Signal Transduction: An Exciting Field of Investigation for Small Molecule Drug Discovery

Worldwide Head of Chemistry, Serono Pharmaceutical Research Institute, Geneva, Switzerland
Molecules 2003, 8(4), 349-358; https://doi.org/10.3390/80400349
Submission received: 1 January 2003
/
Published: 30 April 2003
(This article belongs to the Special Issue Selected Papers from the International Symposium on Frontiers in Molecular Science (ISFMS 2002))
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Introduction
Most protein functions associated with pathological conditions are mediated by protein conformational changes and protein-protein interactions. In terms of drug discovery, what counts is to find ways to modulate a target protein function, not to hit the protein itself. This means that partner proteins (upstream or downstream), which are able to control the function of a target protein, become themselves exciting targets for drug discovery. Understanding the complex processes of protein crosstalk becomes therefore essential, in particular to discover new individual target proteins belonging to well-known classes of therapeutic targets (enzymes, for example).
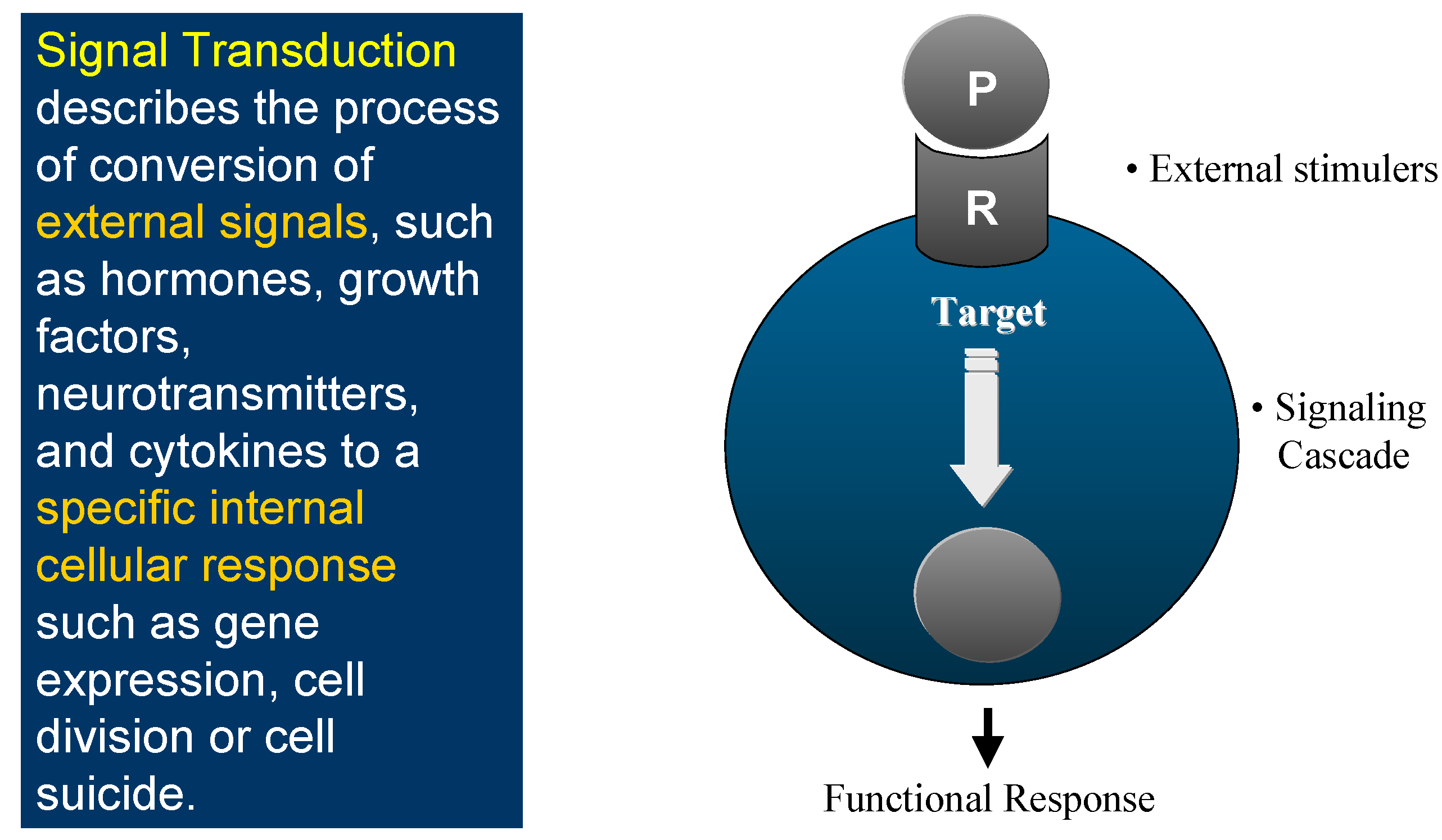
Figure 1.
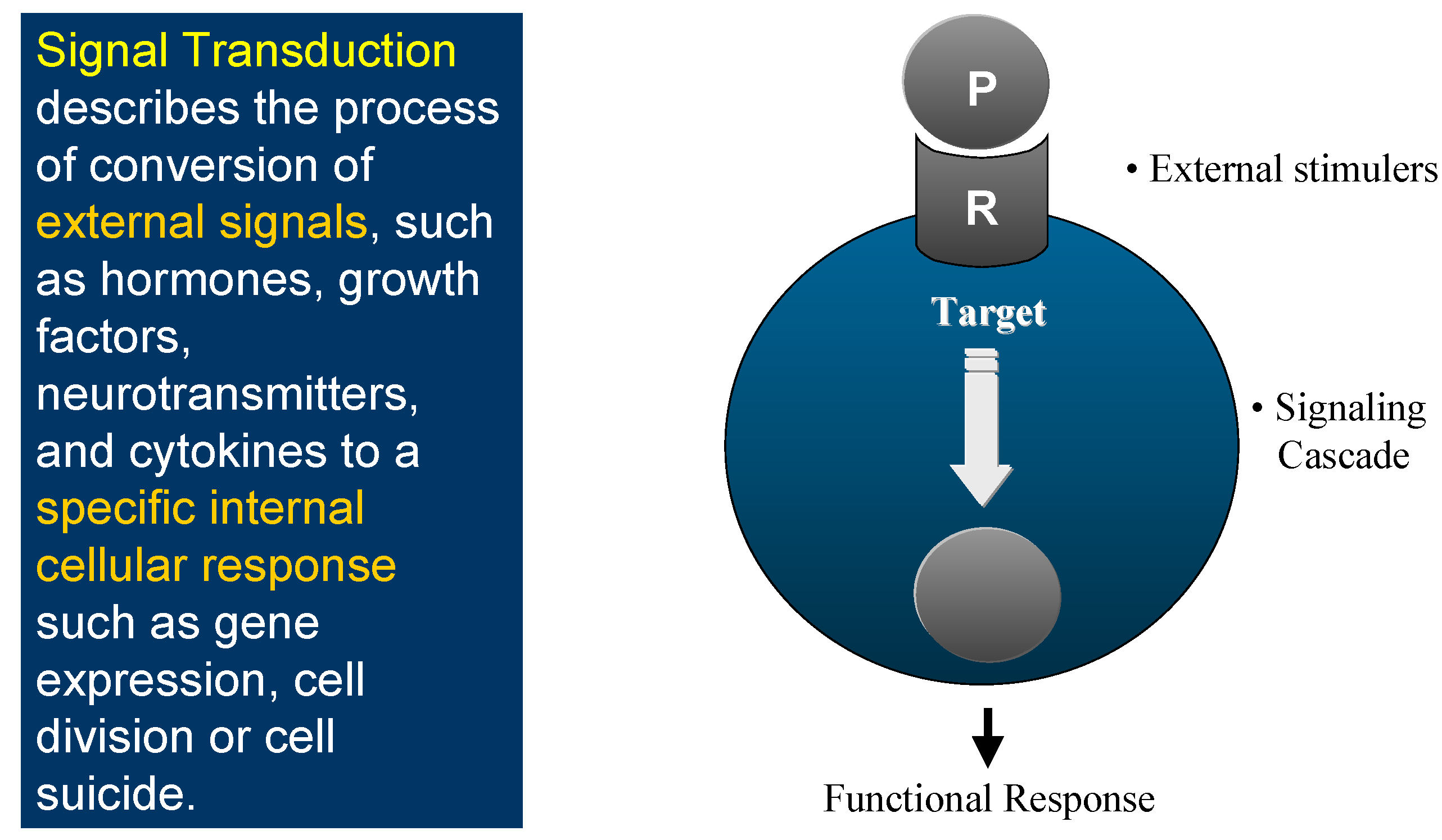
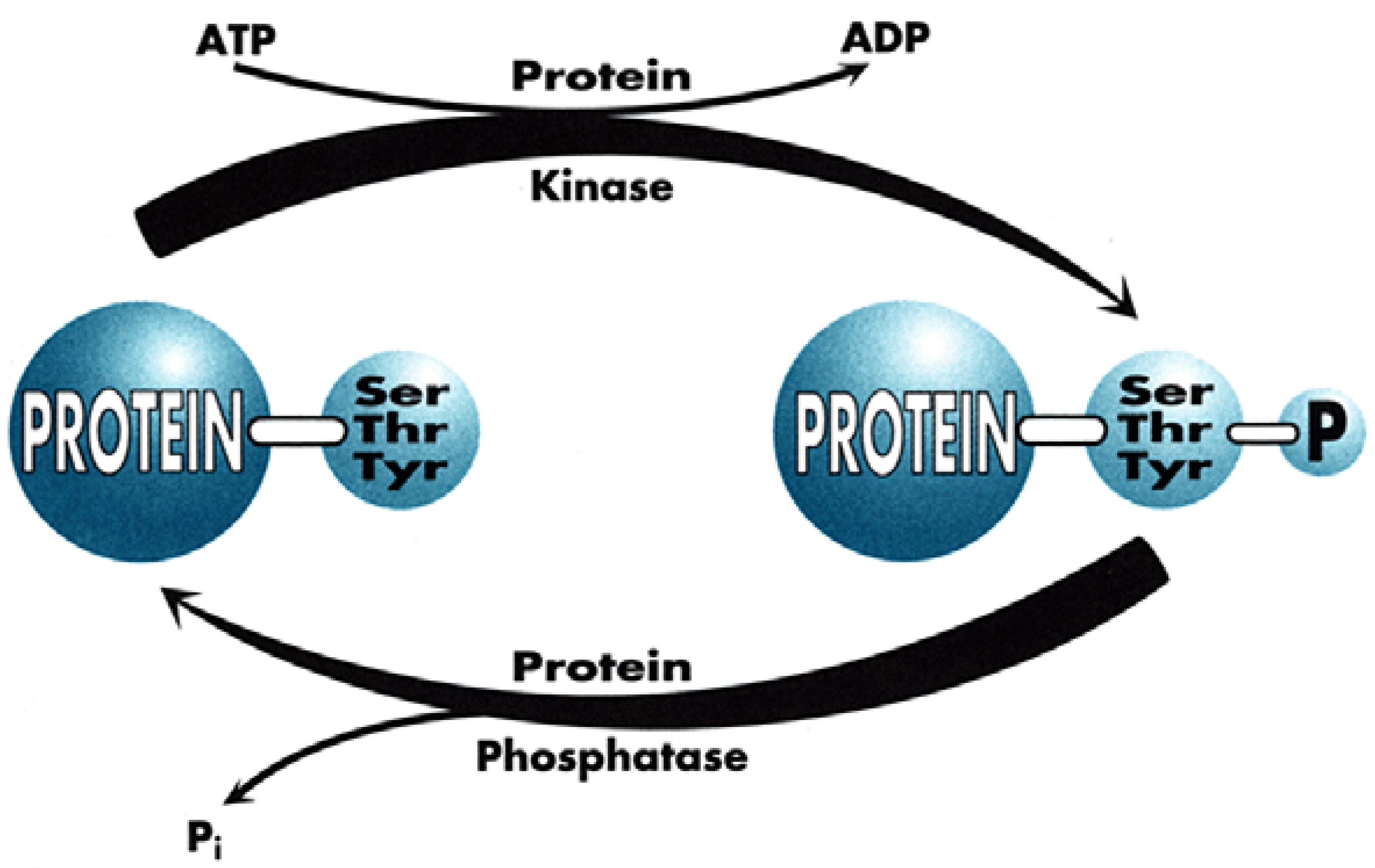
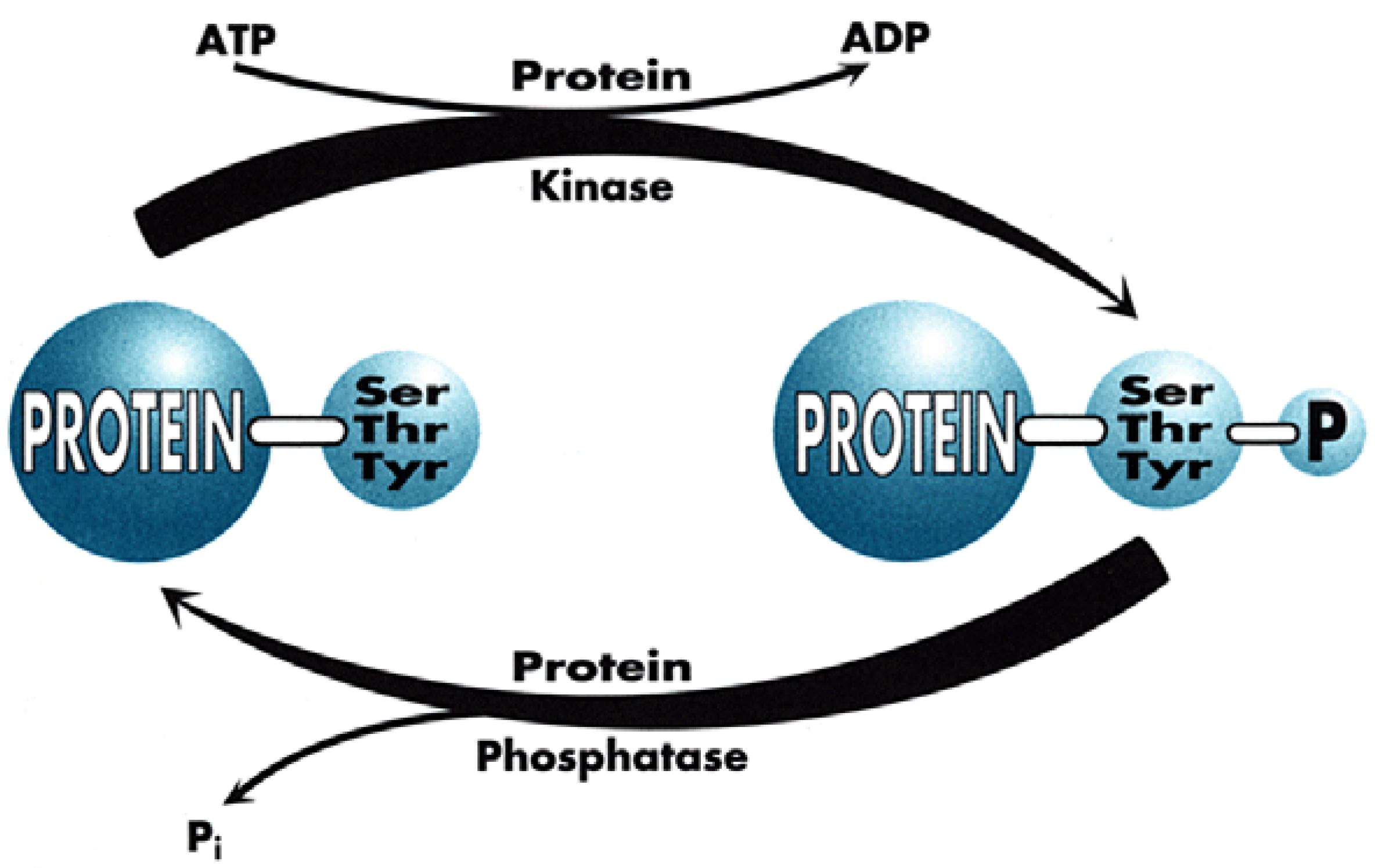
This is the world of Signal Transduction (Figure 1), which for chemists has tremendous advantages for proposing new targets for drug discovery within the scope of current knowledge and know-how. Indeed, plenty of Signal Transduction molecular mechanisms involve protein conformational changes, which are under the control of kinases and phosphatases, two enzyme classes that are