Regioselective Protection of the 4-Hydroxyl of 3,4-Dihydroxy-benzaldehyde

University of Northern British Columbia, Department of Chemistry, 3333 University Way, Prince George, British Columbia, Canada
*
Author to whom correspondence should be addressed.
Molecules 2002, 7(9), 697-705; https://doi.org/10.3390/70900697
Submission received: 30 July 2002
/
Revised: 12 September 2002
/
Accepted: 14 September 2002
/
Published: 30 September 2003
Abstract
:The regioselective protection of the 4-hydroxyl group of 3,4-dihydroxy-benzaldehyde was accomplished with seven different protecting groups (benzyl, p-methoxybenzyl, o-nitrobenzyl, 2,6-dichlorobenzyl, 3,4-dichlorobenzyl, vinyl and propargyl) in yields ranging between 67-75%.
Introduction
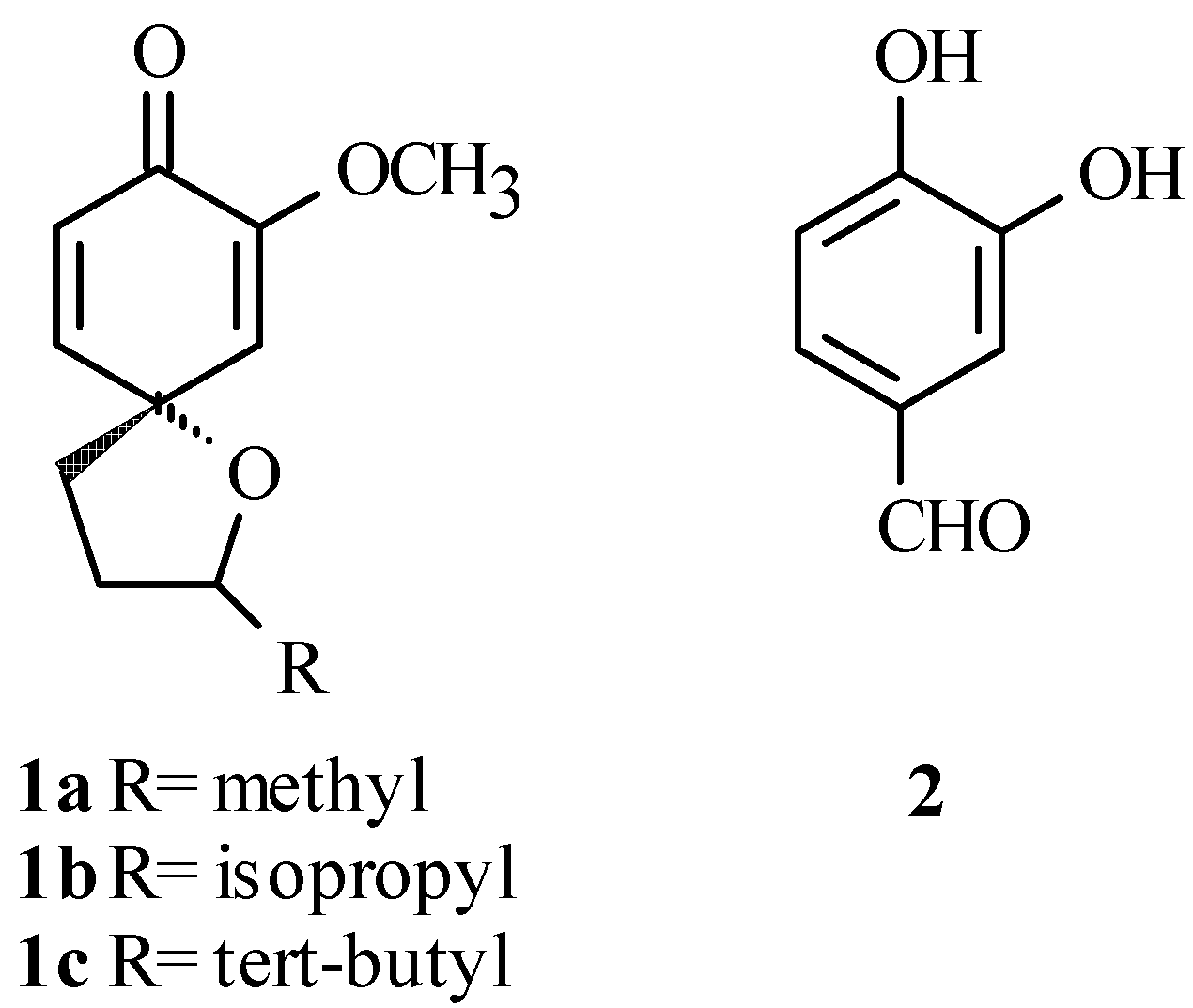
We recently reported our study of the diastereoselective spiroannulation of simple phenols producing spiroethers 1a-c shown in Figure 1 [1]. In a continuation of this work, we needed to replace the methoxy group with other substituents in order to study the effect these groups would have on the spiroannulation reaction. To accomplish this task, we first needed to selectively protect the 4-hydroxyl group of 3,4-dihydroxybenzaldehyde 2 (Figure 1). Unfortunately, a literature survey showed that little had been done in terms of selective protection of this type of catechol. We found a few examples dealing with the selective protection of 2,4- and 2,5-dihydroxybenzaldehydes [2,3], 2,5-dihydroxyacetophenone [4], and methyl 2,5-dihydroxybenzoate [5], with protective groups such as methyl [4], benzyl [2,5], and t-butyldimethylsilyl [3]. More recently, we reported the protection of 2,4-dihydroxybenzaldehyde using the methoxymethyl ether (MOM) protecting group [6]. The selectivity in these cases is normally attributed to the intramolecular H-bonding that exists between the 2-hydroxyl and the carbonyl group preventing the 2-position from being alkylated. In another example, the pivaloyl protecting group was used to selectively protect the 5-hydroxyl group of 1-t-butyl-2,5-dihydroxy-benzene with the selectivity being controlled by steric hindrance [7]. In the case of catechols such as 3,4-dihydroxyacetophenone or 3,4-dihydroxybenzaldehyde, where the acidity of the phenolic hydroxyls controls the selectivity, we found only a few examples of selective protection of the 4-hydroxyl position. One example used the methyl protecting group in the protection of the 4-hydroxyl group of 3,4-dihydroxyacetophenone [8]. However, we considered this protecting group inappropriate for our purpose because Lewis acids would usually be required to remove it from the targeted product. Examples of selective protection of 3,4-dihydroxybenzaldehyde were also found, but typically these reactions provided low yields of the 4-protected products. For instance, the protection of 3,4-dihydroxybenzaldehyde was accomplished in ~ 40% using the allyl protecting groups [9,10], while the benzyl and p-methoxybenzyl protecting groups gave only between 30-57% yield of the 4-protected benzaldehyde [11,12]. Hence, there was a need to find a better method to introduce a protecting group that can be easily removed from the target molecules under mild conditions. We now wish to report our findings on the successful regioselective protection of the 4-hydroxyl group of 2 using seven different protecting groups.
