Synthesis of New Pyrazole and Pyrimidine Steroidal Derivatives
by
Daniel G. Rivera
1,
Klaus Peseke
2,*, 
Isabel Jomarrón
1,
Alina Montero
3,
Reinaldo Molina
3 and
Francisco Coll
1 1
Centro de Investigaciones de Producto Naturales, Facultad de Quimica, Universidad de La Habana, Zapata y G, Vedado, 10400 Habana, Cuba
2
Fachbereich Chemie der Universität Rostock, D-18051 Rostock, Germany
3
Centro Bioactivos Quimicos, Universidad Central de Las Villas, Santa Clara, Cuba
*
Author to whom correspondence should be addressed.
Molecules 2003, 8(5), 444-452; https://doi.org/10.3390/80500444
Submission received: 16 April 2003
/
Revised: 8 May 2003
/
Accepted: 9 May 2003
/
Published: 31 May 2003
Abstract
:The synthesis of steroidal heterocycles containing the pyrazole and pyrimidine ring fused to the 16,17-position of the steroid nucleus is reported. Androstenolone acetate (1) reacted with carbon disulfide, iodomethane and sodium hydride to furnish 3β-acetoxy-16-[bis(methylthio)methylene]-5-androst-5-en-17-one (2). The reactions of 2 with hydrazine hydrate and methylhydrazine afforded the 5’-methylthio- pyrazolo[4’,3’:16,17]androst-5-en-3β-ols 3a and 3b, respectively. Treatment of 2 with amidinium, guanidinium, and isothiuronium salts in the presence of sodium methoxide yielded the 6’-methoxy- pyrimido[5’,4’:16,17]androst-5-en-3β-ols (4a-4e).
Introduction
Steroidal heterocycles containing anellated rings in the steroidal moiety have prompted a great interest in the biological study of many heterocyclic compounds. Specially, in the last two decades new compounds were synthesized in which the androstane ring A was condensed with a great variety of heterocyclic rings [1,2,3,4,5,6,7,8,9,10,11,12,13]. As should be expected, the fusion of heterocycles to steroids often led to a change in their physiological activities and the appearance of new interesting biological behavior. Thus, several steroidal heterocycles have been obtained exhibiting activity as potential inhibitors of cytochrome P450 enzyme aromatase [14,15], with their subsequent clinical application in the treatment of estrogen-dependent breast cancers, while others which have shown moderate binding affinity for the benzodiazepine receptor [16].
