Spectroscopic and Theoretical Studies of Quantum and Electronic Confinement Effects in Nanostructured Materials

1
Department of Chemistry, Nankai University, Tianjin 300071, P. R. China
2
Present address: College of Life Sciences, Nankai University, Tianjin 300071, P. R. China
*
Author to whom correspondence should be addressed.
Molecules 2003, 8(1), 207-222; https://doi.org/10.3390/80100207
Submission received: 1 January 2003
/
Published: 31 January 2003
(This article belongs to the Special Issue Selected Papers from the International Symposium on Frontiers in Molecular Science (ISFMS 2002))
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Nanostructured materials have become the central subject of materials research during the last decade in the past 20th century owing to the novel electronic, optical, and catalytic properties observed in such materials. The unusual properties of these nanostructured materials can be attributed to two main microscopic effects: quantum confinement and electronic confinement. These two effects have dominated the variations of molecular properties in a wide variety of nanostructured materials, ranging from inorganic compounds to organic molecules. The recent advances have focused on host-guest complex systems that have resulted in a deeper understanding of the changes in electronic structures when being confined. In this article we cover some of the key advances in the study of quantum and electronic confinement effects, especially in host-guest systems.
Introduction
Nanostructured materials are a novel form of compounds of theoretical interest and with the potential to develop as an important class of materials for the electronics and photonics industries in the 21th century [1, 2, 3]. The research field of nanostructured materials has already been widely recognized as one of the most promising and rapidly emerging research areas in nowadays materials science. A new era in the development of novel nanostructured materials has been opened since the discovery of the periodic nanoporous inorganic material known as Mobil Catalytic Material 41 (MCM-41) for confining close-packed, functionalized organic molecules within an inorganic host [4]. Since then, a wide range of nanoporous host materials has been prepared, including alumina, zirconia, titania, niobia, tantalum oxide, manganese oxide, and even metals [5]. Meanwhile, numerous papers have then been published on the preparation of nanocomposite materials of novel chemical compositions and on the fundamental understanding of physics of materials at the nanometer scale [6, 7]. Encapsulation of conjugated polyaniline filaments with mobile charge carriers in the nanoporous channels of MCM-41 was a milestone in the history of nanocomposite materials, representing a step toward the design of nanometer electronic devices [8].
The study of nanocomposite materials has become our particular interest and these host-guest complex systems can be best described as being organized into spatially identifiable domains of an organic and an inorganic component. They are generally produced using various methodologies of “soft” inorganic chemistry in liquid or sol/gel media, exploiting either self-assembly mechanisms under the influence of the template agents, or directed assembly of nano building blocks in solution or at liquid/solid interfaces. A general classification has been proposed distinguishing “class I materials”, in which the inorganic and organic components interact only weakly through hydrogen bonding, van der Waals contacts, or electrostatic forces, from “class II materials”, in which the constituents are more strongly linked through ionic/covalent bond formation [9]. Following this classification, the nanocomposite materials involved in this present contribution can be best sorted into the former class.
The spatial organization of dissimilar and commonly incompatible components in these hybrid materials produces a wealth of novel structural features, physical properties, and complex functions, which arise from the synergistic interaction of the individual constituents. Further harnessing the potential of nanocomposite materials requires fine-tuning of the sizes, topologies, and spatial assembly of individual domains and their interfaces. This, in turn, relies on the improved fundamental understanding of the intermolecular interactions in these complex systems, especially those involved weak electron interactions. In the following two sections, we will discuss these electron interactions in great details with the aim to facilitate our appreciation of the quantum and electronic confinement effects.
Quantum Confinement Effect
Concept Definition
Molecules are, in general, localized entities. In a typical molecular solid, the intermolecular interactions are much weaker than the intramolecular bonding energies so the bulk properties of a molecular solid can usually be analyzed as the sum of individual molecular contributions, with small perturbations from the intramolecular forces. Such weak intermolecular interactions rarely extend beyond the nearest neighbors and the electronic structures of a molecular crystal are usually independent of the size of the crystal. Inorganic semiconductors and metals, on the other hand, consist of a network of ordered atoms with no discernible molecular unit. For a semiconductor crystal, electronic excitation consists of a loosely bounded electron-hole pair (the Mott-Wannier exciton), usually delocalized over a length much longer than the lattice constant [10]. As the diameter of the semiconductor crystalline approaches this exciton Bohr diameter, its electronic properties start to change. This is the so-called quantum confinement effect, which can be observed as a blue shift in the band gap or exciton energy [11, 12].
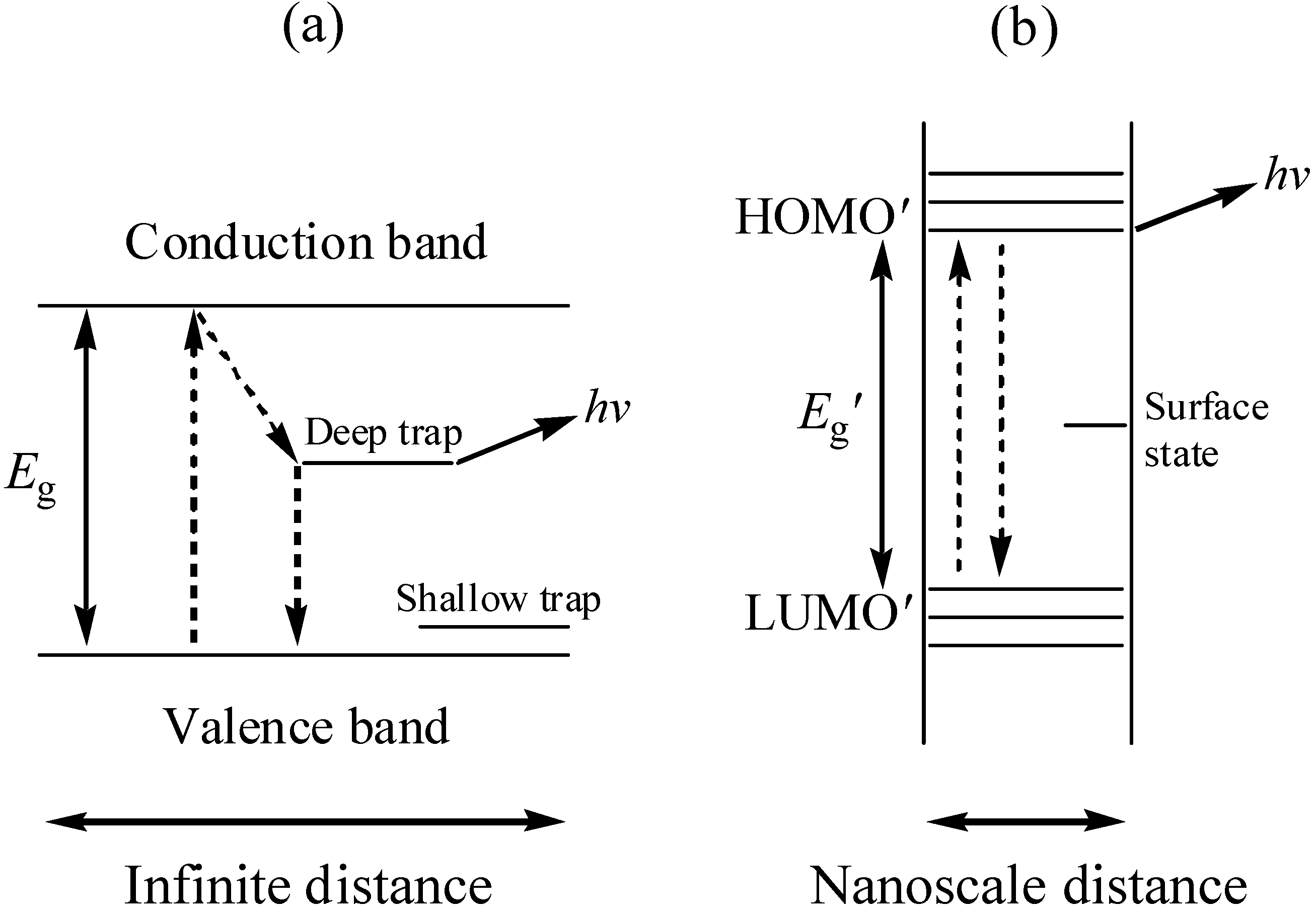
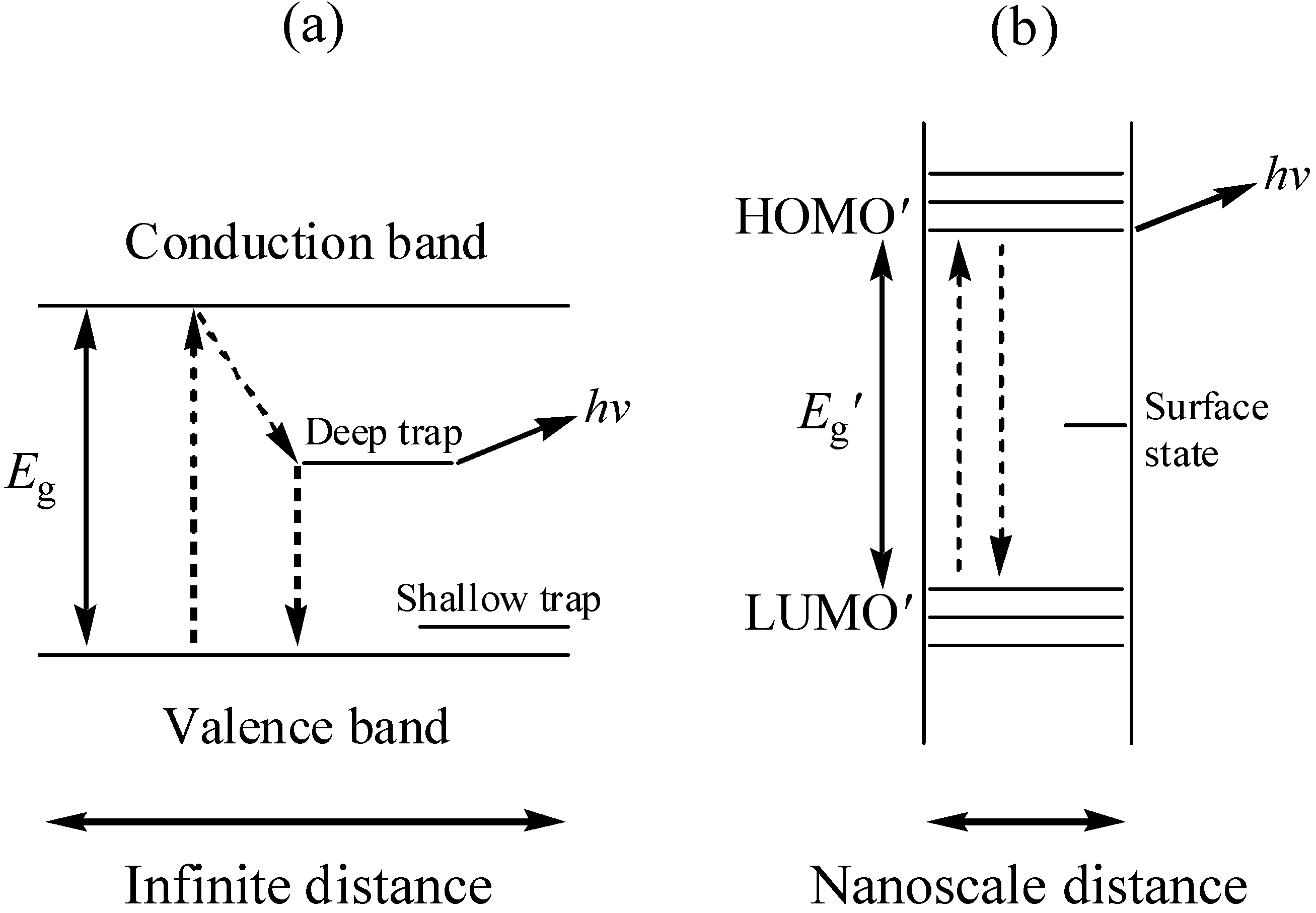
Typically, bulk sample of CdS, irrespective of their size once > ca. 20 nm, will absorb all electromagnetic radiation with an energy greater than the band gap (hν > 2.42 eV); which is classified as direct in this case. However, as particles become smaller, their electronic structure changes. Eventually, continuous bands breaks down and there are discrete bonding and antibonding orbitals in this material [13]. The electronic properties of such small particles are more like those of a molecule than an extended solid [14]. A spatial electronic state diagram showing the quantum confinement effect is shown in Figure 1. As the cluster properties are intermediate between molecules and bulk semiconductors, the quantum size effect can best be explained with hybrid molecular and semiconductor languages.
