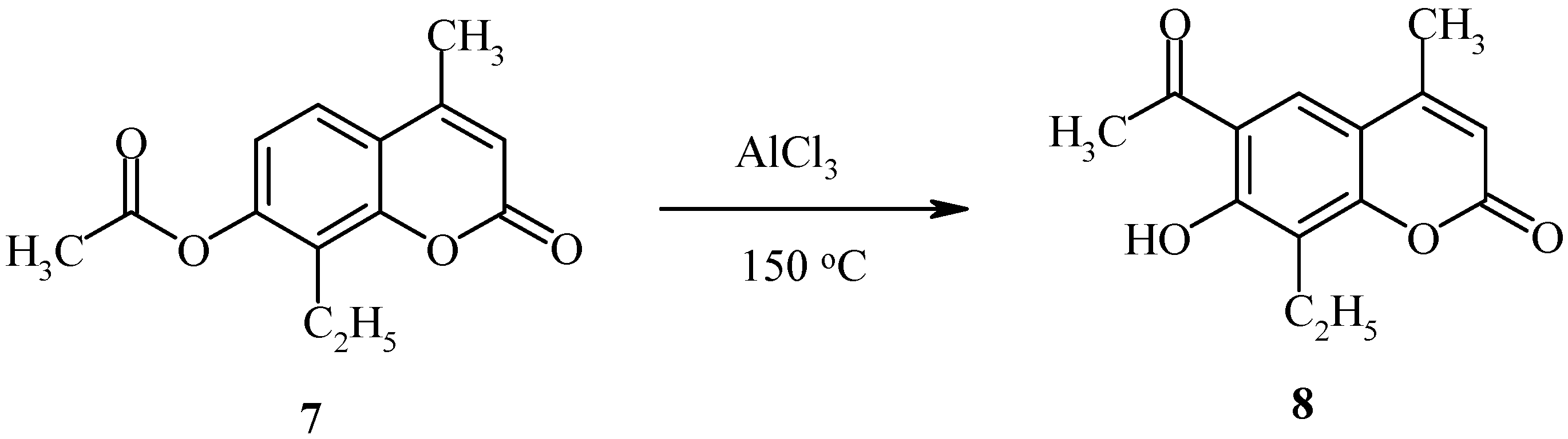
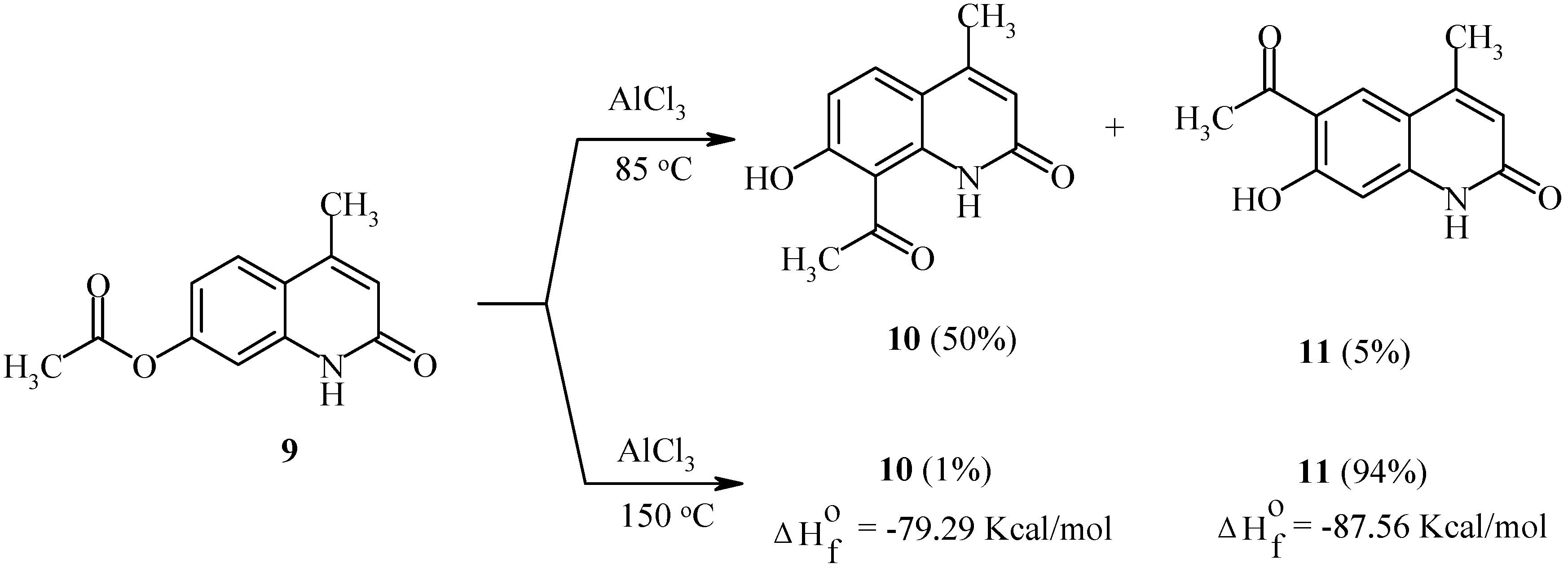
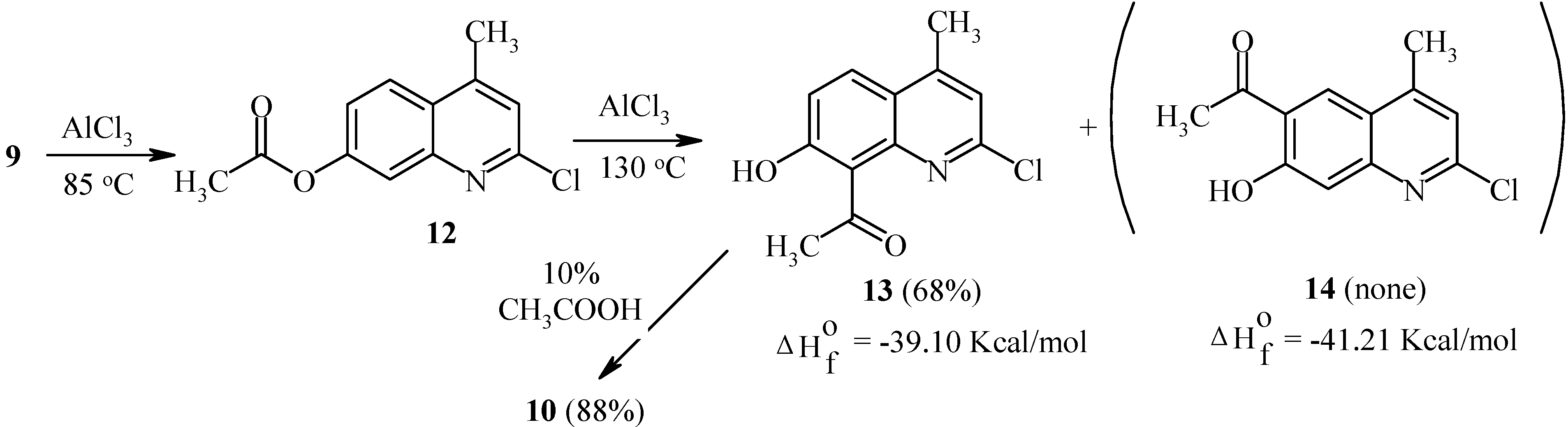
Rearrangement of 7-acetoxycoumarins thus provides intermediates for both angular and linear furocoumarins. We have found the regioselectivity of the Fries rearrangement of acyloxyheteroarenes to be very dependent on the structure of the heteroarene [23]. Regioselectivity data of rearrangements of acyloxycoumarins, bis(acyloxy)coumarins, acyloxyquinolin-2-ones, acyloxy-2-chloroquinolines and their analogs are shown below. In contrast to the reactions of acyloxycoumarins, the regioselectivity of Fries rearrangement of 7-acyloxyquinolin-2-one 9 is very dependent on the temperature.
Scheme 4.

At relatively low temperature (85°C), 8-acetyl-7-hydroxy-4-methylquinolin-2-one (10) is the predominant product of the rearrangement: 50% of 8-isomer 10 and 5% of 6-isomer 11 have been found in the final reaction mixture. However, an increase in temperature to 150°C results in the formation of the 6-isomer 11 as the major product (Scheme 4).
It should be noted that isomers 10 and 11 possess different calculated thermodynamic stabilities. As can be seen, a high rearrangement temperature results in predominant formation of the thermodynamically more stable isomer 11. By contrast, isomer distributions of products at lower temperatures seem to follow a kinetic trend. Substitution of a chlorine atom for the oxo-function in quinolinone 9 greatly affects the regioselectivity of the Fries rearrangement. Thus, 7-acetoxy-2-chloro-4-methylquinoline (12) undergoes the rearrangement with exclusive formation of the 8-acetyl-isomer 13 independent of the temperature (Scheme 5). A possible isomer 14 was not found. Heating of 13 in aqueous acetic acid furnished quinolone 10.
Scheme 5.
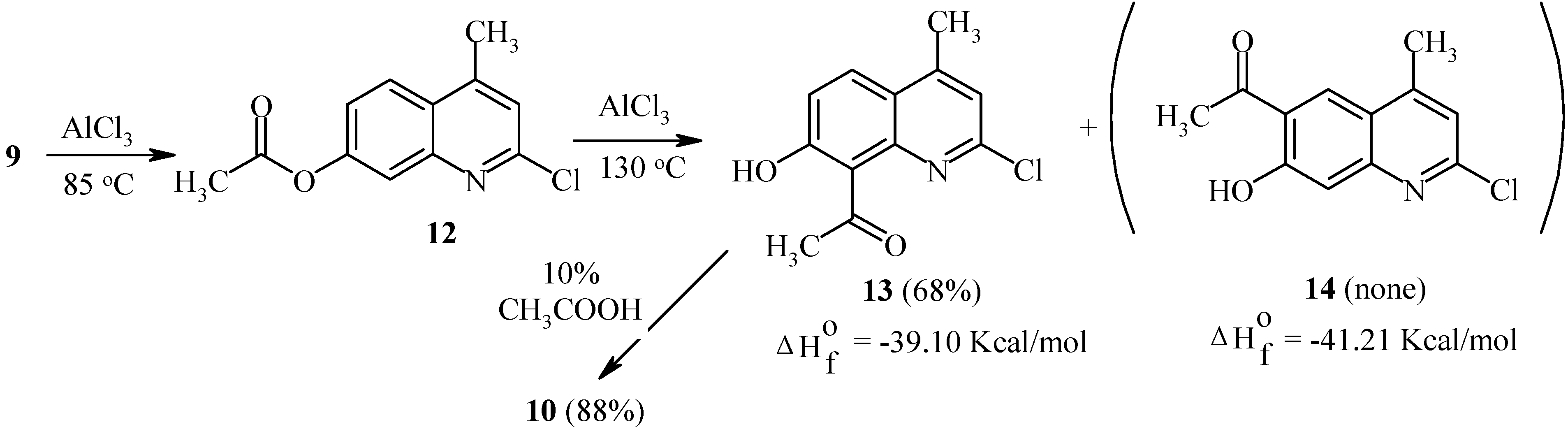
As can be seen from Scheme 5, isomers 13 and 14 exhibit similar calculated thermodynamic stabilities. Therefore, the exclusive formation of 13 appears to be a result of a kinetically controlled rearrangement. As already noted, a similar selectivity has been observed for acetoxycoumarin 1.
The following conclusions concerning regioselectivity of the acyloxyheteroarenes Fries rearrangement agree with the given results. If two isomers, the products of the rearrangement, have different thermodynamic stabilities, one can see a definite effect of the reaction temperature on the regioselectivity. If two isomeric products of the rearrangement possess similar thermodynamic stabilities, then the regioselectivity seems to be independent of the reaction temperature.
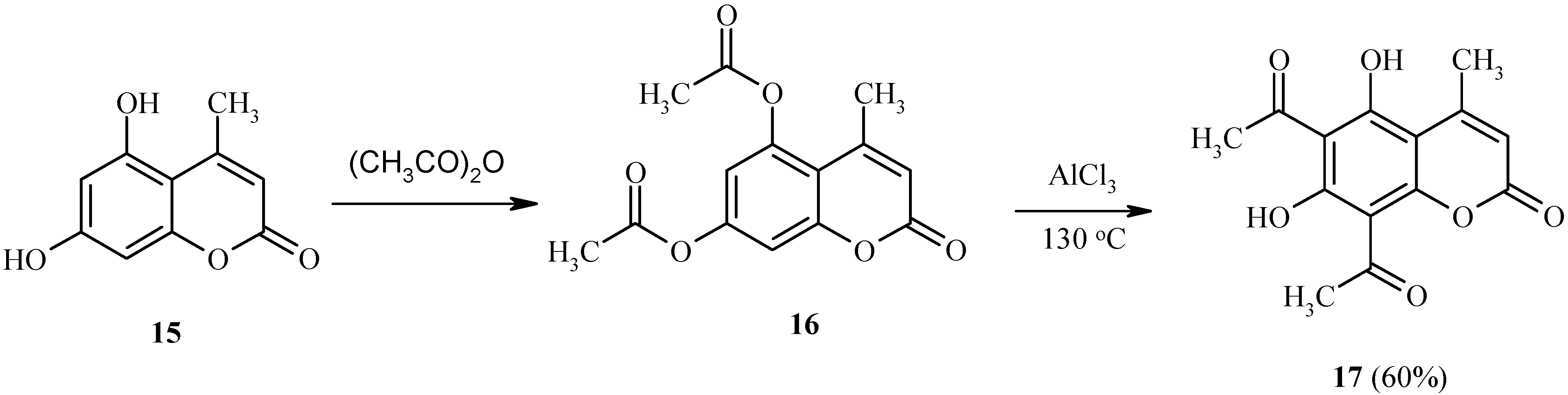
An interesting example of the Fries rearrangement is provided by the transformation of 5,7-bis(acetoxy)-4-methylcoumarin (16), derived from dihydroxycoumarin 15 (Scheme 6). In the rearrangement of 16, two acetoxy functions are simultaneously rearranged in the same molecule to give 17. In summary, the Fries reaction seems to be a convenient method for the synthesis of heteroarenes with adjacent acyl and hydroxyl functions.
Scheme 6.