tractable targets for drug design. Protein kinases catalyse the phosphorylation of Ser, Thr or Tyr residues of proteins (Figure 2), leading to phosphoproteins which generally adopt a different conformation, allowing them to interact with different partner proteins compared to the native, un-phosphorylated ones. Phosphatases, on the contrary, catalyse the de-phosphorylation of these phosphoproteins.
It is important to note that both enzyme types function through very distinct mechanisms since kinases are all characterised by a well-preserved ATP-binding site, close to the catalytic site (since kinases transfer one phosphate residue to proteins, generating ADP as a by-product) while phosphatases work in a way totally independent of ATP, generating inorganic phosphate upon the cleavage of the phospho-protein bond.
Figure 2.
Since the pioneering work of Ed Fisher and Ed Krebs (who were awarded the Nobel Prize in 1992), the reversible phosphorylation of proteins has been widely recognised as central to the regulation of most aspects of cell function and cell physiology. Increasing numbers of human diseases are known to involve mutations, over-expression, genetic association or malfunctioning of protein kinases and phosphatases, and their regulators and effectors. Signal Transduction, mediated by protein phosphorylation is extremely complex and our understanding of signalling mechanisms and their implication in diseases is still in its infancy. However, it represents a fantastic field of investigation to explore where small molecules will play a critical role, both in helping understand cell biology mechanisms to gain knowledge and progress, but also, hopefully, in proposing therapeutic solutions to unmet medical needs.