There is no doubt that the main procedure for the synthesis of steroidal heterocycles has been the reaction of α-hydroxymethylene-oxo-steroids with N,N’-dinucleophiles and N,O-dinucleophiles. We wish to describe in this paper another procedure for the preparation of anellated steroids. The reaction of cyclic and acyclic ketones with carbon disulfide in the presence of a base and an alkylating agent to give α-oxoketene dithioacetals is known [17,18]. These push-pull activated alkenes were reacted with dinucleophiles to yield heterocyclic compounds [19,20,21]. Furthermore, the reactions with carbon disulfide have been widely applied to deoxyuloses in order to obtain the corresponding α-oxoketene dithioacetals with a sugar moiety [22,23,24,25,26,27,28,29,30]. These sugars proved to be very useful consequently in anellation reactions with dinucleophiles.
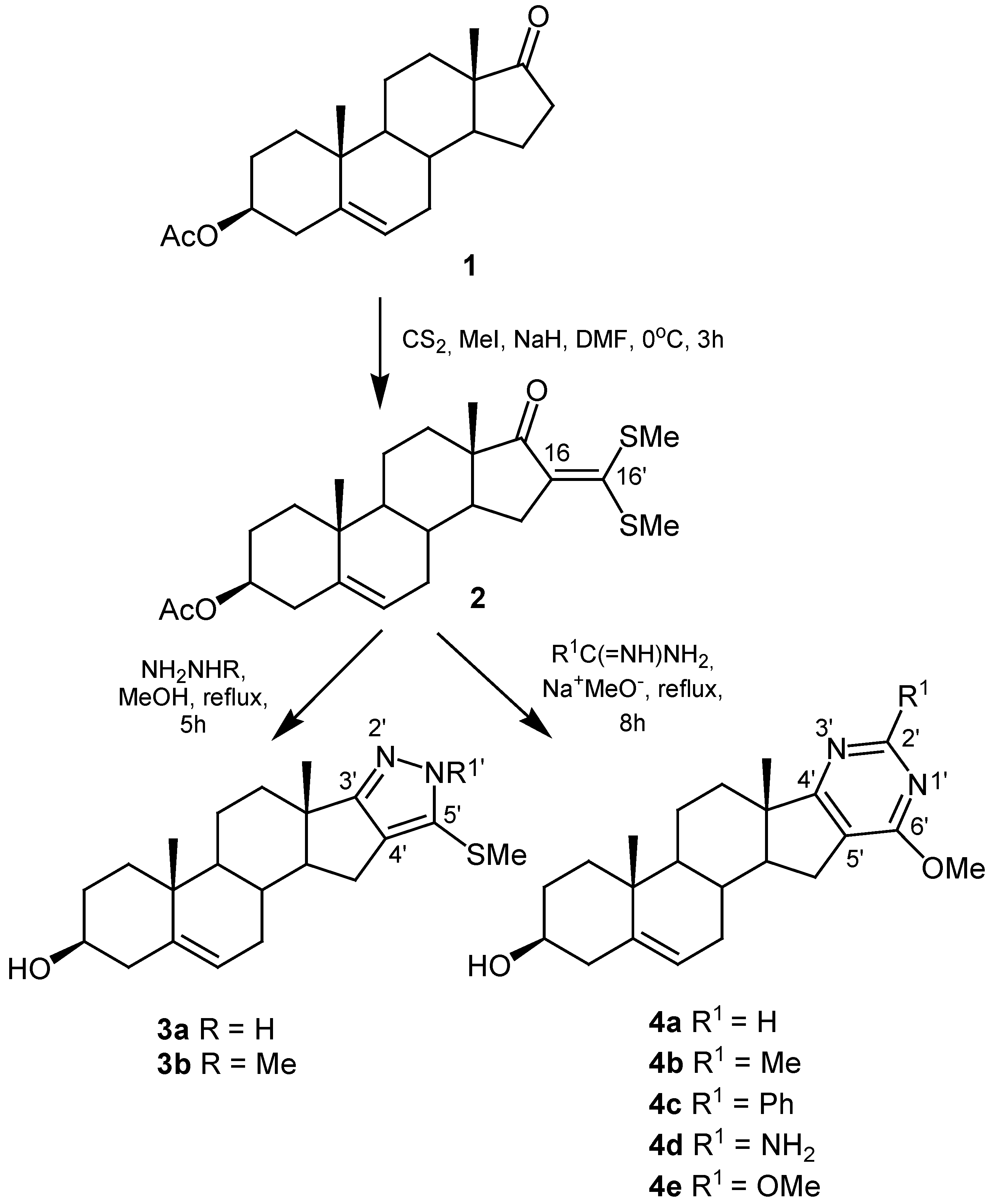
On this basis, we used 3β-acetoxy-16-[bis(methylthio)methylene]androst-5-en-17-one (2) as precursor in a new approach of steroidal heterocycle synthesis consisting in the fusion between nitrogen heterocycles and the steroid ring D of androstenolone acetate (1) at C-16 and C-17. Moreover, in all obtained compounds the Δ5-3β-hydroxy function remains unaffected. Therefore, these molecules are interesting analogues of related hormone 4-en-3-one steroids.
Results and Discussion
The syntheses started with androstenolone acetate (1). This inexpensive compound was chosen because of its synthetic utility and easier accessibility to ring D. Compound 1 was reacted with carbon disulfide, iodomethane and sodium hydride in dimethylformamide as solvent at 0 °C to furnish the corresponding α-oxoketene dithioacetal 2 in 62 % yield (Scheme 1).
The structure of compound 2 was established by NMR spectra, mass spectral data and elemental analysis. The conclusion from a comparison between the 13C-NMR data of compounds 1 and 2 was that the appearance of the signal at 149.9 ppm for C-16’ and the two corresponding to methylthio groups at δ = 18.5 and δ = 17.6 are clear evidence of formation of the desired ketene dithioacetal group. Moreover, the 1H-NMR spectrum showed two singlets at δ = 2.45 and δ = 2.43 assigned to the methylthio groups, and the singlet belonged to the acetyl group confirming that deprotection of the 3β-hydroxyl did not take place under the reaction conditions. On the other hand, taking in account the “push-pull” features of 2, the analysis of its 13C-NMR spectrum allowed the unequivocal assignment of C-16 at δ = 135.4 and the signal corresponding to C-16’ at δ = 149.9 [31].
Scheme 1.