Unusual Fries rearrangements of chloroacetoxyheteroarenes
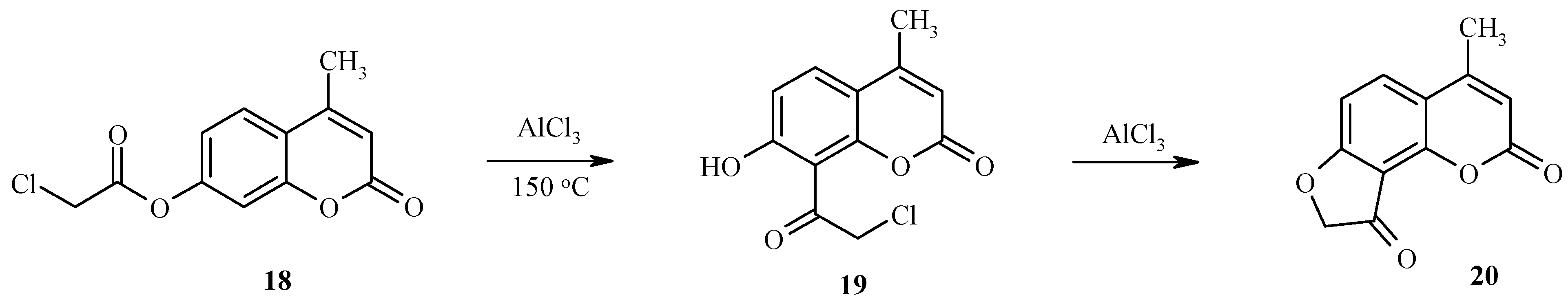
Unusual results have been found for the treatment of 7-chloroacetoxyheteroarenes with excess aluminum trichloride [22]. Unexpectedly, the classical rearrangement is followed by an intramolecular cyclization of an intermediate chloroacetyl hydroxy derivative with formation of a condensed dihydrofuranone. As an example, the rearrangement of 7-chloroacetoxy-4-methylcoumarin (18) to give the final product 20 via the intermediate 19 is shown in Scheme 7.
Scheme 7.
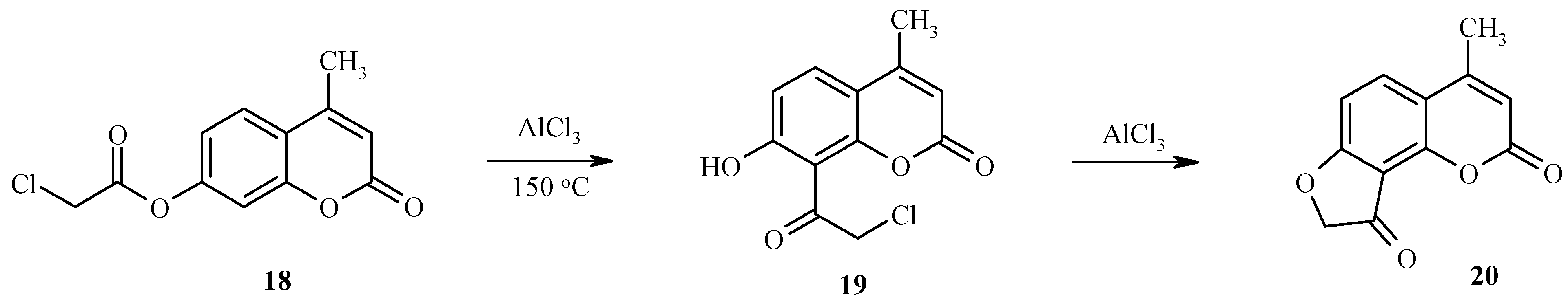
Thus, compound 18 and similar 7-chloroacetoxy substituted coumarins are important substrates for the synthesis of angular furocoumarins.
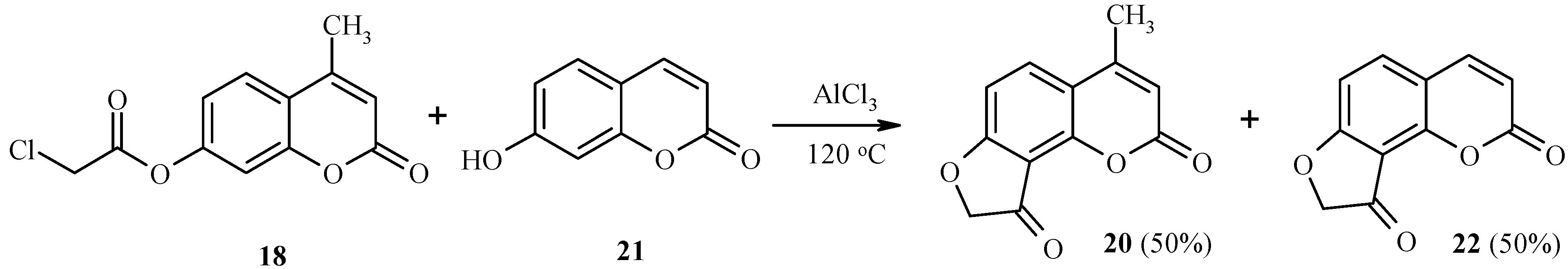
As with 7-acetoxycoumarins, 7-chloroacetoxycoumarins undergo this unusual Fries rearrangement by an intermolecular mechanism as well. Thus, rearrangement of 18 in the presence of 7-hydroxy-4-methylcoumarin (21) gives equimolar amounts of a 4-methyl derivative 20 and its analog 22, lacking the methyl group (Scheme 8) [20].
Scheme 8.

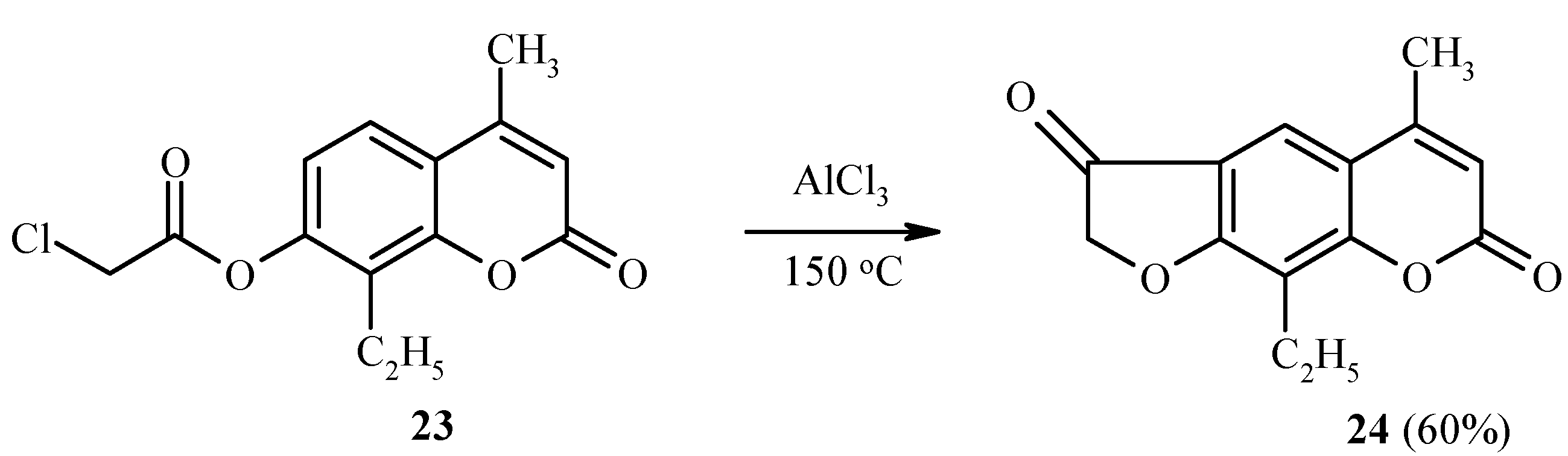
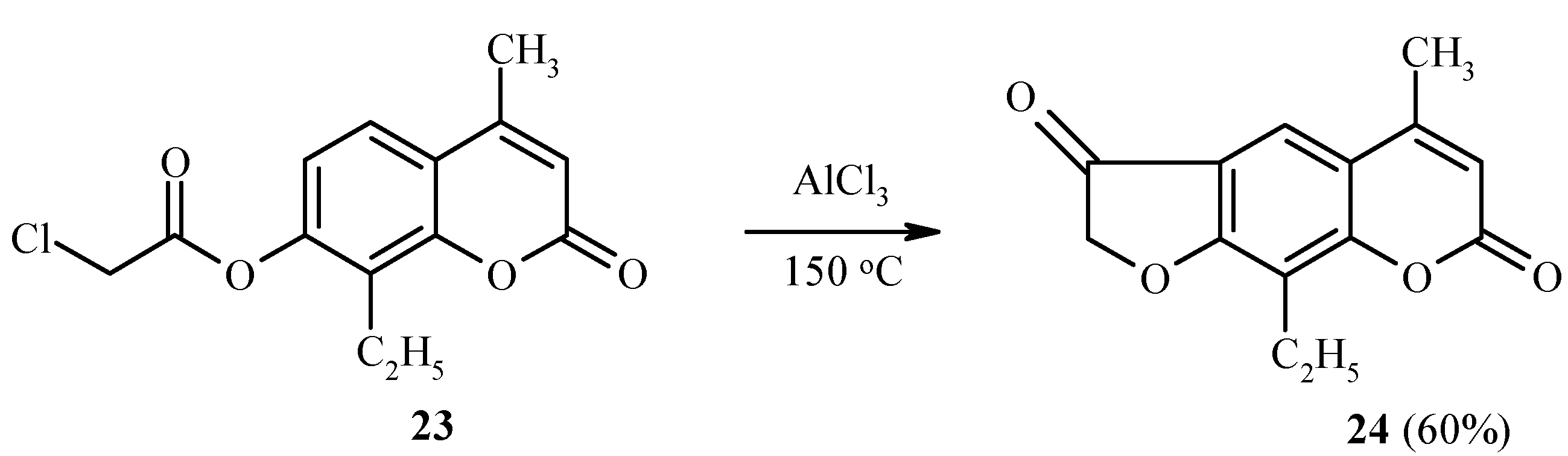
Substrates which have a substituent at position 8 of the coumarin ring system, undergo rearrangement into position 6 [22]. This is illustrated in Scheme 9 by the synthesis of 24 from 23.
Scheme 9.
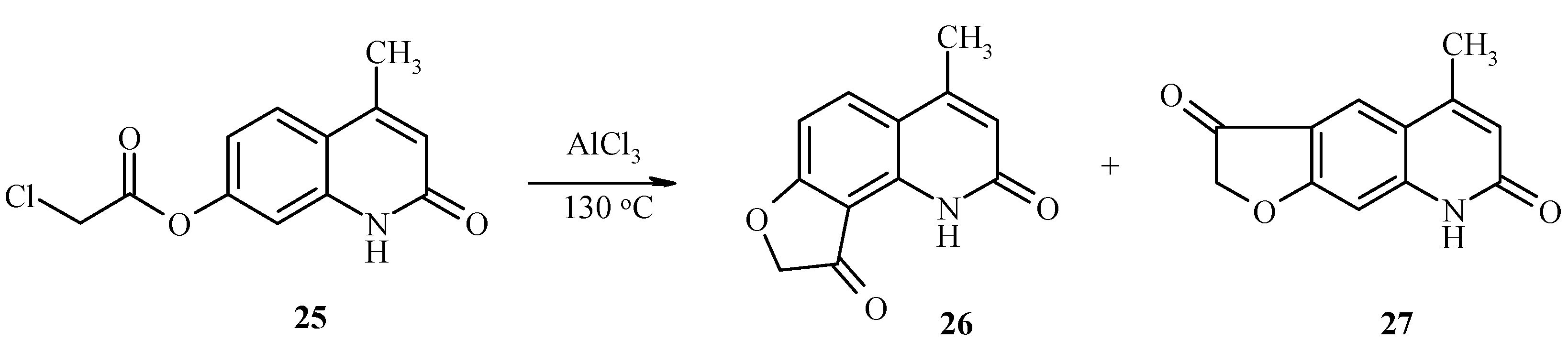
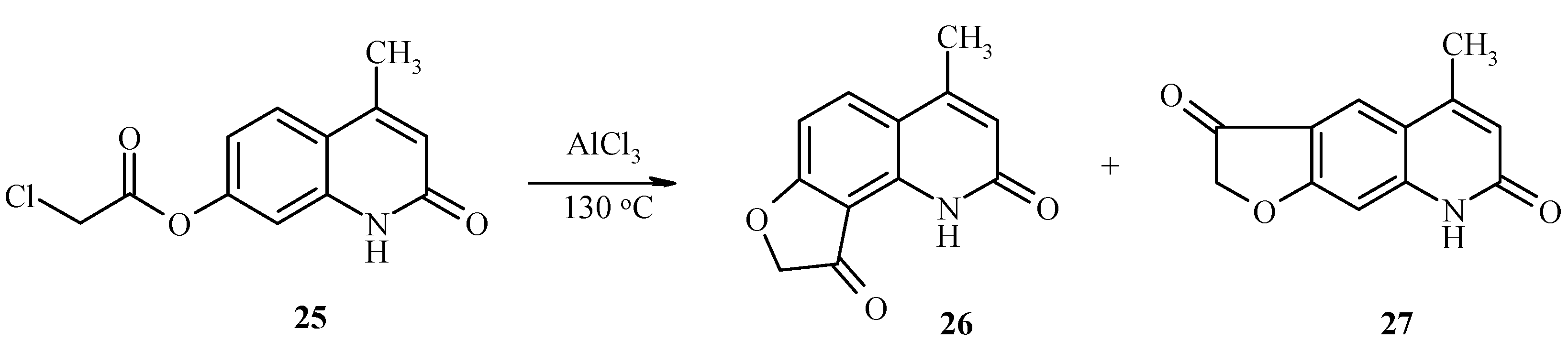
7-Chloroacetoxy-4-methylquinolin-2-one (25) also undergoes the a Fries rearrangement that is followed by an intramolecular cyclization to give a mixture of furoquinolines 26 and 27 (Scheme 10). Due to low regioselectivity, this reaction does not have much synthetic value.
Scheme 10.
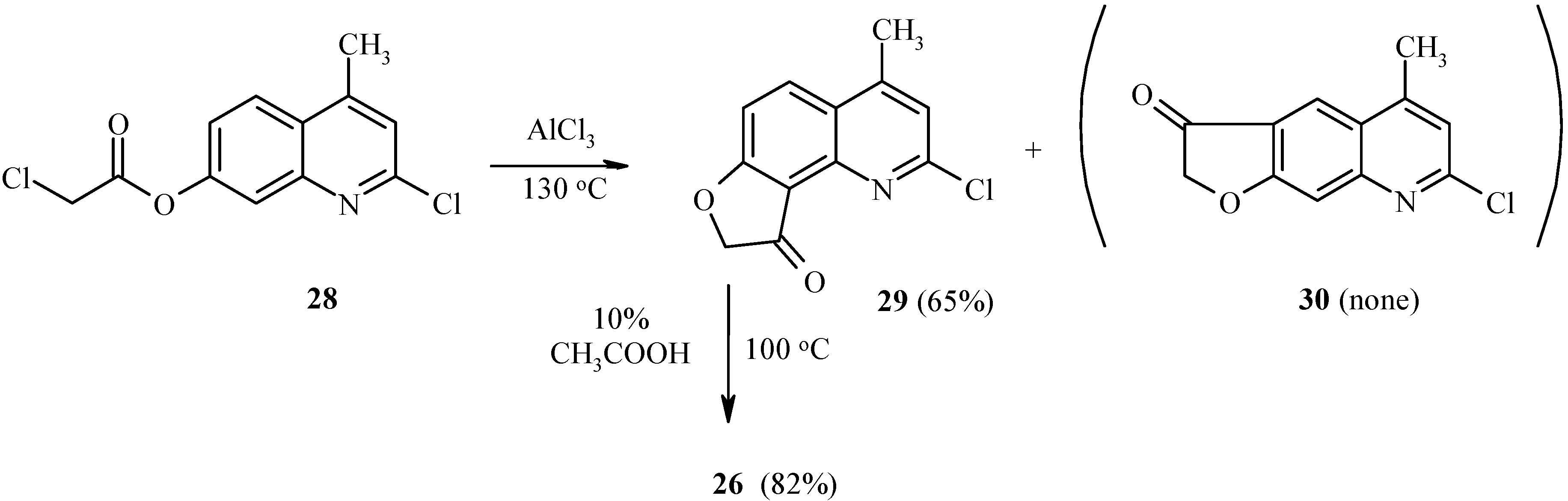
As with 7-acetoxy derivatives, regioselectivity of the Fries rearrangement of 7-chloroacetoxy-substituted substrates is much affected by substitution of the chlorine atom for the 2-oxo function. Exclusive formation of 29 without traces of isomer 30 by rearrangement of 28 can be noted (Scheme 11). The chlorine atom in 29 can be replaced by an oxo or amino function, as exemplified by hydrolysis of 29 to furoquinolinone 26.