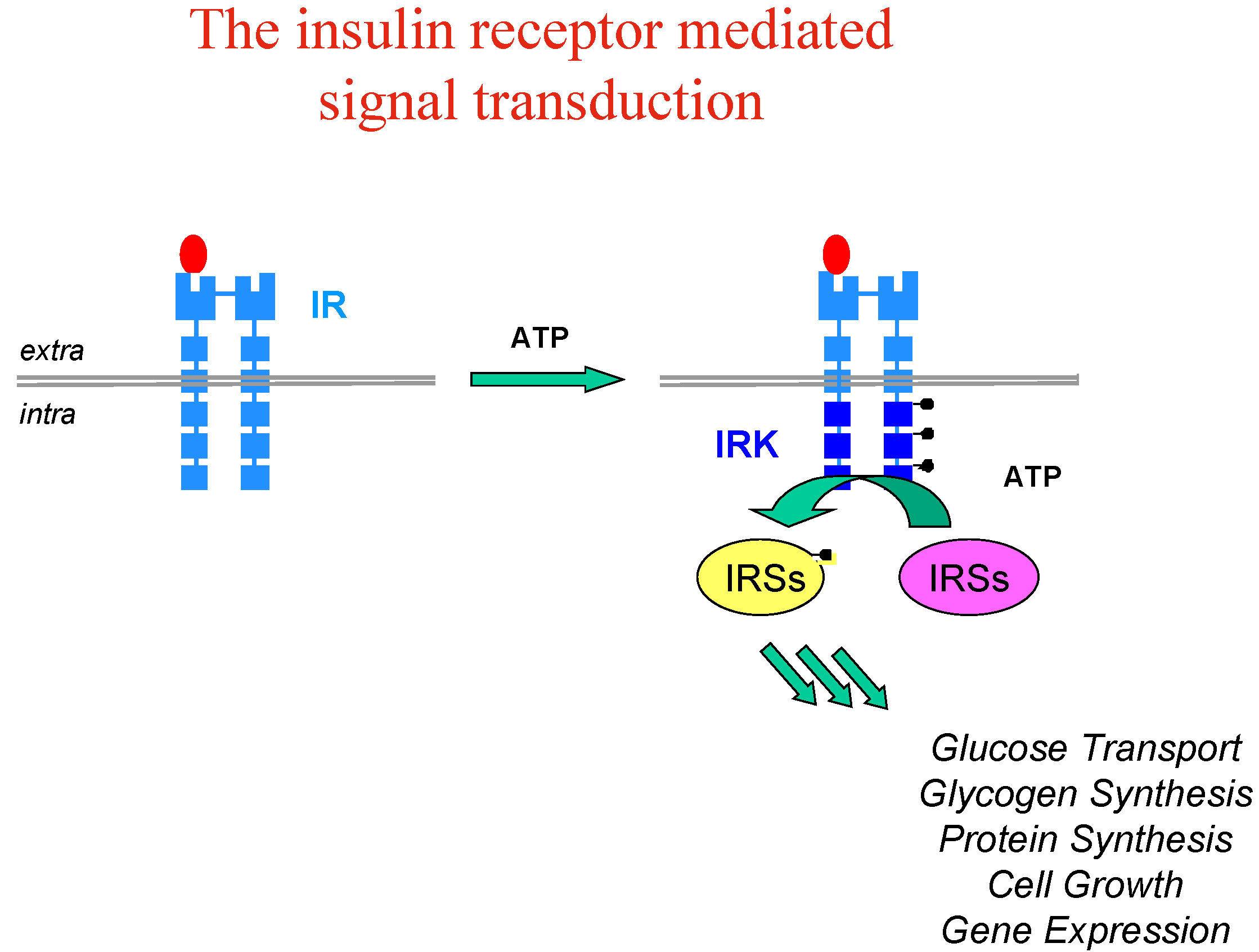
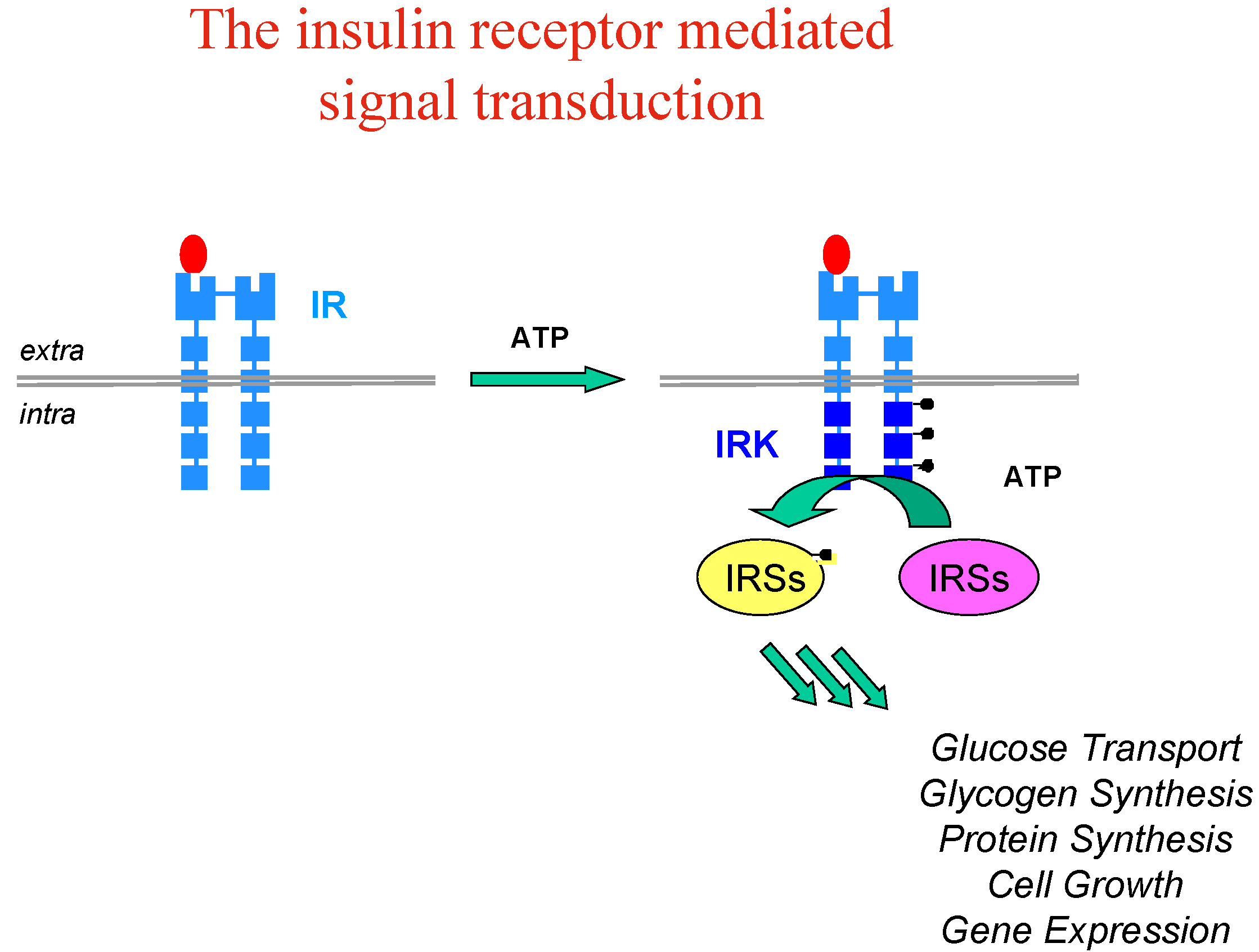
Let’s take the example of insulin, whose implication and therapeutic usefulness is well-known in diabetes type 1, while diabetes type 2 is characterised by insulin-resistance and impaired glucose tolerance with dramatic consequences for patients (blindness, kidney failure, amputations and heart diseases). Insulin resistance appears to involve an early process in insulin signal transduction which is initiated through the binding of insulin to the membrane insulin tyrosine kinase receptor (IR), which causes auto-phosphorylation of the receptor kinase and activates phosphorylation of other protein substrates, including IR substrate proteins (IRS-1, IRS-2, etc…) leading to a cascade of intracellular events which mediate the biological effect of insulin (Figure 3).
Figure 3.
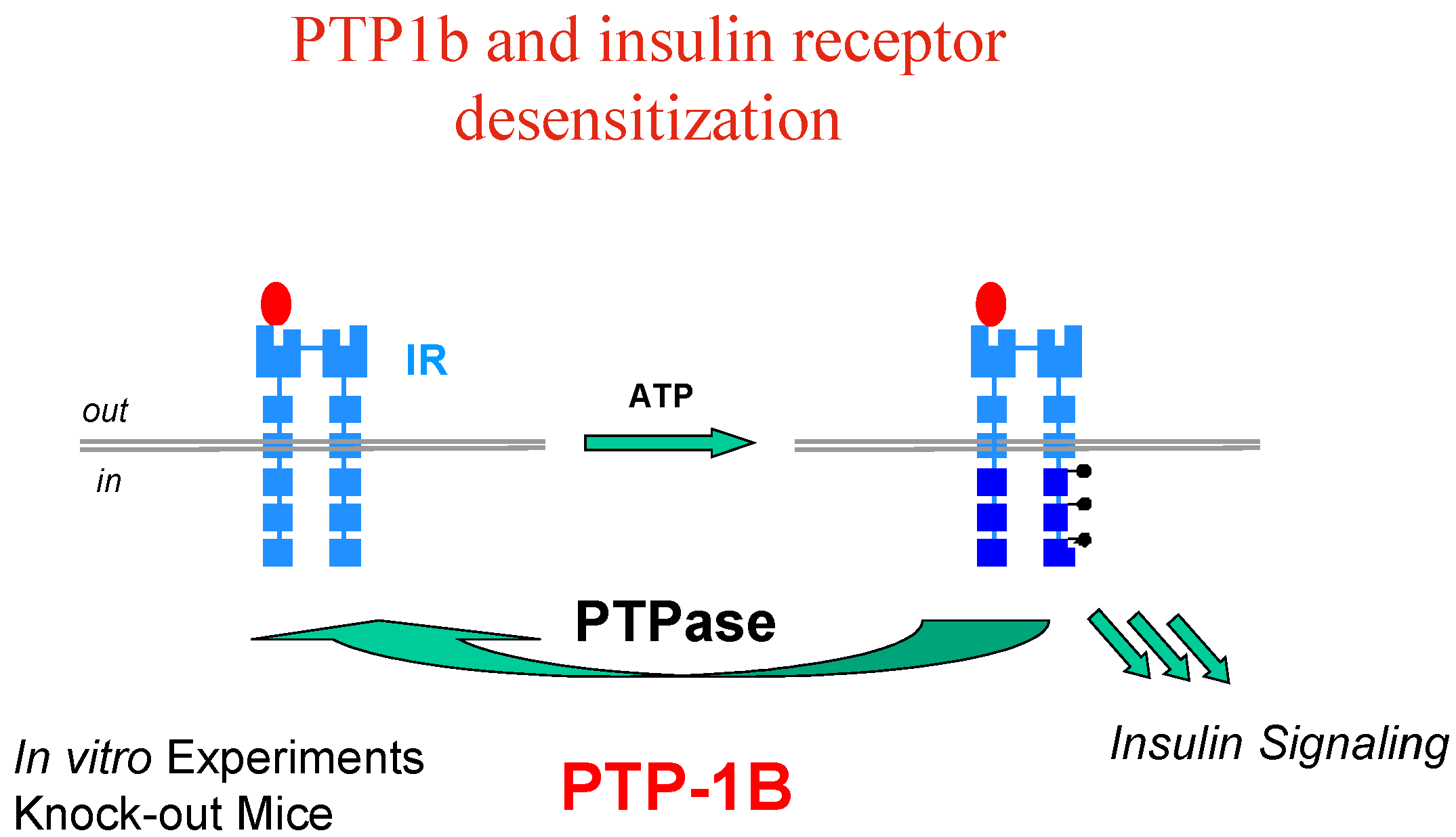
Activation (phosphorylation) of the IR by insulin is reversed by the action of protein tyrosine phosphatases (PTP’s), among which PTP1B has been clearly identified as a major player, based on in vitro experiments and knock-out mice models (Figure 4).
Figure 4.