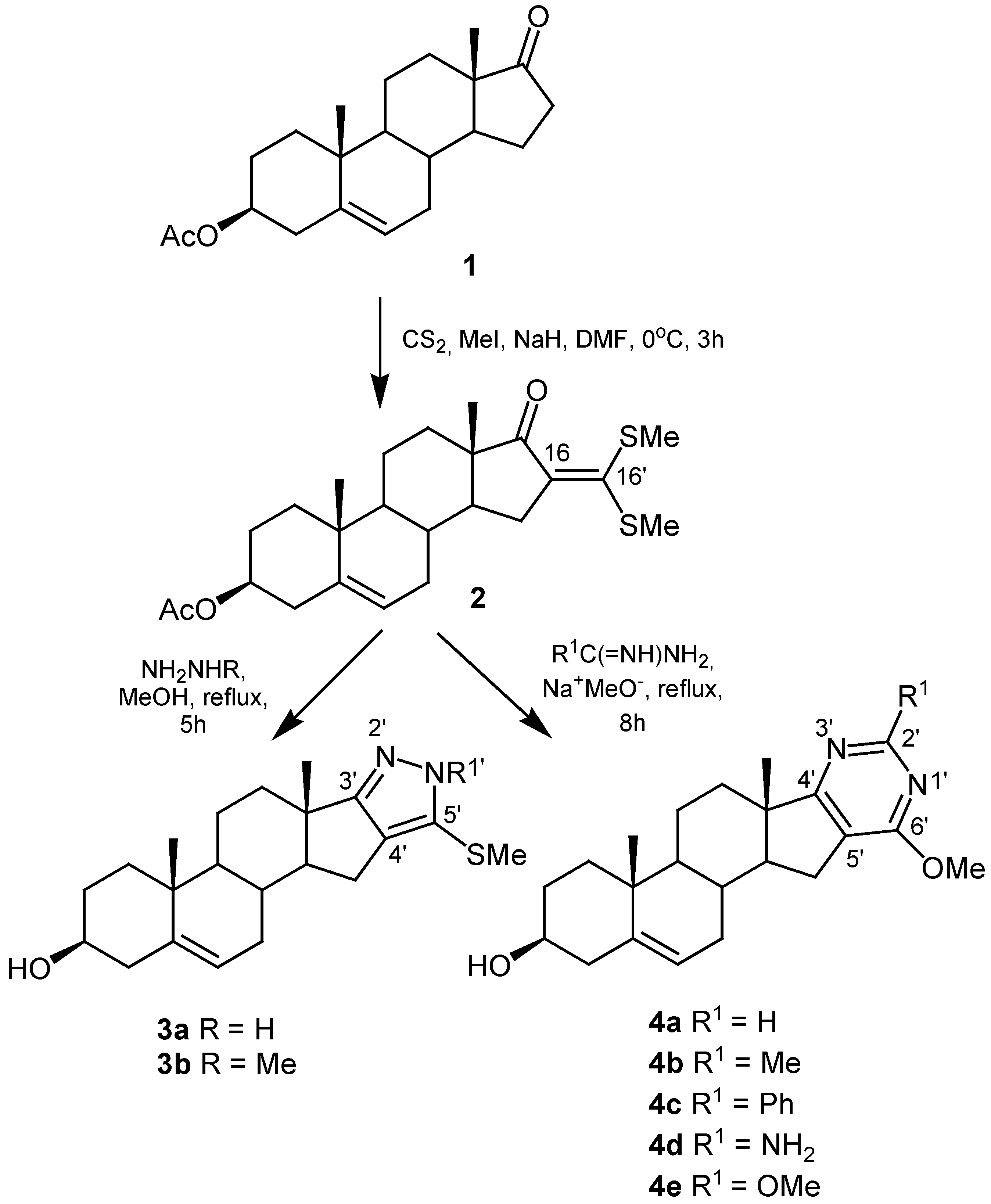
With the aim to obtain the pyrazolo anellated androstenolones, compound 2 was allowed to react with 80% hydrazine hydrate and methylhydrazine, both in boiling methanol, affording compounds 3a and 3b with yields of 70% and 66%, respectively. The pyrimido-anellated androstenolone derivatives 4 were obtained by reaction of α-oxoketene dithioacetal 2 with a variety of amidinium, guanidinium and thiuronium salts in dry methanol in the presence of sodium methoxide at reflux. The C-2’ substituents in fused pyrimidine rings were selected for their different natures, which allow one to provide the heterocycle moiety with either hydrophobic or hydrophilic features. It can be noted that reaction times for obtaining pyrimidine derivatives are larger than those for pyrazole derivatives; this fact is justified by the known low reactivity of α-oxoketene dithioacetals toward amidines. Cleavage of acetyl group occurred due to the strong reaction conditions used in the synthesis of both pyrazole and pyrimidine derivatives. In the second case an additional nucleophilic substitution of a methylthio group occurred because of the high concentration of sodium methoxide required in this synthetic procedure.
Two important spectroscopic features in the 1H- and 13C-NMR spectra of the pyrazole derivatives 3a and 3b were the appearance of only one signal, assigned to the methylthio group, and the absence of the classical acetyl signal confirming the expected substitution of one methylthio and the deprotection of the 3β-hydroxy group, respectively. In the case of compound 3a it was not possible to determine which of the two pyrazole tautomers had been formed, although typically an equilibrium exists between them in solution.