Figure 1.
The spatial electronic state diagram showing the quantum confinement effect in bulk semiconductors (a) and nanoparticles (b).
Figure 1.
The spatial electronic state diagram showing the quantum confinement effect in bulk semiconductors (a) and nanoparticles (b).
Model Design
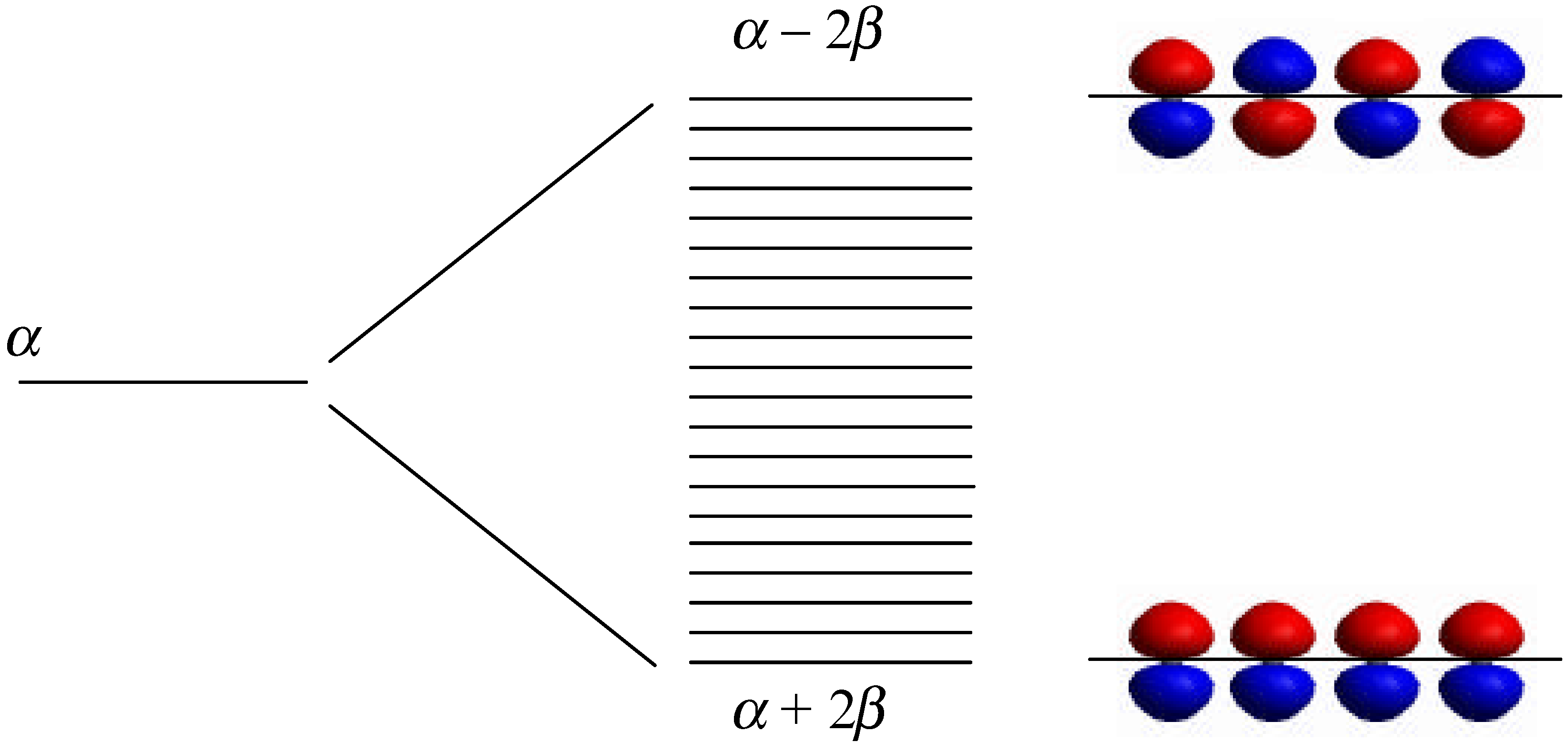
The linear combination of atomic orbitals-molecular orbitals (LCAO-MO) method [15] provides a natural framework to understand the evaluation of clusters from molecules to bulk and the size dependence of the lowest excited-state energy (band gap). An infinite chain of carbon atoms, separated by distance d, each carrying one pπ orbital (polyene, an idealized polyacetylene chain), provides a simple one-dimensional analogue of the bulk solid (Figure 2). The infinite chain is equivalent to a N-annulene where N is infinite. The essential concepts behind the quantum confinement effects of semiconductor clusters can be understood by studying the length dependence of the N-polyene. In the absence of any interactions, an N-atom polyene has N degenerate orbitals, ϕi, each with on-site energy (Coulomb integral) α, <ϕi|H|ϕi>, where H is the electronic Hamiltonian. By turning on the interaction between ϕi and ϕj, represented by the resonance integral β, <ϕi|H|ϕj>, the degeneracy is removed (Figure 3). The lowest energy orbital is at α + 2β and is bonding all neighboring atoms. The highest energy orbital is at α − 2β and is antibonding between all neighboring all neighbor ing atoms. In the middle at energy α, there is a nonbonding orbital.
Figure 2.
N-polyene, a simple one-dimensional analogue of the bulk solid as the designed model.
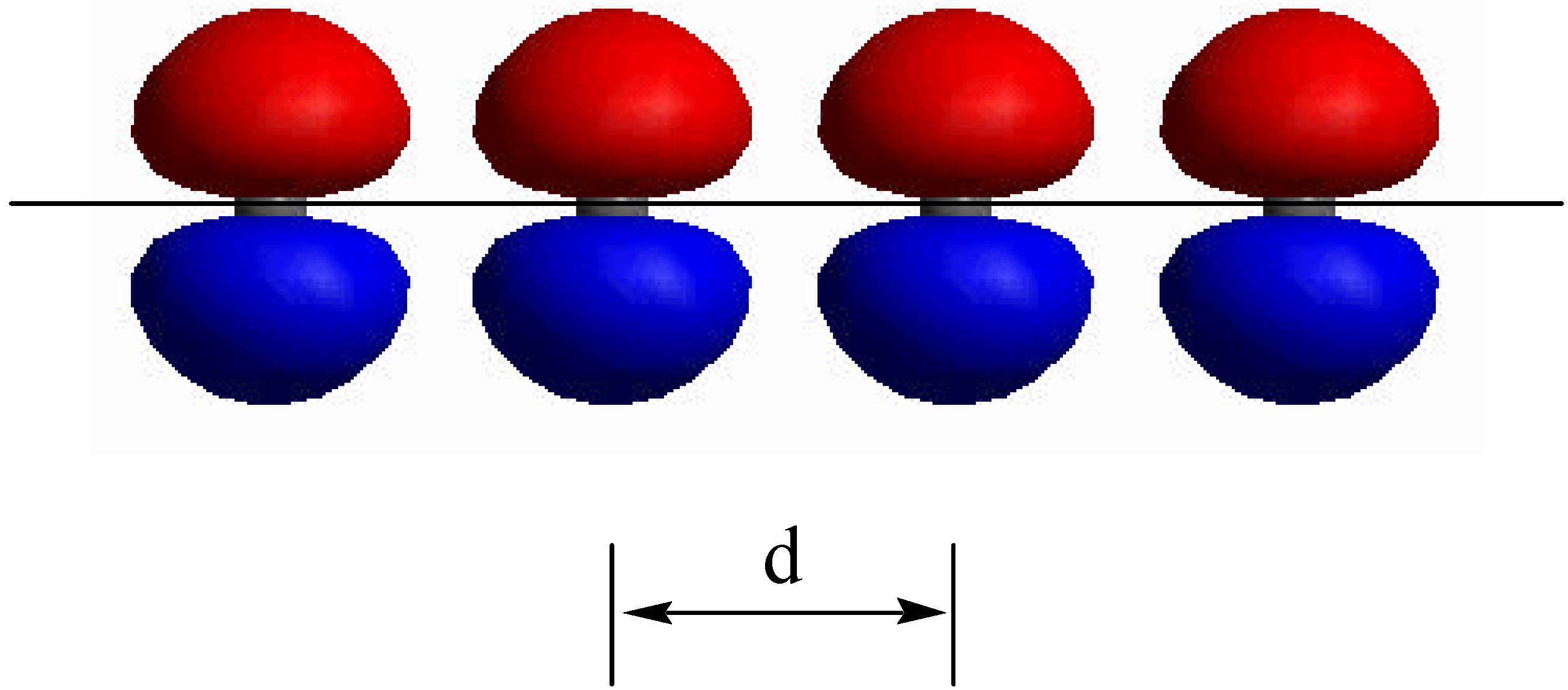
Figure 3.
Molecular orbital diagram for the N-polyene.
The energy levels of these orbitals, Ej, can be easily obtained on the Hückel level as
Ej = α + 2βcos(jπ / N + 1), j = 1, 2, … N
For an infinite chain, the energy band is continuous from α + 2β to α − 2β with a bandwidth of 4β. For a chain of finite atoms, the eigenvalues are discrete. In the case of a linear polymer, there is no degeneracy. So, one can easily prove, by filling electrons into these orbitals, that the HOMO-LUMO gap increases with decreasing chain length, N.
The LCAO-MO method outlined above provides a simple framework to understand many important solid-state concepts. The premier thing here is that as the size of the N-polyene decreases, its energy band becomes discrete and separation between the eigenvalues, and the HOMO-LUMO gaps increase. This is in essence the quantum confinement effect, i.e., the increase of the excited-state energy with decreasing cluster size.
