An Improved Three Step Synthesis of (-)-3β-Hydroxycarvone from (-)-Carvone

1
Departamento de Química, Universidade Federal do Espírito Santo, 29060-900, Vitória, ES, Brazil
2
Departamento de Química, Universidade Federal de São Carlos, Caixa Postal 676, 13565-905, São Carlos, SP, Brazil
*
Author to whom correspondence should be addressed.
Molecules 2002, 7(2), 129-134; https://doi.org/10.3390/70200129
Submission received: 23 May 2001
/
Revised: 15 January 2002
/
Accepted: 31 January 2002
/
Published: 28 February 2002
{kind=link}
Abstract
:(-)-Carvone (3) has been efficiently transformed into (-)-3β-hydroxycarvone (1), which is expected to be a useful synthon or chiral template in the synthesis of natural molecules. This short and efficient synthesis of compound 1 involves regioselective and stereoselective α-hydroxylation of carvone via the trimethylsilyl-dienyl-ether derivative.
Introduction
Carvone (3) is an important chiron for the synthesis of complex natural products [1]. However, its versatility could be greatly enhanced by the development of efficient and selective methodologies for the synthesis of 3-oxy-derivatives. trans-3-Hydroxycarvone (1) has many structural features found in several naturally occurring compounds [2], and therefore can be considered a useful synthon provided that it can be obtained from readily available natural sources such as carvone (3).
In connection with the synthesis of a bisabolane sesquiterpene isolated from Senecio lividus [3], we required multi-gram quantities of 3β-hydroxycarvone (1) for use as our starting material and needed to develop an effective and short method to synthesise this compound.
A literature survey revealed three different approaches to the synthesis of the title compound. Hosokawa et al [4] described the stereoselective preparation of trans-3-hydroxycarvone (1), by oxidation of carvone silyl-dienyl ether with O2 catalysed by PdCl2(MeCN)2/CuI in HMPA. Schulz et al [5] reported the hydroxylation of carvone titanium enolate using a mixture of tert-butylhydroperoxide and MgO, resulting in a mixture of 3β- and 3α-hydroxycarvone (1) and (2), with 40% d.e. Finally, Nagaoka et al [6] described the synthesis of 3-hydroxycarvone from (+)-carvone, in 45% overall yield, via oxidation of its silyl-dienyl ether with m-chloroperbenzoic acid.
These methodologies all provided the desired compound in poor yield. In Hosokawa and Schulz’s methods the yields were only 11 and 20% of trans-3-hydroxycarvone and mixture of diastereomers respectively [4,5]. Furthermore, from the spectroscopic data in Schulz’s paper, it seems that the minor diastereomer should be assigned as cis-isomer (5S,6R) and not (5S,6S) that correspond to the trans isomer. Nagaoka et al. [6], have improved the synthesis yield of 3-hydroxycarvone to 45%. However, the product was probably a mixture of diastereoisomers, since no assignment has been made nor were spectroscopic data and complete experimental details included.
We report herein a short improved methodology based on oxidation of silyl-dienyl ether to prepare (-)-3β-hydroxycarvone (1) by an easy experimental procedure starting from (R)-(-)-carvone (3). This methodology allowed the full characterization of the diastereomeric products formed.