Figure 1.
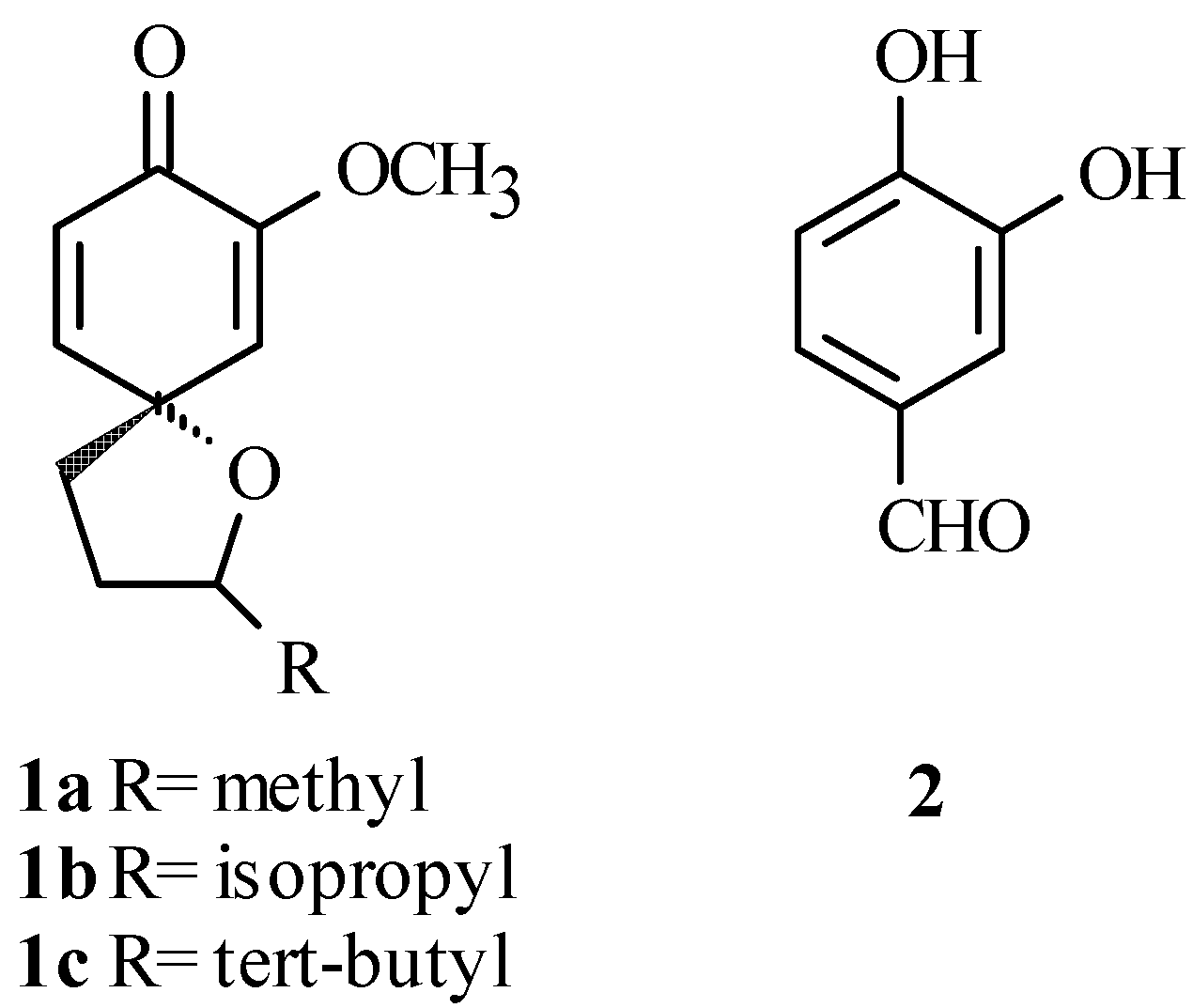
Results and Discussion
Since we had success in the protection of 2,4-dihydroxybenzaldehyde with the MOM protecting group [6], we decided to begin this investigation with that protecting group. Following our reported procedure [MOMCl, K2CO3, acetone, rt, 20h], compound 2 gave primarily one product by thin layer chromatography (tlc). Flash chromatography of the crude reaction mixture provided a colourless solid (87%), but 1H-NMR suggested that two aldehydes (δ 9.84 and 9.81) were present in a 8/2 ratio. GC‑MS analysis of this mixture confirmed the ratio of these two products, and further indicated that the two products were a mixture of the 4-monoprotected and the 3-monoprotected isomers, since each product showed m/z 182 [M+]. Changing the base to the weaker NaHCO3 in an attempt to deprotonate only the 4-hydroxyl group did not succeed, and only 2 was recovered in a quantitative amount after stirring either at room temperature for 2 days, or for 20h at reflux. Under the conditions used by Wymann for the methyl protecting group [Li2CO3, DMF, 60oC, 20h] [8], only benzaldehyde 2 was recovered when MOMCl was used as the alkylating agent. We also carried out these reactions with MEMCl as the alkylating agent and obtained the same results, i.e a 8/2 mixture of isomeric monoprotected aldehydes using the conditions we previously reported for 2,4-dihydroxybenzaldehyde [MEMCl, K2CO3, acetone, rt, 20h] [6], and complete recovery of the starting material under the conditons used by Wymann [MEMCl, Li2CO3, DMF, 60oC, 20h] [8].