Furthermore, as is evident from analysis of 13C-NMR data of compounds 3 and 4, chemical shifts of carbon atoms in rings A, B and C have similar values because of their structural differences are located in ring D. The most relevant chemical shifts of steroidal and heterocycle moieties are listed in Table 1.

{kind=link}
| 3a | 3b | 4a | 4b | 4c | 4d | 4e | |
|---|---|---|---|---|---|---|---|
| C-3 | 71.5 | 71.5 | 71.5 | 71.5 | 71.5 | 71.6 | 71.5 |
| C-5 | 141.1 | 141.2 | 141.2 | 141.2 | 141.2 | 141.2 | 141.2 |
| C-6 | 121.0 | 121.0 | 121.0 | 121.0 | 121.0 | 120.9 | 121.0 |
| C-2’ | - | - | 169.8 | 166.5 | 165.5 | 165.7 | 169.8 |
| C-3’ | 168.1 | 167.3 | - | - | - | - | - |
| C-4’ | 123.9 | 124.9 | 181.8 | 181.8 | 182.4 | 181.6 | 181.8 |
| C-5’ | 131.3 | 129.3 | 113.4 | 115.1 | 116.2 | 118.8 | 113.4 |
| C-6’ | - | - | 162.8 | 165.3 | 163.2 | 159.9 | 163.1 |
| C-18 | 17.9* | 18.1* | 16.8 | 16.8 | 16.9 | 16.8 | 16.8 |
| SCH3 | 17.8* | 17.9* | - | - | - | - | - |
| OCH3 | - | - | 53.2 | 53.3 | 53.5 | 53.5 | 53.6 |
* Assignments that could be interchanged.
The loss of the “push-pull” behavior could be certainly corroborated by the δ changes at C-16 and C-16’ during the formation of these heterocycle rings. In the 13C-NMR data the shift changes noted at C-2’ in the pyrimidine ring of compounds 4 are in agreement with the expected differences. In the 1H-NMR spectrum of compound 4e the lack of the singlet signal for methylthio groups and the appearance of a new singlet at δ = 3.95–4.00 which is typical of methoxy groups confirmed the postulated structure.
Acknowledgments
We are grateful to the Deutscher Akademisher Austauschdienst for scholarships
Experimental
General
Melting points were determined on a Boëtius apparatus and are uncorrected. IR spectra were obtained on a Nicolet 205 FT-IR spectrometer. 1H-NMR and 13C-NMR were recorded on a Bruker ACF-250 spectrometer at 250.13 MHz using TMS as internal standard. Chemical shifts are given in ppm and coupling constant (J) values are in Hz. The multiplicity of the signals on the 13C-NMR spectra was determined using the Distorsionless Enhancement Polarization Transfer (DEPT) sequence. Elemental analyses were performed in a Leco CHNS-932 instrument. The mass spectra were recorded in a AMD 402/3 spectrometer AMD Intectra GmbH. Reactions were monitored by thin-layer chromatography (TLC) on precoated plates with silica gel 60 GF254 (Merck) and spots were visualized with UV light or a spray of vanillin in perchloric acid 1 % w/v and subsequent heating. Flash column chromatography was performed on silica gel 60 (Merck, 70-230 mesh). “Usual work-up” refers to dilution with water, extraction with an organic solvent, washing the extract with HCl (5%) and/or KHCO3 (5%) and water, drying over anhydrous MgSO4 and removal of the solvent under reduced pressure. The solvents were purified and dried according to recommended procedures.