In a bulk semiconductor, the electron and hole are bound together by a screened Coulomb interaction to form a so-called Mott-Wannier exciton [10]. This electron-hole interaction has to be induced for a more qualitative treatment of the quantum confinement effects. By assuming the energy band to be parabolic near the band gap (i.e., the effective mass approximation), the size-dependent shift (with respect to the bulk band gap) in the exciton energy of a small cluster (cluster radius ~ exciton radius) can be derived as
![Molecules 08 00207 i001]() where R is cluster radius, 1/μ = 1/me* + 1/mh*, me* is the electron effective mass, mh* is the hole effective mass, ε is the dielectric constant, and
where R is cluster radius, 1/μ = 1/me* + 1/mh*, me* is the electron effective mass, mh* is the hole effective mass, ε is the dielectric constant, and ![Molecules 08 00207 i002]() is the effective Rydberg energy, e4/2ε2ℏ2(me*-1 + mh*-1). The first term in the above equation is the band gap of the bulk materials, the second represents the particle-in-a-box quantum localization energy and has a simple 1 / R2 dependence, the third term the Coulomb energy with a 1 / R dependence, and the last term is the result of the spatial correlation effect. This last size-independent term is usually small but can become significant for semiconductors with small dielectric constant. Therefore, the cluster radius can be easily determined according to the above formula based on the absorption spectra.
is the effective Rydberg energy, e4/2ε2ℏ2(me*-1 + mh*-1). The first term in the above equation is the band gap of the bulk materials, the second represents the particle-in-a-box quantum localization energy and has a simple 1 / R2 dependence, the third term the Coulomb energy with a 1 / R dependence, and the last term is the result of the spatial correlation effect. This last size-independent term is usually small but can become significant for semiconductors with small dielectric constant. Therefore, the cluster radius can be easily determined according to the above formula based on the absorption spectra.

 is the effective Rydberg energy, e4/2ε2ℏ2(me*-1 + mh*-1). The first term in the above equation is the band gap of the bulk materials, the second represents the particle-in-a-box quantum localization energy and has a simple 1 / R2 dependence, the third term the Coulomb energy with a 1 / R dependence, and the last term is the result of the spatial correlation effect. This last size-independent term is usually small but can become significant for semiconductors with small dielectric constant. Therefore, the cluster radius can be easily determined according to the above formula based on the absorption spectra.
is the effective Rydberg energy, e4/2ε2ℏ2(me*-1 + mh*-1). The first term in the above equation is the band gap of the bulk materials, the second represents the particle-in-a-box quantum localization energy and has a simple 1 / R2 dependence, the third term the Coulomb energy with a 1 / R dependence, and the last term is the result of the spatial correlation effect. This last size-independent term is usually small but can become significant for semiconductors with small dielectric constant. Therefore, the cluster radius can be easily determined according to the above formula based on the absorption spectra.Equation (2), although containing the basic physics of the quantum confinement effect, cannot be expected to be quantitatively correct, especially for very small clusters. This is because for small clusters the eigenvalues of the lowest excited states are located in a region of the energy band that is no longer parabolic (the breakdown of the effective mass approximation). It may be mentioned here that the variations of molecular properties of very small clusters can be interpreted by the electronic confinement effect, which will be discussed in the next section.
Case Study
The search of functional nanostructured materials for information storage has stimulated great interest in nowadays nano-science and technology [16]. The study of nanosized metal sulfide semiconductor materials doped with impurities has been an area of intense activity in recent years [17, 18]. Zinc sulfide is the best host for luminescent materials with the band gap of 3.7 eV. Since the rare-earth ions are excellent luminescence centers, nanosized ZnS doped with them is becoming a new kind of solid-state luminescent material though the radii of rare-earth ions is larger than that of zinc ions. We present here the synthesis of Eu-doped ZnS semiconductor nanoparticles, together with their photoluminescence behavior [19].
ZnS:Eu nanoparticles 1 and 2 were prepared by two methods, A and B, respectively. In method A, Na2S serves as the inorganic sulfur donor and S2- ions from Na2S react with Zn2+ ions immediately. In method B, Zn2+ ions react with S2- ions from the organic sulfur donor CH3CSNH2 gradually. In order to characterize the particle size, the diffuse reflectance absorption spectrum of ZnS:Eu nanoparticles 1 was measured. Comparin g to the absorption maximum of ZnS bulk materials (3.7 eV), the absorption maximum of ZnS nanoparticles blueshifted to 320 nm (3.9 eV). Estimated from the Eq. (2), it is obtained that the grain size of the ZnS:Eu nanoparticle (sample 1) is 6.8 nm. The powder X-ray diffraction of ZnS:Eu semiconductor nanoparticles 1 is also measured. All peaks can be ascribed to a zinc blende crystal structure without extra phases and non-crystalline state. The peaks are markedly broadened, which is characteristic of the nanoparticles. The average grain size of ZnS:Eu nanoparticles can be estimated by Scherrer equation
where the symbols have their usual meanings. The average grain size of the nanoparticles is 6.9 nm estimating from Eq. (3), which agrees well with the result of the diffuse reflectance spectrum.
Dh,k,l = kλ/(B0−b0)cosθ
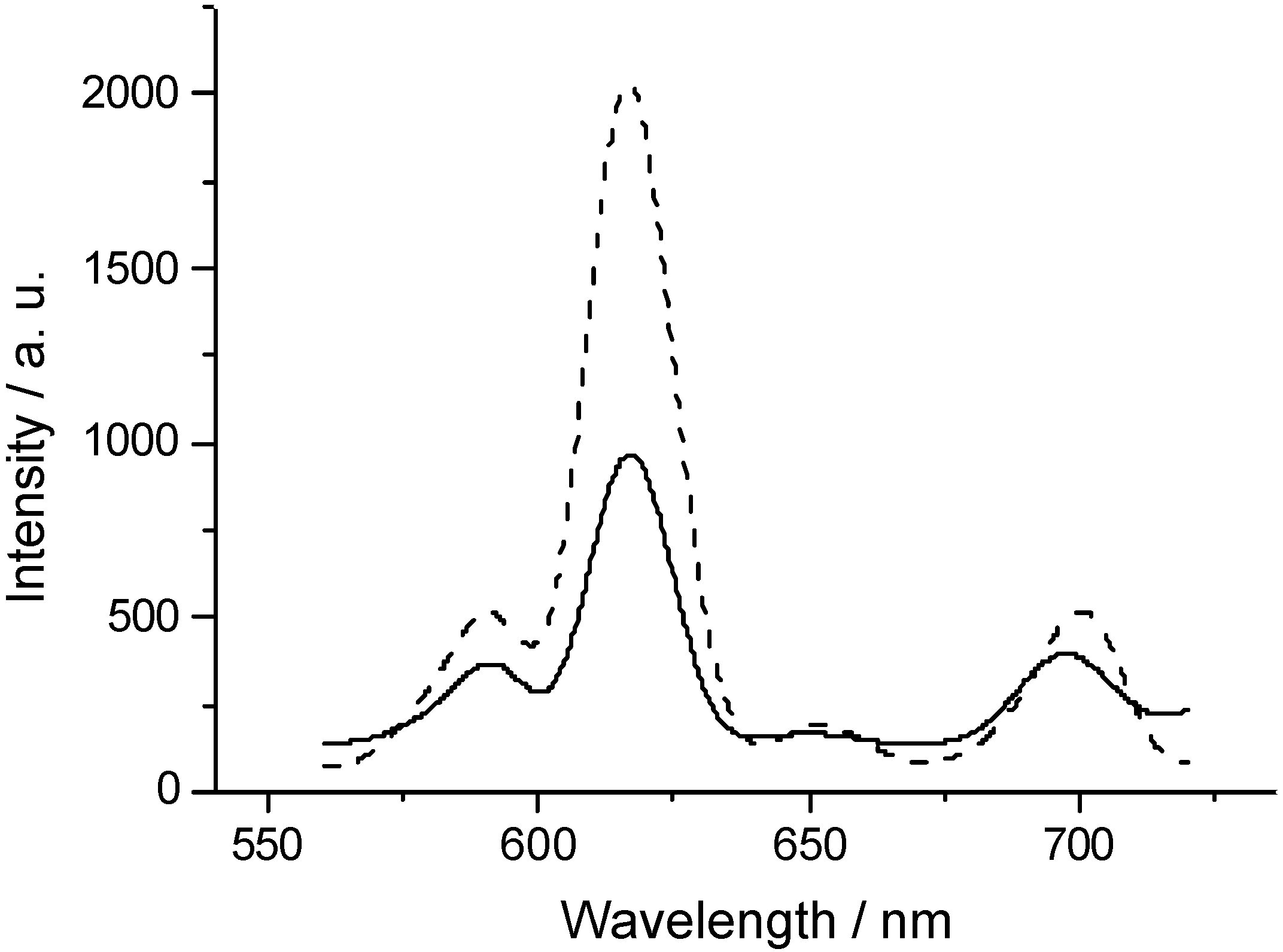
Figure 4.
The photoluminescence spectra of the sample 1 (solid line) and 2 (dashed line). λexc = 396 nm.
Figure 4.
The photoluminescence spectra of the sample 1 (solid line) and 2 (dashed line). λexc = 396 nm.

The photoluminescence spectra of 1 and 2 excited at 396 nm are shown in Figure 4. No further splitting of the emission peaks is observed indicating that the site symmetry of Eu3+ belongs to Oh group (zinc blende crystal belongs to Oh group). If the site symmetry of Eu3+ belongs to Oh group, the emission at 590 nm originated from magneto-dipole allowed 5D0 → 7F1 is dominant. When the site symmetry of Eu3+ is reduced, the intensity of the emission band at 616 nm, which is the electro-dipole allowed 5D0 → 7F2 transition, should increase. So it is evident that two emission peaks of Eu3+ centered at 590 and 616 nm are sensitive to site symmetry and can be used to study site symmetry, i.e., to study the microstructure around Eu3+ and the defects caused by doping of Eu3+ in the lattice of ZnS. As the ion size of Eu3+ is much larger than that of Zn2+, the defects and the deformation of the lattice must appear when Eu3+ gets into the lattice of ZnS, and the site symmetry of doping Eu3+ ion must be decreased also. Therefore, it seems plausible to deduce that ZnS:Eu nanoparticles are doped by higher dopant concentration of Eu3+ in 2 than in 1, which is related to the difference in the preparation procedures.
Electronic Confinement Effect
Concept Definition
During the past ten years investigation in nanocomposite materials, scientists in this field have shown a growing interest in the variations of the molecular properties of the guest organic species [20]. Encapsulation of organic molecules in the nanoporous channels host produces significant changes of the molecular properties of the guest species. Some of these changes such as the variations in the band gap and fluorescence lifetimes of the guest molecules in these nanocomposite materials, have been mentioned in previous studies [6, 7, 21, 22]. These host-guest effects observed in cation exchanged aluminosilicate sieves are even significant, which is related to the exchanged cations and associated fields in the pores [23, 24, 25]. However, if one wants to study exclusively the host-guest effects avoiding the interference of electrostatic effects due to the exchanged cations, it is necessary to use samples of pure silicates, such as MCM-41. In this paper, it is our intention to showcase a new concept, which can contribute to explain these significant changes in the organic-inorganic nanocomposite materials. This new concept, electronic confinement effect, which was first introduced by A. Corma and co-workers [26, 27], is indeed realized in larger cavities, such as nanoporous MCM-41.