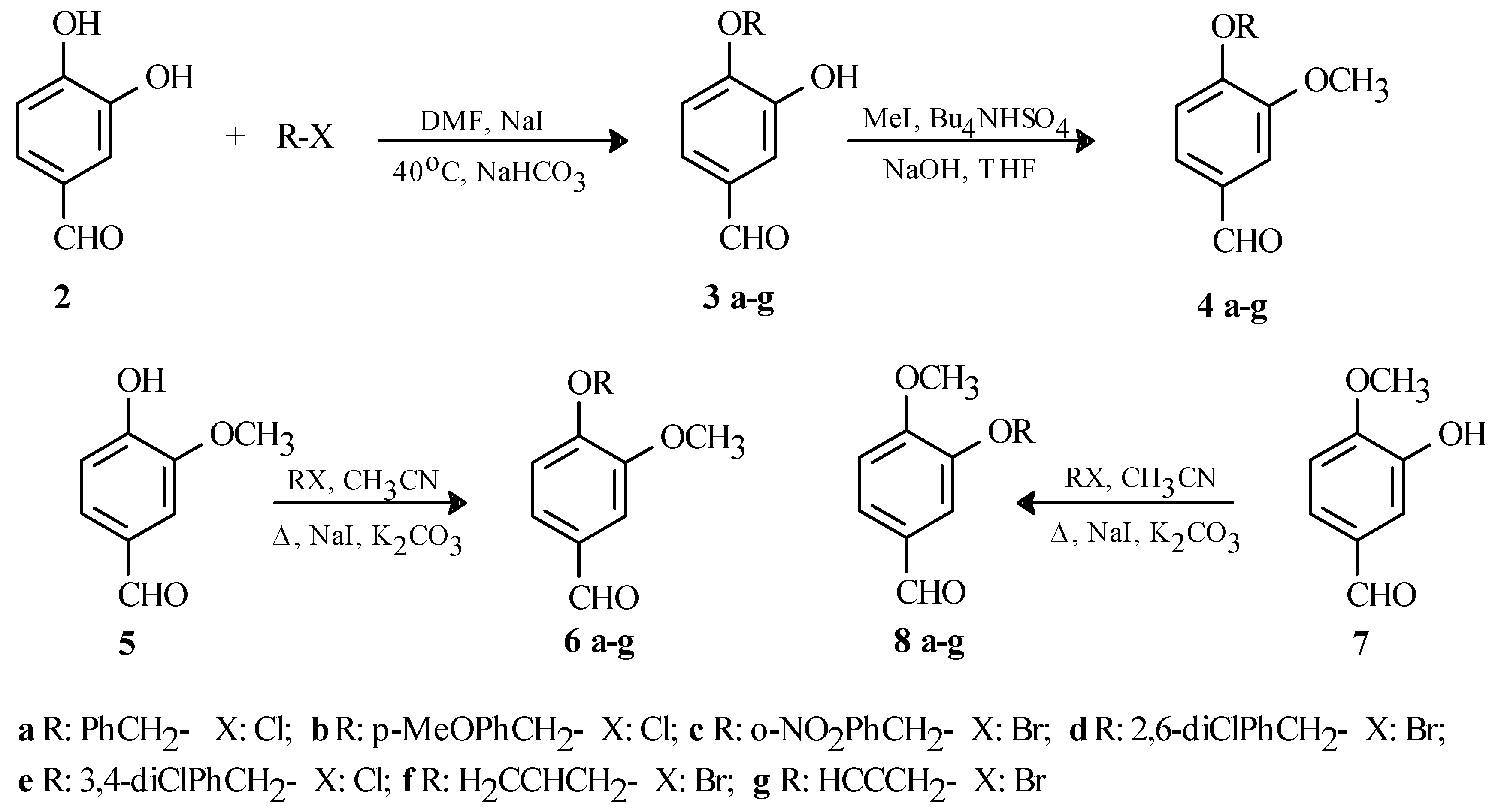
Having no success with the MOM and MEM protecting groups, we looked at more reactive alkyl halides and tried using benzyl chloride. Under the first set of conditions [benzyl chloride, K2CO3, NaI, acetone, rt, 20h], we obtained a mixture of products consisting of both monoprotected aldehydes, as well as the diprotected product. However, with the second set of conditions [benzyl chloride, K2CO3, DMF, NaI], 2 produced a mixture of the monoprotected products (δ 9.80 and 9.76) in 67% yield in a 9/1 ratio when stirred for 6 days at room temperature. Changing the base to NaHCO3 and heating the reaction mixture [benzyl chloride, DMF, NaHCO3, NaI, 40oC, 20h] as shown in Scheme 1 had a positive effect on the yield, as well as the purity of the product. In this case, we were able to isolate only one monoprotected aldehyde (1H-NMR in CDCl3: δ 9.80; GCMS m/z: 228 [M+]) in 71% yield as a colourless solid (mp: 118-120oC).