Results and Discussion
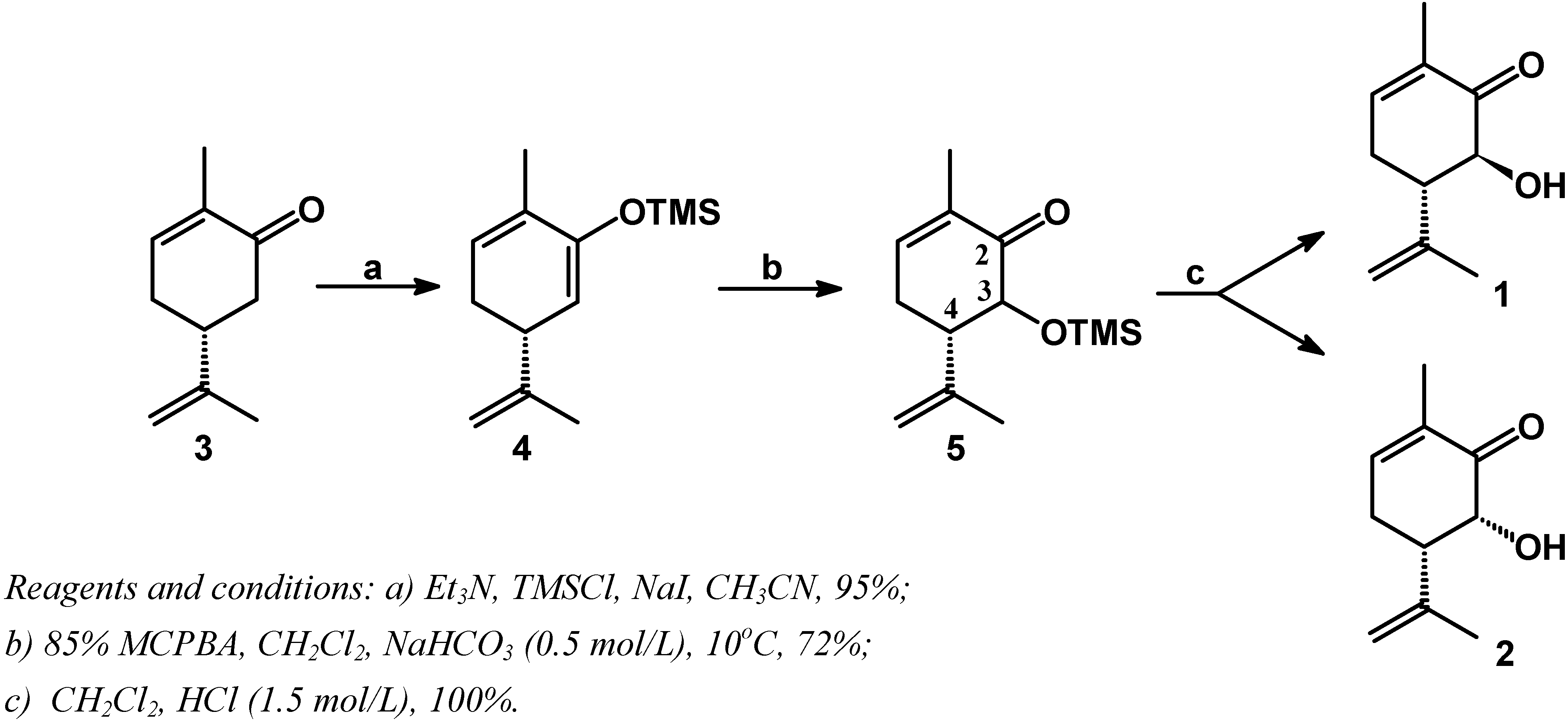
Commercially available (R)-(-)-carvone (3) was kinetically deprotonated with triethylamine [7,8], followed by silylation of the enolate with iodotrimethylsilane to afford the trimethylsilyl-dienyl ether (4) in 95% yield (Scheme 1).
Scheme 1.