It is generally accepted that the structures of channels and cavities with molecular sizes in a few nanometers have a strong influence on diffusion and solvation effects of reactant molecules, and they can become determinant for the catalytic activity of these materials. Diffusion, which involves the dynamics of the molecules into the channels, is described by a statistic mechanical model which correlates well with experimental results [28]. Solvation or cavity effects that involve physiochemical interactions between the host inorganic pore and the guest organic molecules are not well qualified and are still a matter of discussion [29, 30, 31]. The types of interactions normally considered are Coulombic effects, coordination effects, and weak electron interactions. This last kind of interactions accounts for forces of the van der Waals type, which could produce the “docking” of molecules into the cavities of the molecular sieve. In this sense, it has been proposed that sorbate molecules in the nanoporous solids, such as MCM-41, tend to optimize their van der Waals interaction with the surroundings. These van der Waals interactions are amplified by the surface curvature of the pore walls, which interact with the sorbed molecules.
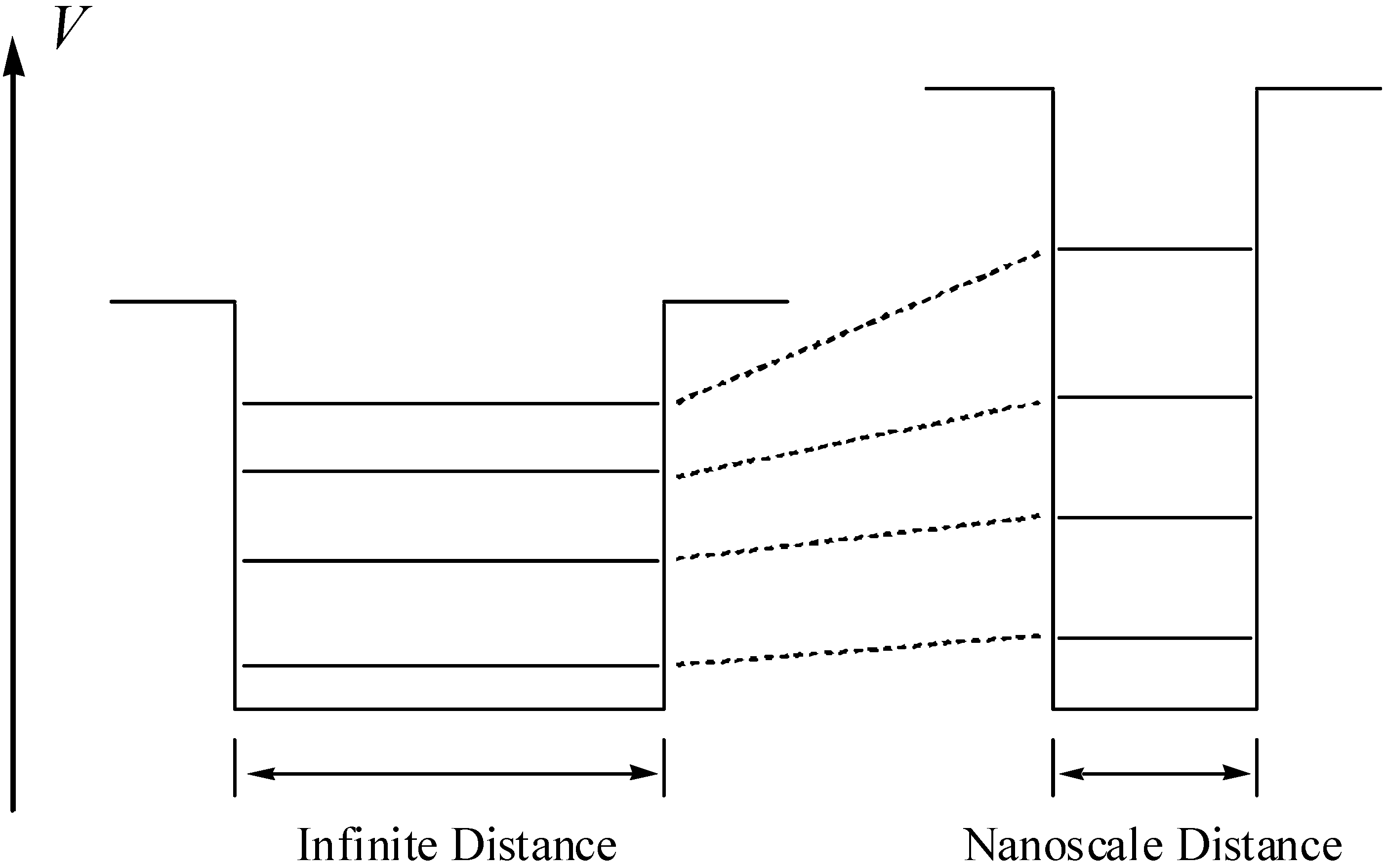
Considering the partial covalent character of the aluminosilicate crystals, electrons are not localized on the framework atoms, but they are partially delocalized through the bulk. This causes the density of the electrons (i.e., the probability for finding the electrons) of the guest molecules to suddenly drop to nearly zero when reaching the walls of the cavity as a consequence of the short-range repulsion with the delocalized electronic clouds of the lattice. This indicates that a contradiction of the orbitals of the guest molecules will occur, with the consequent changes in its energy levels (Figure 5), which implies a preactivation of the molecule when residing in the pore or cavity.
Figure 5.
Change of the energy levels of the particle when confining the microscopic particle. On the left-hand side the energy levels for the unconfined system are depicted and on the right-hand side, the corresponding levels for the confined one.
Figure 5.
Change of the energy levels of the particle when confining the microscopic particle. On the left-hand side the energy levels for the unconfined system are depicted and on the right-hand side, the corresponding levels for the confined one.
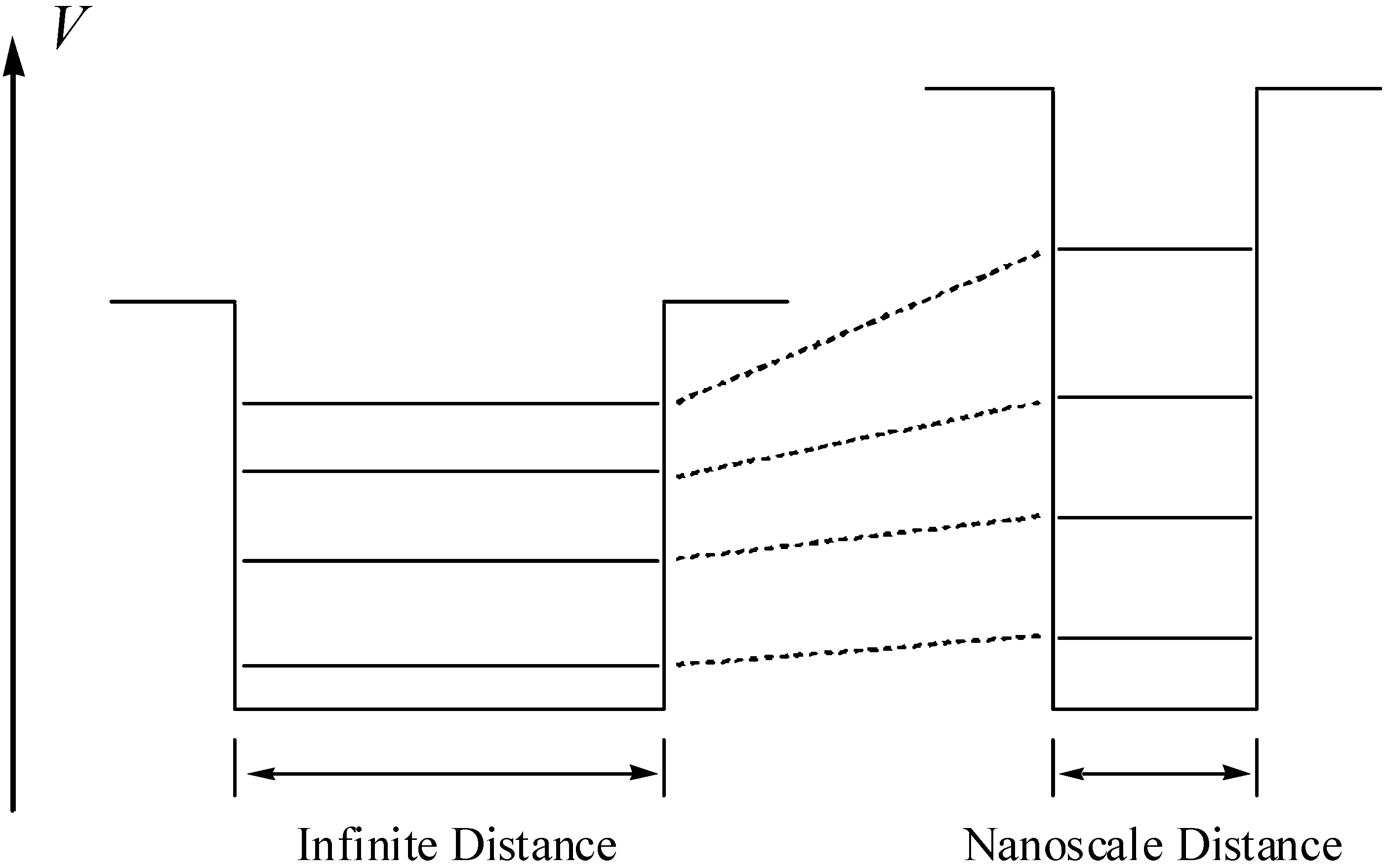
On can now notice that electronic confinement is associated with the previous described quantum confinement, despite the electronic confinement is induced by the intrachannel surface, not by the exciton Bohr diameter. Moreover, quantum confinement effect cannot be expected to be quantitatively correct for very small clusters; in contrast, the electronic confinement effect focuses on both inorganic compounds and organic molecules within nanopores. Hence these clusters, loaded in the inorganic host, must be extremely small, and may be even treated as single molecules in some cases. The new phenomena for such small clusters should be explained by the concepts derived from electronic confinement theory.
Model Design
For the sake of a better understanding of the electronic confinement, we present here a model study by using the aromatic compounds as the guest organic molecules [32, 33]. HMO theory is a method that allows one to obtain a chemical characterization of conjugated π systems without need of involved quantum chemical calculations. One can easily notice that its goal is to give an adequate description of π molecular orbitals (their energy and atomic orbital composition) by only two empirical parameters, namely, the Coulomb integral α and the resonance integral β.