Scheme 1.
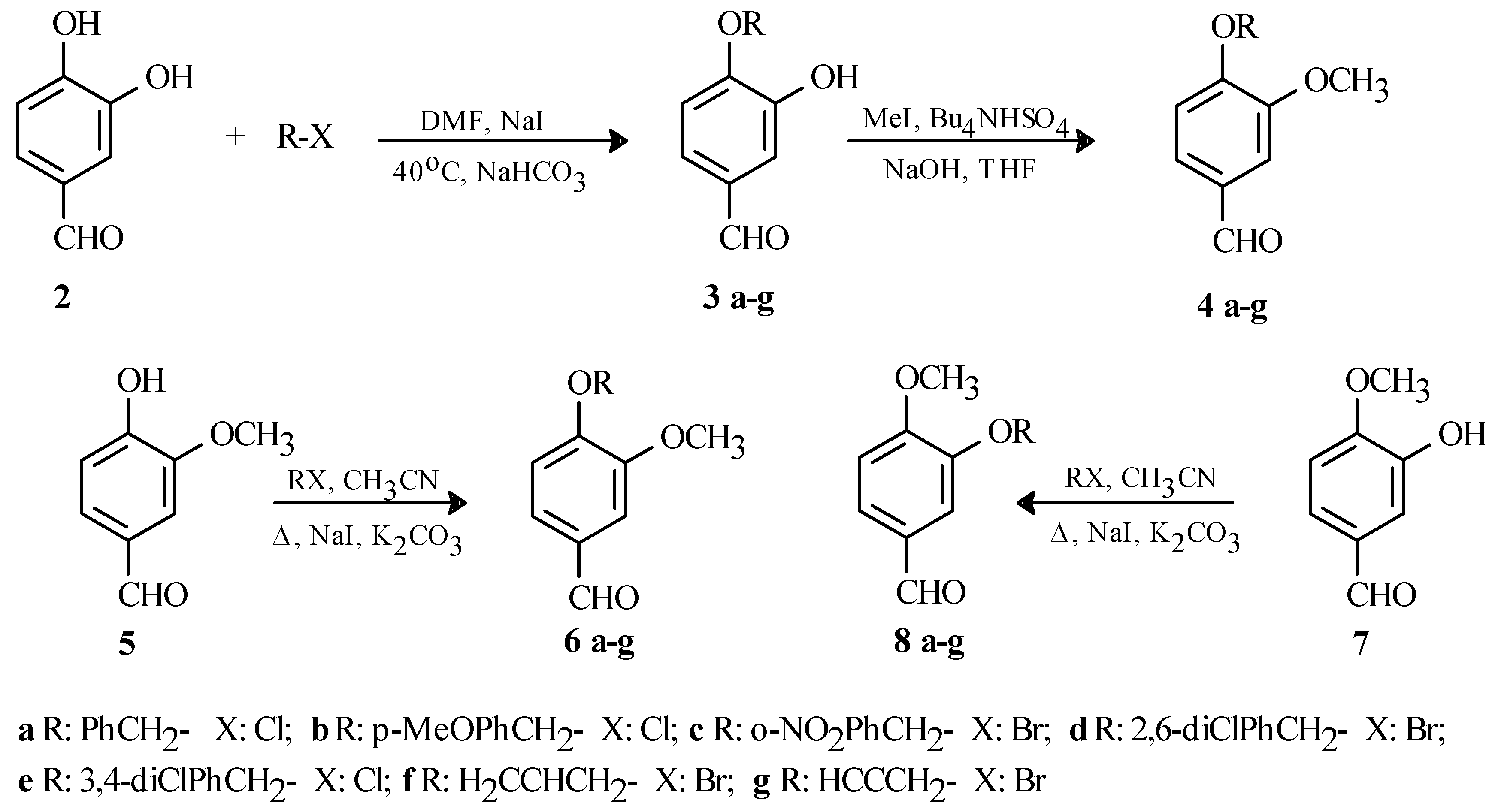
Alkylations of 2 with other alkyl halides using these reaction conditions were also successful and we were able to prepare six other monoprotected benzaldehydes in 67-75% yields as shown in Scheme 1. 1H-NMR data for the aldehyde signal as well as the GC-MS data confirmed the presence of only one monoprotected product in all cases: 1H-NMR in CDCl3: δ 9.84 (3b), 9.86 (3c), 9.87 (3d), 9.86 (3e), 9.85 (3f), 9.86 (3g); GCMS m/z: 258 [M+] (3b), 273 [M+] (3c), 296 [M+] (3d), 296 [M+] (3e), 178 [M+] (3f), 176 [M+] (3g). The yields and melting points of all the compounds shown in Scheme 1 are summarized in Table 1.

