Amino Acid Based Synthesis of Chiral Long Chain Diamines and Tetramines

Laboratory of Organic Chemistry, Department of Chemistry, University of Athens, Panepistimiopolis, Athens 15771, Greece
*
Author to whom correspondence should be addressed.
Molecules 2002, 7(10), 767-776; https://doi.org/10.3390/71000767
Published: 31 October 2002
{kind=link}
{kind=link}
{kind=link}
Abstract
:A method for the synthesis of long chain diamines and tetramines starting from natural α-amino acids is reported. Diamines and tetramines were prepared through the Wittig olefination reaction of N-protected amino aldehydes obtained from phenylalanine and lysine. A 1,2,17,18-tetramine was synthesized using (2S)-1-azido-2-[bis(tert-butoxycarbonyl)-amino]-5-oxopentane as key-intermediate compound.
Introduction
In recent years compounds containing amine functionalities have attracted much attention, because of their interesting biological properties. The naturally occurring polyamines putrescine, spermidine and spermine, as well as their synthetic analogues, are involved in various important biological functions [1]. Compounds incorporating the 1,2-diamine functionality are currently the topic of studies conducted in several fields, e.g. in chemotherapy and in stereoselective organic synthesis [2].
Natural α-amino acids may be used as starting materials for the synthesis of chiral amines through modification of the α-carboxy group. For example, enantiopure α-methyl amines have been prepared from various α-amino acids [3], while 1,2-diamines and triamines have been made from glutamic acid and lysine respectively [4]. C2-Symmetric and pseudo C2-symmetric based diols, epoxides and dideoxy derivatives of HIV protease inhibitors containing the 1,4-diamine functionality have been synthesized starting from L-phenylalanine and L-tyrosine [5]. C2-Symmetrical chiral 1,4-and 1,5-diamines with stereogenic centers adjacent to the nitrogen atom have been prepared by diastereoselective alkylations of bisoxazolidines derived from (R)-phenylglycinol [6]. The total synthesis of N-alkyl and acylpolyamine derivatives, based on the coupling of N-tritylamino acids with amines and subsequent reduction, has also been described [7]. We have recently shown that long chain 1,2-diamines exhibit interesting antiinflammatory [8] and cytotoxic activity [9,10,11]. Furthermore, we have studied the interactions of polyamines, lipidic 1,2-diamines and aminoglycosides with nucleic acids [12,13]. Within our research program focused on synthesis and study of polyamines, we present here a methodology for the synthesis of chiral long chain diamines and tetramines starting from natural α-amino acids.
Results and Discussion
Our strategy to synthesize chiral long chain diamines and tetramines was based on Wittig olefination of N-protected α-amino aldehydes with alkylidene triphenylphosphoranes and bis(triphenyl-phosphoranes). Such aldehydes may be prepared either by reduction of an amino acid carboxy derivative or by oxidation of 2-amino alcohols [14]. We decided to prepare α-amino aldehydes by oxidation of 2-amino alcohols, using NaOCl in the presence of a catalytic amount of a TEMPO derivative, a method which appears superior to the reductive methods in terms of preservation of the enantiomeric purity [15]. Boc-protected amino alcohols 1a,b, easily prepared from N-tert-butoxycarbonyl-L-phenylalanine and Nα,Nε-di(tert-butoxycarbonyl)-L-lysine by reduction of either their mixed anhydrides [16] or their acyl fluorides with NaBH4 [17], were chosen as enantiopure starting materials.