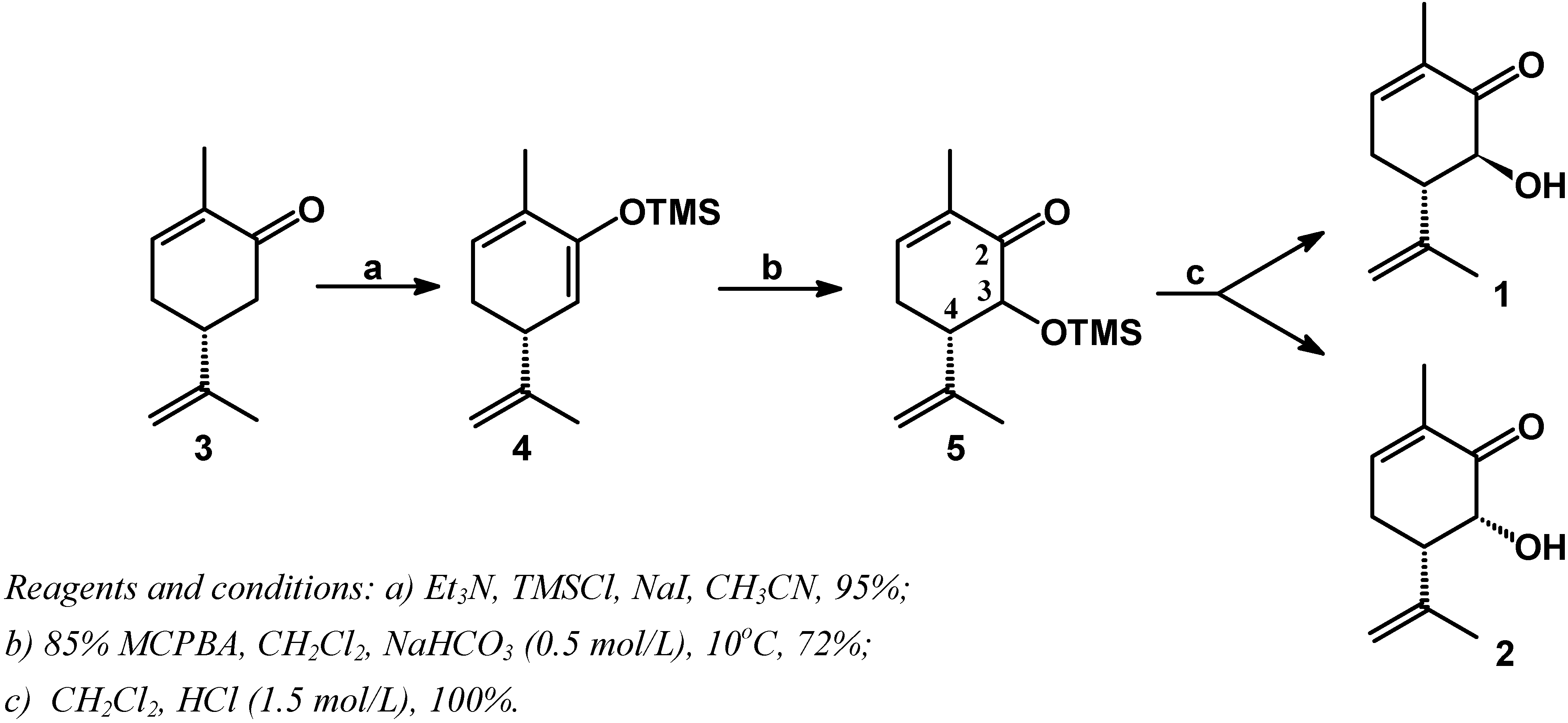
The freshly purified dienyl-trimethylsilyl ether (4) was immediately oxidized with 85% m-chloroperbenzoic acid [9,10,11] in dichloromethane and 0.5 mol/L aqueous NaHCO3 solution at 10oC. It is known that dienyl-trimethylsilyl ethers are readly transformed into α-hydroxyenones by the use of oxidants like hydroperoxides such m-chloroperbenzoic acid [9,10,11] or tert-butylhydroperoxide [12]. The reaction proceeds via epoxidation followed by rearrangement leading to α-silyloxyenones, and acid work up results in the formation of α-hydroxyenones.
Use of aqueous NaHCO3 solution results in a better yield due to quenching of m-chlorobenzoic acid generated during the reaction. Apparently, this by-product causes hydrolysis of the dienyl-trimethylsilyl ether (4) prior to epoxidation. In agreement with this, use of 50% m-chloroperbenzoic acid lead to a decreased yield of 3-hydroxycarvone as well as recovery of carvone.
Treatment of the crude reaction mixture with 1.5 mol/L HCl aqueous solution give a mixture (85:15, respectively) of the two diastereomers (1) and (2), in 72% of yield from (4). 3β-Hydroxy-carvone (1) and 3α-hydroxycarvone (2) were easily and cleanly separated by radial thin-layer chromatography.