{kind=link}
{kind=link}
| Series | R | Yielda | Melting Point (oC)b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 6 | 8 | 3 | 4 | 6 | 8 | ||
| a | benzyl | 71 | 83 | 89 | 82 | 118-120 | 58-60 | 60-61 | 59-60 |
| b | p-methoxybenzyl | 75 | 100 | 90 | 78 | 125-127 | 105-107 | 106-107 | 80-81 |
| c | o-nitrobenzyl | 67 | 85 | 83 | 86 | 147-149 | 126-127 | 127-129 | 138-139 |
| d | 2,6-dichlorobenzyl | 69 | 85 | 80 | 80 | 125-126 | 124-125 | 125-126 | 167-168 |
| e | 3,4-dichlorobenzyl | 68 | 85 | 86 | 93 | 153(d) | 113(d) | 115(d) | 82(d) |
| f | allyl | 72 | 91 | 94 | 86 | 55-56 | 24-26 | 24-25 | n/ac |
| g | propargyl | 69 | 94 | 86 | 88 | 77-78 | 84-85 | 85-86 | 71-72 |
a isolated yields after column chromatographyb melting points were recorded on a hot stage instrument and are uncorrected c 8 is an oil at room temperature
In order to conclusively identify the major product produced in these reactions, we methylated [2M NaOH, THF, CH3I, Bu4NHSO4] the products 3a-g to give the derivatives 4a-g as shown in Scheme 1. We also prepared compounds 6a-g and 8a-g from vanillin (5) and isovanillin (7) respectively, and compared the spectroscopic data of these compounds with that of 4a-g, looking for spectroscopic evidence that would confirm the identity of these compounds. As previously mentioned, we already knew from GC-MS that only monoprotected benzaldehydes were produced. We compared the 1H‑NMR and IR spectra of 4a-g to those of 6a-g and 8a-g, but we did not find any significant difference between any of these spectra that would help in confirming the structures of the monoprotected benzaldehydes 3a-g. However, careful analysis of the 13C-NMR spectra for the same compounds showed one significant difference in all cases. We noticed that the signals for the aromatic carbons C2 and C5 were further apart in the case of 4a-g and 6a-g (C2 ~112.5ppm and C5 ~109.5ppm), while the signals for the same carbons almost collapse to only one line for compounds 8a-g (C2 ~111.5 ppm and C5 ~111.0 ppm). The results shown in Table 2 suggest that compounds 4a-g and 6a-g are identical based on the chemical shifts of these two carbons. Further evidence can be observed when comparing the entire 13C-NMR spectra. When comparing 4a-g and 6a-g, the spectra appear to be duplicates of one another. However, when comparing 4a-g and 8a-g differences other that those for C2 and C5 can be observed, although much less significant, confirming that 4a-g and 8a-g are different compounds. Mass spectral analysis and mixed melting point experiments also indicated that 4a-g and 6a-g were identical compounds.
| Series | R | 2 | 4 | 6 | 8 | ||||
|---|---|---|---|---|---|---|---|---|---|
| C2 | C5 | C2 | C5 | C2 | C5 | C2 | C5 | ||
| a | benzyl | 114.6 | 111.7 | 112.6 | 109.5 | 112.5 | 109.4 | 111.4 | 110.9 |
| b | p-methoxybenzyl | 114.6 | 111.6 | 112.0 | 109.4 | 112.0 | 109.3 | 110.9 | 110.8 |
| c | o-nitrobenzyl | 115.1 | 112.1 | 112.6 | 109.6 | 112.6 | 109.6 | 112.2 | 111.2 |
| d | 2,6-dichlorobenzyl | 114.8 | 112.1 | 113.2 | 110.0 | 113.2 | 110.1 | 112.6 | 111.3 |
| e | 3,4-dichlorobenzyl | 115.0 | 111.7 | 112.5 | 109.6 | 112.5 | 109.6 | 111.3 | 111.1 |
| f | allyl | 114.6 | 111.6 | 112.0 | 109.4 | 112.0 | 109.3 | 110.9 | 110.8 |
| g | propargyl | 115.0 | 112.0 | 112.6 | 109.5 | 112.6 | 109.5 | 111.9 | 111.0 |
a 13C-NMR data were obtained in CDCl3 on a Bruker AMX300 spectrometer at 75.4 MHz using CHCl3 as the internal standard. Chemical shifts are expressed in δ units.
We also attempted the regioselective protection of 2 with two other alkyl halides, phenacyl bromide and α-bromoacetonitrile. In the case of phenacyl bromide [DMF, NaI, NaHCO3, 40oC, 20h], we obtained a mixture of products (8 spots by tlc) that were unseparable and not identified. Under the other set of conditions [K2CO3, acetone, rt, 20h] the reaction did not show any selectivity and the 1H‑NMR spectrum of the crude reaction mixture showed that both possible monoprotected aldehydes were obtained along with some of the diprotected product. In the case of α-bromoacetonitrile, only benzaldehyde 2 was recovered in quantitative yield under either set of conditions.