Scheme 11.

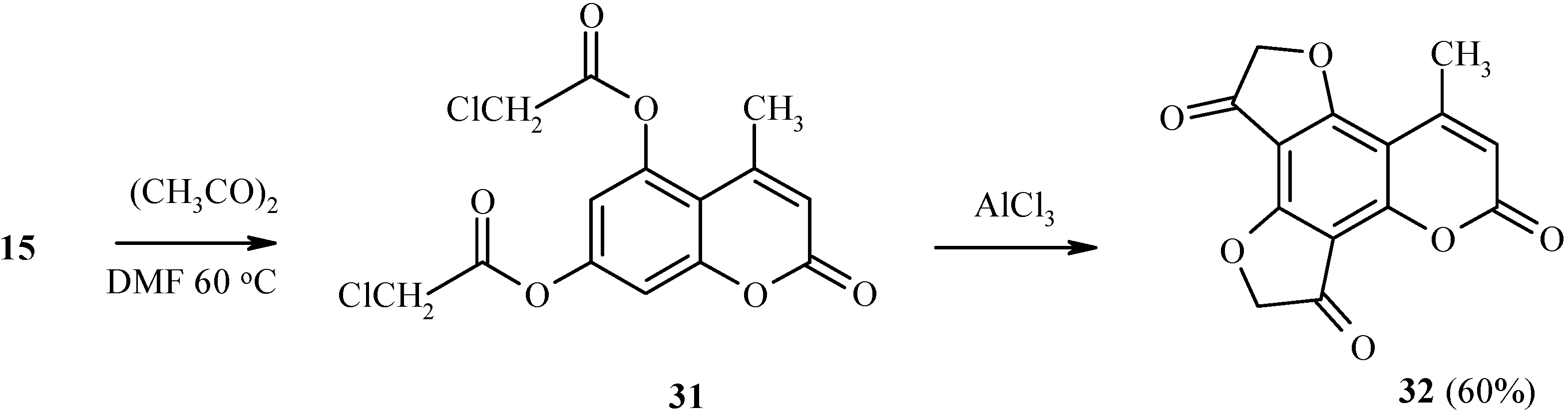
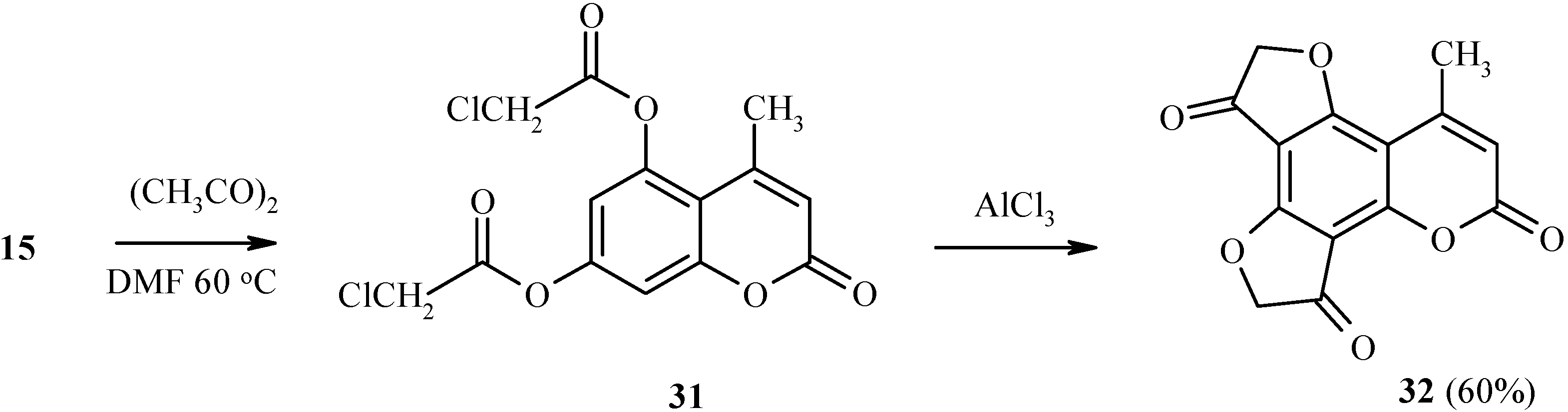
Two dihydrofuranone rings in the same heteroarene molecule can be constructed under Fries rearrangement as well (Scheme 12). For example, 5,7-bis(chloroacetoxy)coumarin 31 has been prepared by chloroacetylation of compound 15. Heating of 31 with a large excess of aluminum trichloride gave 32 in good yield.
Scheme 12.
Base-catalyzed condensation of acetylhydroxyheteroarenes with α-halocarbonyl compounds
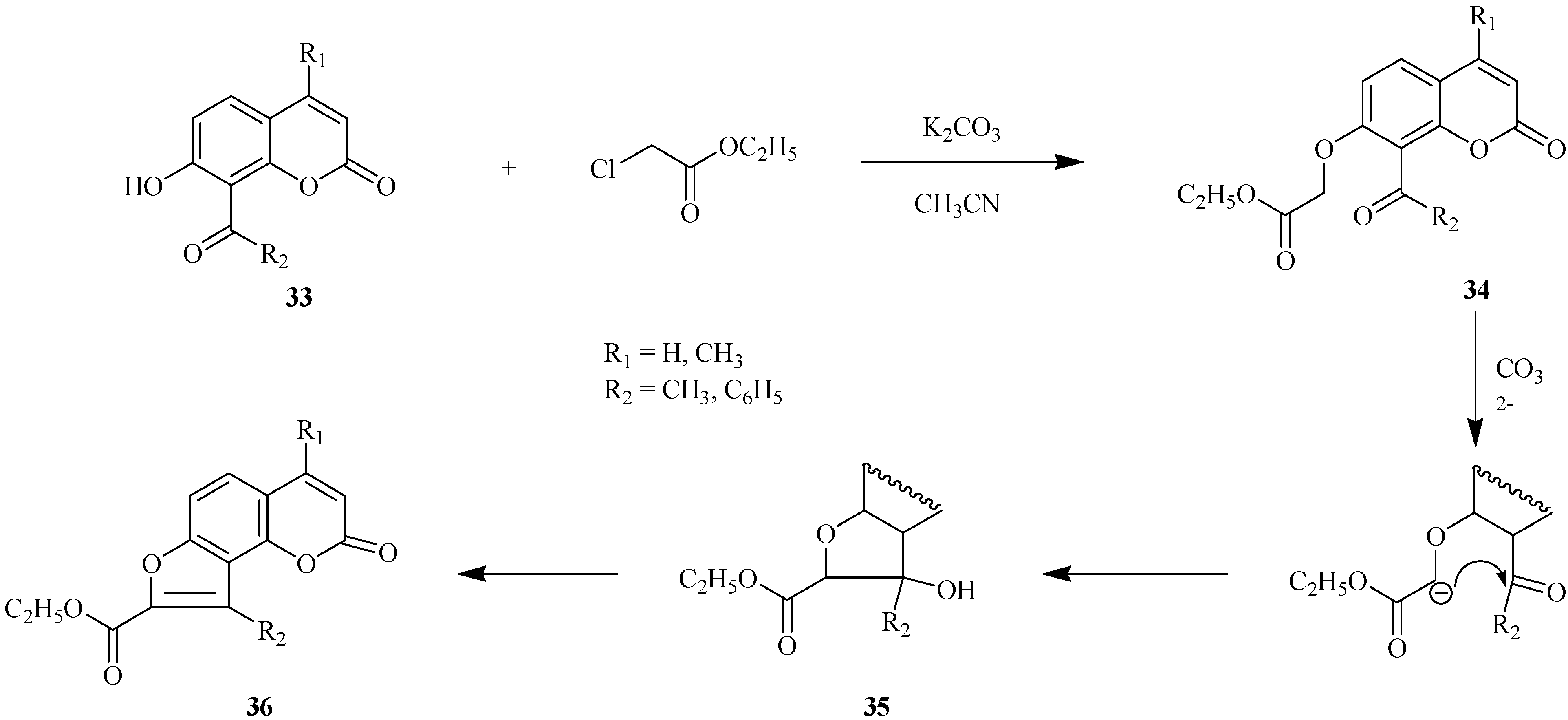
Base-catalyzed condensation of acetylhydroxyheteroarenes with α-halocarbonyl compounds is a preparative method for the construction of a furan ring in condensed heteroarenes [23,24]. This approach is shown in Scheme 13 for condensation of 8-acyl-7-hydroxycoumarins 33 with ethyl chloroacetate.
Scheme 13.
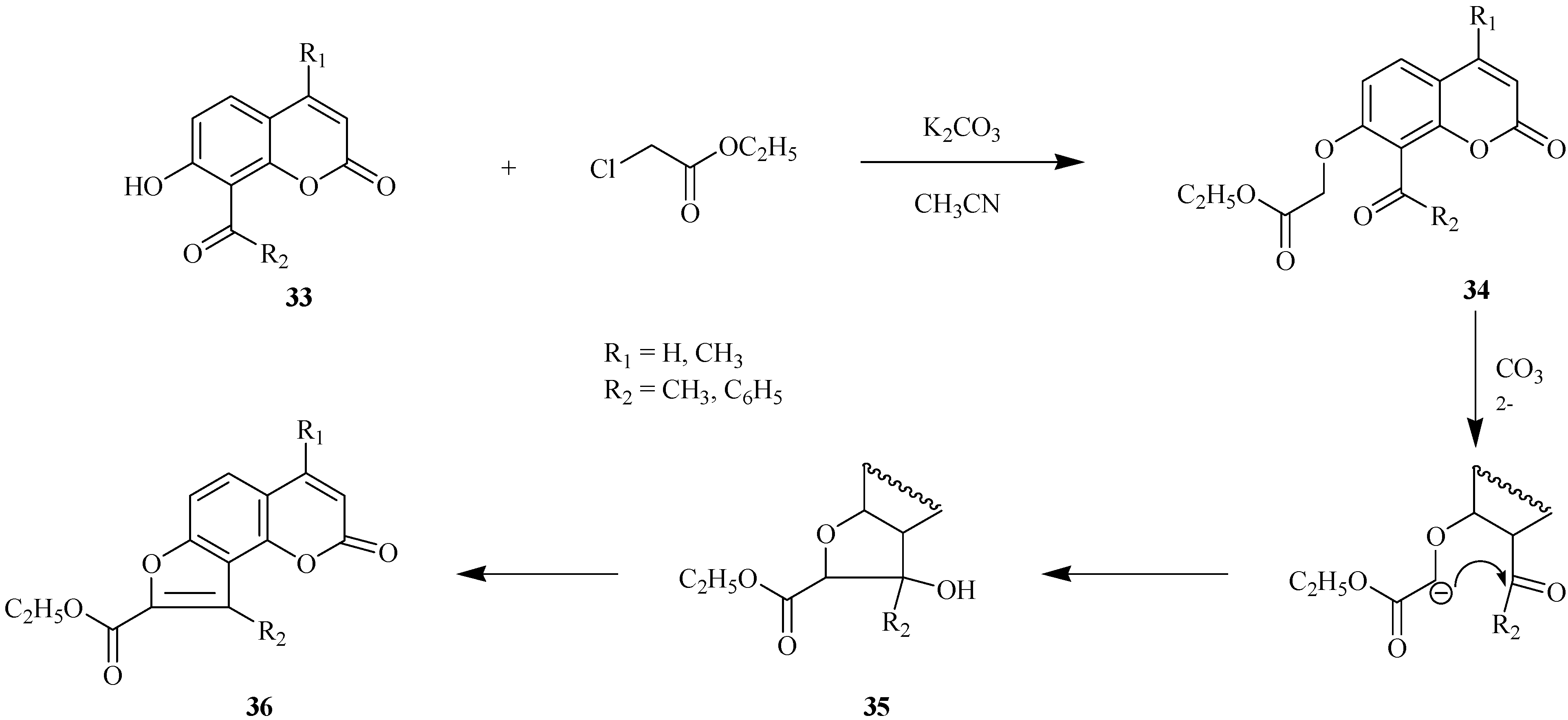
Ethyl coumarinyloxyacetate 34 is formed in the first step of the reaction. This step is followed by ionization of the oxymethylene group and intramolecular cyclization with successive formation of intermediate 35. Compound 35 undergoes dehydration to 8-(ethoxycarbonyl)furo[2,3-h]coumarin 36 which is an angelicine analog. These transformations provide a convenient way for preparation of furocoumarins with hydrophilic functional groups at the furan ring of the final furoheteroarene.