The stereochemistry of the stereogenic center at C-3 of (1) and (2) was assigned on the basis of their 1H-NMR spectra and are consistent with the published data [4,5]. The 1H-NMR spectrum of isomer (1) showed a coupling constant of J3-4 = 12.5 Hz for the carbinolic hydrogen, characteristic of a trans relative configuration. For the minor isomer (2), the 1H-NMR spectrum showed a coupling constant of J3-4 = 5.9 Hz for the carbinolic hydrogen, characteristic of a cis relative configuration.
Conclusions
A short and simple strategy has been developed for the synthesis of (-)-3β-hydroxycarvone (1). This modified methodology improved effectively the total yield [from (-)-carvone to (-)-3β-hydroxycarvone (1)] from 45% (ref. [6], cis and trans mixture) to 68% as a mixture and 58% as a single isomer. The success of the approach is achieved due to an easy and inexpensive experimental procedure, which consequently should be amenable to extrapolation to a much larger scale.
Experimental
General
Optical rotations were taken on a Perkin-Elmer polarimeter model 241. Column chromatography was performed on silica gel 60 (70-230 mesh ASTM Merck). Radial thin-layer chromatography was carried out on a Chromatotron 8924 (silica gel 60PF254 Merck). Infrared spectra were recorded either with a Bomen Michelson model 102 FTIR or with a Bomem Hartman & Braun MB-Series. 1H- and 13C-NMR spectra were recorded either on a Bruker ARX-200 (200 MHz), a Bruker ARX-400 (400 MHz) or a Varian FT-80A (80 MHz) spectrometer in CDCl3 with TMS as an internal standard. Microanalyses were performed on a Fisons EA 1108 CHNS-0 Analyzer, at the Department of Chemistry, UFSCar. Solvents were distilled prior to use: triethylamine was refluxed over CaH2, distilled and stored over KOH; acetonitrile and dichloromethane were refluxed over P2O5, distilled and stored over molecular sieves. Chlorotrimethylsilane was distilled over CaH2. NaI was dried for 24 h under vacuum at 140oC and stored under nitrogen.
(R)-3-(1-methylethenyl)-6-methyl-1,5-cyclohexadienyl trimethylsilyl ether (4).
To a solution of (R)-(-)-carvone (3) (5.0 g; 33.3 mmol) and triethylamine (6.3 mL; 45 mmol) under nitrogen was added chlorotrimethylsilane (5.7 mL; 45 mmol) and the resulting mixture was stirred vigorously for 10 min. Then a solution of NaI (6.75 g; 45 mmol) in dry acetonitrile (45 mL) was added dropwise and the reaction mixture was further stirred at room temperature for 1 h. The mixture was diluted with ice-water (250 mL) and hexane (100 mL). The aqueous layer was extracted with cold hexane (3x100 mL). The organic layer and hexane extracts were combined, washed with cold water (3x100 mL) and dried over MgSO4. The solvent was evaporated under reduced pressure, and the oily residue was purified by silica gel column chromatography (98:2 hexane-acetone as eluent) to afford 7.03 g (95% yield) of 4; IR νmax. (film): 3100, 2820, 1662, 1655, 1646, 1595, 1250, 1180 cm-1; 1H-NMR (80 MHz) δ: 0.23 [s, 9H, (CH3)3Si-]; 1.74 [br. s, 6H, =C-CH3 x 2]; 2.00-2.25 [m, 2H, CH2]; 2.80-3.18 [m, 1H, (=C)2CHCH2]; 4.60-4.75 [m, 3H, =CH2 and CH=C(OTMS)]; 5.40-5.55 [m, 1H, CH2-CH=C(CH3)]; 13C-NMR (20 MHz) δ: 0.4 [(CH3)3-Si]; 16.9 [CH3-C=]; 20.1 [CH3-C=]; 28.6 [CH2]; 41.8 [(=C)2CHCH2]; 105.3 [HC=C(OTMS)]; 109.8 [=CH2]; 122.7 [CH2-CH=C(CH3)]; 131.6 [(TMSO)C-C=CH]; 147.9 [H2C=CCH3]; 149.7 [=C-OTMS].
(5S,6S)-(-)-6-hydroxy-2-methyl-5-(1-methylethenyl)-2-cyclohexen-1-one (1).