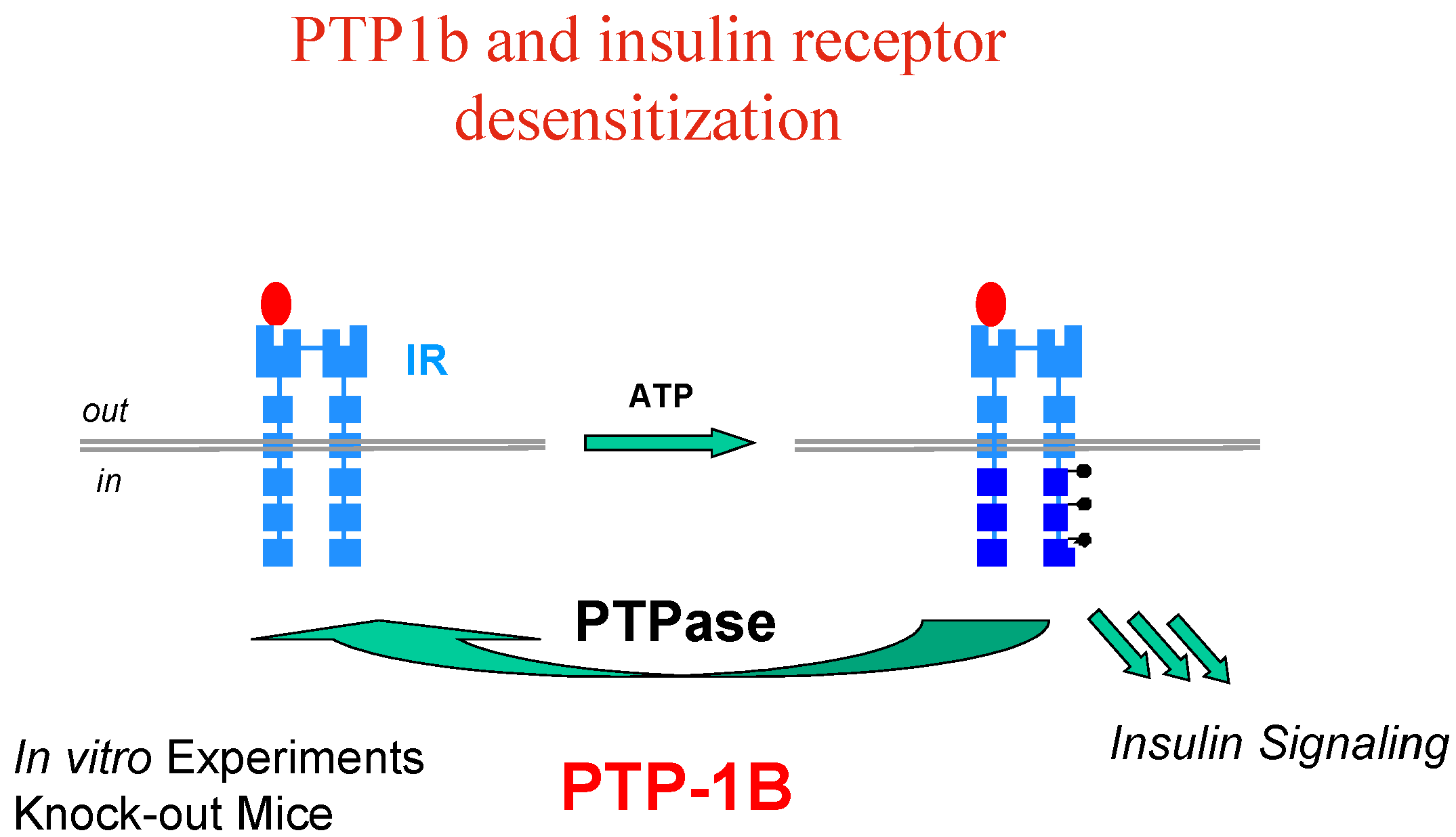
Interestingly enough, it has also been found that PTP1B is involved in regulating the leptin-signalling pathway as well, and resistance to the leptin hormone is a hallmark of obesity. Thus, PTP1B is thought to function as a negative regulator of insulin and leptin signal transduction; PTP1B inhibitors have therefore the potential to restore insulin/leptin sensitivity and should be effective in treating type 2 diabetes and obesity, which are linked diseases.
The search for PTP1B inhibitors is currently extensively pursued by several pharma and biotech companies. Our efforts in Serono, based on rational design, (screening of focused libraries around proprietary scaffold design with the help of PTP1B X-ray structures and in-house SAR studies), followed by drug-like properties optimisation led us to identify PTP1B inhibitors which increase glucose uptake in C2C12 muscle cells and decrease plasma glucose and insulin levels in genetically modified obese mice (db/db) upon oral administration in a dose dependant manner, with no sign of toxicity. Efforts are currently pursued to move the most promising PTP1B inhibitors to the clinic and evaluate their therapeutic potential in diabetic or obese patients.
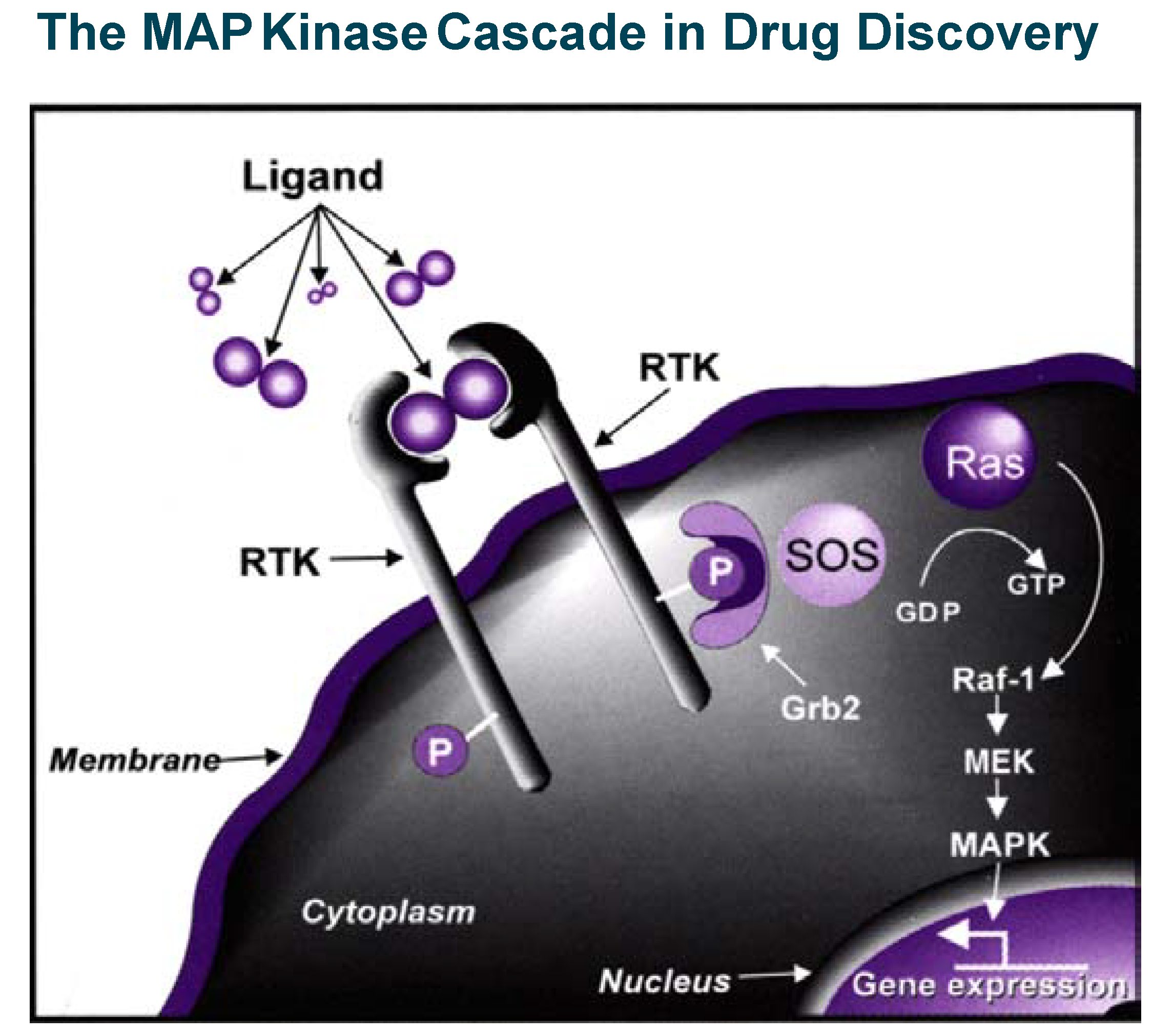
The insulin/PTP1B example illustrates in a powerful way that blocking phosphatases can stimulate a signal transduction cascade and therefore mimic or replace a secreted protein which action is beneficial in pathological conditions. Inhibition of kinases (which, most of the time positively relay signalling events) is, on the contrary, a powerful way to inhibit or control signal transduction cascades, which become particularly relevant in drug discovery when pathological conditions are associated with stimulation of intra-cellular signalling (see the example of MAP kinase cascade in Figure 5). This is the case in important therapeutic areas like cancer, inflammation and apoptosis-related disorders where efforts to find selective kinase inhibitors have literally exploded in the last five years. A landmark event occurred in May 2001, when GleevecTM (Glivec® outside the U.S.), the first important drug targeting specifically a protein kinase (ABL) was approved for clinical use to treat chronic myeloid leukaemia.
Figure 5.