3β-acetoxy-16-[bis(methylthio)methylene]androst-5-en-17-one (2)
Sodium hydride (60%, 303 mg, 7.6 mmol), carbon disulfide (0.46 mL, 7.6 mmol) and methyl iodide (0.76 mL, 12.1 mmol) were added to a stirred solution of androstenolone acetate 1 (1.000 g, 3.03 mmol) in anhydrous DMF (60 mL). The mixture was stirred for 3 hours at 0 °C, poured into ice-water (200 mL) and extracted with chloroform (3 x 100 mL). The combined extracts were washed with water (3 x 20 mL), dried over anhydrous MgSO4 and the solvent was evaporated. The crude product was purified by column chromatography with 10:1 toluene/ethyl acetate elution and crystallized from acetone to furnish 2 (0.8165 g, 62 %) as yellow needles, mp: 165.7–168.0 °C; IR (KBr, cm–1): 1739 (C=O, Ac), 1682 (C=O), 1479 (C=C); 1H-NMR (CDCl3): δ = 0.91 (s, 3H, Me-19), 1.04 (s, 3H, Me-18), 2.03 (s, 3H, Ac), 2.44 (s, 3H, SMe), 2.45 (s, 3H, SMe), 4.59 (m, 1H, H-3α), 5.40 (d, 1H, J = 4.9 Hz, H-6); 13C-NMR (CDCl3): δ = 14.3 (Me-18), 17.6 (SMe), 18.5 (SMe), 19.3 (Me-19), 20.3 (C-11), 21.4 (CH3CO), 27.7 (C-2), 30.8 (C-12), 30.9 (C-8), 31.8 (C-7), 32.9 (C-15), 36.7 (C-10), 36.8 (C-1), 38.0 (C-4), 49.7 (C-13), 50.1 (C-14), 50.1 (C-9), 73.7 (C-3), 121.8 (C-6), 135.4 (C-16), 140.0 (C-5), 149.9 (C-16’), 170.5 (CH3CO), 205.0 (C-17); MS (EI): m/z (%): 434 (65.8, M+.), 374 (100); Calc for C24H34O3S2: C 66.32, H 7.88, S 14.75; Found: C 66.32, H 7.99, S 14.67.
5’-Methylthio- pyrazolo[4’,3’:16,17]androst-5-en-3β-ol (3a)
Compound 2 (200 mg, 0.46 mmol) was dissolved in anhydrous methanol (50 mL), hydrazine hydrate (80%, 0.8 mL) was added and the mixture was stirred at reflux for 4 hours until all the starting material had disappeared as indicated by TLC (disappearance of yellow color). Then the reaction mixture was concentrated and usual work-up (CHCl3) yielded a crude product, which was purified by column chromatography with 3:1 chloroform/ethyl acetate elution to give 117.8 mg (66 %) of 3a, mp (ethanol): 176.2–178.0 °C; IR (KBr, cm–1): 3354 (OH), 3168 (NH), 1454, 1436, 1408, 1373 (C=C, C=N); 1H-NMR (CDCl3): δ = 1.02 (s, 3H, Me-19), 1.07 (s, 3H, Me-18), 2.42 (s, 3H, SMe), 3.53 (m, 1H, H-3α), 4.27 (broad signal, NH), 5.38 (d, 1H, J = 5.20 Hz, H-6); 13C-NMR (CDCl3): δ = 17.8 (SMe), 17.9 (Me-18), 19.4 (Me-19), 20.4 (C-11), 30.3 (C-12), 30.8 (C-8), 31.5 (C-7), 32.1 (C-2), 33.7 (C-15), 36.7 (C-10), 37.1 (C-1), 40.9 (C-13), 42.2 (C-4), 50.5 (C-9), 62.1 (C-14), 71.5 (C-3), 121.0 (C-6), 123.9 (C-4’), 131.3 (C-5’), 141.1 (C-5), 168.1 (C-3’); MS (EI): m/z (%): 358 (21.7, M+.), 287 (100); Calc. for C21H30N2OS: C 70.35, H 8.43, N 7.81, S 8.94; Found: C 70.08, H 8.69, N 8.02, S 8.78.
1’-Methyl-5’-methylthio- pyrazolo[4’,3’:16,17]androst-5-en-3β-ol (3b)
Compound 2 (200 mg, 0.46 mmol) was dissolved in anhydrous methanol (50 mL), methyl hydrazine (1 mL) was added and the mixture was stirred at reflux for 5 hours until all the starting material had disappeared as indicated by TLC. Then the reaction mixture was concentrated and usual work-up (CHCl3) yielded a crude product, which was purified by column chromatography with 4:1 chloroform/ethyl acetate elution to afford 129.4 mg (70 %) of 3b, mp (ethanol): 181–183 °C; IR (KBr, cm–1): 3349 (OH), 1455, 1371, 1356, 1349 (C=C, C=N); 1H-NMR (CDCl3): δ = 1.00 (s, Me-19), 1.06 (s, Me-18), 2.36 (s, 3H, SMe), 3.53 (m, 1H, H-3α), 3.80 (s, 3H, NMe), 5.38 (d, 1H, J = 4.9 Hz, H-6); 13C-NMR (CDCl3): δ = 17.9 (SMe), 18.1 (Me-18), 19.3 (Me-19), 20.5 (C-11), 30.3 (C-12), 30.8 (C-8), 31.5 (C-7), 32.1 (C-2), 33.7 (C-15), 36.3 (NMe), 36.7 (C-10), 37.1 (C-1), 40.8 (C-13), 42.2 (C-4), 50.5 (C-9), 61.8 (C-14), 71.5 (C-3), 121.0 (C-6), 124.9 (C-4’), 129.3 (C-5’), 141.2 (C-5), 167.3 (C-3’); MS (EI): m/z (%): 372 (38.6, M+.), 327 (100); Calc. for C22H32N2OS: C 70.92, H 8.66, N 7.52, S 8.61; Found: C 70.5 8, H 8.74, N 7.53, S 8.49.