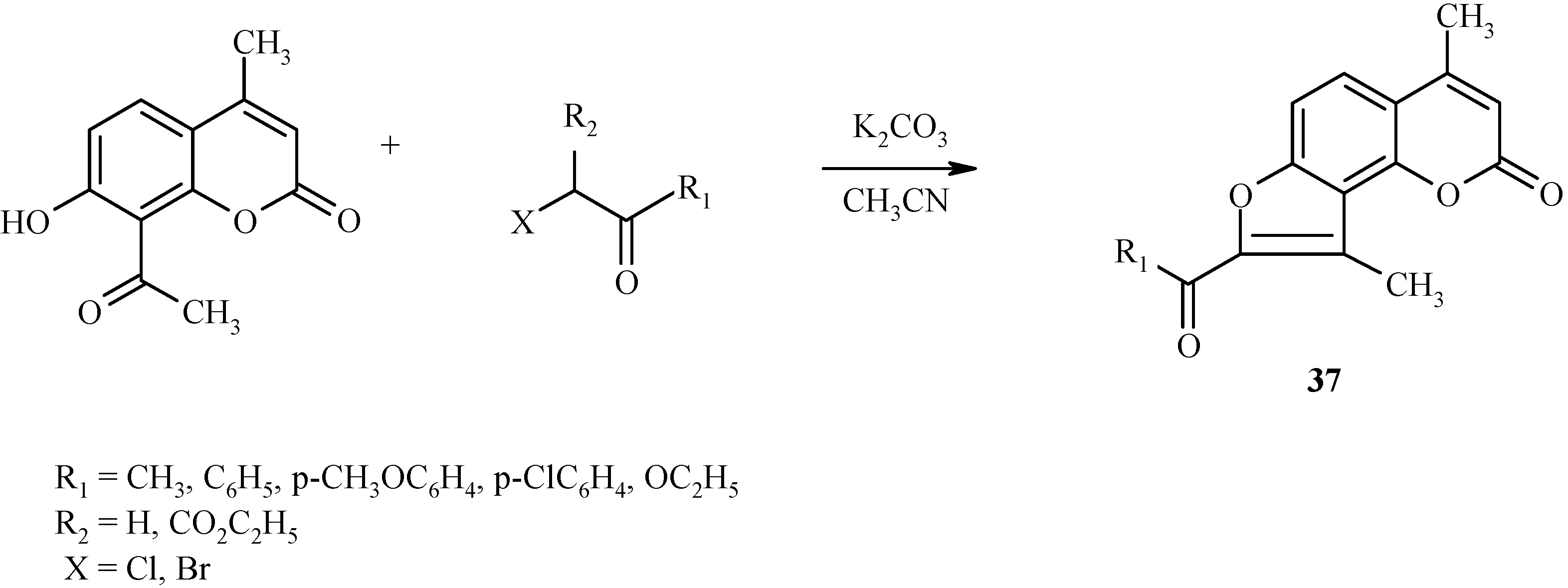
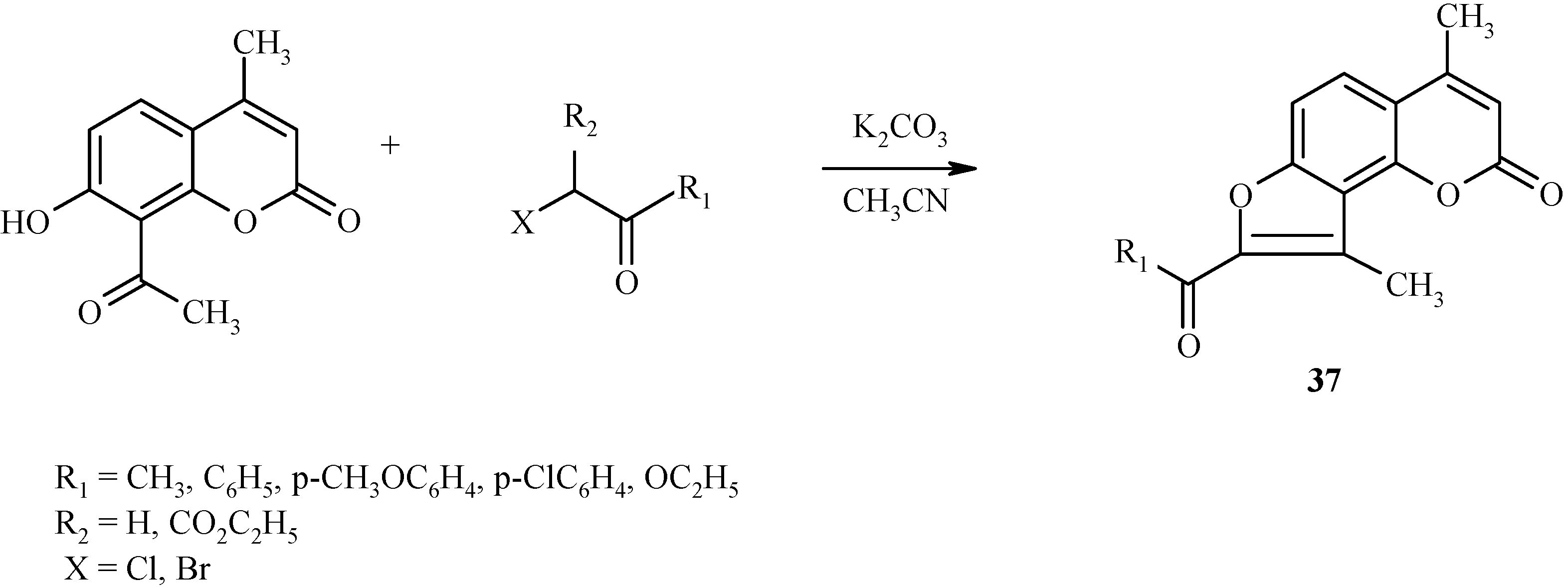
A similar condensation takes place with α-haloketones, as shown in Scheme 14 for the preparation of angelicine derivative 37.
![Molecules 09 00050 g014]() Base-catalyzed condensation of acylhydroxycoumarins with ethyl chloroacetate is also a useful reaction for preparation of psoralen derivatives, as exemplified in Scheme 15 by the synthesis of a substituted psoralen 38.
Base-catalyzed condensation of acylhydroxycoumarins with ethyl chloroacetate is also a useful reaction for preparation of psoralen derivatives, as exemplified in Scheme 15 by the synthesis of a substituted psoralen 38.
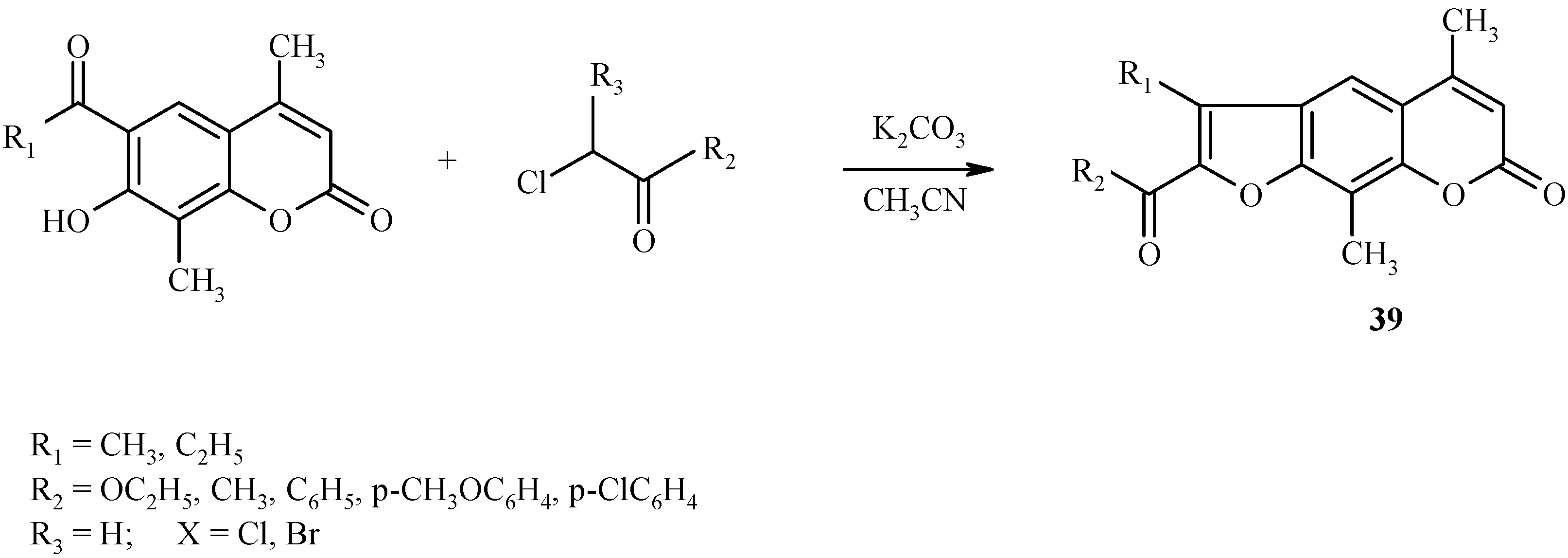
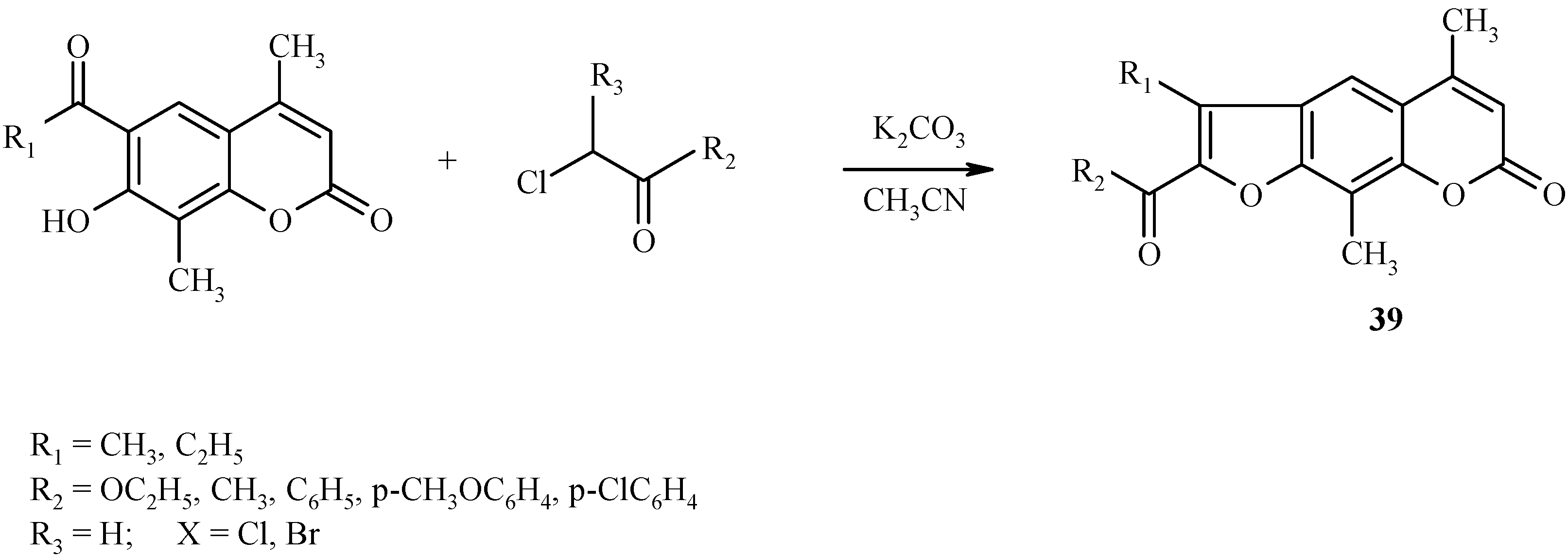
![Molecules 09 00050 g015]() Similar condensations leading to other psoralen derivatives 39 are shown in Scheme 16.
Similar condensations leading to other psoralen derivatives 39 are shown in Scheme 16.
Scheme 14.
Scheme 15.
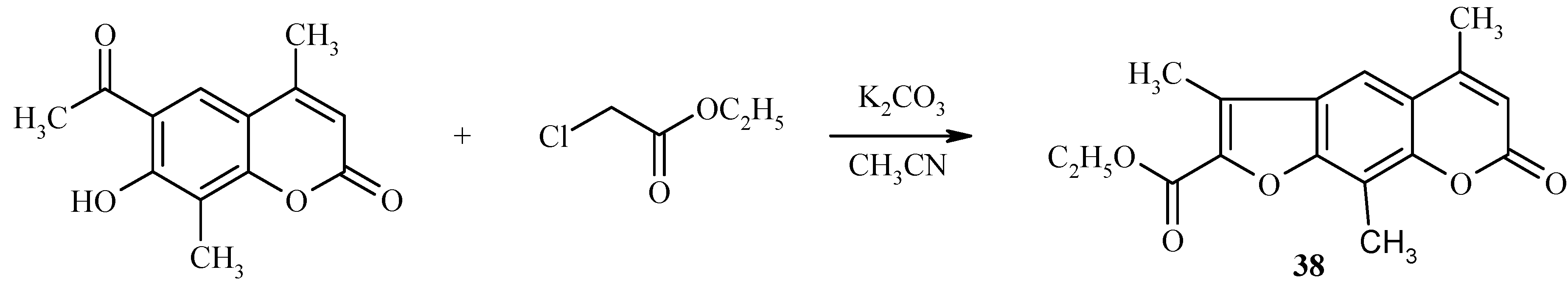
Scheme 16.