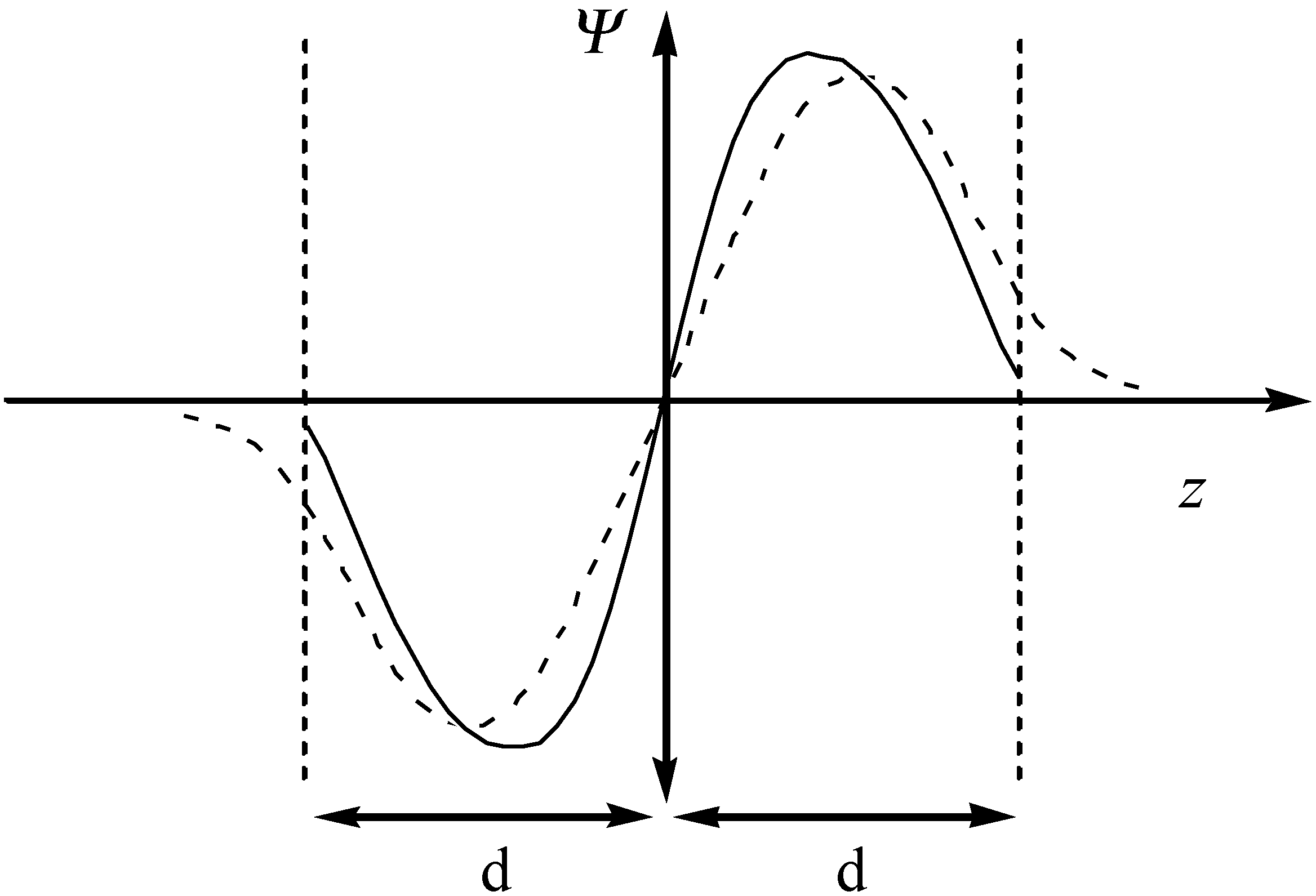
A confined model system can be built by locating the conjugated molecules parallel to two surfaces, for simplicity, may be simulated by two infinite planes located at a certain distance. In this case, the atomic orbitals can still have as the normal 2pz orbitals a node at the molecular plane but they are adapted to the confinement, which makes them vanish beyond the limit of the plane. The schematic representation of the Ψ function of an aromatic molecule in unconfined and confined spaces is shown in Figure 6.
Figure 6.
The schematic representation of the Ψ function of the conjugated molecule in unconfined (dashed line) and confined (solid line) spaces.
Figure 6.
The schematic representation of the Ψ function of the conjugated molecule in unconfined (dashed line) and confined (solid line) spaces.
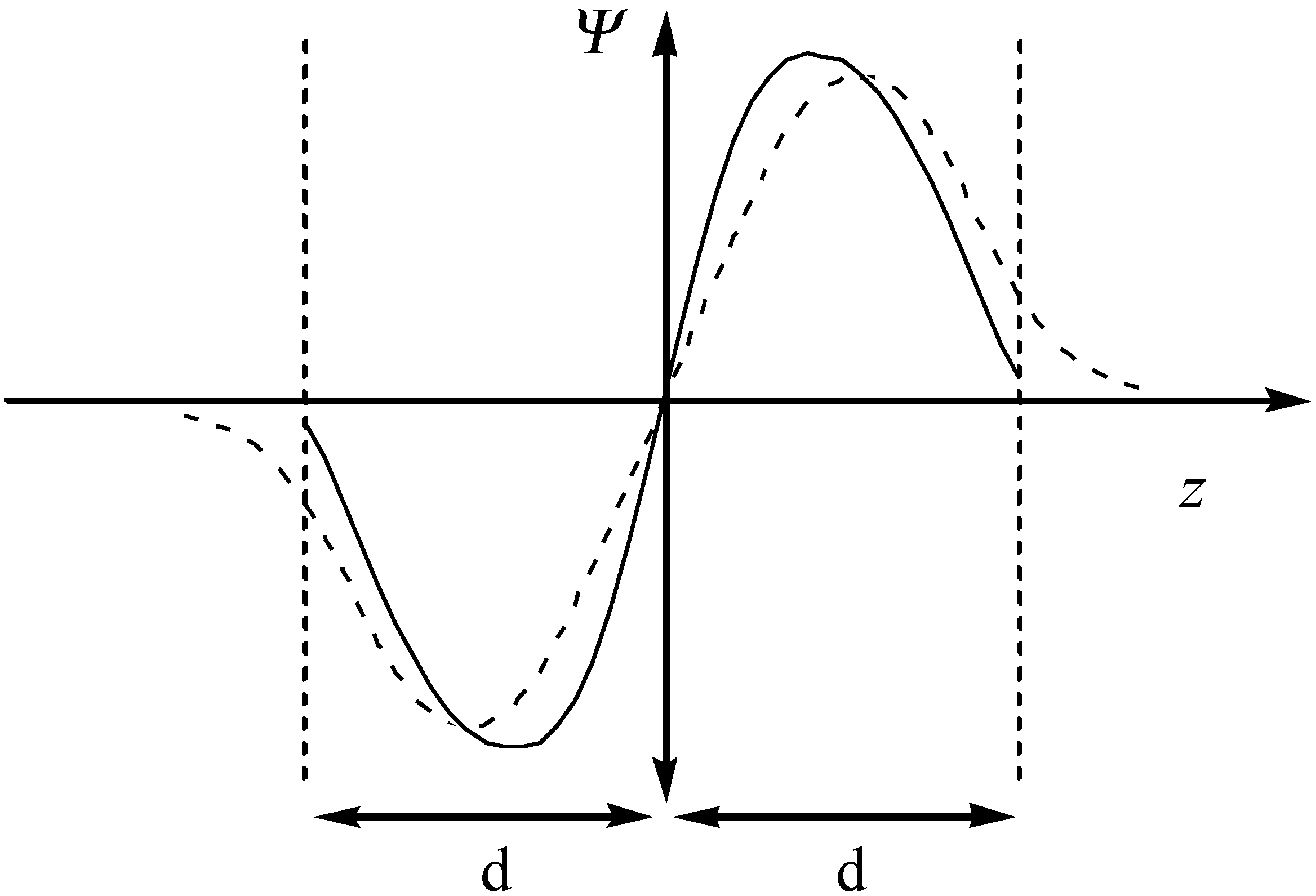
It is evident from the figure that the density of the confined 2pz orbitals suddenly drop to nearly zero when reaching the infinite plane as a consequence of the short-range repulsion with the delocalized electronic clouds. This implies that a contraction of the molecular orbitals of the guest molecules will occur, with the consequent changes in its energy levels. Hence, the energy changes of an aromatic compound may be expressed as
where Δα and Δβ are (in atomic units)
![Molecules 08 00207 i003]()
![Molecules 08 00207 i004]()
ΔEHOMO = EHOMO’ − EHOMO = (α’ + xβ’) − (α + xβ) = Δα + Δβ
ΔELUMO = ELUMO’ − ELUMO = (α’ − xβ’) − (α − xβ) = Δα − Δβ


In this expression a(x,y,z) and b(x,y,z) are the 2pz atomic orbitals on the neighboring carbon atoms a and b, respectively, while a’(x,y,z) and b’(x,y,z) are the equivalent confined atomic orbitals. Since in real situations in real situations we do not know the actual expression of the atomic orbitals in the confined system, we can state that Δα and Δβ are always positive quantities. Therefore, we obtain the following conditions for the Coulomb and resonance integrals of the confined system based on Eq. (5)
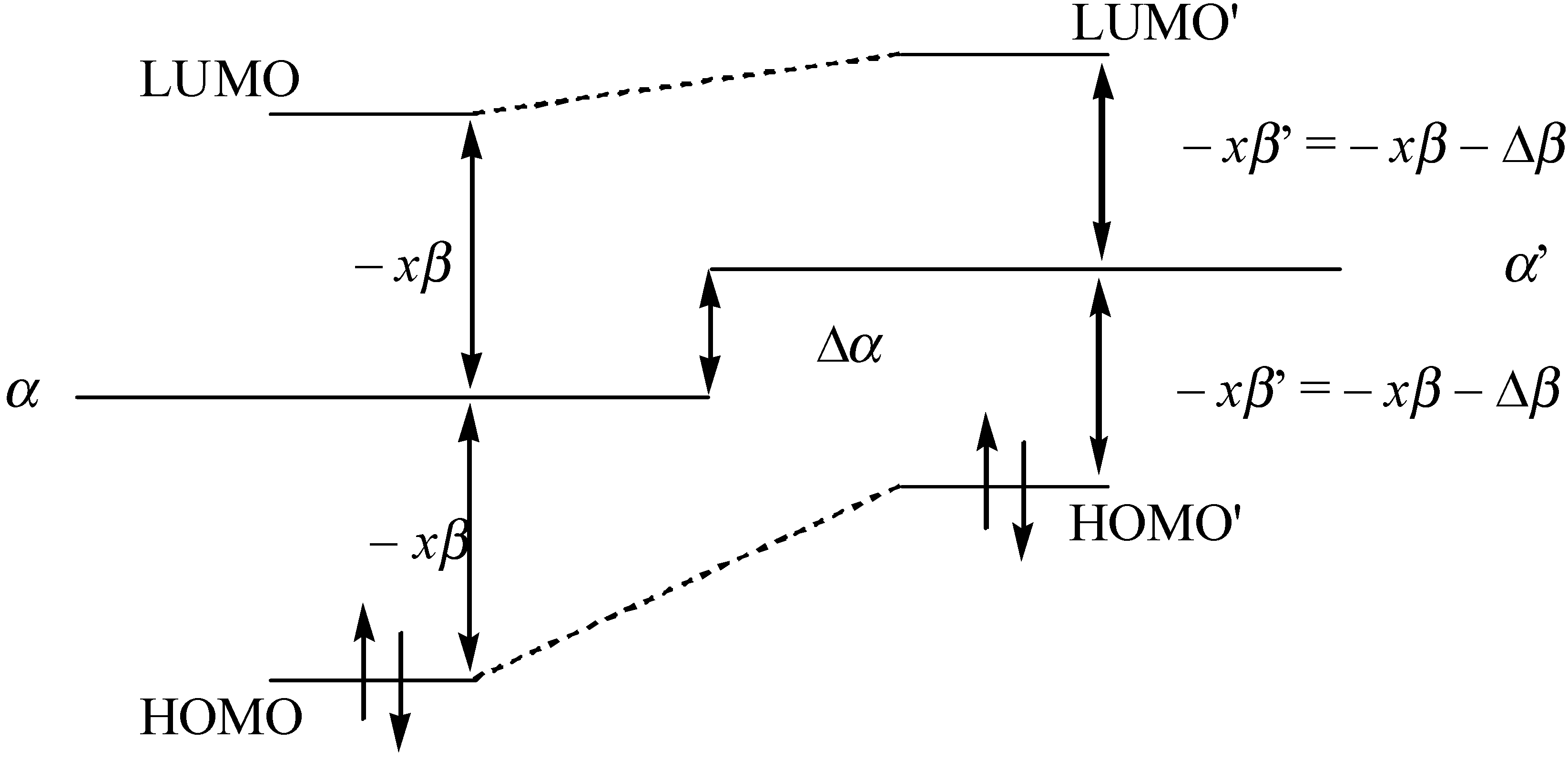
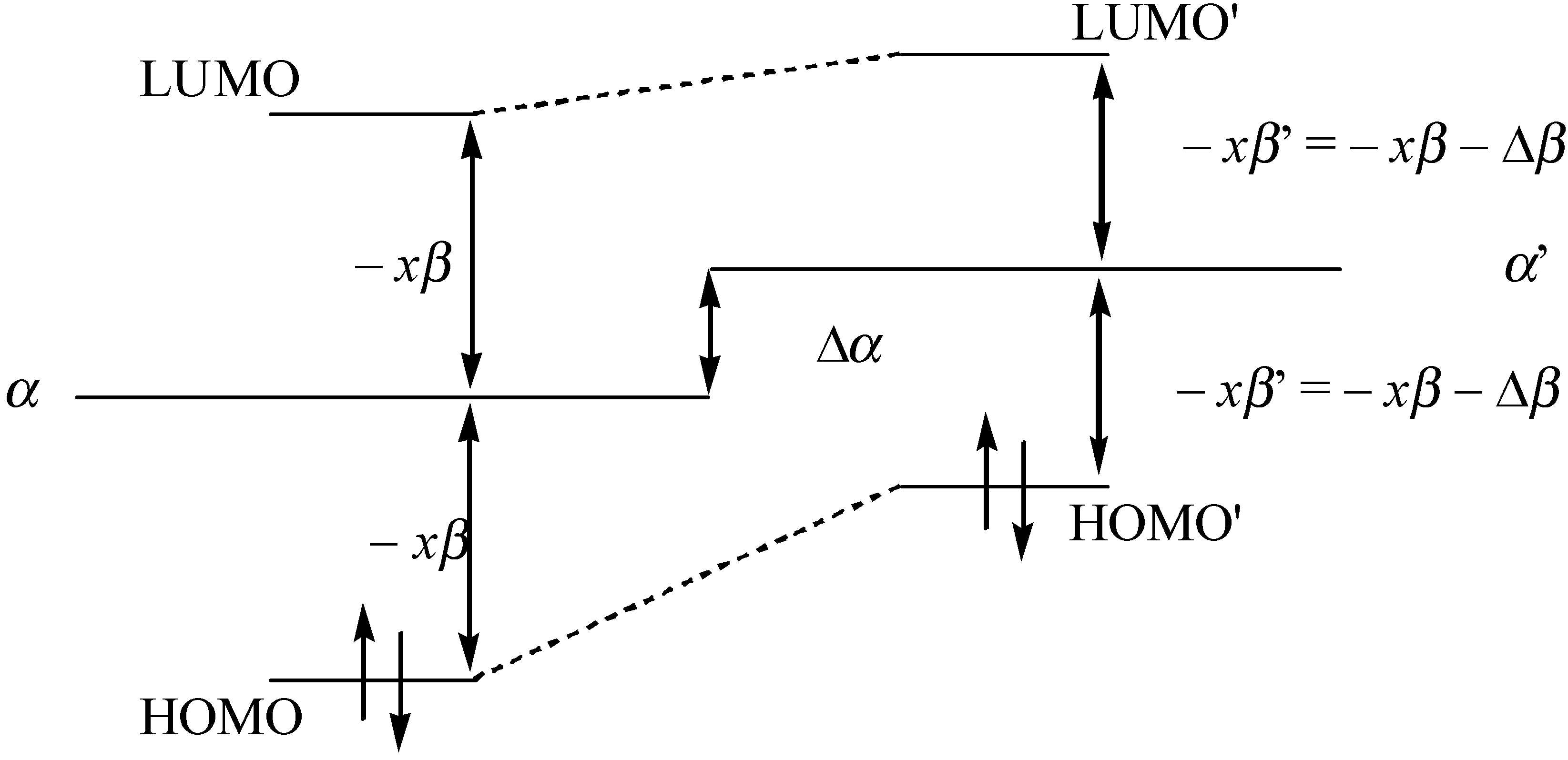