6’-Methoxy- pyrimido[5’,4’:16,17]androst-5-en-3β-ol (4a)
Compound 2 (100 mg, 0.23 mmol) was dissolved in a solution of sodium (26.5 mg, 1.15 mmol) in anhydrous methanol (50 mL), formamidinium acetate (120 mg, 1.15 mmol) was added and the mixture was stirred at reflux for 8 hours. Usual work-up (CHCl3) yielded a crude product, which was purified by column chromatography to furnish 50.74 mg (68 %) of 4a, mp (acetone): 178–180 °C; IR (KBr, cm–1): 3450 (OH), 1593, 1558 (C=C, C=N); 1H-NMR (CDCl3): δ = 0.98 (s, 3H, Me-19), 1.08 (s, 3H, Me-18), 3.53 (m, 1H, H-3α), 3.98 (s, 3H, OMe), 5.38 (d, 1H, J = 5.20 Hz, H-6,), 8.60 (s, 1H, H-2’); 13C-NMR (CDCl3): δ = 16.8 (Me-18), 19.4 (Me-19), 20.5 (C-11), 26.5 (C-15), 30.6 (C-8), 31.6 (C-7), 31.3 (C-12), 32.9 (C-2), 36.8 (C-10), 37.1 (C-1), 42.2 (C-4), 46.0 (C-13), 50.6 (C-9), 53.5 (MeO), 55.6 (C-14), 71.6 (C-3), 118.8 (C-5’), 121.0 (C-6), 141.2 (C-5), 159.9 (C-6’), 165.7 (C-2’), 181.6 (C-4’); MS (EI): m/z (%): 354 (74.22, M+.), 243 (100); Calc. for C22H30N2O2: C 74.54, H 8.53, N 7.90; Found: C 74.28, H 8.31, N 7.68.
6’-Methoxy-2’-methyl- pyrimido[5’,4’:16,17]androst-5-en-3β-ol (4b)
Compound 2 (100 mg, 0.23 mmol), sodium (26.5 mg, 1.15 mmol) in anhydrous methanol (50 mL) and acetamidinium hydrogen chloride (110 mg, 1.15 mmol) was reacted as described for preparation of 4a to furnish 58.4 mg (75 %) of 4b, mp (acetone): 210–212 °C; IR (KBr, cm–1): 3363 (OH), 1591, 1565 (C=C, C=N); 1H-NMR (CDCl3): δ = 0.96 (s, Me-19), 1.07 (s, Me-18), 2.60 (s, Me-2’), 3.53 (m, 1H, H-3α), 3.95 (s, OMe), 5.38 (d, 1H, J = 5.20 Hz, H-6,); 13C-NMR (CDCl3): δ = 16.8 (Me-18), 19.4 (Me-19), 20.5 (C-11), 25.8 (Me-2’), 26.1 (C-15), 30.6 (C-8), 31.2 (C-12), 31.6 (C-7), 32.9 (C-2), 36.7 (C-10), 37.1 (C-1), 42.2 (C-4), 46.0 (C-13), 50.6 (C-9), 53.2 (MeO), 55.7 (C-14), 71.5 (C-3), 115.1 (C-5’), 121.0 (C-6), 141.2 (C-5), 165.3 (C-6’), 166.5 (C-2’), 181.8 (C-4’); MS (EI): m/z (%): 368 (76.95, M+.), 353 (100); Calc. for C23H32N2O2: C 74.96, H 8.75, N 7.60; Found: C 74.45, H 8.62, N 7.33.
6’-Methoxy-2’-phenyl- pyrimido[5’,4’:16,17-c]androst-5-en-3β-ol (4c)
Compound 2 (100 mg, 0.23 mmol), sodium (26.5 mg, 1.15 mmol) in anhydrous methanol (50 mL) and benzamidinium hydrogen chloride (180 mg, 1.15 mmol) was reacted as described for preparation of 4a to furnish 57.12 mg (62 %) of 4c, mp (acetone): 241–243 °C; IR (KBr, cm–1): 3396 (OH), 1593, 1552 (C=C, C=N); 1H-NMR (CDCl3): δ = 1.02 (s, Me-19), 1.10 (s, Me-18), 3.54 (m, 1H, H-3α), 4.09 (s, OMe), 5.38 (d, 1H, J = 5.20 Hz, H-6), 7.43–7.46 (m, 3H, Ph), 8.43–8.47 (m, 2H, Ph); 13C-NMR (CDCl3): δ = 16.9 (C-18), 19.4 (C-19), 20.5 (C-11), 26.4 (C-15), 30.7 (C-8), 31.3 (C-12), 31.6 (C-7), 33.1 (C-2), 36.8 (C-10), 37.1 (C-1), 42.3 (C-4), 46.1 (C-13), 50.8 (C-9), 53.3 (MeO), 55.8 (C-14), 71.6 (C-3), 116.2 (C-5’), 121.1 (C-6), 128.2, 128.3, 130.0, 138.3 (Ph), 141.2 (C-5), 163.2 (C-6’), 165.5 (C-2’), 182.4 (C-4’); MS (EI): m/z (%): 430 (100, M+.); Calc. for C28H34N2O2: C 78.10, H 7.96, N 6.51; Found: C 77.93, H 7.82, N 6.34.