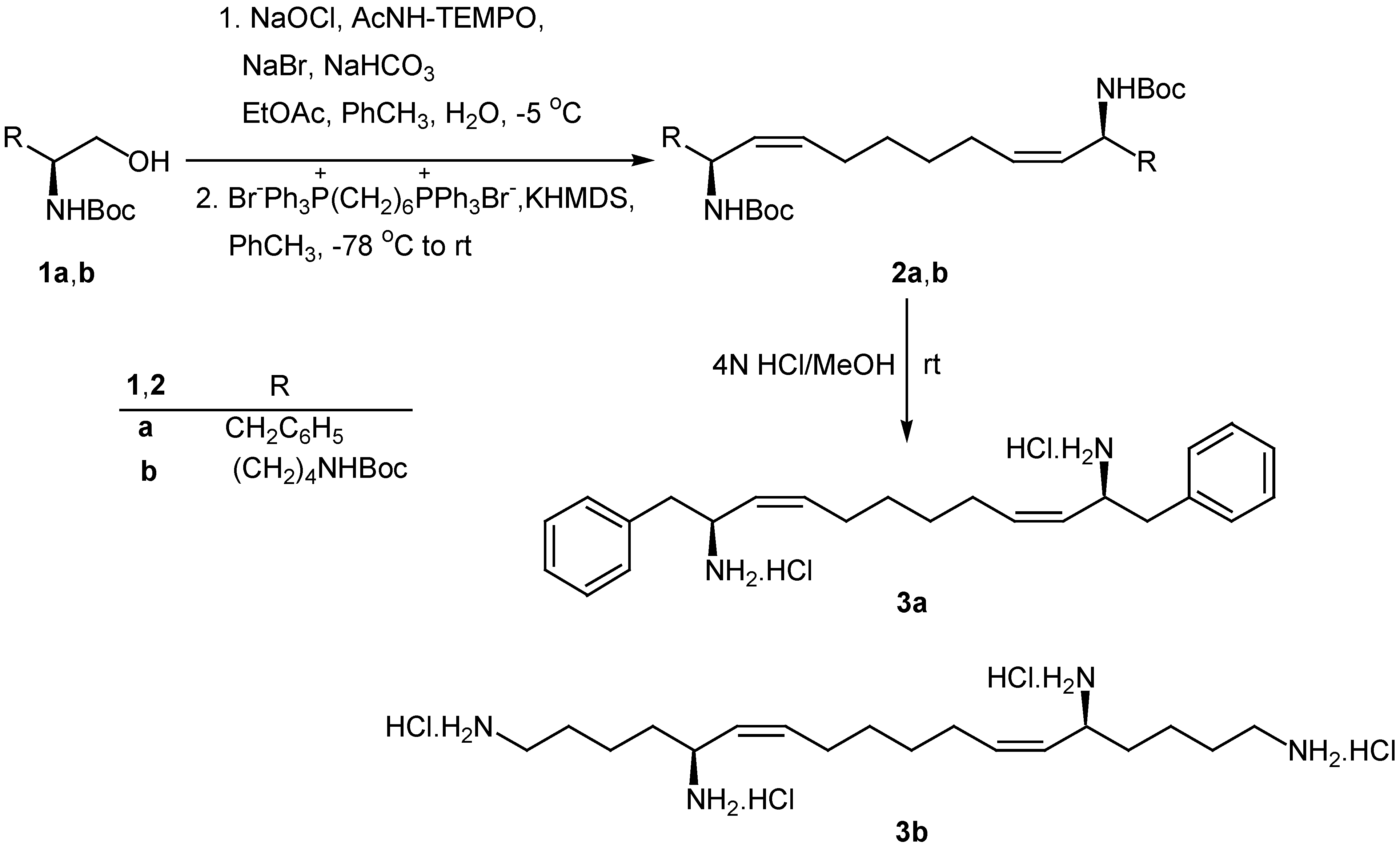
Scheme 1.