Conclusions
We have successfully protected the 4-hydroxyl group of 3,4-dihydroxybenzaldehyde in 70% yield with active halides such as benzyl chlroride derivatives, allyl bromide and propargyl bromide. The reaction showed higher regioselectivity than with other alkyl halides such as MOMCl, MEMCl, phenacyl bromide or α-bromoacetonitrile. Given the number of available benzyl halide derivatives, this method should be a valuable tool for the protection of other catechols bearing an electron-withdrawing substituent para to one of the hydroxyl groups. It may also find usefulness in industry, for example in the synthesis of alkaloids such as galanthamine [13], as well as in the synthesis of antimycotic agents (benzylidene thiazolidinediones) which requires the selective protection of the 4-hydroxyl group of 3,4-dihydroxybenzaldehyde [12].
Experimental
Melting points were determined on a hot stage instrument and are uncorrected. Infrared spectra were recorded either as KBr pellets or neat on a Perkin Elmer System 2000 FTIR. 1H-NMR spectra were recorded on a Bruker AMX300 spectrometer at 300MHz and chemical shifts are expressed in ppm using TMS as internal standard. 13C-NMR spectra were recorded on a Bruker AMX300 spectrometer at 75.4MHz and chemical shifts are expressed in ppm using chloroform as internal standard. Mass spectra were recorded on a Hewlett Packard 5898B spectrometer.
General procedure for the selective protection of 3,4-dihydroxybenzaldehyde:
To a solution of 3,4-dihydroxybenzaldehyde 2 (~1.0 mmol) in DMF (5 mL) was added sodium bicarbonate (~1.5 mmol), the alkyl halide (~2.0 mmol) and sodium iodide (~0.3 mmol). The resulting mixture was stirred at 40oC for 24h. 10% Aqueous HCl (10 mL) was added and the solution was extracted with ethyl acetate (3 x 10 mL). The organic fractions were combined, washed with brine (10 mL), dried (anhydrous MgSO4) and the solvent was evaporated in vacuo to give a brown liquid. Subsequent chromatography on silica gel using the solvent systems indicated afforded a colourless solid in all cases.
4-Benzyloxy-3-hydroxybenzaldehyde (3a). From 137 mg of 2, we obtained 3a (160 mg, 71%) after chromatography (20% EtOAc/hexanes); Mp: 118-120oC. Spectral data were identical to those previously reported [12].
3-Hydroxy-4-(p-methoxybenzyloxy)-benzaldehyde (3b). From 116 mg of 2, we obtained 3b (163 mg, 75%) after chromatography (15% acetone/hexanes); Mp: 125-127oC. Spectral data were identical to those previously reported [12].
3-Hydroxy-4-(o-nitrobenzyloxy)-benzaldehyde (3c). From 115 mg of 2, we obtained 3c (153 mg, 67%) after chromatography (25% EtOAc/hexanes); Mp: 147-149oC; IR (KBr) cm-1: 3410 (OH), 1685 (CO); 1H-NMR (CDCl3) δ: 5.68 (s, 2H, OCH2), 5.82 (s, 1H, exchangeable with D2O, OH), 6.97 (d, 1H, J=8.3Hz, H5), 7.57 (m, 5H, aromatic Hs), 8.22 (d, 1H, J=8.2Hz, benzyl Ar-H3), 9.86 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 68.3 (OCH2), 112.1 (C2), 115.1 (C5), 124.6 (benzyl Ar-C3), 125.7 (C6), 128.7 (benzyl Ar-C6), 129.5 (benzyl Ar-C4), 131.5 (C1), 132.1 (benzyl Ar-C1), 134.5 (benzyl Ar-C5), 146.4 (C3), 147.3 (benzyl Ar-C2), 150.4 (C4), 191.1 (CO); MS m/z (relative %): 273 [M+] (2), 119 (100), 92 (93), 64 (63), 63 (31).
4-(2,6-Dichlorobenzyloxy)-3-hydroxybenzaldehyde (3d). From 112 mg of 2, we obtained 3d (166 mg, 69%) after chromatography (20% EtOAc/hexanes); Mp: 125-126oC; IR (KBr) cm-1: 3419 (OH), 1681 (CO); 1H-NMR (CDCl3) δ: 5.45 (s, 2H, OCH2), 5.79 (s, 1H, exchangeable with D2O, OH), 7.29 (m, 6H, aromatic Hs), 9.87 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 66.4 (OCH2), 112.1 (C5), 114.8 (C2), 124.4 (C6), 128.9 (benzyl Ar-C3), 131.0 (C1), 131.4 (benzyl Ar-C4), 131.5 (benzyl Ar-C2), 137.2 (benzyl Ar-C1), 146.8 (C3), 150.9 (C4), 191.2 (CO); MS m/z (relative %): 296 [M+] (7), 254 (100), 253 (77), 207 (46).
4-(3,4-Dichlorobenzyloxy)-3-hydroxybenzaldehyde (3e). From 125 mg of 2, we obtained 3e (181 mg, 68%) after chromatography (15% EtOAc/hexanes); Mp: 153oC (decomposed); IR (KBr) cm-1: 3423 (OH), 1683 (CO); 1H-NMR (CDCl3) δ: 5.16 (s, 2H, OCH2), 5.77 (s, 1H, exchangeable with D2O, OH), 6.99 (d, 1H, J=8.3Hz, H5), 7.42 (m, 5H, aromatic Hs), 9.86 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 70.0 (OCH2), 111.7 (C5), 115.0 (C2), 124.5 (C6), 127.2 (benzyl Ar-C6), 129.9 (benzyl Ar-C2), 131.2 (C1), 131.4 (benzyl Ar-C5), 133.2 (benzyl Ar-C3), 133.4 (benzyl Ar-C4), 135.6 (benzyl Ar-C1), 146.4 (C3), 150.6 (C4), 191.1 (CO); MS m/z (relative %): 296 [M+] (4), 254 (63), 253 (27), 207 (100).