2’-Amino-6’-methoxy- pyrimido[5’,4’:16,17]androst-5-en-3β-ol (4d)
Compound 2 (100 mg, 0.23 mmol), sodium (26.5 mg, 1.15 mmol) in anhydrous methanol (50 mL) and guanidinium nitrate (140 mg, 1.15 mmol) was reacted as described for preparation of 4a to furnish 49.98 mg (64 %) of 4d, mp (acetone): 218–221 °C; IR (KBr, cm–1): 3472 (OH), 3318, 3209 (NH2), 1602, 1568 (C=C, C=N); 1H-NMR (CDCl3): δ = 1.00 (s, Me-19), 1.06 (s, Me-18), 3.06 (s, 2H, NH2), 3.53 (m, 1H, H-3α), 5.38 (d, 1H, H-6, J = 4.9 Hz); 13C-NMR (CDCl3): δ = 16.8 (C-18), 19.4 (C-19), 20.5 (C-11), 26.1 (C-15), 30.6 (C-8), 31.2 (C-12), 31.6 (C-7), 32.9 (C-2), 36.7 (C-10), 37.1 (C-1), 42.2 (C-4), 46.0 (C-13), 50.6 (C-9), 53.2 (MeO), 55.7 (C-14), 71.5 (C-3), 113.4 (C-5’), 121.0 (C-6), 141.2 (C-5), 162.8 (C-6’), 169.8 (C-2’), 181.8 (C-4’); MS (EI): m/z (%): 369 (100, M+.); Calc. for C22H31N3O2: C 71.51, H 8.46, N 11.37; Found: C 71.38, H 8.64, N 11.13.
2’,6’-dimethoxy- pyrimido[5’,4’:16,17]androst-5-en-3β-ol (4e)
Compound 2 (100 mg, 0.23 mmol), sodium (26.5 mg, 1.15 mmol) in anhydrous methanol (50 mL) and S-methylisothiuronium sulfate (320 mg, 1.15 mmol) was reacted as described for preparation of 4a, to furnish 52.11 mg (59 %) of 4e, mp (acetone): 214–216° C; IR (KBr, cm–1): 3463 (OH), 1591, 1559 (C=C, C=N); 1H-NMR (CDCl3): δ = 0.96 (s, Me-19), 1.07 (s, 3 Me-18), 3.53 (m, 1H, H-3α), 3.95 (s, OMe), 4.08 (s, OMe ), 5.38 (d, 1H, J = 5.20 Hz, H-6); 13C-NMR (CDCl3): δ = 16.8 (C-18), 19.4 (C-19), 20.5 (C-11), 26.4 (C-15), 30.7 (C-8), 31.3 (C-12), 31.6 (C-7), 33.1 (C-2), 36.8 (C-10), 37.1 (C-1), 42.3 (C-4), 46.1 (C-13), 50.8 (C-9), 53.2 (MeO), 53.6 (MeO), 55.8 (C-14), 71.6 (C-3), 113.4 (C-5’), 121.1 (C-6), 141.2 (C-5), 169.8 (C-2’), 181.8 (C-4’), 163.1 (C-6’); MS (EI): m/z (%): 384 (100 M+.); Calc. for C23H32N2O3: C 71.84, H 8.39, N 7.29; Found: C 71.67, H 8.44, N 7.18.
References
- Newman, H. C.; Potts, G. O.; Ryan, W. T.; Stonner, F. W. J. Med. Chem. 1970, 13, 948–951.
- Newman, H. C. J. Med. Chem. 1971, 14, 1246–1246.
- Newman, H. C.; Stonner, F. W. Helv. Chim. Acta 1972, 55, 2014–2017.
- Campbell, M. M.; Craig, R. C. J. Chem. Soc. Perkin Trans.1 1980, 766–774.
- Bajwa, J. S.; Sykes, P. J. J. Chem. Soc. Perkin Trans. 1 1980, 1019–1024.