Since α and β are both negative quantities, the above equations indicates that the values of α and β increase when confining the molecule. In Figure 7 we have shown a qualitative correlation energy spectrum of the aromatic molecule when it is confined.
α’ = α + Δα > α
xβ’ = xβ + Δβ > xβ
Figure 7.
Qualitative description of the frontier orbital energy spectrum for the conjugated molecule.
Figure 7.
Qualitative description of the frontier orbital energy spectrum for the conjugated molecule.
Case Study
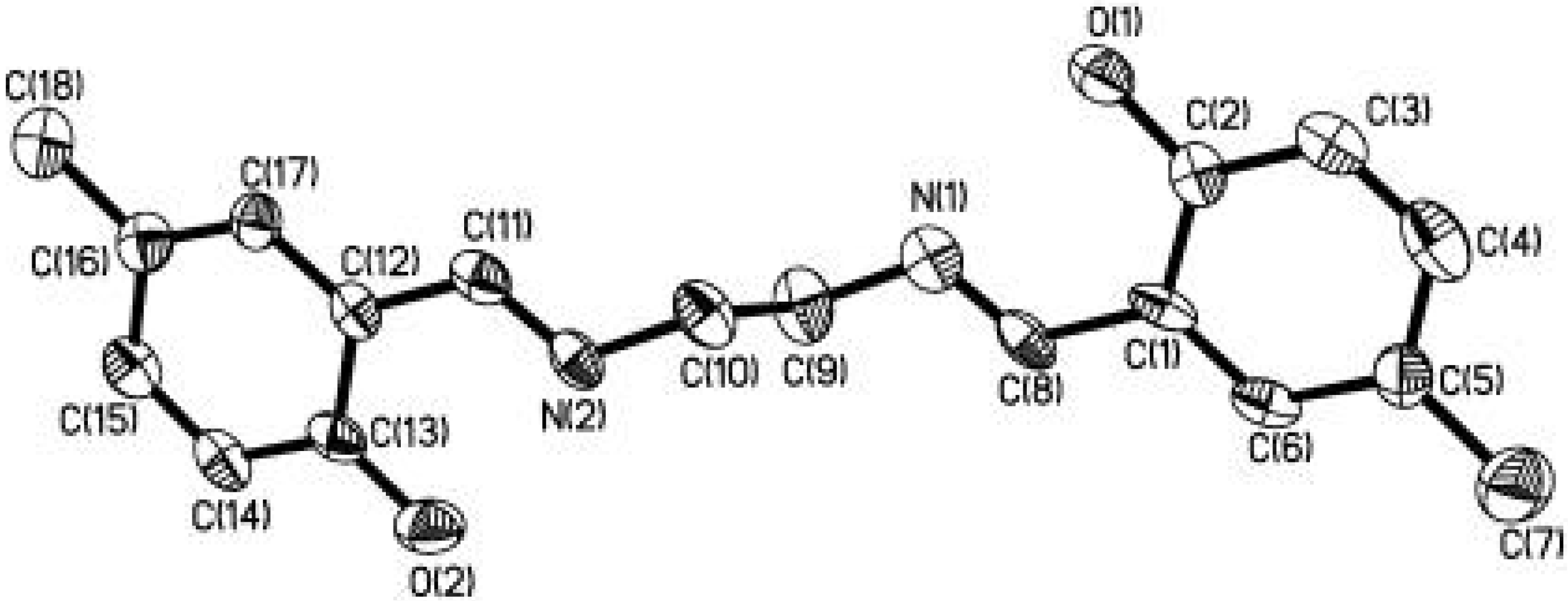
The salicylaldehyde-derived Schiff base molecule, which is one class of the most extensively studied organic molecules in molecular spectroscopy, is found to be a suitable guest molecule for this purpose [34, 35]. By means of self-assembly, we have encapsulated a new Shiff base molecule N,N'- bis(2-hydroxy-5-methylbenzylidene)-1,2-ethanediamine (3) in the nanoporous channels of MCM-41 host [36]. The molecular structure diagram of 3 is shown in Figure 8. The C(2)-O(1) bond length (1.348(9) Å) is longer than 1.30 Å indicating the ground state of 3 is enol form in the crystal.
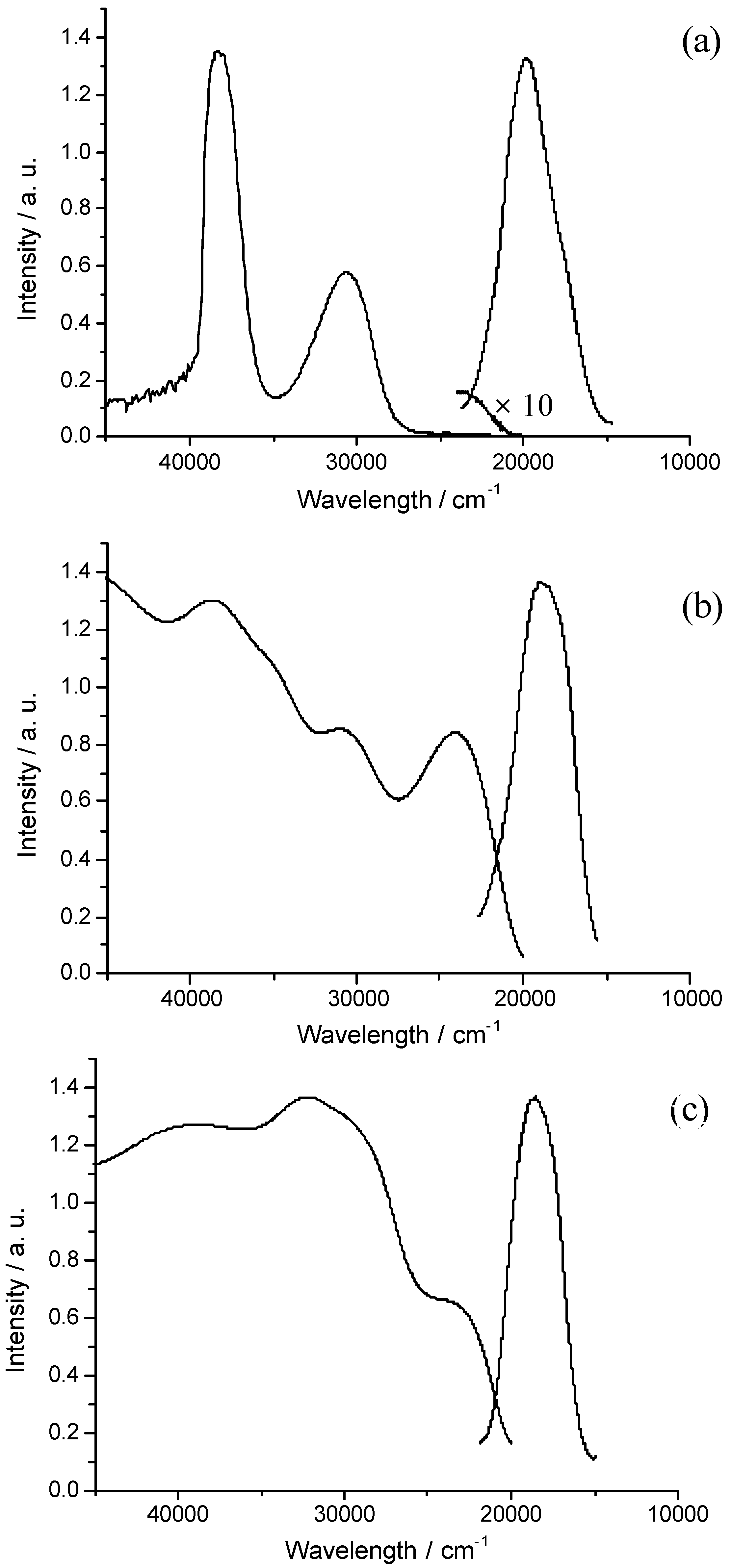
Figure 9 shows the corresponding UV-Visible absorption and emission spectra of 3 at room temperature in dimethyl sulphoxide (DMSO) solution (1.0 × 10-4 M), in nanoporous MCM-41 and in pure crystals, respectively. The emissions of 3 result from strictures due to excited-state intramolecular proton transfer reaction. In DMSO the 0-0 transition of 3 appears at 23041 cm-1 (2.86 eV) and is remarkably shifted via 21413 cm-1 (2.66 eV) of 3 in MCM-41, to 20921 cm-1 (2.60 eV) of 3 in pure crystals. Given that the main contribution to the S0 → S1 transition is due to the HOMO and LUMO, this bathochromic shift can be correlated with a decreasing of the HOMO-LUMO band gap. The shift of the 0-0 transitions from 3 in MCM-41 to 3 pure crystals may result from the change of lattice state due to the increase in the surface area. It is likely that the increase in surface area causes lattice softening, and therefore the Coulombic interaction energies between molecules become smaller, leading to wider band gap [37].