4-Allyloxy-3-hydroxybenzaldehyde (3f). From 125 mg of 2, we obtained 3f (116 mg, 72%) after chromatography (20% EtOAc/hexanes); Mp: 55-56oC. Spectral data were identical to those previously reported [9].
3-Hydroxy-4-propargyloxybenzaldehyde (3g). From 112 mg of 2, we obtained 3g (99 mg, 69%) after chromatography (20% EtOAc/hexanes); Mp: 77-78oC; IR (KBr) cm-1: 3422 (OH), 2129 (alkyne), 1679 (CO); 1H-NMR (CDCl3) δ: 2.63 (s, 1H, propargyl CH), 4.87 (s, 2H, OCH2), 5.95 (s, 1H, exchangeable with D2O, OH), 7.10 (d, 1H, J=8.2Hz, H5), 7.45 (d, 1H, J=8.2Hz, H6), 7.48 (s, 1H, H2), 9.86 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 57.0 (OCH2), 76.7 (propargyl CH), 77.3 (propargyl C2), 112.1 (C5), 115.0 (C2), 124.3 (C6), 131.5 (C1), 146.5 (C3), 149.8 (C4), 191.3 (CO); MS m/z (relative %): 176 [M+] (100), 147 (33), 137 (64), 109 (57), 81 (47).
General procedure for the methylation of derivatives 3a-g
To a solution of monoprotected benzaldehydes 3a-g (~0.5 mmol) in THF (5 mL) was added 2M NaOH solution (5 mL), CH3I (~2 mmol) and Bu4NHSO4 (~100 mg). The resulting yellow solution was stirred at room temperature for 24h. Water (20 mL) was added and the solution was extracted with ethyl acetate (3 x 10 mL). The organic fractions were combined, dried (MgSO4) and the solvent was evaporated in vacuo. In all cases the crude products gave colourless solids after purification by chromatography on silica gel using the indicated solvent systems.
4-Benzyloxy-3-methoxybenzaldehyde (4a). From 150 mg of 3a, we obtained 4a (132 mg, 83%) after chromatography (20% EtOAc/hexanes); Mp: 58-60oC. Spectral data were identical to those obtained from an authentic sample purchased from Aldrich Chemical Co.
4-(p-Methoxybenzyloxy)-3-methoxybenzaldehyde (4b). From 84 mg of 3b, we obtained 4b (88 mg, 100%) after chromatography (20% EtOAc/hexanes); Mp: 105-107oC (lit: 102-103oC) [14]; IR (KBr) cm-1: 1682 (CO); 1H-NMR (CDCl3) δ: 3.84 (s, 3H, benzyl OCH3), 3.93 (s, 3H, OCH3), 5.18 (s, 2H, OCH2), 6.92 (d, 2H, J=8.5Hz, benzyl Ar-H3), 7.01 (d, 2H, J=8.0Hz, H5), 7.39 (m, 4H, aromatic Hs), 9.84 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 55.5 (benzyl OCH3), 56.2 (OCH3), 70.9 (OCH2), 109.4 (C5), 112.5 (C2), 114.3 (benzyl Ar-C3), 126.8 (C6), 128.0 (benzyl Ar-C1), 129.2 (benzyl Ar-C2), 130.3 (C1), 150.2 (C3), 153.9 (C4), 159.8 (benzyl Ar-C4), 191.2 (CO); MS m/z (relative %): 272 [M+] (3), 122 (10), 121 (100), 91 (5).
3-Methoxy-4-(o-nitrobenzyloxy)benzaldehyde (4c). From 99 mg of 3c, we obtained 4c (88 mg, 85%) after chromatography (20% EtOAc/hexanes); Mp: 126-127oC; IR (KBr) cm-1: 1681 (CO); 1H-NMR (CDCl3) δ: 4.00 (s, 3H, OCH3), 5.65 (s, 2H, OCH2), 7.01 (d, 1H, J=8.2Hz, H5), 7.44 (dd, 1H, J=1.8, 8.2Hz, H6), 7.48 (d, 1H, J=1.8Hz, H2), 7.53 (t, 1H, J=7.6Hz, benzyl Ar-H4), 7.72 (t, 1H, J=7.6Hz, benzyl Ar-H5), 7.93 (d, 1H, J=7.9Hz, benzyl Ar-H6) 8.22 (d, 1H, J=8.3Hz, benzyl Ar-H3), 9.86 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 56.3 (OCH3), 67.9 (OCH2), 109.6 (C5), 112.6 (C2), 125.4 (benzyl Ar-C3), 126.9 (C6), 128.5 (benzyl Ar-C6), 128.8 (benzyl Ar-C4), 131.0 (C1), 133.2 (benzyl Ar-C1), 134.5 (benzyl Ar-C5), 146.9 (C3), 150.3 (C4), 153.1 (benzyl Ar-C2), 191.1 (CO); MS m/z (relative %): 287 [M+] (3), 153 (7), 137 (10), 136 (100), 78 (43).