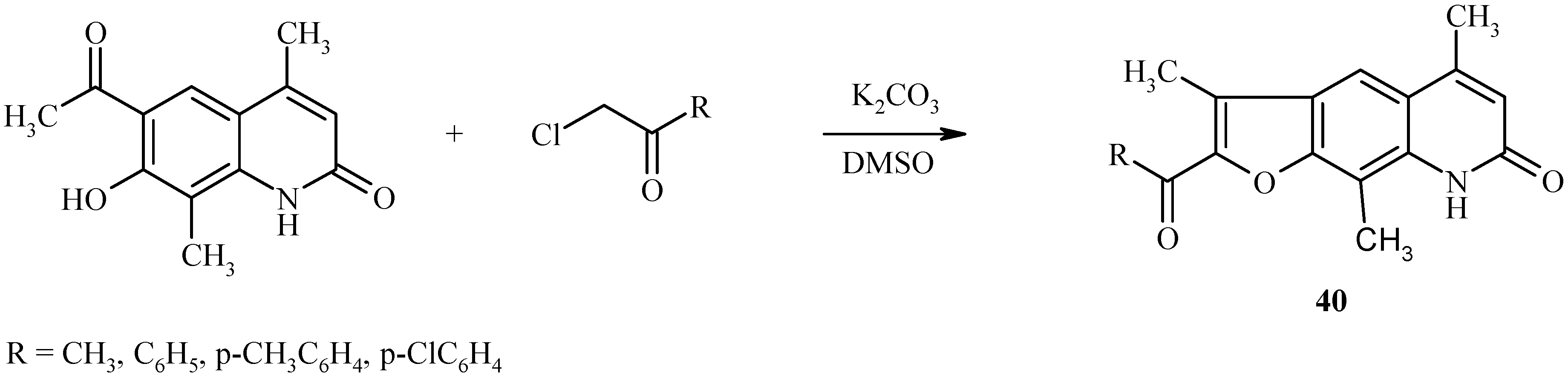
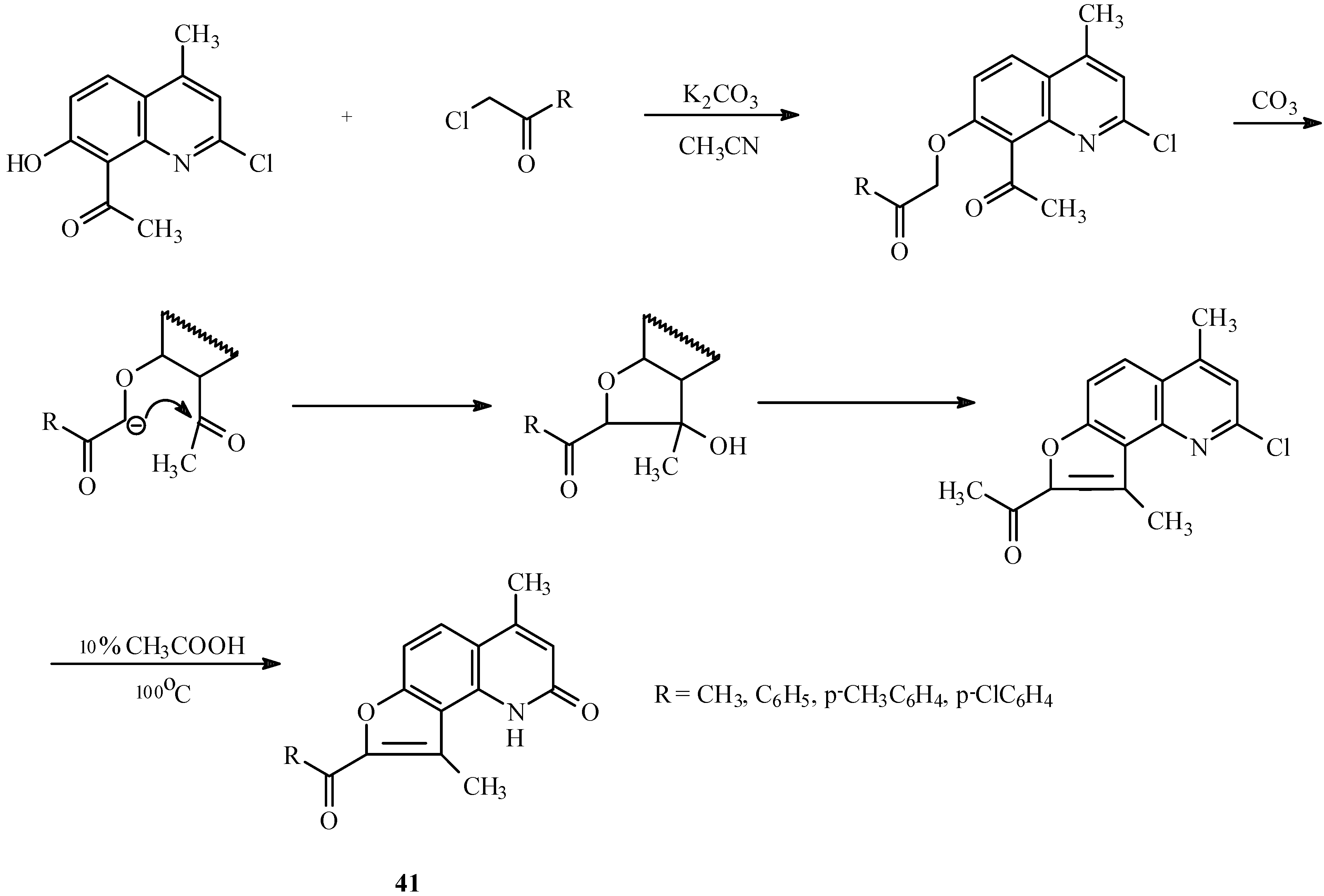
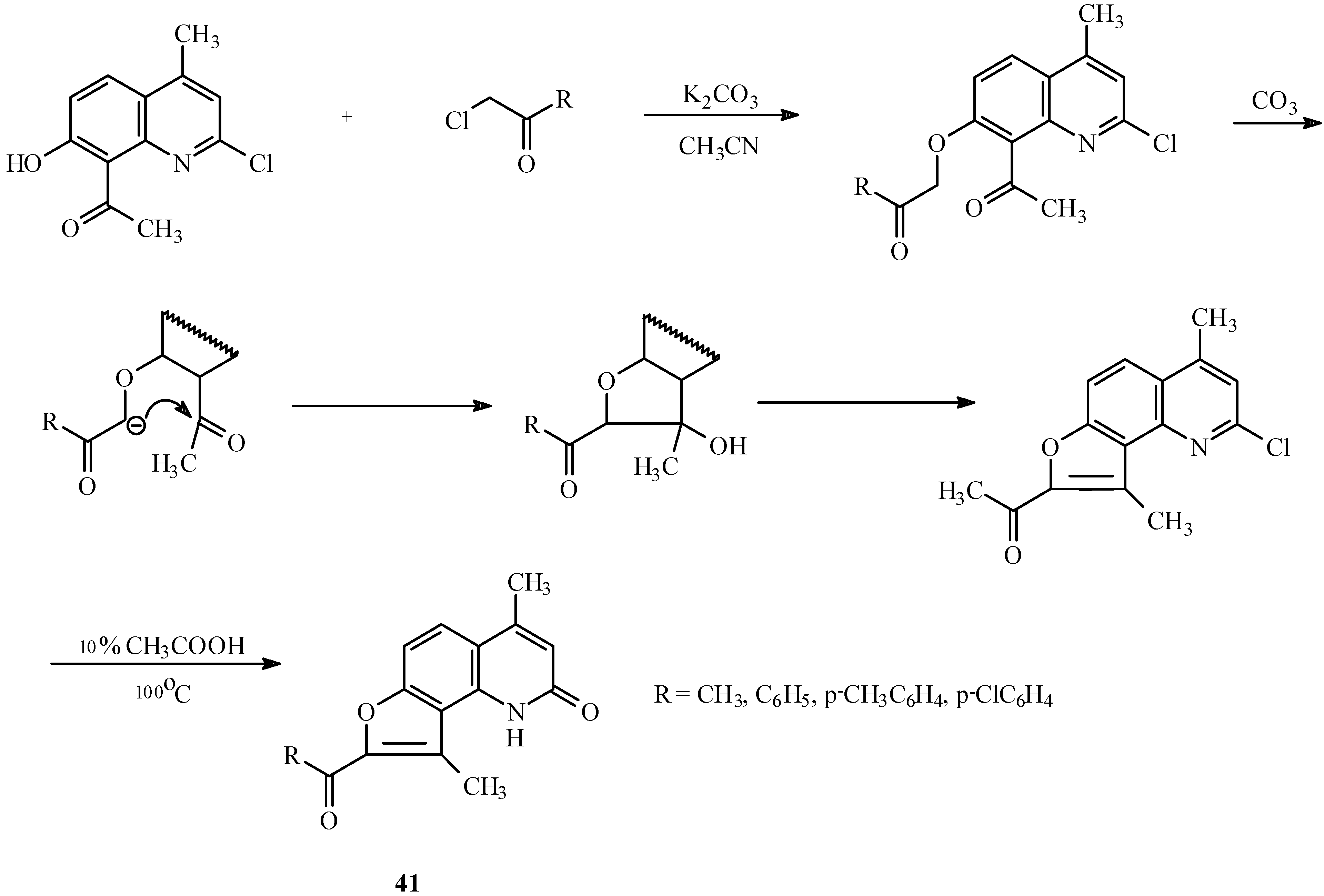
The base-catalyzed condensation of the acyl/hydroxyl functionality with α-halocarbonyl compounds can also be used for the synthesis of other fused furoheteroarenes as well (23, 24). For example, both linear (40, Scheme 17) and angular furoquinolines (41, Scheme 18) have been prepared by this method.
Scheme 17.

Scheme 18.
Dihydrofuroheteroarenes – key intermediates of condensed furoheteroarenes
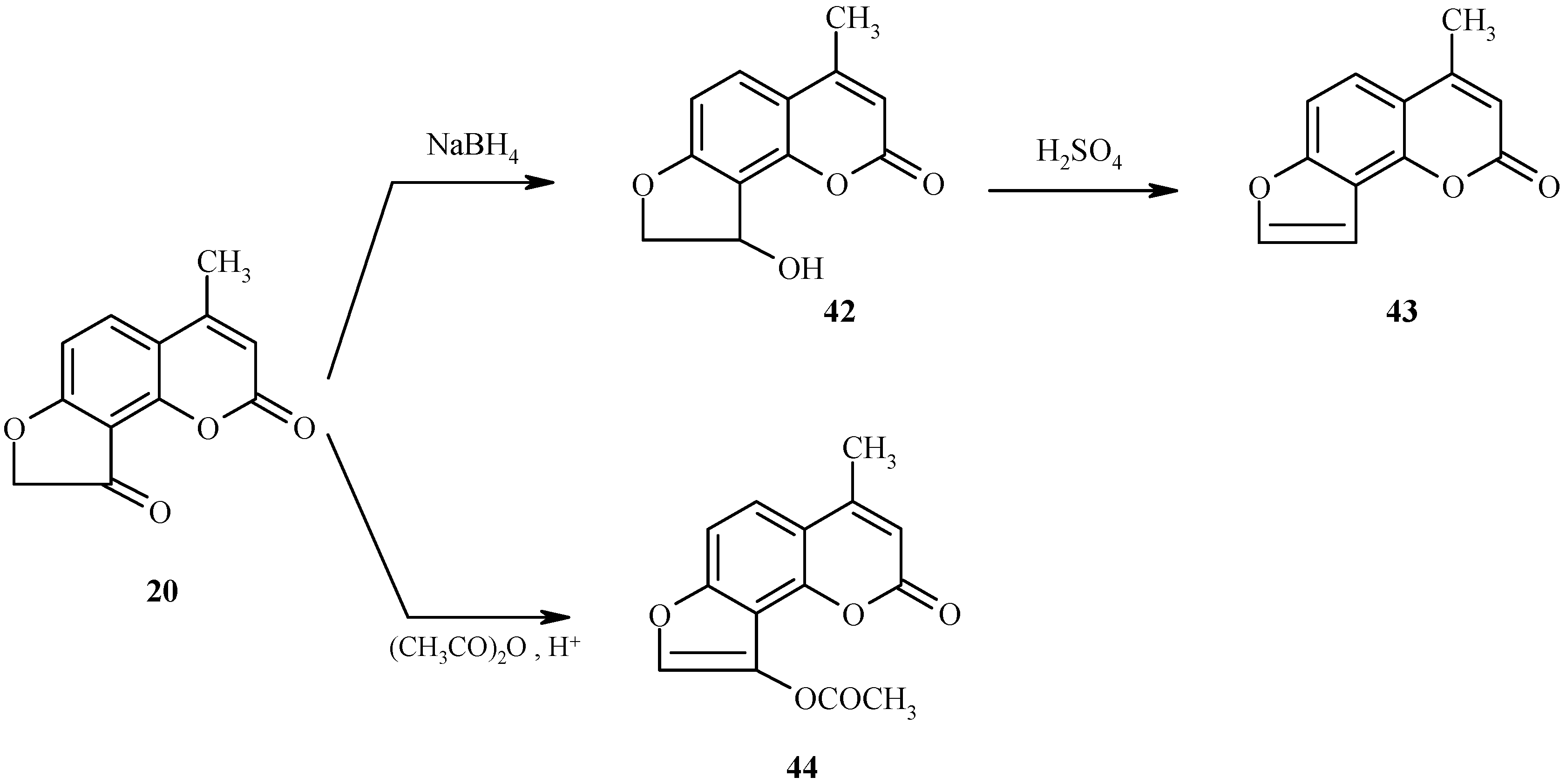
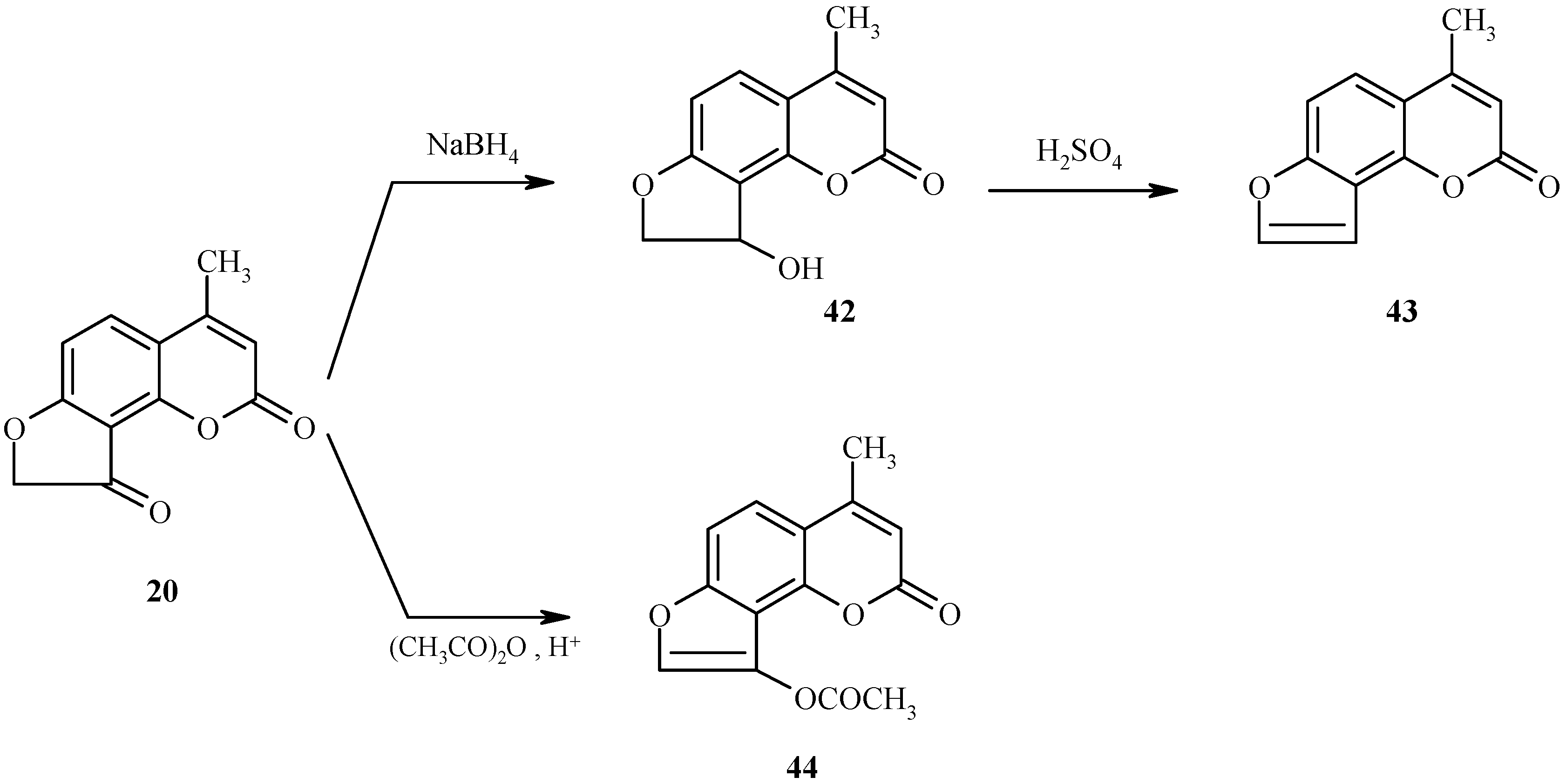
The transformations discussed above proceed with the intermediacy of dihydrofuroheteroarenes (for an example, see 35 in Scheme 13). We have shown that other dihydrofuroheteroarenes can efficiently be aromatized to condensed furoheteroarenes [25]. Two independent examples are shown in Scheme 19. As can be seen, reduction of the keto function in 20 followed by dehydration of the resultant alcohol 42 gives furocoumarin 43. In the second example, the acetoxy-substituted furocoumarin 44 is produced by acetylation of the enol form of 20.
Scheme 19.