Figure 8.
Molecular structure diagram (hydrogens omitted) of 3. Thermal ellipsoids are drawn at 30% probability level.
Figure 8.
Molecular structure diagram (hydrogens omitted) of 3. Thermal ellipsoids are drawn at 30% probability level.
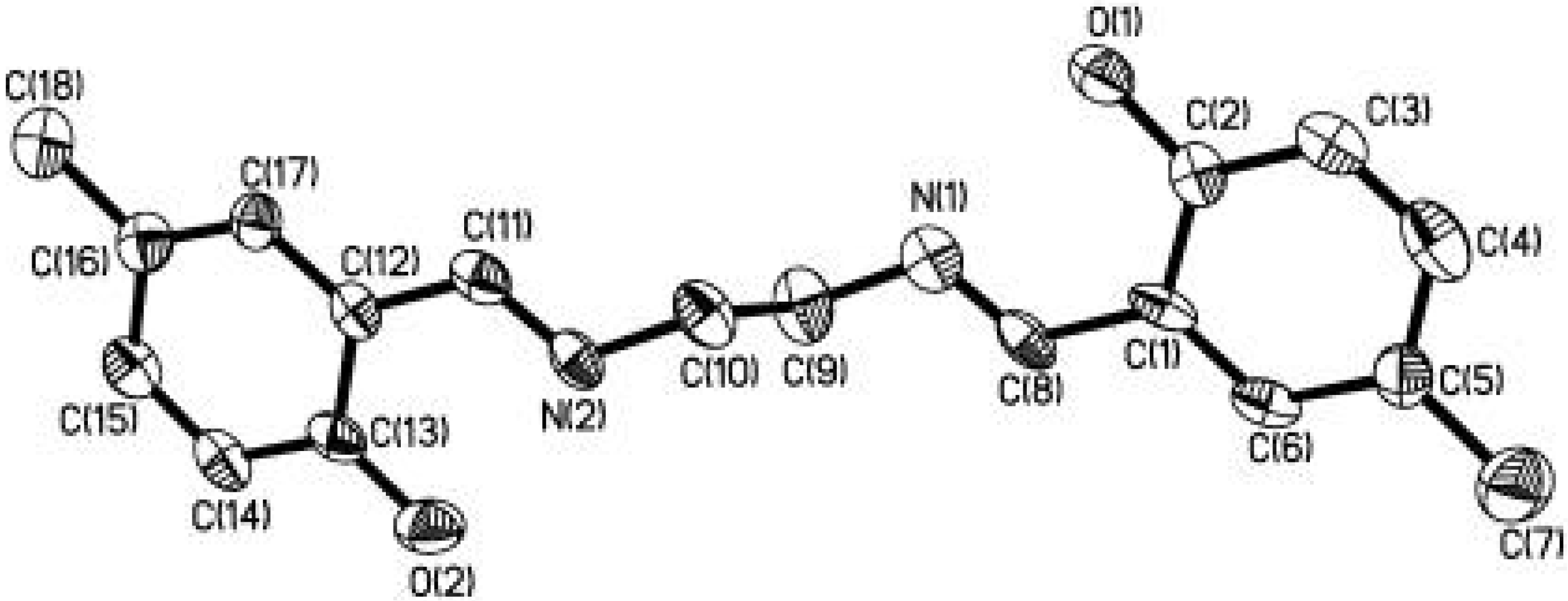
Figure 9.
The normalized absorption and emission spectra of 3 in DMSO (λexc = 25000 cm-1) (a), in nanoporous MCM-41 (λexc = 24100 cm-1) (b) and in pure crystals (λexc = 24100 nm-1) (c) at room temperature.
Figure 9.
The normalized absorption and emission spectra of 3 in DMSO (λexc = 25000 cm-1) (a), in nanoporous MCM-41 (λexc = 24100 cm-1) (b) and in pure crystals (λexc = 24100 nm-1) (c) at room temperature.

Since 3 can be treated as dispersive molecules in both DMSO solution and nanoporous MCM-41, this trend should be rationalized by the electronic confinement theory that all the energy levels of 3 increase in the nanoporous channels host as a result of the confinement. Since the increase for the HOMO has been predicted to be more sensitive than the LUMO, the overall effect is a reduction on the band gap of the frontier orbitals.
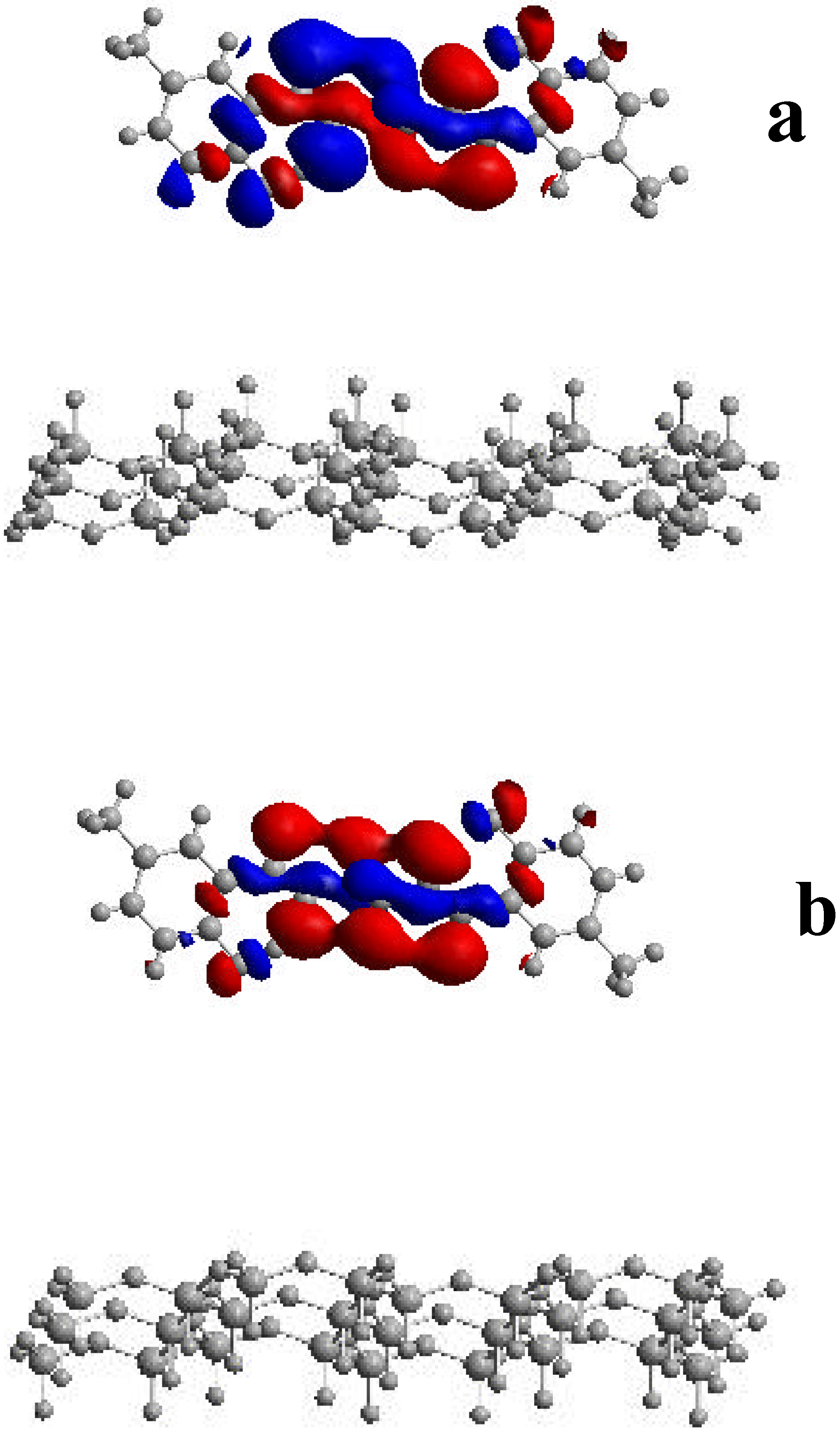
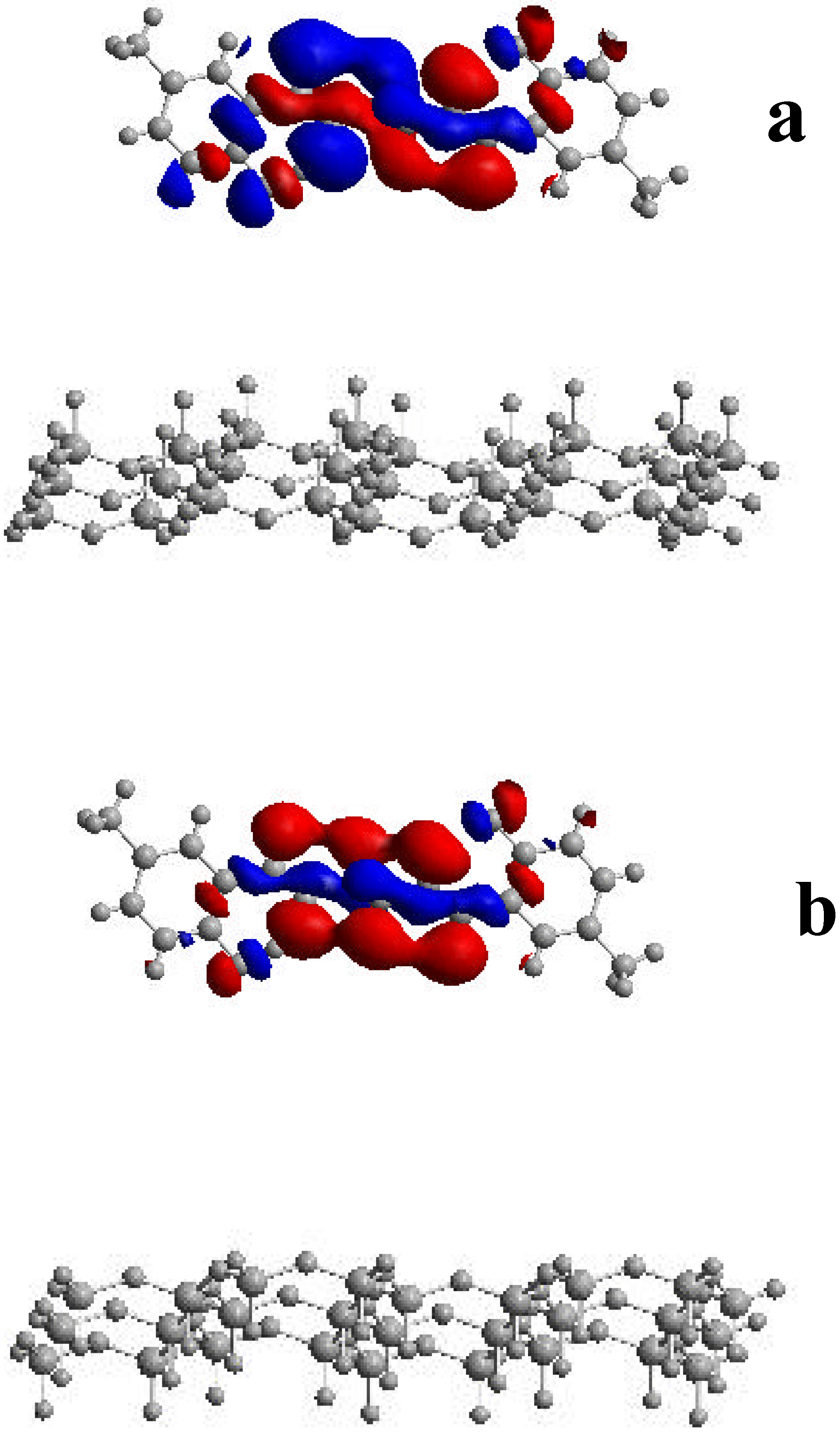
To analyze the origin of this energy increase we have studied the composition of the HOMO orbitals. Three-dimensional plots of the surfaces of the HOMO orbitals for 3 in gas phase and confined within two mica sheets separated by 3.0 nm are shown in Figure 10. As can be seen from the figure, the main components of the HOMO orbitals of the 3-mica system are localized on the methylene and hydroxyl groups of 3, with little contributions on the mica sheets. Since the HOMO orbitals are confined within two mica sheets, they were slightly contracted compared to those in gas phase being this deformation the origin of the increase of the HOMO energy. These results can rationalize the variations in the 0-0 transitions observed here for this complex system.