4-(2,6-Dichlorobenzyloxy)-3-methoxybenzaldehyde (4d). From 166 mg of 3d, we obtained 4d (148 mg, 85%) after chromatography (15% EtOAc/hexanes); Mp: 124-125oC; IR (KBr) cm-1: 1683 (CO); 1H-NMR (CDCl3) δ: 3.90 (s, 3H, OCH3), 5.40 (s, 2H, OCH2), 7.18 (d, 1H, J=8.1Hz, H5), 7.37 (m, 5H, aromatic Hs), 9.89 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 56.4 (OCH3), 66.5 (OCH2), 110.0 (C5), 113.2 (C2), 126.7 (C6), 128.7 (benzyl Ar-C3), 128.9 (benzyl Ar-C2), 130.9 (benzyl Ar-C4), 131.5 (C1), 137.4 (benzyl Ar-C1), 150.6 (C3), 154.0 (C4), 191.2 (CO); MS m/z (relative %): 312 [M++2] (14), 310 [M+] (20), 161 (67), 159 (100), 123 (7).
4-(3,4-Dichlorobenzyloxy)-3-methoxybenzaldehyde (4e). From 105 mg of 3e, we obtained 4e (93 mg, 85%) after chromatography (15% EtOAc/hexanes); Mp: 113oC (decomposed); IR (KBr) cm-1: 1684 (CO); 1H-NMR (CDCl3) δ: 3.97 (s, 3H, OCH3), 5.13 (s, 2H, OCH2), 6.94 (d, 1H, J=8.1Hz, H5), 7.28 (d, 1H, J=8.1Hz, H6), 7.43 (m, 3H, aromatic Hs), 7.55 (s, 1H, H2), 9.85 (s, 1H, CHO); 13C-NMR (CDCl3) δ: 56.2 (OCH3), 69.6 (OCH2), 109.6 (C5), 112.5 (C2), 126.6(C6 and benzyl Ar-C6), 129.3 (benzyl Ar-C2), 130.8 (C1), 130.9 (benzyl Ar-C5), 132.5 (benzyl Ar-C3), 133.1 (benzyl Ar-C4), 136.4 (benzyl Ar-C1), 150.2 (C3), 153.1 (C4), 191.1 (CO); MS m/z (relative %): 312 [M++2] (9), 310 [M+] (13), 161 (70), 159 (100), 123 (7).
4-Allyloxy-3-methoxybenzaldehyde (4f). From 268 mg of 3f, we obtained 4f (262 mg, 91%) after chromatography (20% EtOAc/hexanes); Mp: 24-25oC. Spectral data were identical to those previously reported [15].
3-Methoxy-4-propargyloxybenzaldehyde (4g). From 96 mg of 3g, we obtained 4g (97 mg, 94%) after chromatography (20% EtOAc/hexanes). Mp: 84-85oC. Spectral data were identical to those previously reported [16].
Acknowledgements
The authors would like to thank the University of Northern British Columbia for the financial support of this work.
References
- Plourde, G.L. Tetrahedron Lett. 2002, 43, 3597.
- Mendelson, W.L.; Holmes, M.; Dougherty, J. Synth. Commun. 1996, 26, 593.
- Liu, A.; Dillon, K.; Campbell, R.M.; Cox, D.C.; Huryn, D.M. Tetrahedron Lett. 1996, 37, 3587.
- Vyas, G.N.; Shah, N.M. Org. Synt., Collect. Vol. IV 1963, 836.
- Kotecha, N.R.; Ley, S.V.; Montégani, S. Synlett 1992, 395.
- Plourde, G.L.; Fisher, B.B. Molecules 2002, 7, 315.
- Lam, L.K.T.; Farhat, K. Org. Prep. Proced. Int. 1978, 10, 79.
- Wymann, W.E.; Davis, R.; Patterson, J.W., Jr.; Pfister, J.R. Synth. Commun. 1988, 18, 1379.
- Reitz, A.; Avery, M.A.; Verlander, M.S.; Goodman, M. J. Org. Chem. 1981, 46, 4859.
- Kessar, S.V.; Gupta, Y.P.; Mohammad, T.; Goyal, M.; Sawal, K.K. J. Chem. Soc. Chem. Comm. 1983, 400.
- Markey, S.P.; Powers, K.; Dubinsky, D.; Kapin, I.J. J. Labbelled Comp. and Radiopharm. 1979, 17, 103.
- Orchard, M.G.; Neuss, J.C.; Galley, C.M.S. PCT Int. Appl. 2002, 99, WO 02/17915 A1.
- Kuenburg, B.; Czollner, L.; Frohlich, J.; Jordis, U. Org. Process R & D 1999, 3, 425.
- Vig, O.P.; Bari, S.S.; Sharma, A.; Sattar, M.M. Ind. J. Chem. 1990, 29B, 284.
- Ayer, W.A.; Craw, P.A. Can. J. Chem. 1991, 69, 1909.
- Ishii, H.; Ishikawa, T.; Wada, H.; Miyazaki, H.; Kaneko, Y.; Harayama, T. Chem. Pharm. Bull 1992, 40, 2614.
- Sample availability: all compounds are available from MDPI.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Plourde, G.L.; Spaetzel, R.R. Regioselective Protection of the 4-Hydroxyl of 3,4-Dihydroxy-benzaldehyde. Molecules 2002, 7, 697-705. https://doi.org/10.3390/70900697
AMA Style
Plourde GL, Spaetzel RR. Regioselective Protection of the 4-Hydroxyl of 3,4-Dihydroxy-benzaldehyde. Molecules. 2002; 7(9):697-705. https://doi.org/10.3390/70900697
Chicago/Turabian StylePlourde, Guy L., and Randy R. Spaetzel. 2002. "Regioselective Protection of the 4-Hydroxyl of 3,4-Dihydroxy-benzaldehyde" Molecules 7, no. 9: 697-705. https://doi.org/10.3390/70900697