Synthesis of diamine 3a and tetramine 3b
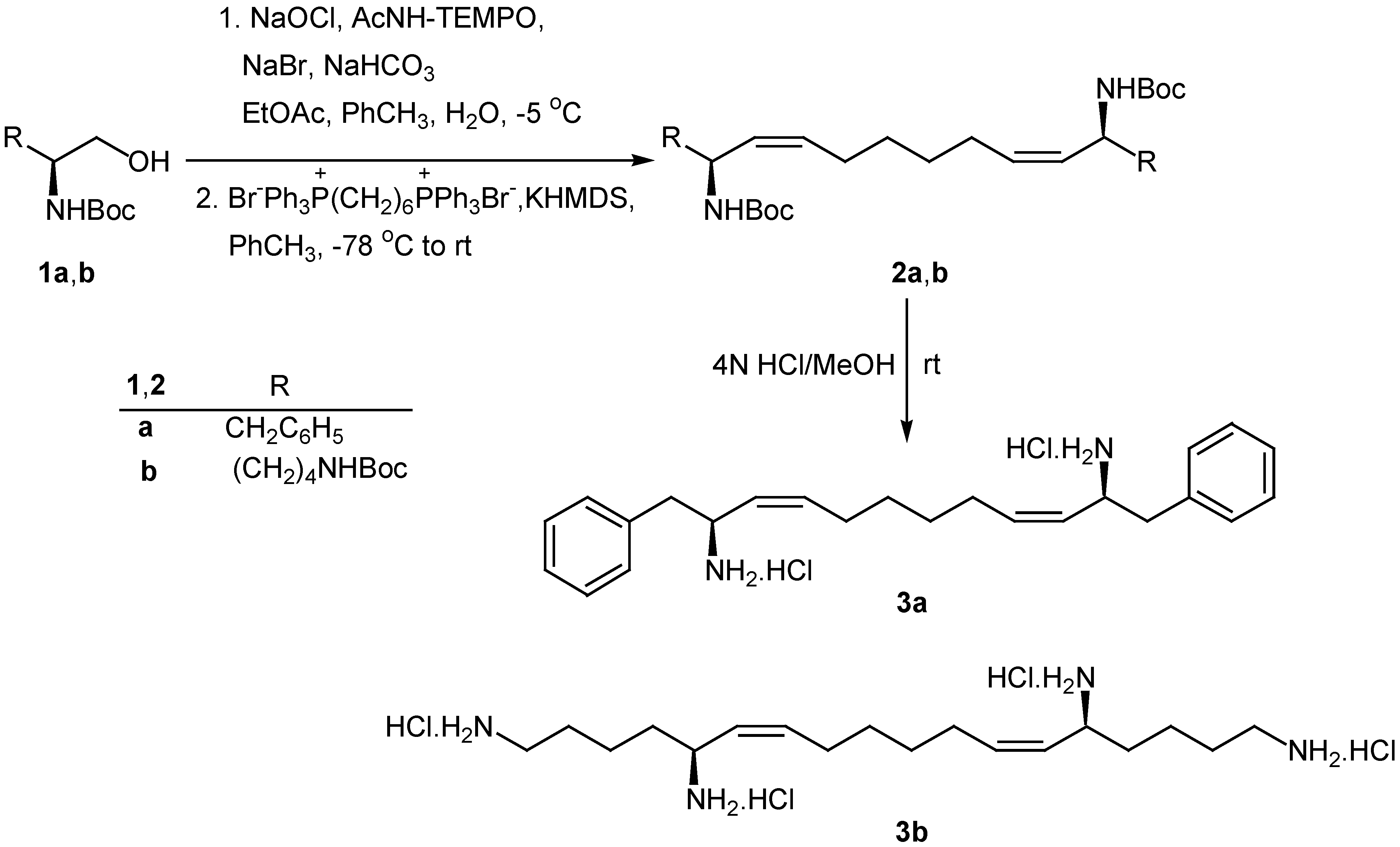
Oxidation of 1a,b using NaOCl in the presence of a catalytic amount 4-acetamido-2,2,6,6-tetramethyl-1-piperidinyloxy free radical (AcNH-TEMPO) [18,19] afforded the corresponding N-protected α-amino aldehydes, which were directly used for the Wittig reaction with Ph3P=CH(CH2)4CH=PPh3 without any additional purification (Scheme 1). The bis(phosphonium) ylide was generated from the corresponding bis(triphenylphosphonium) salt with KHMDS in toluene at 0 °C and the Wittig reaction was carried out at –78 °C to produce N-protected amines 2a,b. The geometry of the double bonds was Z (>95%), based on NMR data. It is known that the use of KHMDS for the generation of non-stabilized ylides under such experimental conditions leads to high Z-selectivity [20,21]. Free diamine 3a and tetramine 3b were obtained from 2a,b by treatment with HCl in MeOH.