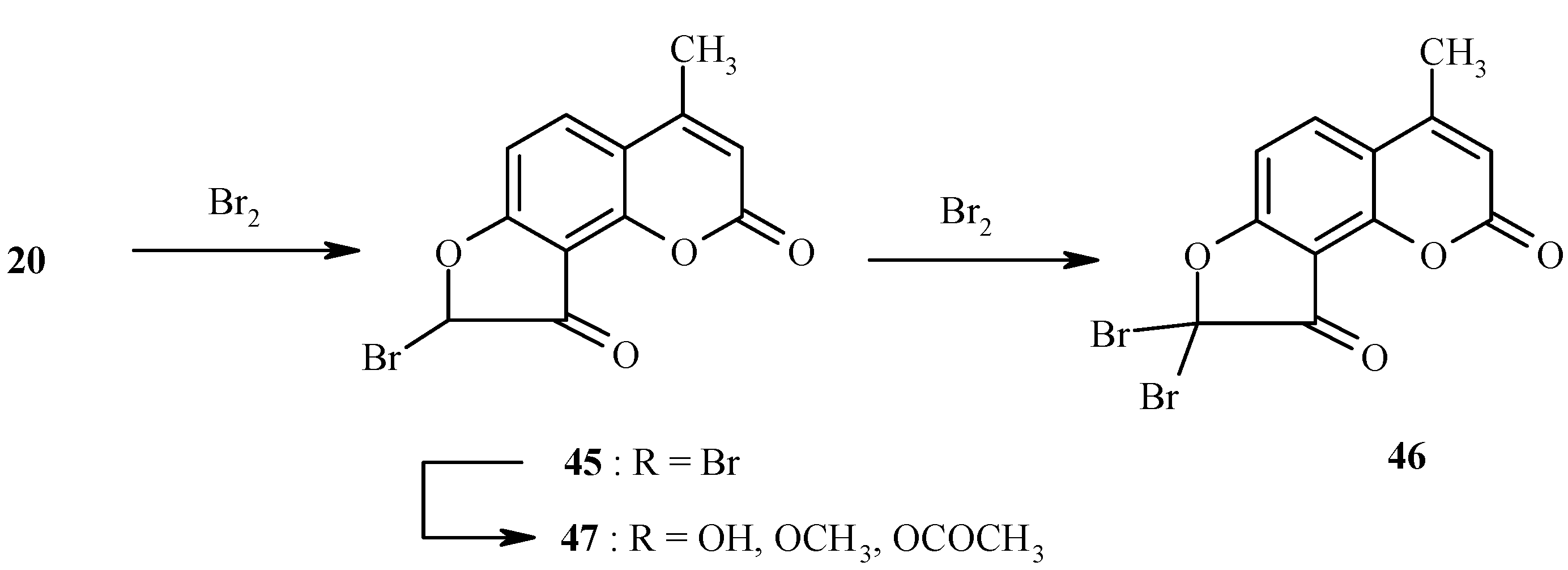
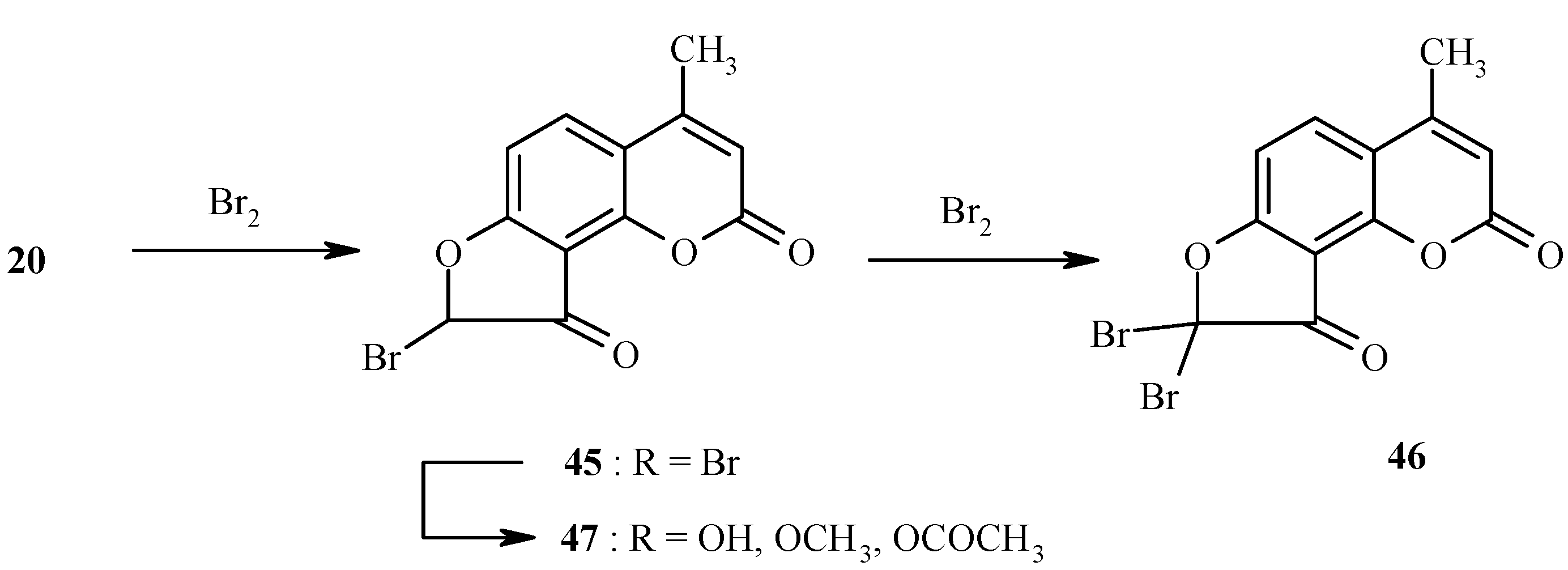
Ketones, such as 20, are easily halogenated at the position α- to the carbonyl group. Chlorination and bromination have been performed [26]. For example, bromination of 20 to give 46 through the intermediacy of 45 is quantitative in dry dioxane (Scheme 20). Depending on the reaction conditions, compound 45 can also be isolated in high yield. Electrophilic formylation of dihydrofurocoumarinone 20 has been carried out as well. The bromine atom in 45 is highly reactive in nucleophilic displacement reactions (Scheme 20) [26].
Scheme 20.
To a large extent, the reactivity of dihydrofurocoumarinones such as 20, 45 or 47 is based on their keto-enol tautomerism (Scheme 21). Tautomeric transformations of these compounds have been studied by spectral and computational methods [27,28].
Scheme 21.
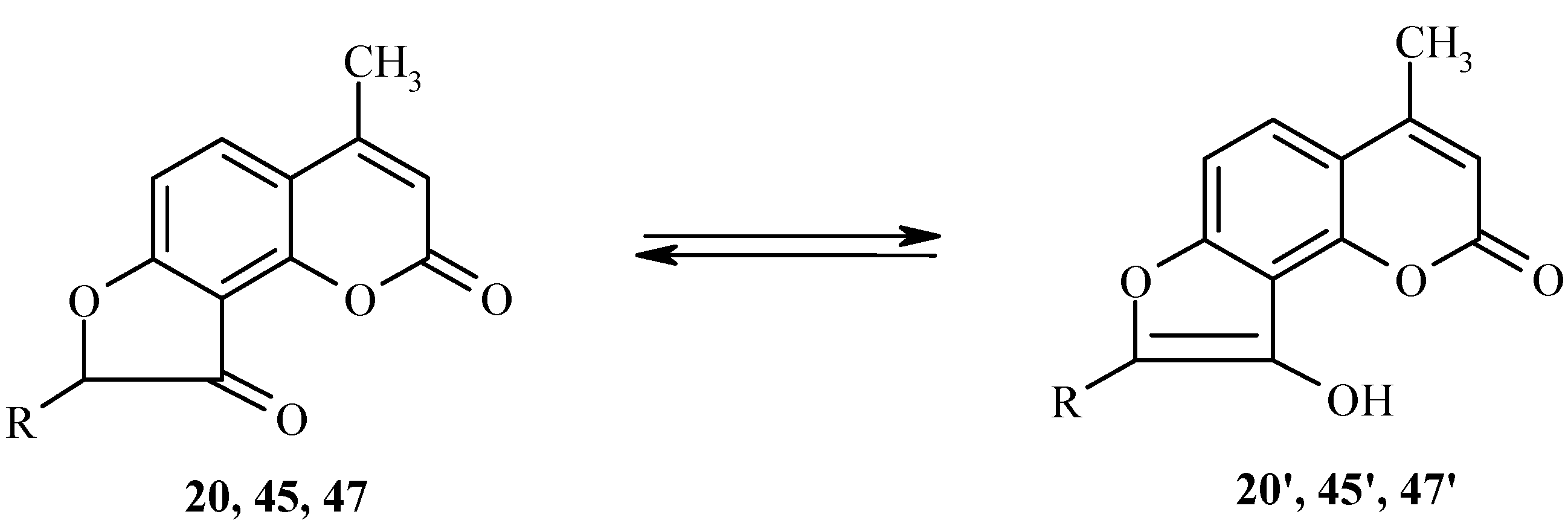
For example, the longest-wavelength absorption band in the UV spectrum of 20 is at 330 nm in methanol and at 285 nm in carbon tetrachloride. The bands at 330 nm and 285 nm have been assigned to the enol form 20’ and the keto form 20, respectively. Both semiempirical and nonempirical quantum mechanical calculations are fully consistent with these assignments.
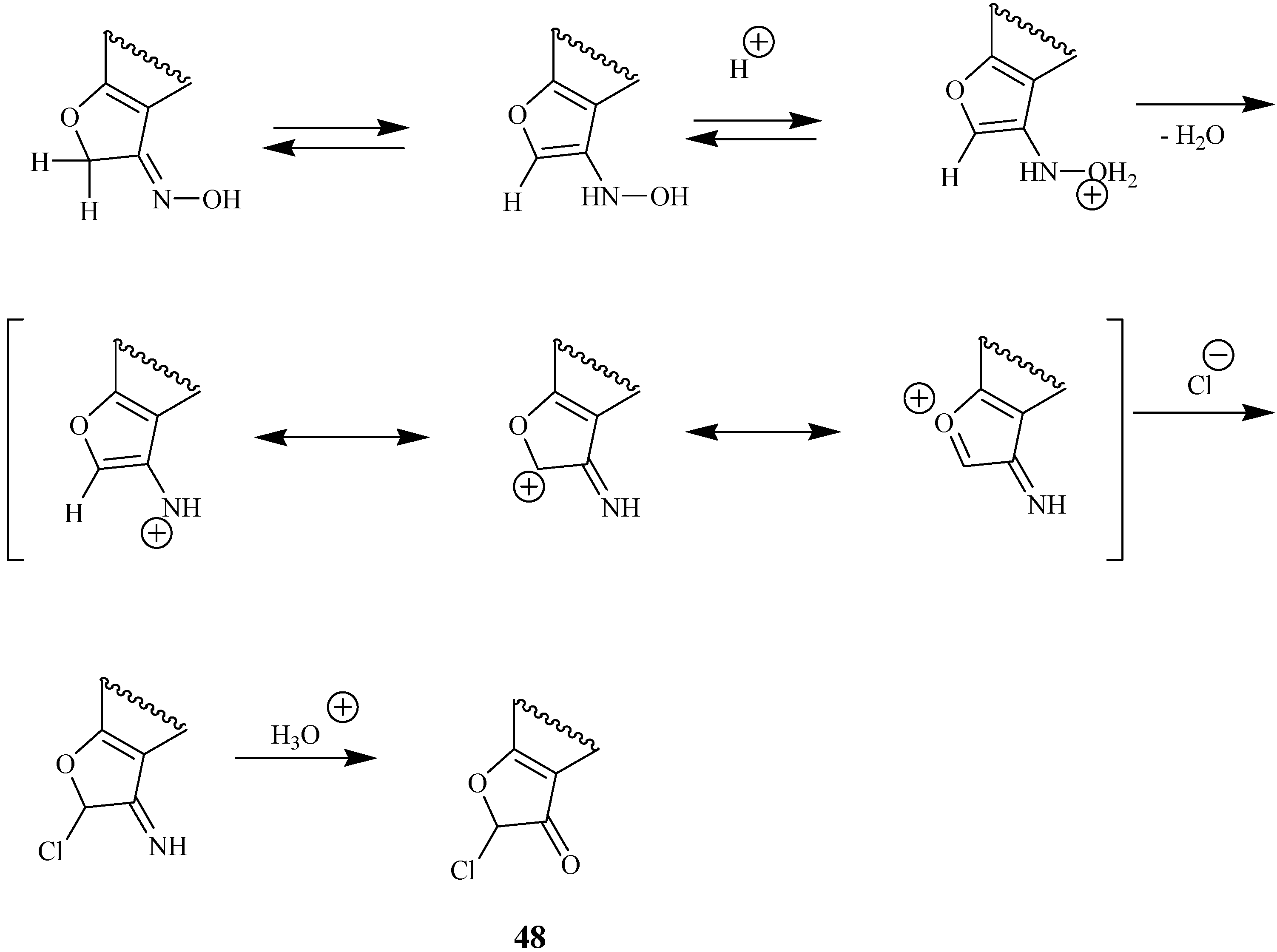
Tautomeric transformations of the oxime of 20 are the key steps of its halogenation in the presence of hydrogen chloride or hydrogen bromide in acetic acid [29,30,31]. The chlorination which furnishes α-chloroketone 48 is shown in Scheme 22.
Scheme 22.

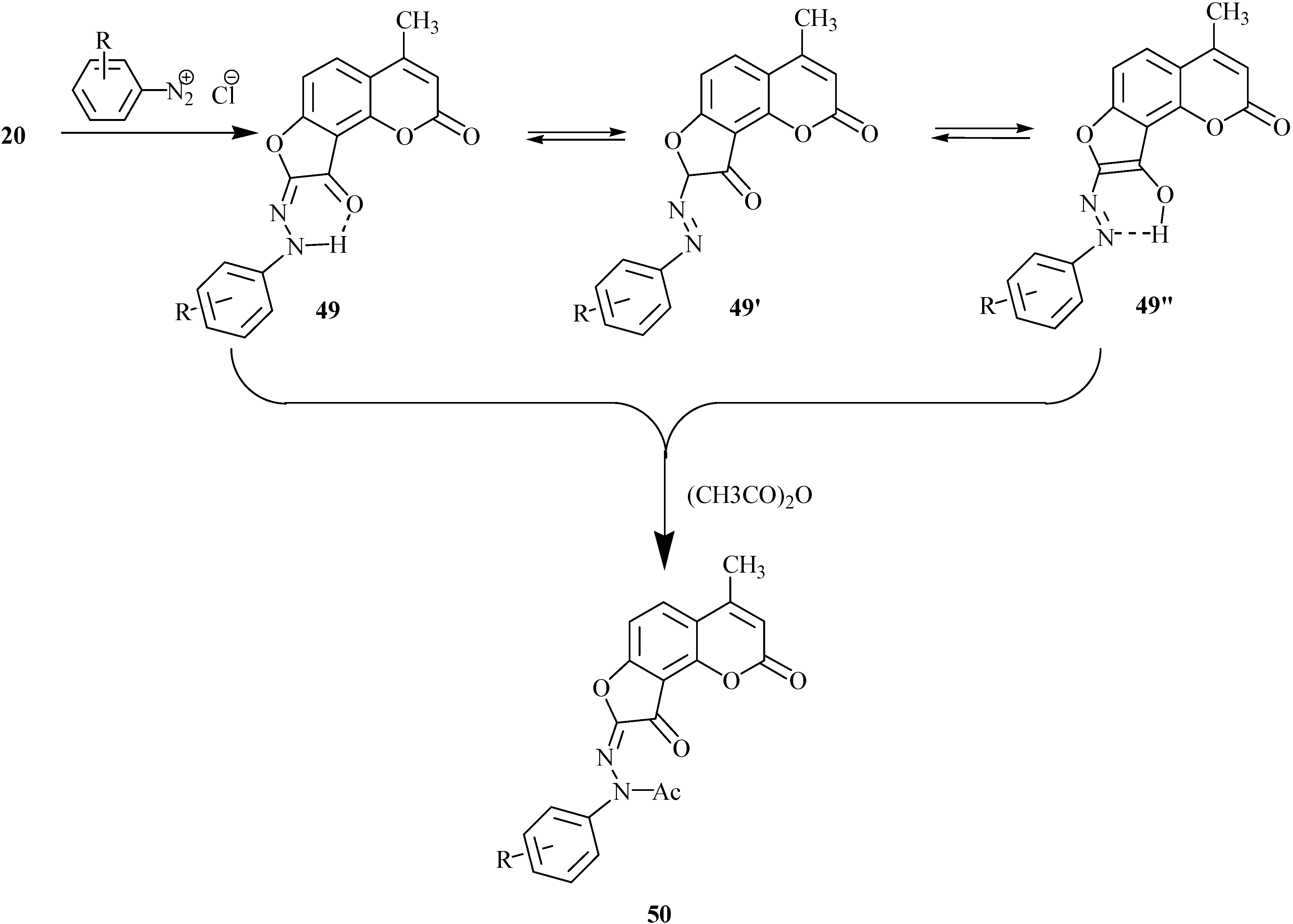
One of the useful synthetic transformations of dihydrofurocoumarinone 20 is the coupling reaction with arenediazonium salts [32,33]. Depending on the solvent and pH conditions, the substitution takes place at the 8 or 6 positions. The coupling in an acetic acid-dioxane mixture in the presence of a weak base (pyridine, sodium acetate) involves the 8 position and yields product 49 as a major tautomer (Scheme 23). This conclusion was derived from spectral studies and supported by quantum mechanical calculations. Acetylation of the tautomeric mixture 49, 49’ and 49’’ gave a single acetyl derivative 50.