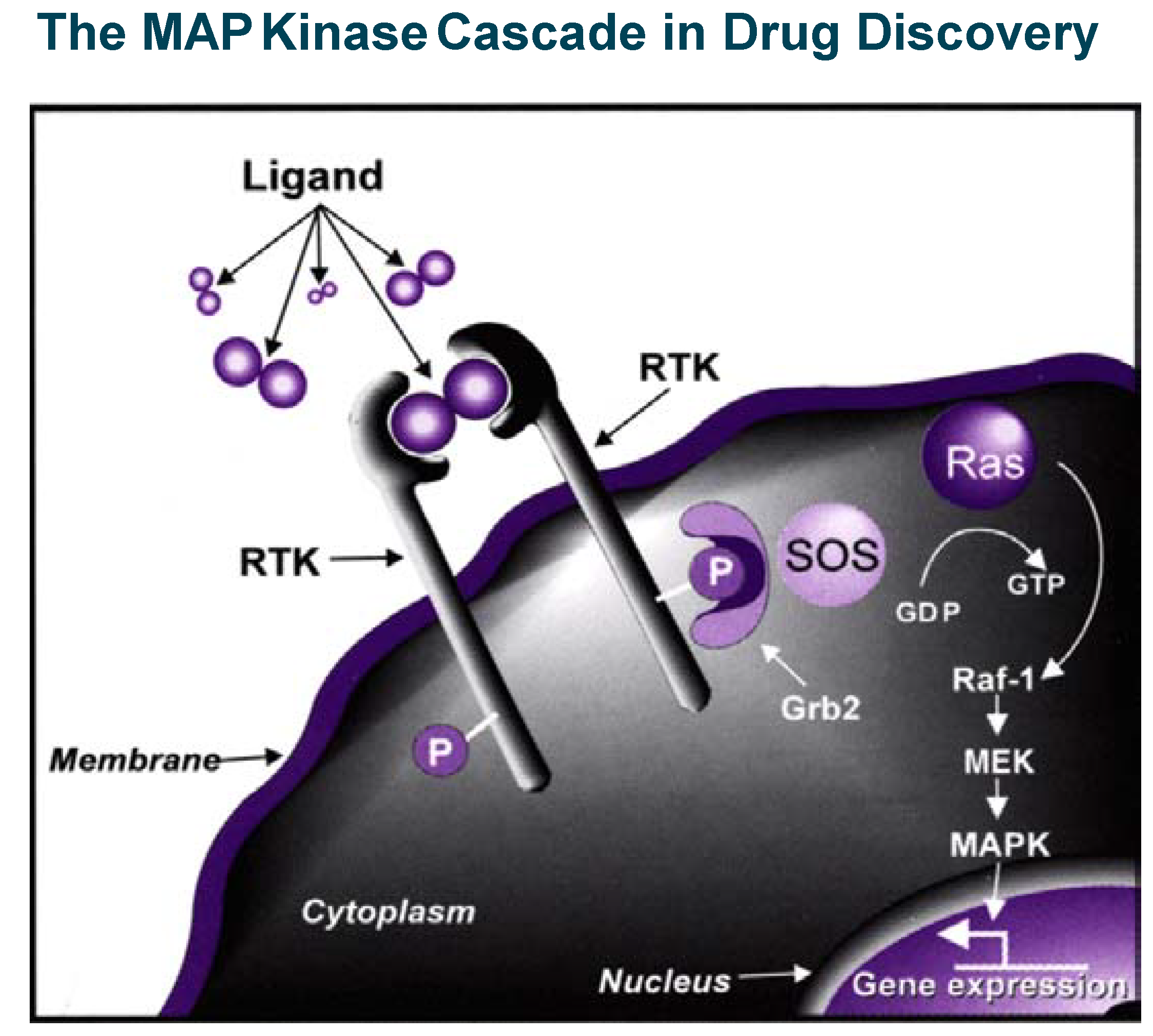
The human genome encodes for around 500 different kinases, which control most cellular events, directly or indirectly. This is what makes kinases attractive as a drug target class; however this also raises immediately two important questions: What kinase for what disease? And how to design selective inhibitors for specific kinases of therapeutic interest?
What kinase for what disease?
Validation of individual kinases for drug discovery (which means obtaining enough convincing evidence that inhibitors of such kinases will demonstrate a therapeutic effect in patients during phase II clinical trials) is not trivial and remains one of the most challenging aspects of post-genomic drug discovery. Dissection of signal transduction mechanisms is certainly a good starting point, generally followed by knock-out animal experiments and/or pharmacological studies with known inhibitors. However, none of these approaches is fully satisfactory and new ones are currently under investigation based on chemical genetics, as for example the Analog-Sensitive Kinase Allele (ASKA) technology where chemists have engineered modified kinases and inhibitors (by generating functionally active kinase mutants which are specifically inhibited by chemically modified inhibitors), thus allowing to study specific responses in vivo in knock-in animals, (and therefore validate the kinase for a given disease) prior to start Med Chem. Such mouse disease models with functionally intact, specifically and pharmacologically inhibitable kinase targets will also provide invaluable information with respect to therapeutic index linked to the target kinase inhibition, new substrates identification and biomakers discovery. This is a beautiful example where a multidisciplinary approach (chemistry, together with structural biology, molecular biology, genomics an pharmacology) is providing new breakthough tools which will allow drug discovery to progress very rapidly.
Designing selective kinase inhibitors
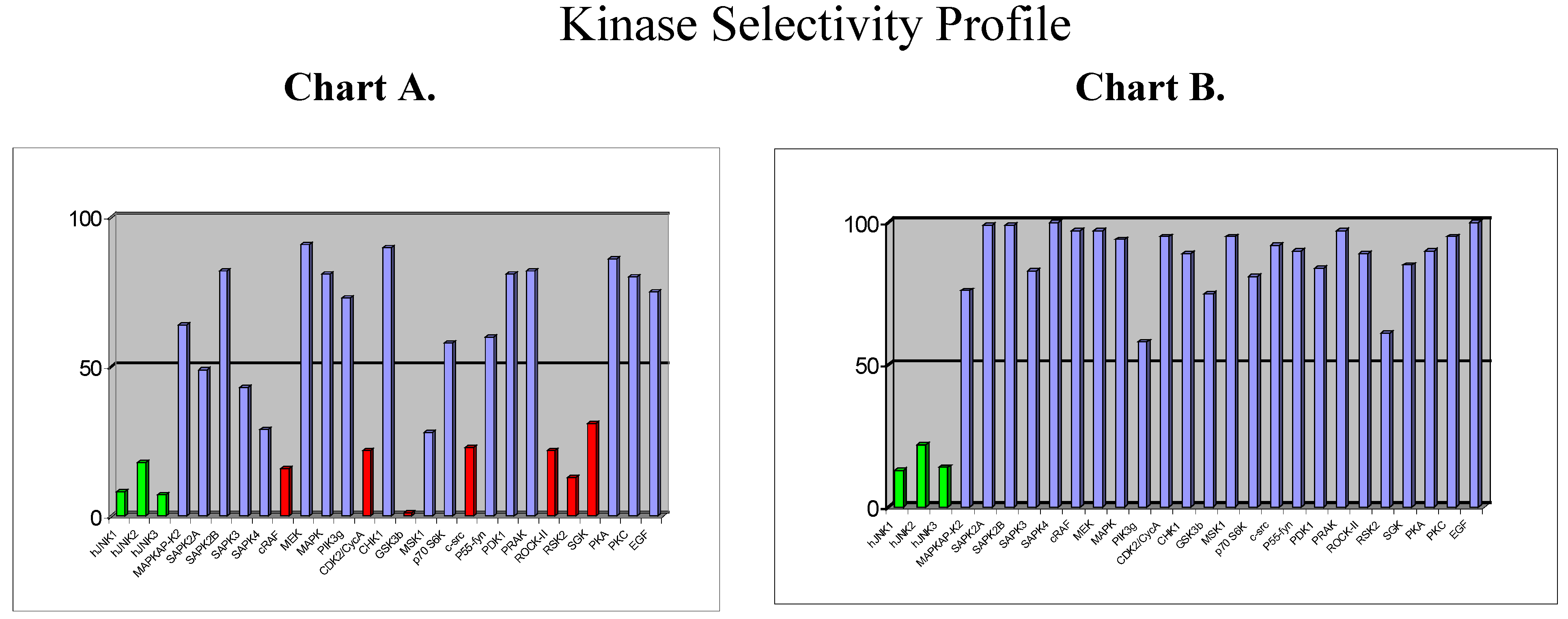
Beyond the choice of the kinase, the next challenge will be to design selective inhibitors. So far, the vast majority of kinase inhibitors reported to date are ATP competitive, which means that they bind to the ATP binding site, which is common for all kinases and which is highly conserved in terms of amino acid sequence. It is therefore not surprising that plenty of ATP-competitive inhibitors are poorly selective (as for example staurosporine). But, … is it possible to design selective ATP-competitive inhibitors? As part of our programme directed toward the discovery of JNK inhibitors in inflammation/apoptosis related disorders, we have discovered a new chemical class of ATP-competitive inhibitors derived from the benzothiazole scaffold. First compounds from this series which were identified as potent JNK inhibitors also displayed strong activity against a few other kinases, as shown by the selectivity profile depicted in chart A below (Figure 6).
Figure 6.