- Ramadas, S. R.; Krishna, M. V. Indian. J. Chem. Sect. B 1983, 1093–1095.
- Bajwa, J. S.; Sykes, P. J. J. Chem. Soc. Perkin Trans.1 1980, 481–486.
- Marini-Bettolo, R.; Tsai, C. S.; Tsai, T. Y.; Wiesner, K. Heterocycles 1981, 15, 305–308.
- Ramadas, S. R.; Krishna, M. V. Heterocycles 1981, 16, 2169–2171.
- Singh, H.; Yadav, M. R.; Garg, S. P.; Sharma, R.; Paul, D. Heterocycles 1985, 23, 2931–2938.
- Pradhan, S. K.; Akamanchi, K. G. Heterocycles 1989, 28, 813–839.
- Charalambos, C. J. Heterocycl. Chem. 1996, 33, 539–558.
- Shamsuzzaman, A. S.; Akram, M. K. Heterocycles 1998, 48, 329–334.
- Li, S.; Parish, E. J.; Rodriguez-Valenzuela, C.; Brodie, A. M. H. Bioorg. Med. Chem. 1998, 6, 1525–1529.
- Fink, B. E.; Mortensen, D. S.; Stauffer, S. R.; Aron, Z. D.; Katzenellenbogen, J. A. Chem. Biol. 1999, 6, 205–219.
- Grandolini, G.; Ambrogi, V.; Perioli, L. Farmaco 1996, 51, 203–207.
- Dieter, R. K. Tetrahedron 1986, 42, 3029–3096.
- Tominaga, Y. J. Heterocycl. Chem. 1989, 26, 1167–1204.
- Borrmann, D. Houben-Weyl, Methoden der Organischen Chemie; Müller, E., Ed.; Thieme: Stuttgart, 1969; Band VII/4; p. 404. [Google Scholar]
- Schaumann, E. Houben-Weyl, Methoden der Organischen Chemie; Klamann, D., Ed.; Thieme: Stuttgart, New York, 1985; Band E 11; p. 232. [Google Scholar]
- Peseke, K.; Feist, H.; Quincoces, J. Targets in Heterocyclic Sytems– Chemistry and Properties; Vol. 5, Attanasi, O.A., Ed.; Societa Chimica Italiana: Rome, 2001; pp. 299–326. [Google Scholar]
- Peseke, K.; Feist, H.; Cuny, E. Carbohydr. Res. 1992, 230, 319–325.
- Peseke, K.; Feist, H.; Köll, P. Carbohydr. Res. 1993, 247, 315–322.
- Gómez, M.; Quincoces, J.; Peseke, K.; Reinke, H. J. Carbohy. Chem. 1999, 18, 57–68.
- Peseke, K.; Thiele, G.; Michalik, M. Liebigs Ann. 1995, 1633–1636.
- Gómez, M.; Quincoces, J.; Kuhla, B.; Peseke, K.; Reinke, H. J. Carbohydr. Chem. 1998, 17, 57–68.
- Methling, K.; Aldinger, S.; Peseke, K.; Michalik, M. J. Carbohydr. Chem. 1999, 18, 429–439.
- Kuhla, B.; Peseke, K.; Thiele, G.; Michalik, M. J. Prakt. Chem. 2000, 342, 240–244.
- Gómez, M.; Peseke, K.; Reinke, H.; Quincoces, J.; Michalik, M. J. Prakt. Chem. 2000, 342, 389–395.
- Herrera, L.; Feist, H.; Michalik, M.; Quincoces, J.; Peseke, K. Carbohydr. Res. 2003, 338, 293–298.
- Michalik, M.; Peseke, K.; Radeglia, R. J. Prakt. Chem. 1985, 327, 103–108.
- Sample availability: Available from MDPI
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
MDPI and ACS Style
Rivera, D.G.; Peseke, K.; Jomarrón, I.; Montero, A.; Molina, R.; Coll, F. Synthesis of New Pyrazole and Pyrimidine Steroidal Derivatives. Molecules 2003, 8, 444-452. https://doi.org/10.3390/80500444
AMA Style
Rivera DG, Peseke K, Jomarrón I, Montero A, Molina R, Coll F. Synthesis of New Pyrazole and Pyrimidine Steroidal Derivatives. Molecules. 2003; 8(5):444-452. https://doi.org/10.3390/80500444
Chicago/Turabian StyleRivera, Daniel G., Klaus Peseke, Isabel Jomarrón, Alina Montero, Reinaldo Molina, and Francisco Coll. 2003. "Synthesis of New Pyrazole and Pyrimidine Steroidal Derivatives" Molecules 8, no. 5: 444-452. https://doi.org/10.3390/80500444