To a solution of compound 4 (0.59 g; 2.7 mmol) in dichloromethane (15 mL) and aqueous NaHCO3 (10 mL of 0.5 mol/L solution) at 10 oC, was added portionwise over several minutes m-chloroperbenzoic acid 85% (0.63 g; 3.65 mmol) and the resulting mixture was stirred for 2 h at room temperature. Then water (10 mL) was added and the layers were separated. The aqueous layer was extracted with dichloromethane (3 x 6 mL). The organic layers were combined, washed with aqueous Na2SO3 (3 x 30 mL), aqueous NaHCO3 (3 x 20 mL), and dried over MgSO4. The solvent was evaporated under reduced pressure, and the oily residue was filtered through Florisil (dichloromethane as eluent) to afford 0.46 g (72% yield) of 5. The crude compound 5 (0.44 g; 1.86 mmol) was dissolved in dichloromethane (5 mL). To this solution was added 1.5M aqueous HCl (4 mL), and the mixture was stirred at room temperature for 3 h. Then the organic layer was separated and washed with 0.1M aqueous NaOH (5 x 5 mL), brine (3 x 5 mL) and dried over MgSO4. The solvent was removed in vacuo to afford 0.310 g (100% yield) of an 85:15 mixture of the two diastereomers, 1 and (5S,6R)-(-)-6-hydroxy-2-methyl-5-(1-methylethenyl)-2-cyclohexen-1-one (2). The diastereomers were separated by radial thin-layer chromatography (85:15 hexane-ethyl acetate as eluent) to provide pure samples of 1 (0.264 g, 85% yield) and 2 (0.05 g, 15% yield).
(1): [α]D24-25.8 (c 1.3, CHCl3); IR νmax. (film): 3473, 3075, 1674, 1441, 1037 cm-1; 1H-NMR (200 MHz) δ: 1.72 [br. s, 6H, =C-CH3 x 2]; 2.25-2.40 [m, 2H]; 2.50-2.70 [m, 1H, CH-COH]; 3.75 [br. s, 1H, OH]; 4.66 [d, J = 12.5 Hz, 1H, CHOH]; 4.81 [br. s, 2H, =CH2]; 6.60-6.70 [m, 1H, -CH=]; 13C-NMR (20 MHz) δ: 14.8 [CH3-C=]; 18.5 [CH3-C=]; 30.4 [CH2]; 50.8 [CHCHOH]; 74.1 [HCOH]; 112.9 [H2C=C(CH3)]; 132.6 [HC=C(CH3)C=O]; 143.9 [H2C=C(CH3)]; 145.1 [HC=C(CH3)C=O]; 200.1 [C=O]; 13C-NMR (J-Modi, 100 MHz) δ: 15.71; 19.12; 31.07; 48.35; 75.62; 114.08; 134.88; 143.89; 145.82; 200.19; Anal. Calcd. for C10H14O2: C, 72.27; H, 8.49. Found: C, 72.35; H, 8.48.
(2): [α]D24-67.7 (c 1.0, CHCl3); IR νmax. (film): 3469, 3087, 1675, 1451, 1031 cm-1; 1H-NMR (400 MHz) δ: 1.70 [dd, J = 0.77 Hz, J = 1.31 Hz, 3H, ipropylidene-CH3]; 1.84 [ddd, J = 1.47 Hz, J = 2.60 Hz, J = 6.8 Hz, 3H, CH3]; 2.50-2.58 [m, 1H]; 2.68-2.78 [m, 1H], 3.17-3.21 [m, 1H, CH-COH]; 3.58 [br. s, 1H, OH]; 4.43 [d, J = 5.9 Hz, 1H, CHOH]; 4.71-4.72 [m, 1H, =CH2]; 4.85-4.87 [m, 1H, =CH2]; 6.67 [ddd, J = 1.31 Hz, J = 2.70 Hz, J = 7.0 Hz, 1H, -CH=]; 13C-NMR (100 MHz) δ: 15.27 [CH3-C=]; 23.08 [CH3-C=]; 29.75 [CH2]; 46.99 [CHCHOH]; 74.69 [HCOH]; 113.82 [H2C=C(CH3)]; 133.82 [HC=C(CH3)C=O]; 143.06 [H2C=C(CH3)]; 144.08 [HC=C(CH3)C=O]; 199.78 [C=O]; 13C-NMR (DEPT-135, 100 MHz) δ: 15.27; 23.08; 29.75; 46.99; 74.69; 113.82; 144.08; Anal. Calcd. for C10H14O2: C, 72.27; H, 8.49. Found: C, 72.08; H, 8.60.