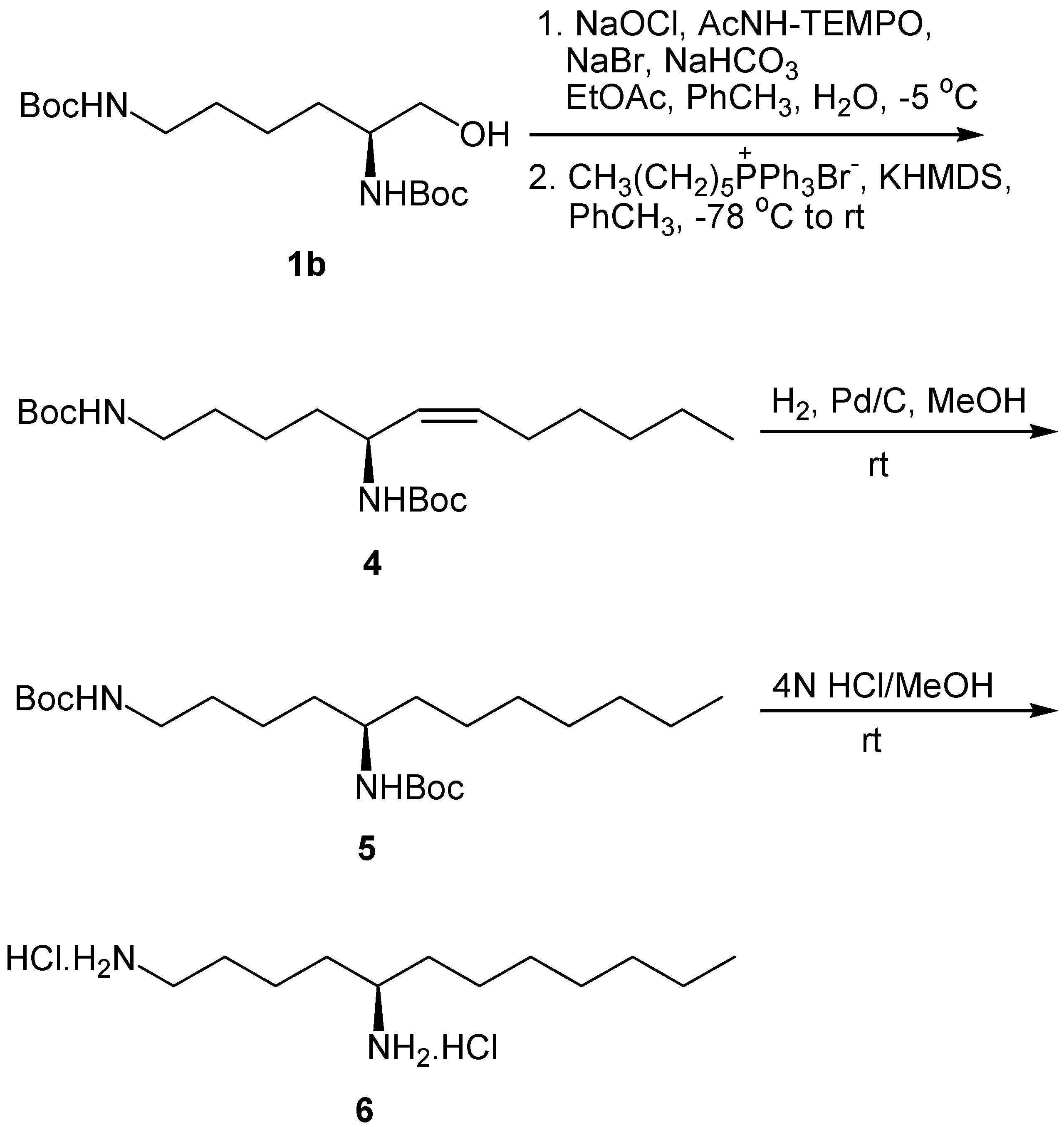
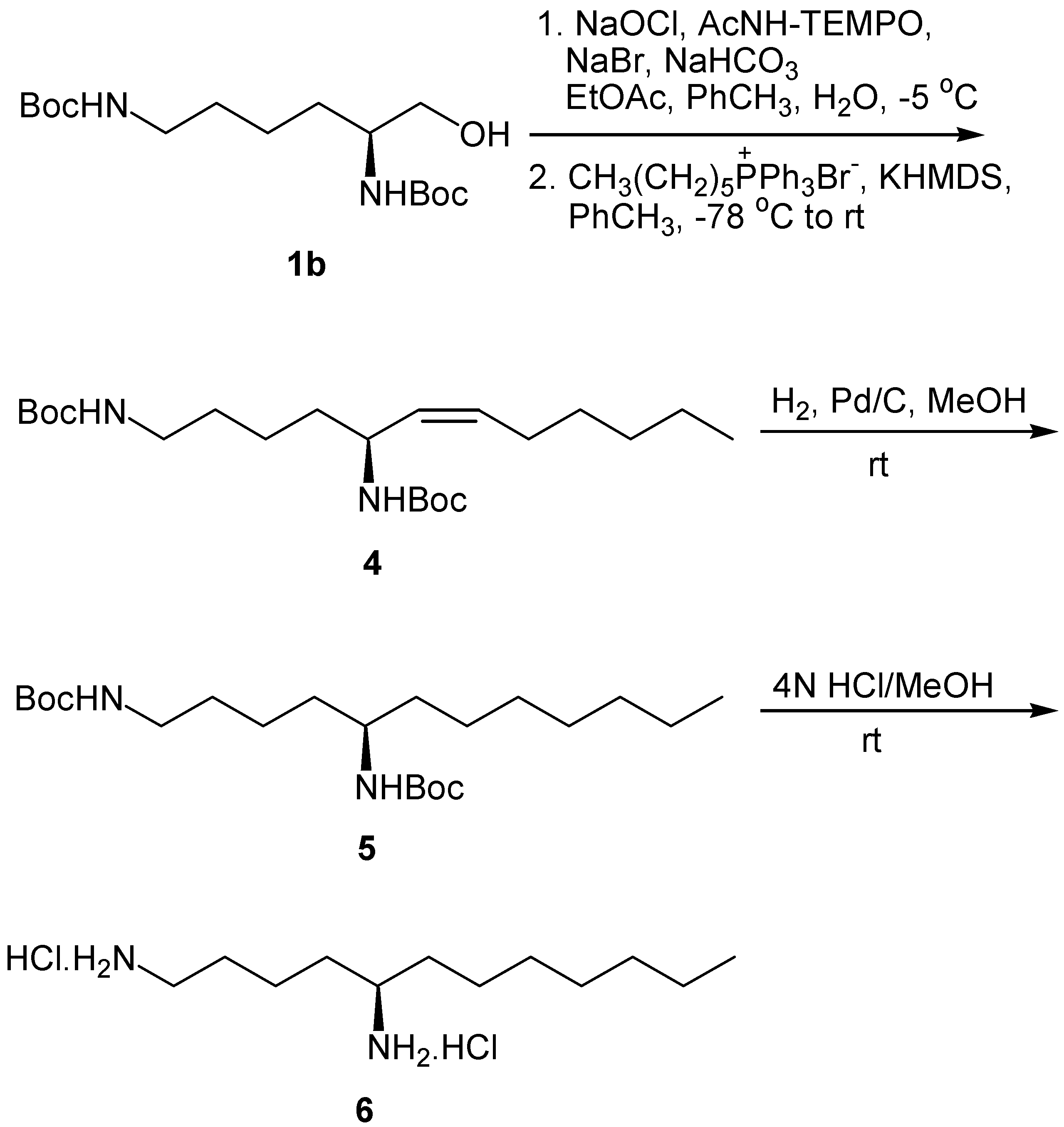
Wittig reaction of the N-protected α-amino aldehyde derived from lysine with C6 ylide under the conditions described for the synthesis of 2a,b, produced the Z-unsaturated N,N-diprotected chiral 1,5-diamine 4 (Scheme 2). Catalytic hydrogenation of 4 resulted to the saturated diamine 5, which was deprotected by treatment with HCl in MeOH to afford 1,5-diaminododecane (6).