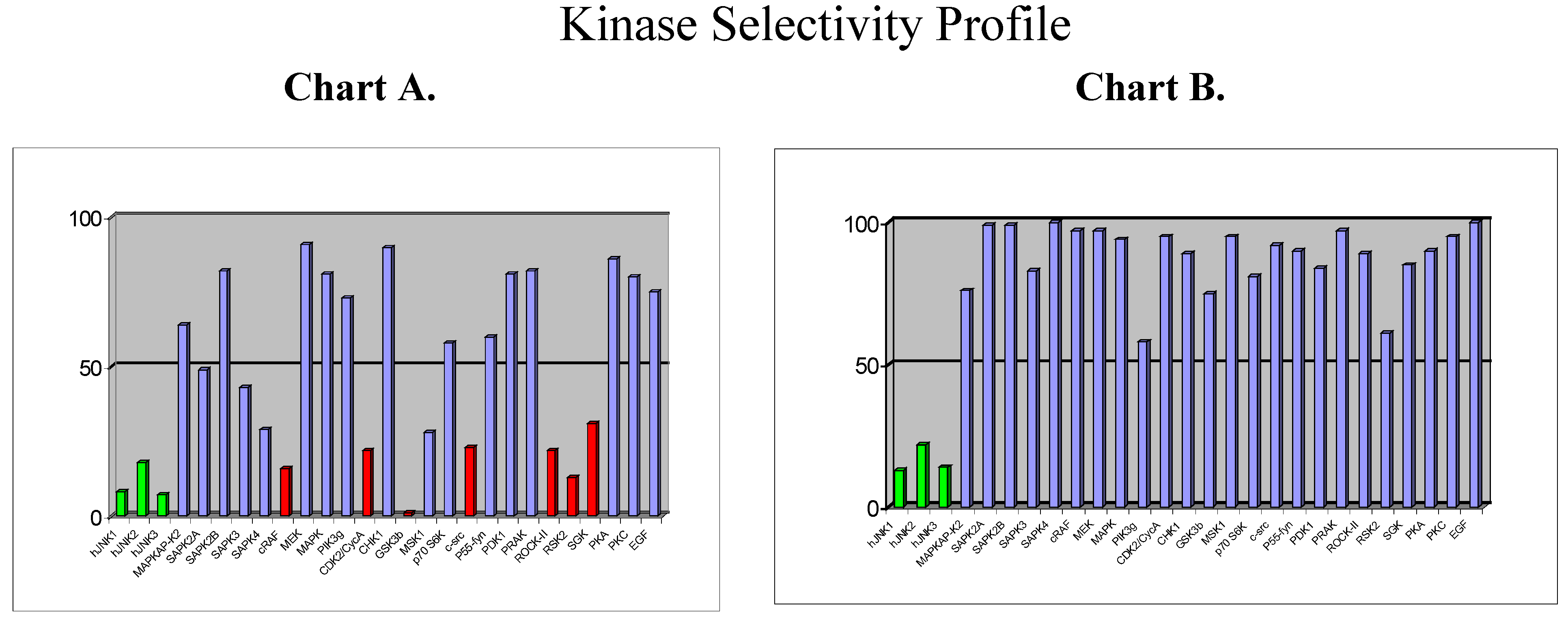
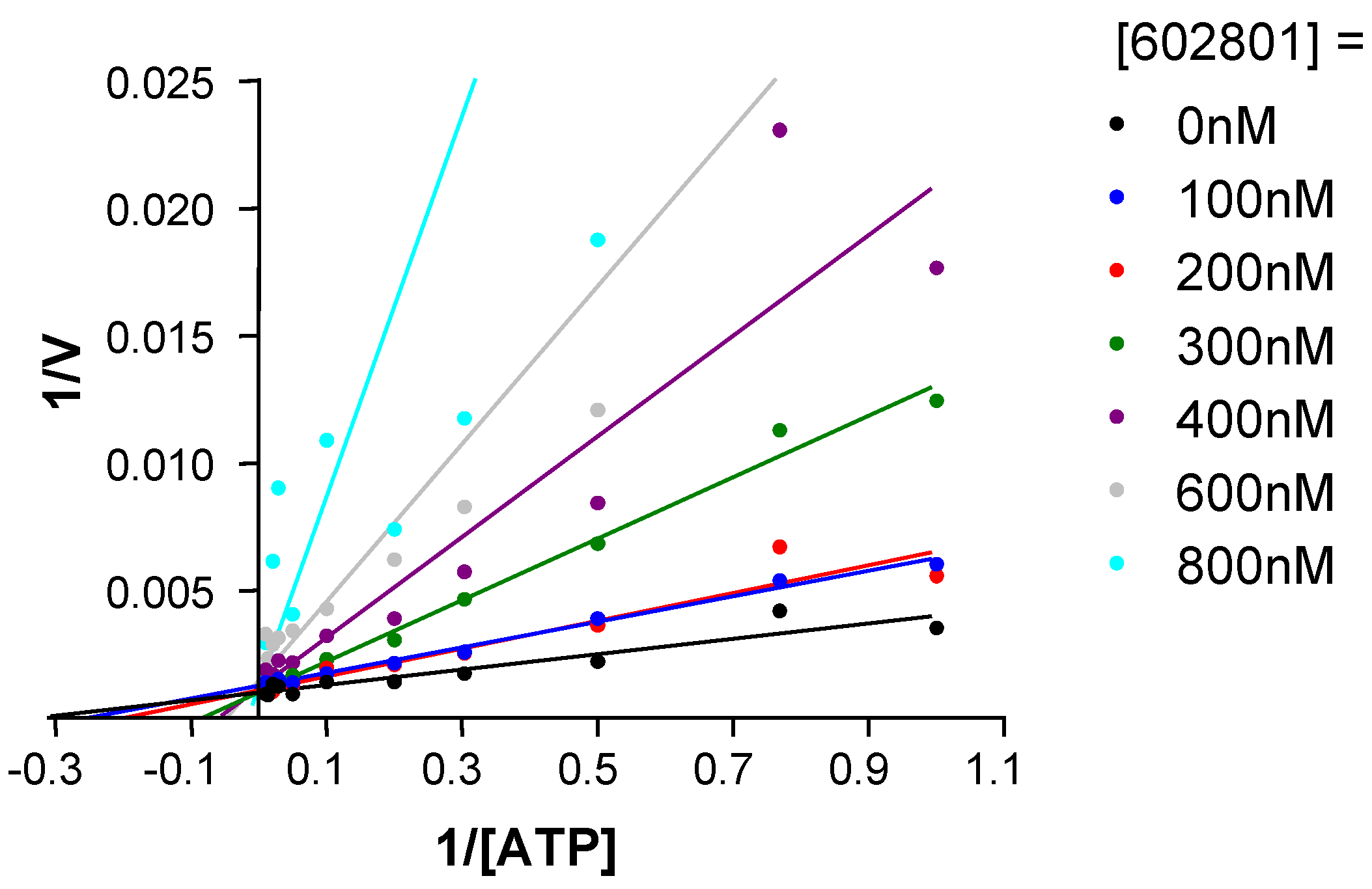
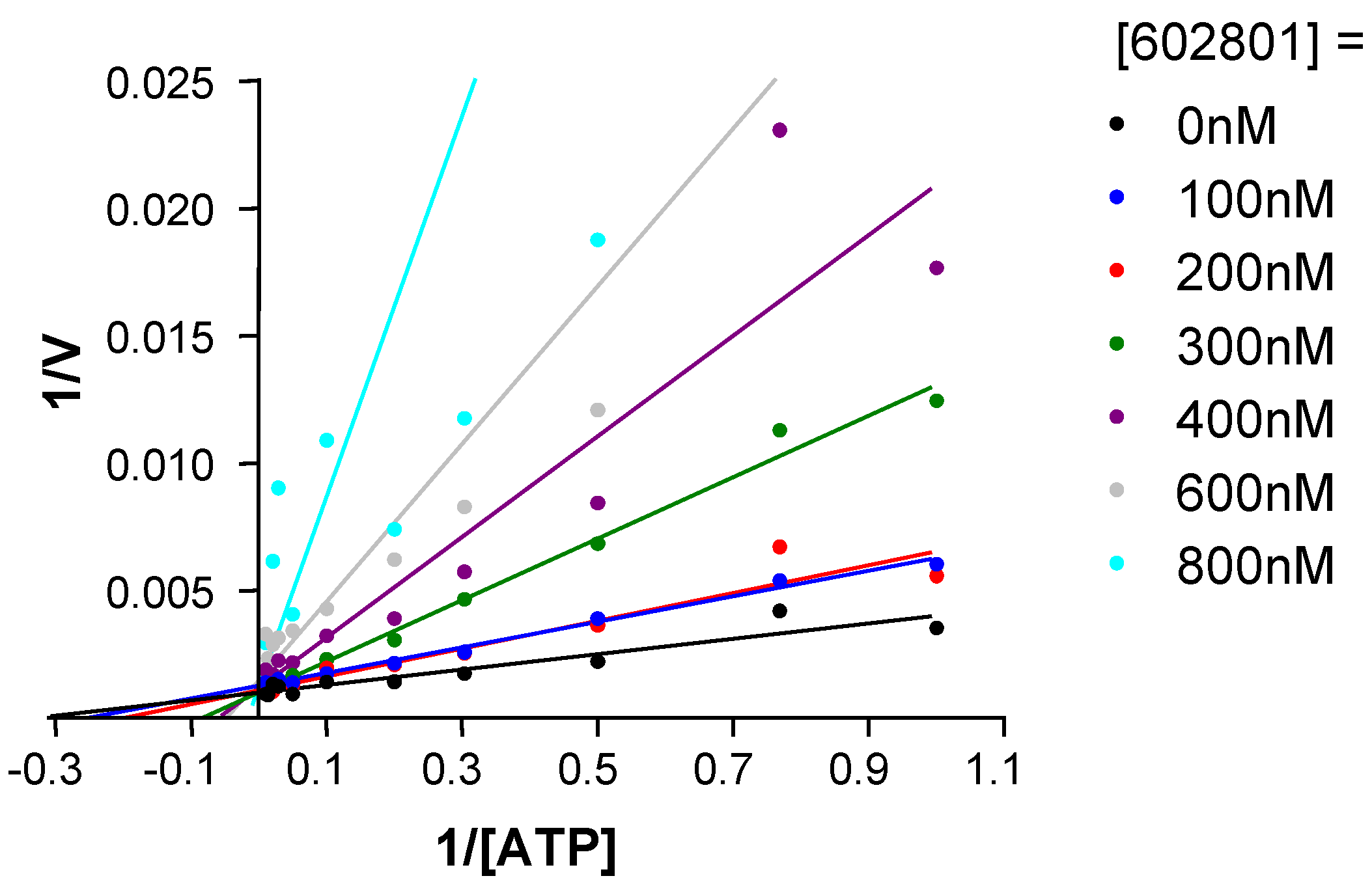
Chemical modifications based on JNK 3D-structure, docking experiments and in-house SAR data provided second generation JNK inhibitors, which were found highly selective versus other kinases, as shown by the selectivity profile depicted in chart B. Kinetic experiments at various ATP and inhibitor concentrations demonstrate that this compound is purely ATP competitive as shown by Lineweaver-Burk representation in Figure 7.
Figure 7.
The best JNK inhibitors identified so far from this project are currently evaluated in pre-clinical studies, based on preliminary promising results in animal models of auto-immune diseases and neuronal apoptosis.
This example shows that it is possible to design new potent selective inhibitors of kinases, which are ATP competitive. The starting point is a generic kinase scaffold (hopefully proprietary) from which selectivity can be achieved through proper substitution driven by rationale design based on the kinase specific sequence and its 3D structure. This is a powerful tool, because it can be applied to different kinases, from the same central scaffold, providing proper information is available to direct chemist’s efforts in the right direction. Forthcoming kinases of therapeutic interest (among the 500 ones encoded by the human genome) will benefit from recent progresses in bioinformatics (in particular prediction of 3D structure based or primary sequence) and high-throughput production of protein constructs associated with new crystallisation technologies, which will provide new kinase 3D models or real structures for in silico design of selective inhibitors.
What about non-ATP competitive inhibitors?