Figure 10.
Plots of the HOMO orbitals of 3 in the gas phase (a) and upon confinement inside two mica sheets separated by 3.0 nm (b).
Figure 10.
Plots of the HOMO orbitals of 3 in the gas phase (a) and upon confinement inside two mica sheets separated by 3.0 nm (b).
Concluding Remarks
Recent advances in the study of quantum and electronic confinement effects in nanostructured materials have opened the doorway for systematic investigations of the electronic structures at the nanoscale. For inorganic semiconductors and metals, when their diameter approaches the exciton Bohr diameter, its electronic properties start to change as indicated by a blue shift in the band gap. This is the quantum confinement effect. Clusters with diameters ranging from 1.0 to 15.0 nm can be routinely synthesized in a variety of media such as nanoporous molecular sieves, zeolites, or as polymers. The variations of molecular properties of the guest species in these complex systems can be elucidated by the electronic confinement effect.
In these nanocomposite materials, an asymmetric confinement will exist by molecules being much closer to one “wall” than to the other one. So, the motion of electrons will be more restricted in one direction (along the wall of the nanoporous channels) than in the other directions. The results presented here may prove useful in the fabrication of optoelectronic devices, such as organic-light-emitting-devices (OLEDs) using electroluminescent nanotube crystals [38, 39]. Since the pathways open for n/p migration-recombination are limited essentially to only one dimension, it may be confer a greater degree of order on electroluminescent processes by utilizing porous, channel-type materials containing light-emitting and electronically interacting guest molecules (Figure 11).
Figure 11.
Schematic drawing of the electroluminescent properties which may arise when confining guest molecules within parallel channels of a porous host structure.
Figure 11.
Schematic drawing of the electroluminescent properties which may arise when confining guest molecules within parallel channels of a porous host structure.

The last decade in the past 20th century have demonstrated the great influence of nanostructured materials on the development of nano-science and technology. Ultimately, the scientific and technological impact of these materials mainly depends on their novel electronic, optical, and catalytic properties. Hence, studies concerning the quantum and electronic confinement observed in these materials are of exceptionally importance. Nanostructured materials have introduced hierarchy into materials science and bode well for the further development of materials and composite with novel properties, new functions, and perceived utilities in a wide range of applications. The future looks very bright for nanostructured materials!
Acknowledgements
The authors wish to express their gratitude to Prof. Guo-Qing Tang (Institute of Modern Optics, Nankai University) for performing parts of the spectroscopic measurements in this work. Financial supports of the National Natural Science Foundation of China and the TRAPOYT of MOE of China are also acknowledged. Lei Z. Zhang thanks the Research Award Program for Undergraduate Students of Department of Chemistry, Nankai University.
References
- Frontiers in Materials Science (Special Report). Science 1997, 277, 1213.
- Nanoscale Materials (Special Issue). Acc. Chem. Res. 1999, 32, 387.
- Organic-Inorganic Nanocomposite Materials (Special Issue). Chem. Mater. 2001, 13, 3059.
- Kresge, C. T.; Leonowicz, M. E.; Roth, W. J.; Vartuli, J. C.; Beck, J. S. Nature 1992, 359, 710.
- Liu, J.; Shiu, Y.; Nie, Z.; Chang, J. H.; Wang, L.-Q.; Fryxell, G. E.; Samuels, W. D.; Exarhos, G. J. J. Phys. Chem. A 2000, 104, 8328.
- Ozin, G. A. Adv. Mater. 1994, 6, 71.Ozin, G. A. Chem. Commun. 2000, 419.
- Stein, A.; Melde, B. J.; Schroden, R. C. Adv. Mater. 2000, 12, 1403.
- Wu, C.-G.; Bein, T. Science 1994, 264, 1757.
- Sanchez, C.; Ribot, F. New J. Chem. 1994, 18, 1007.
- Knox, R. S. Theory of Excitons; New York: Academic Press, 1963. [Google Scholar]
- Brus, L. E. J. Chem. Phys. 1983, 79, 5566.Brus, L. E. J. Chem. Phys. 1984, 80, 4403.
- Wang, Y.; Herron, N. Phys. Rev. B 1990, 42, 7253.Wang, Y.; Herron, N. J. Phys. Chem. 1991, 95, 525.
- Steigerwald, M. L.; Brus, L. E. Acc. Chem. Res. 1990, 23, 183.
- Weller, H. Angew. Chem. Int. Ed. Engl. 1993, 32, 41.
- Burdett, J. K. Prog. Solid State Chem. 1984, 15, 173.
- Bol, A. A.; Meijerink, A. J. Lumin. 2000, 87-89, 315.
- Gong, X.; Chen, W. J.; Wu, P. F.; Chan, W. K. Appl. Phys. Lett. 1998, 73, 2875.
- Bhargava, R. N.; Gallagher, D.; Hong, X.; Nurmikko, A. Phys. Rev. Lett. 1994, 72, 416.
- Wang, Z.-X.; Zhang, L. Z.; Xiong, Y.; Tang, G.-Q.; Zhang, G.-L.; Chen, W.-J. J. Chem. Res. 2002, 7, 348.
- Corma, A. Chem. Rev. 1997, 97, 2373.
- Ganschow, M.; Wark, M.; Wöhrle, D.; Schulz-Ekloff, G. Angew. Chem. Int. Ed. 2000, 39, 161.
- Bockstette, M.; Wöhrle, D.; Braun, I.; Schulz-Ekloff, G. Microporous Mesoporous Mater. 1998, 23, 83.
- Blatter, F.; Frei, H. J. Am. Chem. Soc. 1993, 115, 7501.
- Blatter, F.; Frei, H. J. Am. Chem. Soc. 1994, 116, 1812.
- Sun, H.; Blatter, F.; Frei, H. J. Am. Chem. Soc. 1996, 118, 6873.
- Zicovich-Wilson, C. M.; Corma, A.; Viruela, P. J. Phys. Chem. 1994, 98, 10863.
- Corma, A.; García, H.; Sastre, G.; Viruela, P. M. J. Phys. Chem. B 1997, 101, 4575.
- Cohen de Lara, E.; Kahn, R. Guideline for Mastering the Properties of Molecular Sieves; NATO ASI Series; 1990; Volume 221, p. 169. [Google Scholar]
- Rabo, J. A.; Gajda, G. J. Guideline for Mastering the Properties of Molecular Sieves; NATO ASI Series; 1990; Volume 221, p. 273. [Google Scholar]
- Derouane, E. Guideline for Mastering the Properties of Molecular Sieves; NATO ASI Series; 1990; Volume 221, p. 225. [Google Scholar]
- Van Santen, R. A. Guideline for Mastering the Properties of Molecular Sieves; NATO ASI Series; 1990; Volume 221, p. 201. [Google Scholar]
- Zhang, L. Z.; Cheng, P.; Liao, D.-Z. J. Chem. Phys. 2002, 117, 5959.
- Zhang, L. Z.; Cheng, P. Chin. J. Inorg. Chem. 2003, 19, 7.
- Douhal, A.; Lahmani, F.; Zewail, A. H. Chem. Phys. 1996, 207, 477.
- Cohen, M. D.; Flavian, S. J. Chem. Soc. B 1967, 321.
- Zhang, L. Z.; Xiong, Y.; Cheng, P.; Tang, G.-Q.; Liao, D.-Z. Chem. Phys. Lett. 2002, 358, 278.
- Fu, H.-B.; Yao, J.-N. J. Am. Chem. Soc. 2001, 123, 1434.
- Zhang, L. Z.; Xiong, Y.; Cheng, P.; Tang, G.-Q.; Wang, L.-J.; Liao, D.-Z. J. Mater. Chem. 2001, 11, 2903.
- Langley, P. J.; Hulliger, J. Chem. Soc. Rev. 1999, 28, 279.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Zhang, L.Z.; Sun, W.; Cheng, P. Spectroscopic and Theoretical Studies of Quantum and Electronic Confinement Effects in Nanostructured Materials. Molecules 2003, 8, 207-222. https://doi.org/10.3390/80100207
AMA Style
Zhang LZ, Sun W, Cheng P. Spectroscopic and Theoretical Studies of Quantum and Electronic Confinement Effects in Nanostructured Materials. Molecules. 2003; 8(1):207-222. https://doi.org/10.3390/80100207
Chicago/Turabian StyleZhang, Lei Z., Wei Sun, and Peng Cheng. 2003. "Spectroscopic and Theoretical Studies of Quantum and Electronic Confinement Effects in Nanostructured Materials" Molecules 8, no. 1: 207-222. https://doi.org/10.3390/80100207