Acknowledgements
The authors wish to thank the following Brazilian agencies for financial support: FAPESP, CAPES and CNPq. The (R)-(-)-carvone (3) used as starting material was generously donated by Dragoco Perfumes e Aromas Ltda. R.B.S. expresses the author’s thanks to Prof. Edna F. Medeiros (DQ-UFES) and Prof. Mauricio G. Constantino (USP-Ribeirão Preto) for their comments and suggestions about the manuscript. The authors also express their appreciation to the referees for invaluable contributions, corrections and suggestions to the manuscript.
References
- Ho, T.-L. Enantioselective Synthesis: Natural Products from Chiral Terpenes; John Wiley & Sons: New York, 1995; pp. 123–183. [Google Scholar]
- Fraga, B. M. Nat. Prod. Rep. 1998, 15, 73–92.Fraga, B. M. Nat. Prod. Rep. 2000, 17, 483–504.Stadler, M.; Anke, H.; Sterner, O. Tetrahedron 1994, 50, 12649–12654.Bohlmann, F.; Zdero, C.; Scott, R. Phytochemistry 1987, 26, 1999–2006.Bohlmann, F.; Pathak, V. P.; Grenz, M.; Banerjee, S.; Wolfrum, C.; Barua, R. N.; Jakupovic, J. Phytochemistry 1987, 26, 1049–1052.
- Bohlmann, F.; Cardoso, J. M.; Jakupovic, J. Phytochemistry 1987, 26, 2321–2324.
- Hosokawa, T.; Nakahira, T.; Takano, M.; Murahashi, S-I. J. Mol. Catalysis 1992, 74, 489–498.
- Schulz, M.; Kluge, R.; Schuβler, M.; Hoffmann, G. Tetrahedron 1995, 51, 3175–3180.
- Hirai, Y.; Ito, K.; Nagaoka, H. Heterocycles 1998, 48, 235–238.
- Poirier, J. M.; Hennequin, L. Synth. Commun. 1985, 15, 217–244.
- Cazeau, P.; Duboudin, F.; Moulines, F.; Babot, O.; Dunogues, J. Tetrahedron 1987, 43, 2089–2100.Cazeau, P.; Duboudin, F.; Moulines, F.; Babot, O.; Dunogues, J. Tetrahedron 1987, 43, 2075–2087.
- Rubotton, G. M.; Gruber, J. M. J. Org. Chem. 1978, 43, 1599–1602.
- Rubotton, G. M.; Vazquez, M. A.; Pelegrina, D. R. Tetrahedron Lett. 1974, 4319–4322.
- Pennanen, S. I. Tetrahedron Lett. 1980, 21, 657–658.
- Hosokawa, T.; Inui, S.; Murahashi, S-I. Chem. Lett. 1983, 1081–1082.
- Sample Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
MDPI and ACS Style
Dos Santos, R.B.; Brocksom, T.J.; Zanotto, P.R.; Brocksom, U. An Improved Three Step Synthesis of (-)-3β-Hydroxycarvone from (-)-Carvone. Molecules 2002, 7, 129-134. https://doi.org/10.3390/70200129
AMA Style
Dos Santos RB, Brocksom TJ, Zanotto PR, Brocksom U. An Improved Three Step Synthesis of (-)-3β-Hydroxycarvone from (-)-Carvone. Molecules. 2002; 7(2):129-134. https://doi.org/10.3390/70200129
Chicago/Turabian StyleDos Santos, Reginaldo B., Timothy J. Brocksom, Paulo R. Zanotto, and Ursula Brocksom. 2002. "An Improved Three Step Synthesis of (-)-3β-Hydroxycarvone from (-)-Carvone" Molecules 7, no. 2: 129-134. https://doi.org/10.3390/70200129