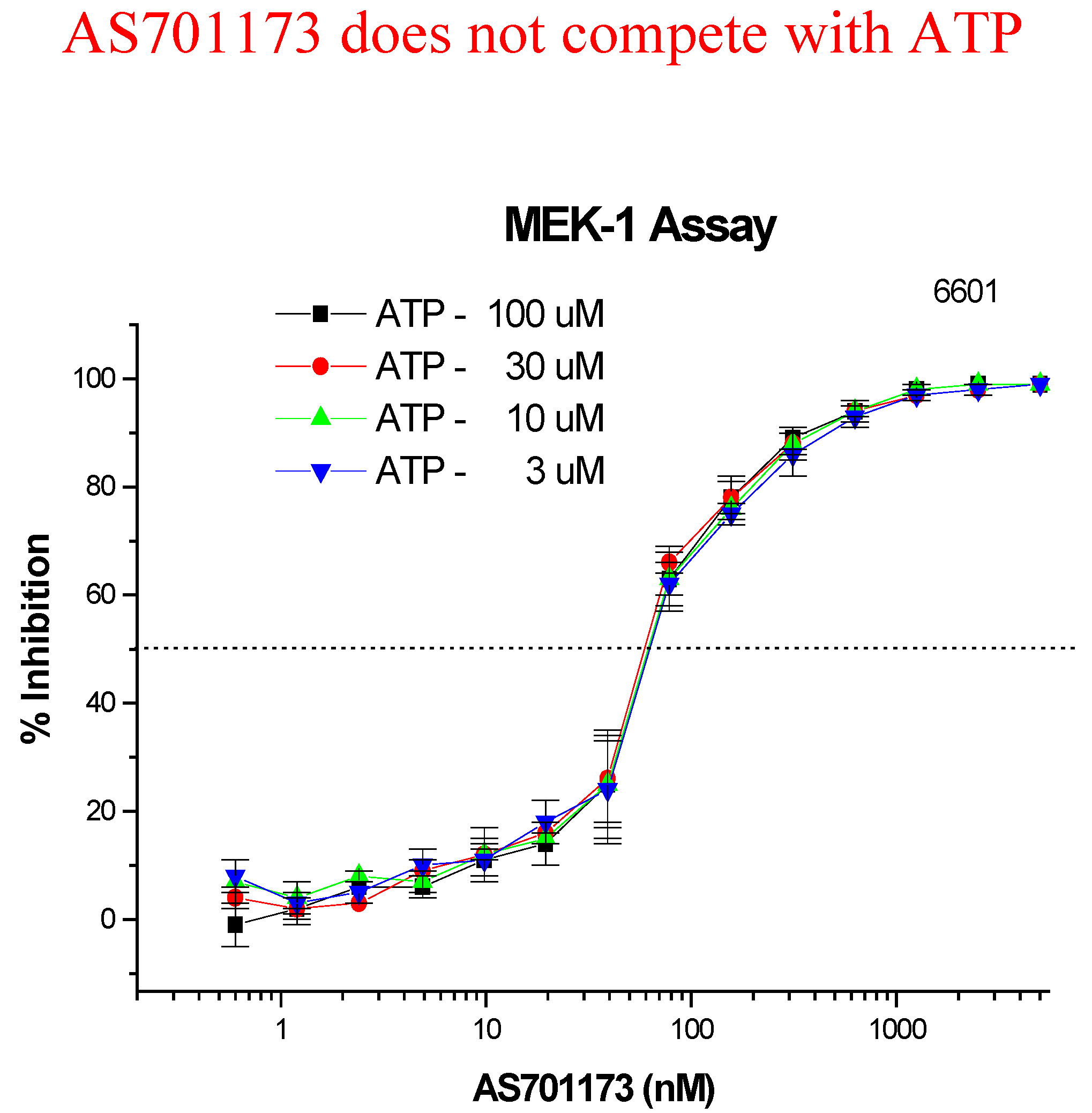
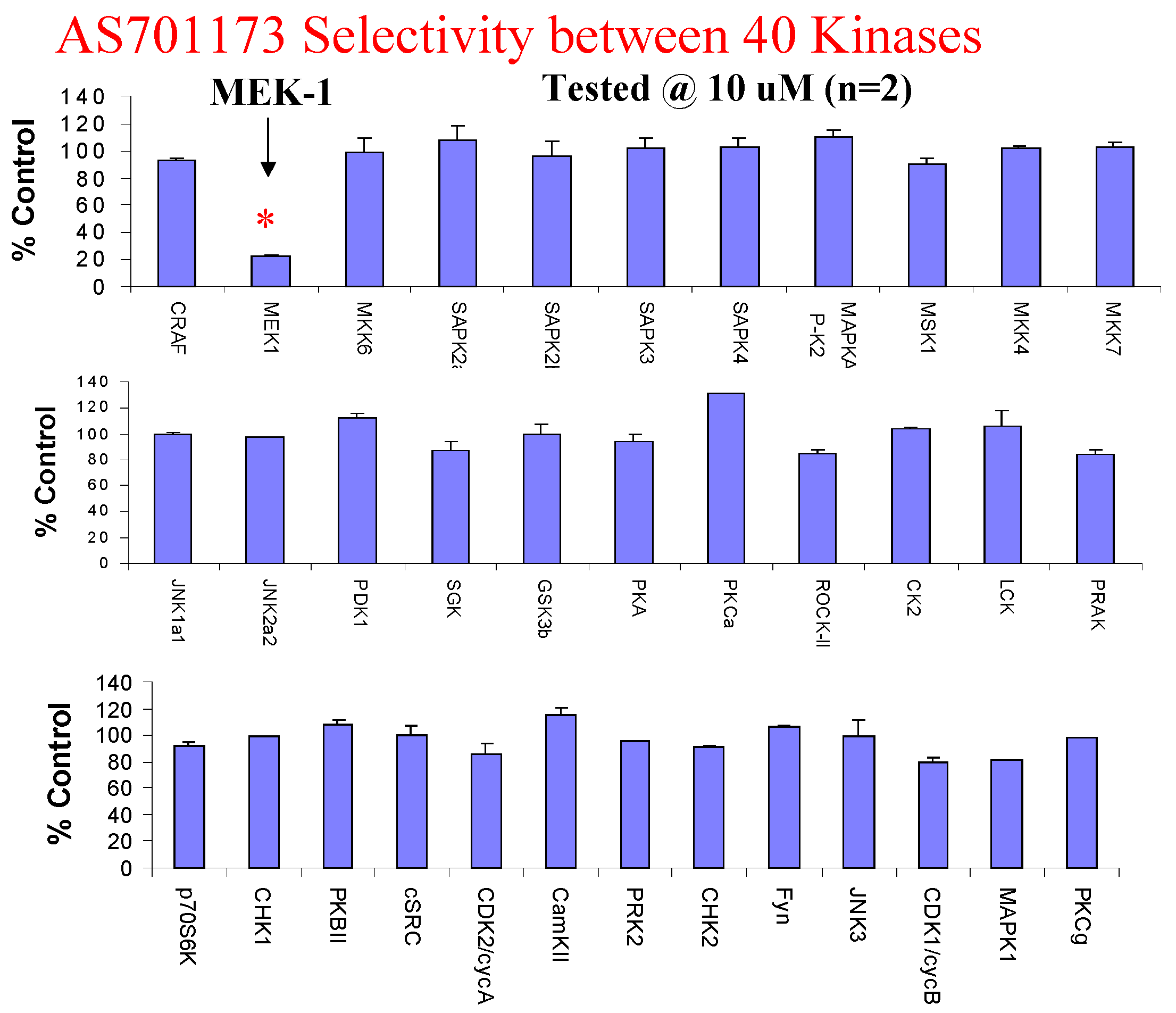
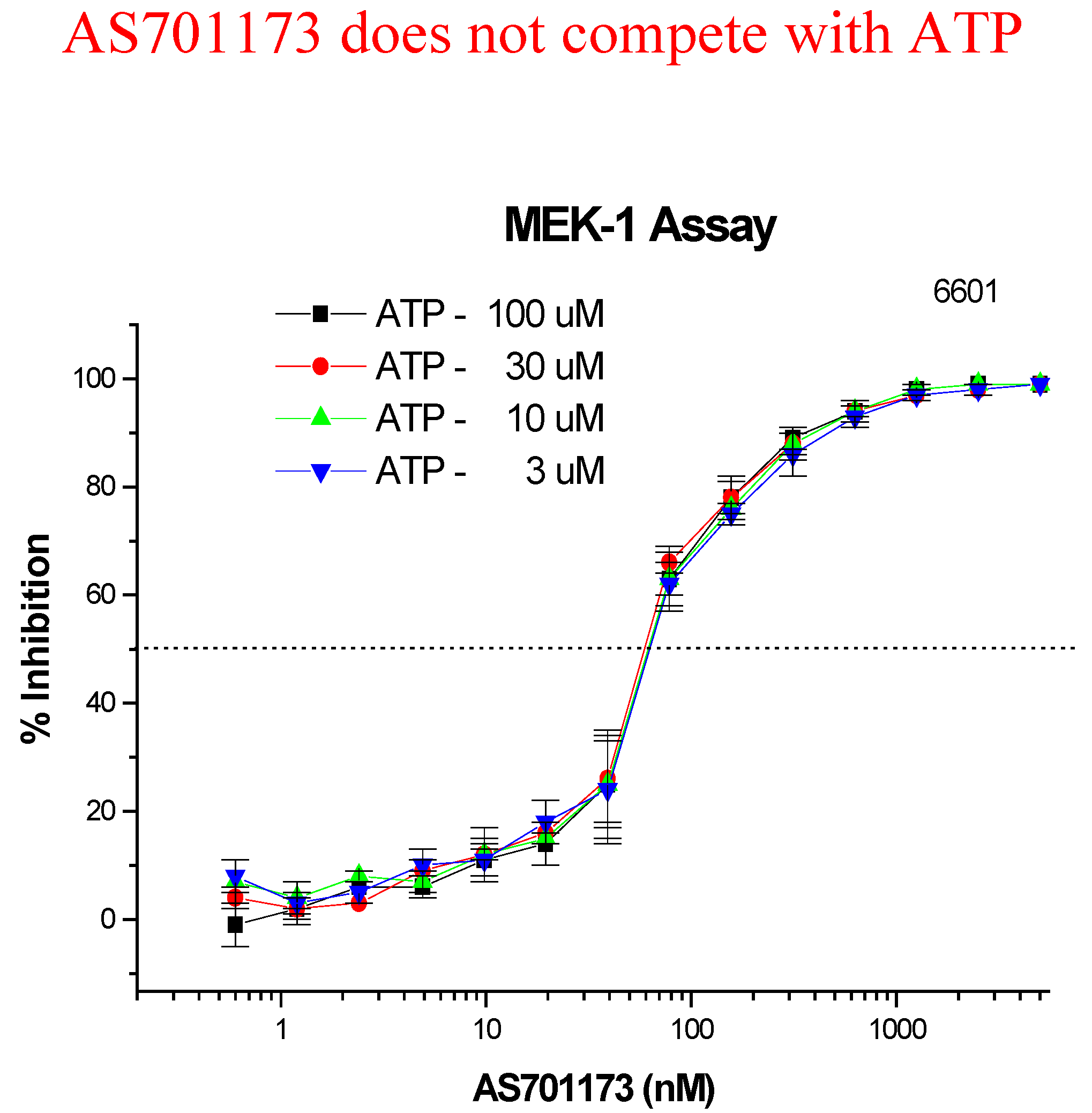
An alternative way to avoid promiscuity in the design of kinase inhibitors is to look for non-competitive inhibitors with respect to ATP, with the aim to hit interactions at kinase sites, which are not commonly found within the enzyme class. Very little is known to really define a strategy based on rationale design for such type of inhibitors and therefore high-throughput screening of highly diverse collection of compounds remains as one of the most promising approach. This is the one we have followed as part of our efforts to find new MEK inhibitors as potential anti-cancer agents. Screening of 50K compounds led to the identification of a few positives among which AS701173 was characterised as a potent (IC50=30nM) non ATP-competitive inhibitor since inhibition of MEK by that particular inhibitor was independent of ATP concentrations used in the assay conditions (Figure 8).