Scheme 23.

Dihydrofurocoumarinones as intermediates of angelicine analogs
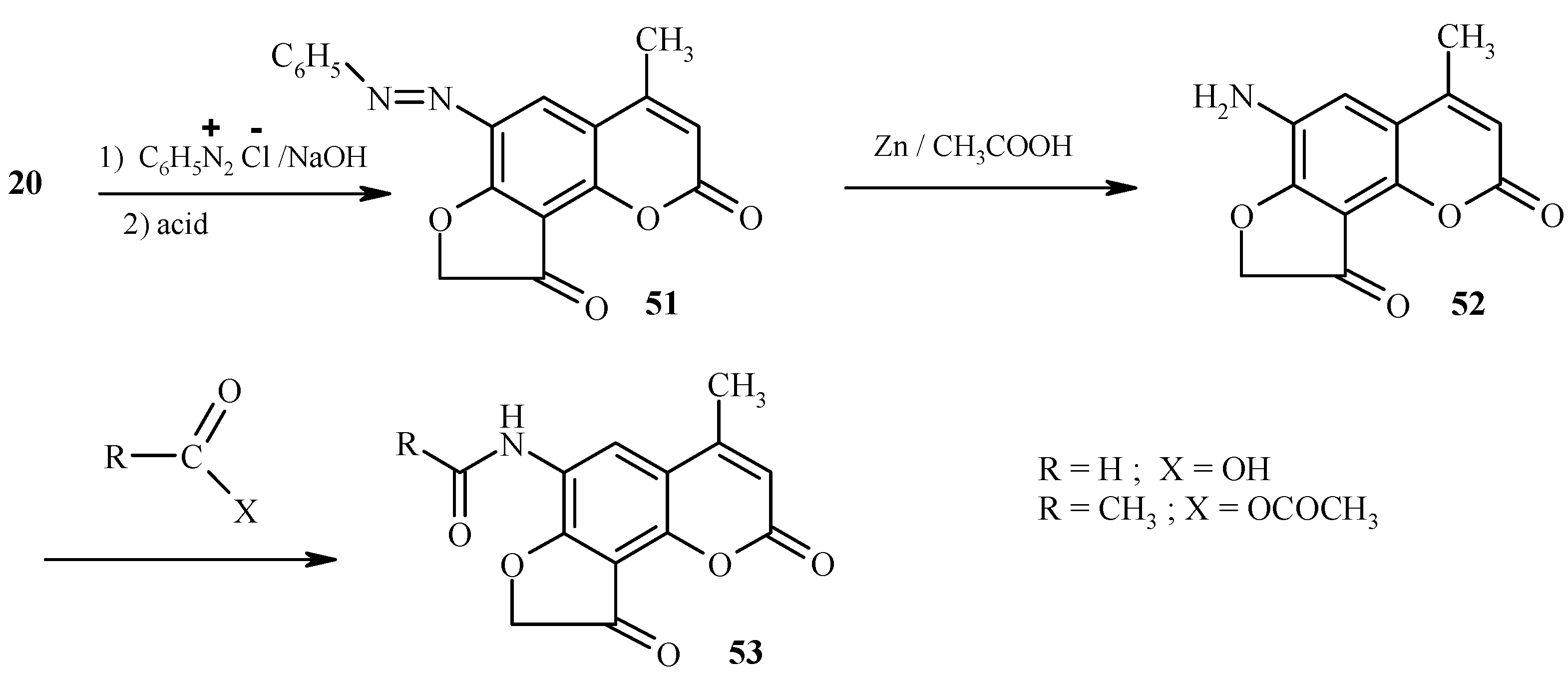
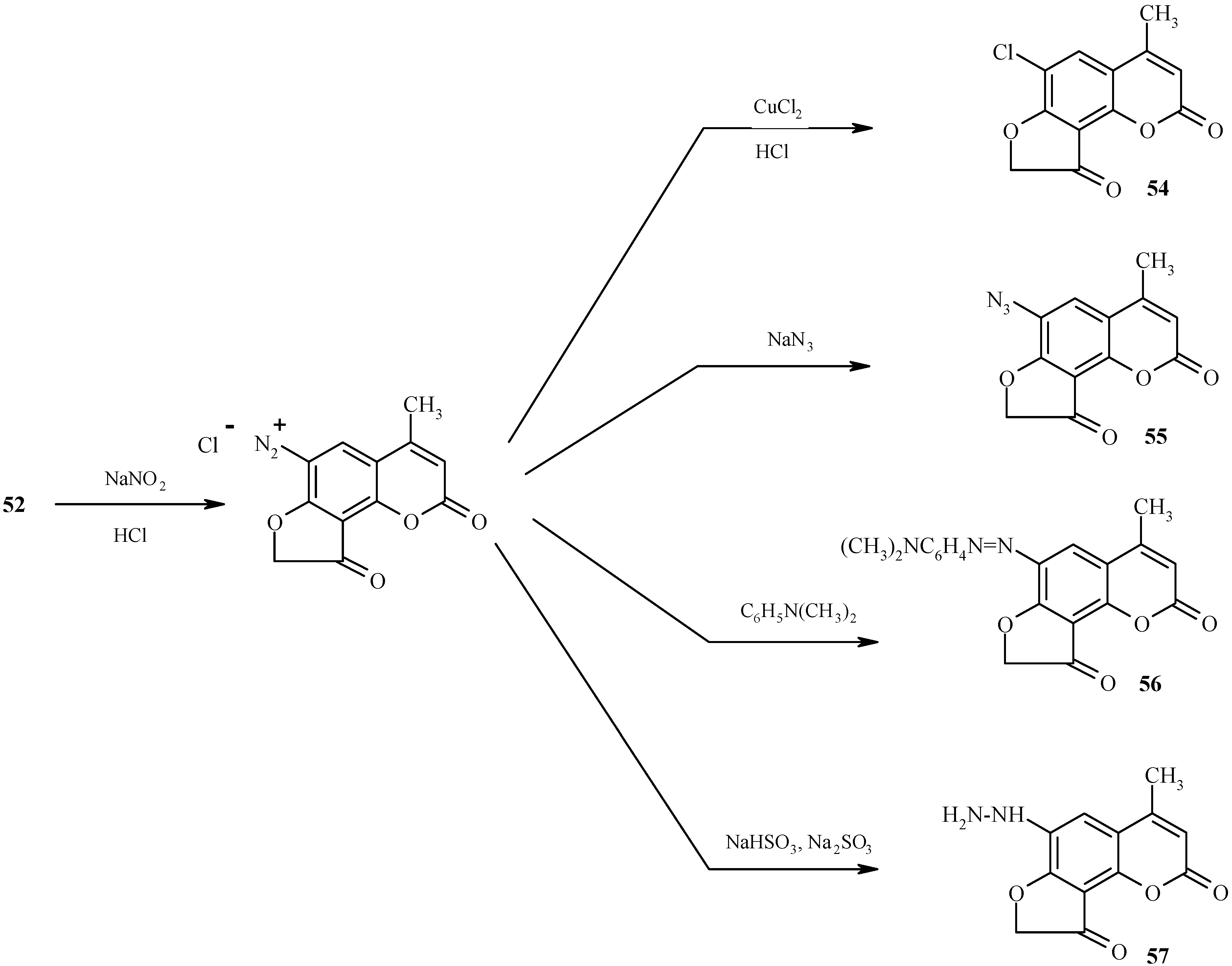
Dihydrofurocoumarinone 21 and analogs are important intermediates in the synthesis of 6-substituted angelicines [34,35]. These intermediate compounds are obtained by coupling of dihydrofurocoumarinones with arenediazonium salts in the presence of sodium hydroxide. In the presence of NaOH the 6-membered lactone undergoes ring opening, thus leaving the 6 position of the coumarin as the only reactive site. The lactone is reinstated upon acidification (Scheme 24). Reduction of the azo-coupled product 51 provides an easy access to the amino derivative 52.
Scheme 24.
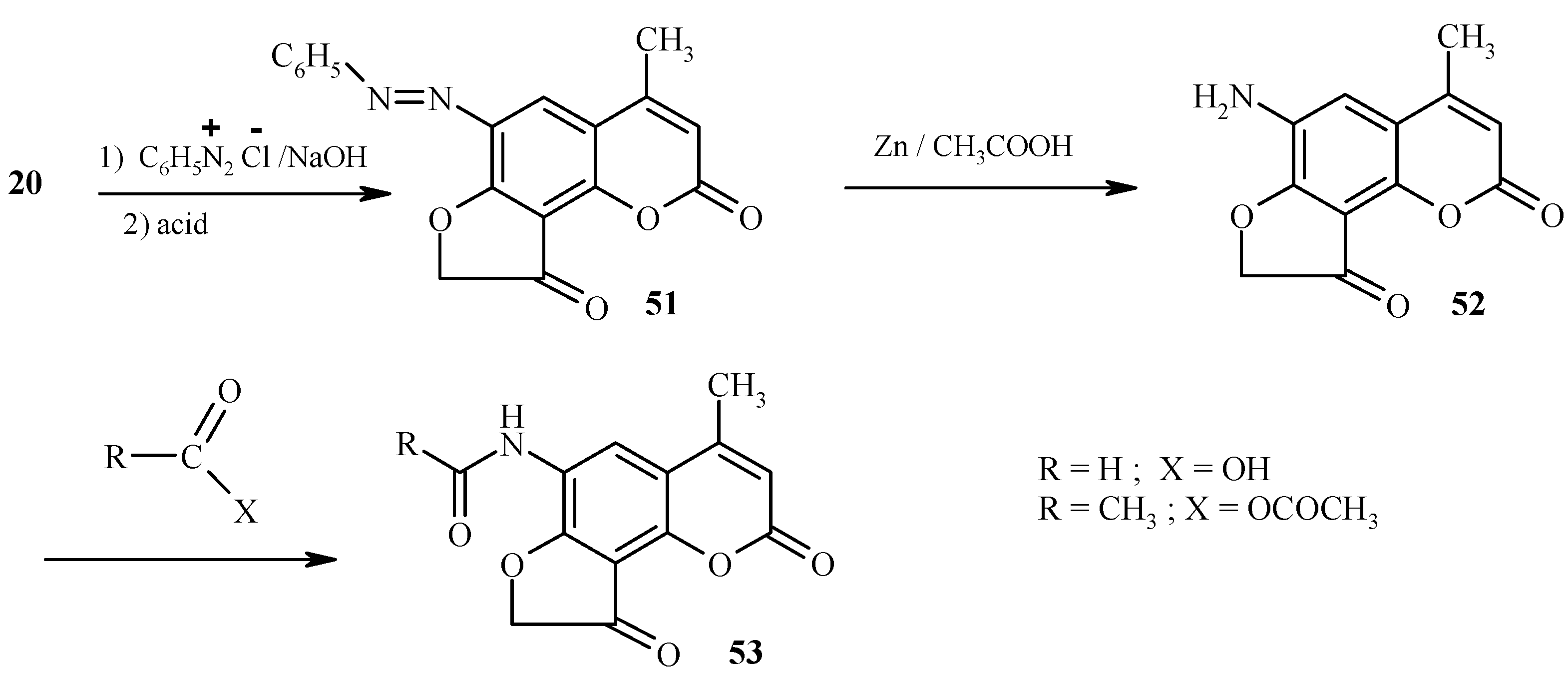
Scheme 25.

As expected, 6-aminodihydrofurocoumarinone 52 possesses properties of an aromatic amine. Its transformations to 53 (Scheme 24) and 54-57 (Scheme 25) are provided for illustration.
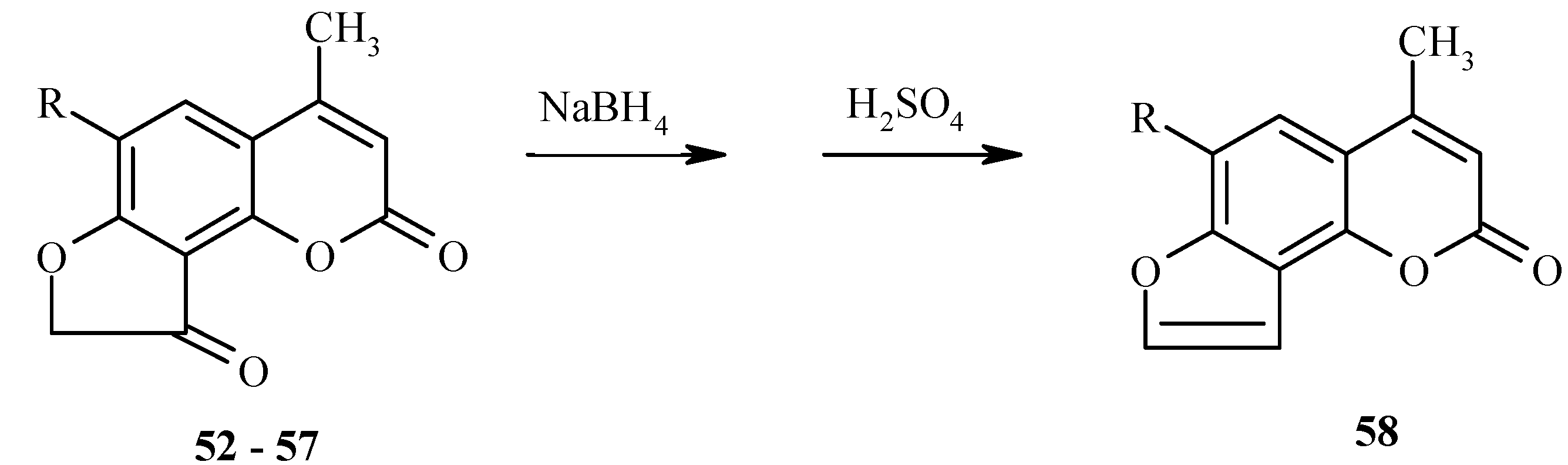
All 6-substituted dihydrofurocoumarinones 52-57 have been transformed into the corresponding 6-substituted angelicines 58, as shown in Scheme 26.
Scheme 26.

New angelicine analogs have been synthesized from 29 (Scheme 27) and 32 (Scheme 28) by using a similar reduction/dehydration methodology. The furocoumarins are fluorescent in water and strongly fluorescent in isopropanol [36].
Scheme 27.
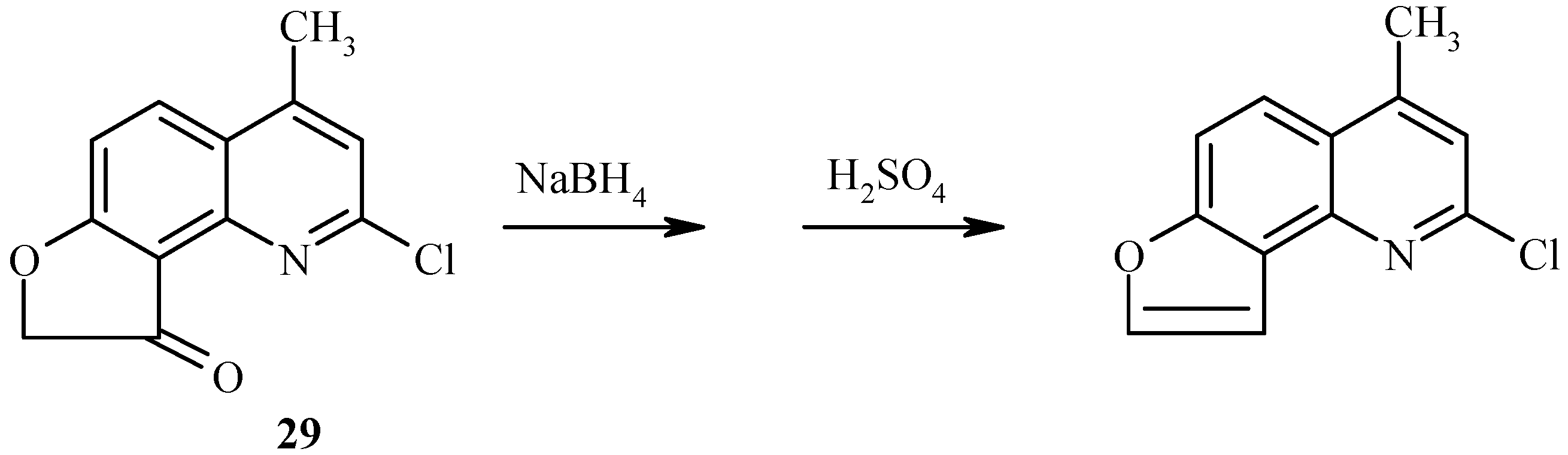
Scheme 28.

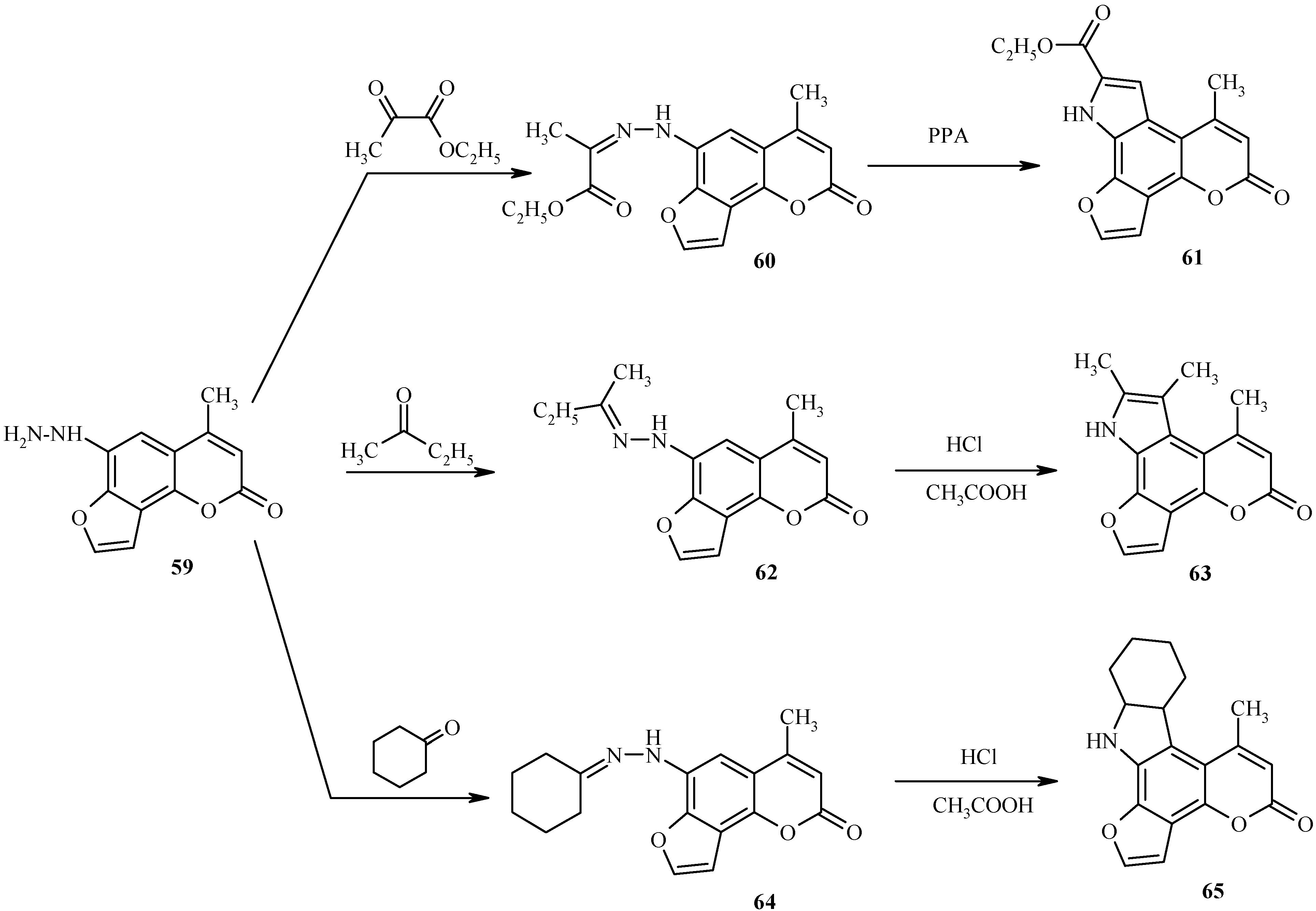
Pyrrolofurocoumarins
6-Hydrazino substituted furocoumarin 59 (Scheme 29) is obtained in good yield upon diazotization of the amine 58 (R = NH2) followed by reduction of the resultant diazonium salt with tin dichloride in concentrated hydrochloric acid [34]. As shown in this scheme, the hydrazine 59 is a practical starting material for high-yield syntheses of pyrrole-fused furocoumarins 61, 63 and 65 through the intermediacy of the respective hydrazones 60, 62 and 64. Since these hydrazones are quite unstable compounds, their Fisher cyclization should be performed immediately after their preparation to ensure high yields of the cyclized products.
Scheme 29.

A success of the Fisher cyclization of hydrazones depends on the structure of hydrazone, acid catalyst, temperature and other factors. As a rule, strong mineral acids are required for the Fisher reaction. We have successfully used several different acid media such as sulfuric acid – acetic acid, hydrogen chloride – acetic acid, thionyl chloride – ethanol, and polyphosphoric acid.
Under optimized conditions, polyphosphoric acid is the best solvent for cyclization of 60 to 61. On the other hand, compounds 63 and 65 are obtained in yields greater than 85% when the Fisher cyclization of the respective hydrazones 62 and 64 are conducted in a mixture of hydrochloric and acetic acids.
Conclusions
Fries rearrangement of acyloxyheteroarenes, condensation of acylhydroxyheteroarenes with α‑carbonyl compounds and transformations of dihydrofuroheteroarenes provide useful synthetic routes to new photosensitive linear and angular furocoumarins, furoquinolones and furoquinolines as well as to their functional and condensed derivatives.
References
- Murray, R.D.H. The Natural Coumarins, Occurrence, Chemistry and Biochemistry; Wiley-Interscience: New York, 1982. [Google Scholar]
- Fahr, E. Pharmazeutische Zeitung 1982, 127, 163.
- Edelson, R.L. J. Photochem. Photobiol., B: Biol. 1991, 10, 165.
- Guiotto, A.; Chilin, A.; Manzini, P.; Dall’Aqua, F.; Bordin, F.; Rodighiero, P. Il Farmaco 1995, 50, 479.
- Saffran, W.A. Psoralen DNA Photobiology; Gasparro, F.P., Ed.; CRC Press, Inc.: Boca Raton, Fl, 1988; vol.11, Chapter 6; p. 73. [Google Scholar]
- Dall’Aqua, F.; Vedaldi, D.; Caffieri, S.; Guiotto, A.; Rodighiero, P.; Carrlassare, F.; Bordin, F. J. Med. Chem. 1981, 24, 178.
- Guiotto, A.; Rodighiero, P.; Manzini, P.; Pfstorini, G.; Carlassare, F.; Vedaldi, D. J. Med. Chem. 1984, 27, 959.
- Bordin, F.; Carlassare, F.; Baccichetti, F.; Guiotto, A.; Rodighiero, P.; Vedaldi, D.; Dall’Aqua, F. Photochem. Photobiol. 1979, 29, 1063.
- Dall’Aqua, F.; Vedaldi, D.; Guiotto, A.; Rodighiero, P.; Carlassare, F.; Baccichetti, F.; Bordin, F. J. Med. Chem. 1981, 24, 806.
- Guiotto, A.; Rodighiero, P.; Pastorini, G.; Manzini, P.; Bordin, F.; Baccichetti, F.; Carlassare, F.; Vedaldi, D.; Dall’Aqua, F. Eur. J. Med., Chem-Chim. Ther. 1981, 16, 489.
- Dall’Aqua, F.; Vedaldi, D.; Bordin; Baccichetti, F.; Carlassare, F.; Tamaro, M.; Guiotto, A.; Rodighiero, P.; Pastorini, G.; Recchia, G.; Cristofolini, M. J. Med. Chem. 1983, 26, 870.
- Vedaldi, D.; Dall’Aqua, F.; Baccichetti, F.; Carlassare, F.; Bordin, F.; Baccichetti, F.; Guiotto, A.; Rodighiero, P.; Manzini, P. Il Farmaco 1991, 46, 1381.
- Demaret, J.-P.; Brunie, S.; Ballini, J.-P.; Cadet, J.; Vigny, P. J. Photochem. Photobiol., B: Biol. 1990, 6, 207.
- Caffieri, S.; Vedaldi, D.; Chilin, A.; Pozzan, A. J. Photochem. Photobiol., B: Biol. 1994, 22, 151.
- Chen, X.; Kagan, J. J. Photochem. Photobiol., B: Biol. 1994, 23, 27.
- Csik, G.; Ronto, G.; Nocentini, S; Averbeck, S; Averbeck, D. J. Photochem. Photobiol., B: Biol. 1994, 24, 129.
- Chen, X.; Kagan, J. J. Photochem. Photobiol., B: Biol. 1994, 22, 51.
- Chilin, A.; Marzano, C.; Guiotto, A.; Manzini, P.; Baccichetti, F.; Carlassare, F.; Bordin, F. J. Med. Chem. 1999, 42, 2936.
- Dallavia, L.; Gia, O.; Magno, S.M.; Santana, L.; Teijeira, M.; Uriarte, E. J. Med. Chem. 1999, 42, 4405.
- Traven, V.F.; Chibisova, T.A. Mendeleev Commun. 1995, 1, 21.
- Traven, V.F.; Chibisova, T.A. Mendeleev Commun. 1997, 3, 249.
- Chibisova, T.A.; Traven, V.F. Zh. Org. Khim. 1999, 35, 924.
- Traven, V.F.; Podhaluzina, N. Ya.; Vasilyev, A.V.; Manaev, A.V. Arkivoc 2000, 1, Part 6, ms0089.
- Traven, V.F.; Chibisova, T.A.; Shorshnev, S. V.; Eliason, R.; Wakefield, D.H. Heterocycl. Commun. 1996, 2, 345.
- Traven, V.F.; Rozhkov, R. V.; Tolmachev, A.Yu.; Kuznezova, N.A.; Podhaluzina, N.Ya.; Carberry, E.A. Heterocycl. Commun. 1997, 3, 339.
- Traven, V.F.; Tolmachev, A. Yu.; Kanevskii, D.S.; Podhaluzina, N.Ya. Heterocycl. Commun. 1999, 5, 183.
- Traven, V.F.; Saharuk, I.I. Khim. Geterotsikl. Soed. 2001, 312.
- Traven, V.F.; Chibisova, T.A. Mendeleev Commun. 1997, 3, 331.
- Safronova, O.B.; Senchenja, I.N.; Traven, V.F. Zh. Obshch. Khim. 2001, 71, 588.
- Traven, V.F.; Manaev, A.V.; Chibisova, T.A. Zh. Obshch. Khim. 2001, 71, 1006.
- Chibisova, T.A.; Traven, V.F. Heterocycl. Commun. 1997, 3, 331.
- Traven, V.F.; Saharuk, I.I. Heterocycl. Commun. 1998, 4, 429.
- Rozhkov, R.V.; Traven, V.F.; Vasilyev, A.V. Heterocycl. Commun. 1998, 4, 567.
- Traven, V.F.; Makarov, I.G.; Saharuk, I.I. Khim. Geterotsikl. Soed. 2001, 501.
- Traven, V.F.; Makarov, I.G.; Saharuk, I.I. Heterocycl. Commun. 1999, 5, 379.
- Tolmachev, A.Yu.; Suslov, V.V.; Gabrdzhiu, A.V.; Traven, V.F. Zh. Org. Khim. 2003, 39, 930.
© 2004 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Traven, V.F. New Synthetic Routes to Furocoumarins and Their Analogs: A Review. Molecules 2004, 9, 50-66. https://doi.org/10.3390/90300050
AMA Style
Traven VF. New Synthetic Routes to Furocoumarins and Their Analogs: A Review. Molecules. 2004; 9(3):50-66. https://doi.org/10.3390/90300050
Chicago/Turabian StyleTraven, Valery F. 2004. "New Synthetic Routes to Furocoumarins and Their Analogs: A Review" Molecules 9, no. 3: 50-66. https://doi.org/10.3390/90300050