Figure 8.
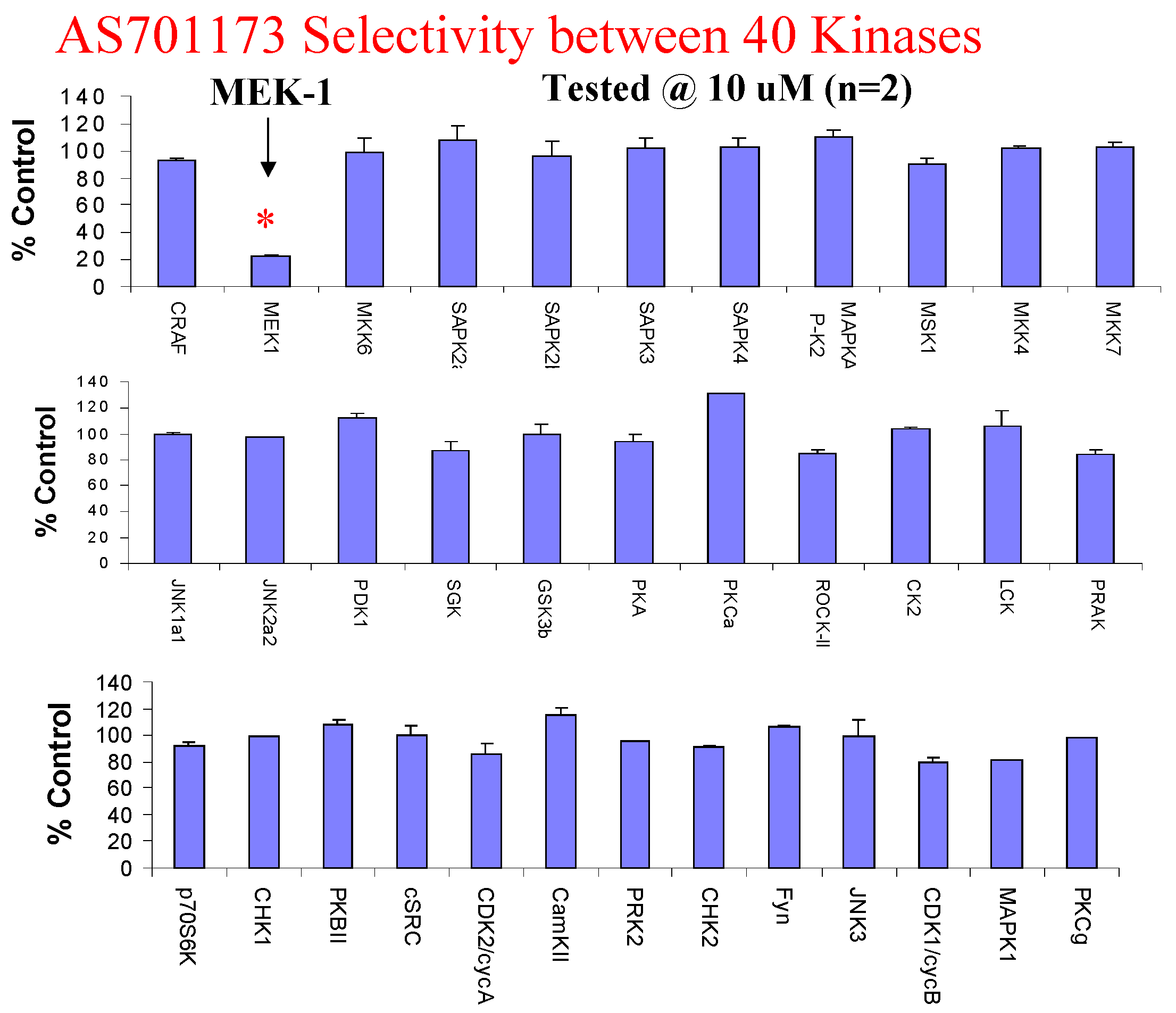
Evaluation of AS701173 against a panel of 40 available kinases show that this compound is highly selective for MEK since none of the other kinase was inhibited by more than 20% at 10μM.
Figure 9.
Lead optimisation based on this new selective MEK inhibitor is in progress and promising preliminary results have recently been obtained with newly found analogs demonstrating potent activity in tumour cell proliferation assays and in in vivo models of cancer.
From a chemist’s perspective, these two examples show that combinatorial chemistry and chemical diversity (cfr MEK selective inhibitors) on one side, as well as “surgical” modifications of molecules in a precise way driven by rationale design (cfr JNK selective inhibitors design) on the other side are complementary tools in drug discovery; both of them can only lead to success if chemists master the art of organic chemistry, and if the art of organic chemistry is continuously offering new methods, reactions, reagents, strategies to solve new emerging challenges.
Kinases and phosphatases inhibitors described above illustrate the potential of signal transduction in drug discovery where understanding of cell biology events has allowed to identify druggable targets which play a critical role in diseases and for which chemists are able to design potent and selective inhibitors with promising therapeutic applications. This drug discovery strategy has enormous potential since, on one hand many proteins that will be identified as key players in pathological conditions will be up-or down-regulated, directly or indirectly by kinases or phosphatases. On the other hand, the design and synthesis of selective, potent, cell permeable drug-like inhibitors of kinases and phosphatases is within the scope of chemist’s current expertise. Synergy between synthetic chemistry, computational chemistry, enzymology, bioinformatics, cheminformatics, cell biology, structural biology and molecular pharmacology will provide the necessary technology platform to speed-up the discovery of inhibitors and improve the quality of drug candidates.
Acknowledgments
| PTP1b: | Rob Hooft, Agnès Bombrun, Dominique Swinnen, Pierre-Alain Vitte. |
| JNK: | Pascale Gaillard, Christian Rommel, Montse Camps, Yolande Chvatchko, Dominique Perrin, Anthony Nichols, Jean-Pierre Gotteland. |
| MEK: | Robert Murray, Sharad Magar, Peter Blume- Jensen |
Leading References
- Graves, J. D.; Krebs, E. G. Protein Phosphorylation and Signal Transduction. Pharmacol. Ther. 1999, 82, 111–121. [Google Scholar]
- Hunter, T. Signaling – 2000 and Beyond. Cell 2000, 100, 113–127. [Google Scholar]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinases signaling. Nature 2001, 411, 355–365. [Google Scholar]
- Johnson, T. O.; Ermolieff, J.; Jirousek, M. R. Protein Tyrosine Phosphatase 1B Inhibitors for Diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar]
- Tobin, J. F.; Tam, S. Recent Advances in the Development of Small Molecule Inhibitors of PTP1b for the Treatment of Insulin Resistance and Type 2 Diabetes. Curr. Op. Drug Disc. Devel. 2002, 5, 500–512. [Google Scholar]
- Hooft van Huijsduijnen, R.; Bombrun, A.; Swinnen, D. Selecting Protein Tyrosine Phosphatases as Drug Targets. Drug Discov. Today 2002, 7, 1013–1019. [Google Scholar]
- Druker, B. J. STI571 (Gleevec) as a Paradigm for Cancer Therapy. Trends Mol. Med. 2002, 8, 514–518. [Google Scholar]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, Imatinib), a Rationally Developed Targeted Anti Cancer Drug. Trends Mol. Med. 2002, 1, 493–502. [Google Scholar]
- Manning, G.; Whyte, D. B.; Martinez, R.; Hunter, T. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1916. [Google Scholar]
- Bishop, A. C.; et al. A Chemical Switch for Inhibitor-Selective Alleles of any Protein Kinase. Nature 2000, 407, 395–401. [Google Scholar]
- Bishop, A. C.; Buzko, O.; Shokat, K. M. Magic Bullets for Protein Kinases. Trends Cell Biol. 2001, 11, 167–172. [Google Scholar]
- Bridges, A. J. Chemical Inhibitors of Protein Kinases. Chem Rev 2001, 101, 2541–2572. [Google Scholar]
- Cohen, T. Protein Kinases – the Major Drug Targets for the Twenty-first Century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar]
- Sebold-Leopold, J. S. Development of Anti-cancer Drugs Targeting the MAP Kinase Pathway. Oncogene 2000, 19, 6594–6599. [Google Scholar]
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Halazy, S. Signal Transduction: An Exciting Field of Investigation for Small Molecule Drug Discovery. Molecules 2003, 8, 349-358. https://doi.org/10.3390/80400349
AMA Style
Halazy S. Signal Transduction: An Exciting Field of Investigation for Small Molecule Drug Discovery. Molecules. 2003; 8(4):349-358. https://doi.org/10.3390/80400349
Chicago/Turabian StyleHalazy, Serge. 2003. "Signal Transduction: An Exciting Field of Investigation for Small Molecule Drug Discovery" Molecules 8, no. 4: 349-358. https://doi.org/10.3390/80400349