Scheme 2.
Synthesis of 1,5-diaminododecane (6)
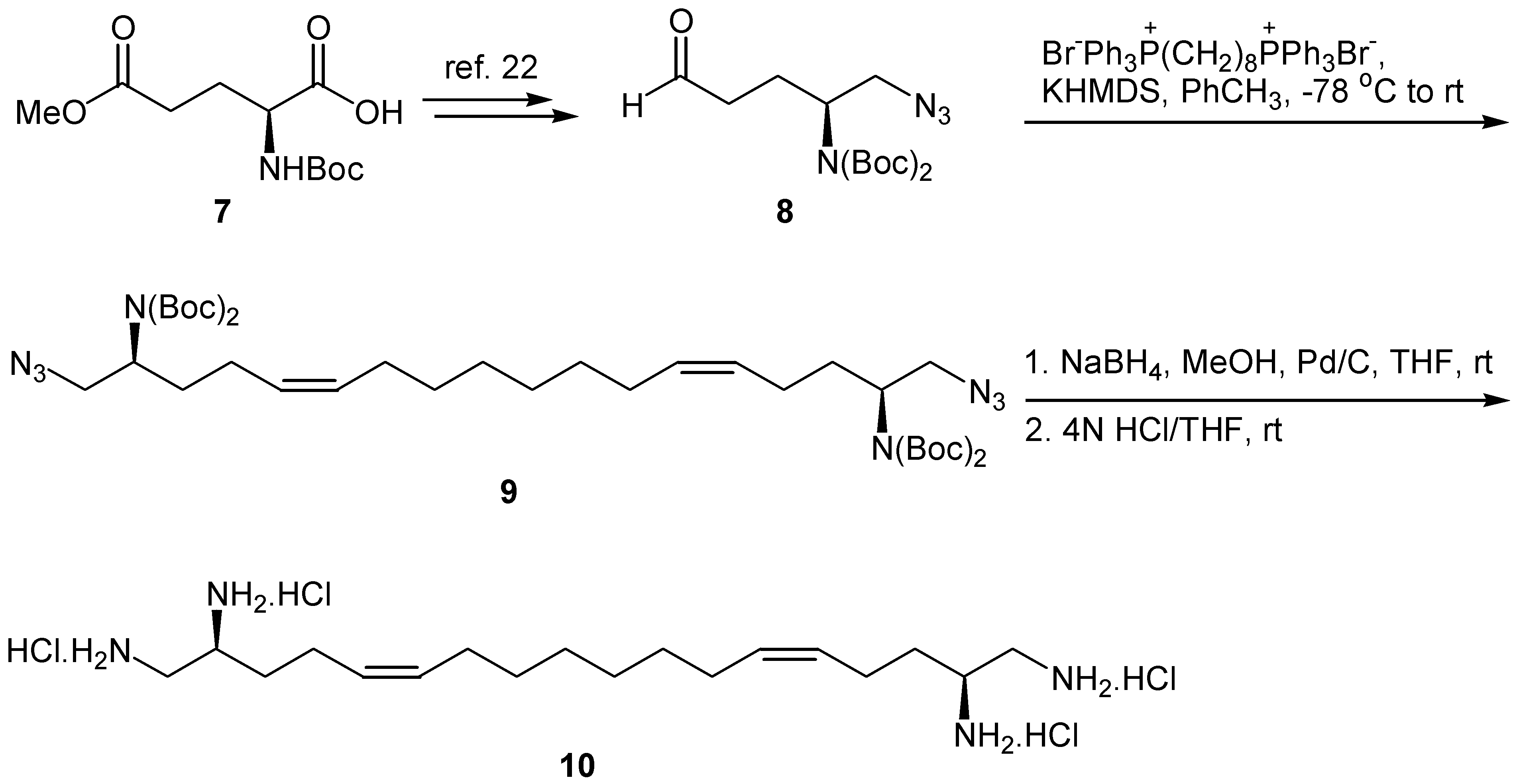
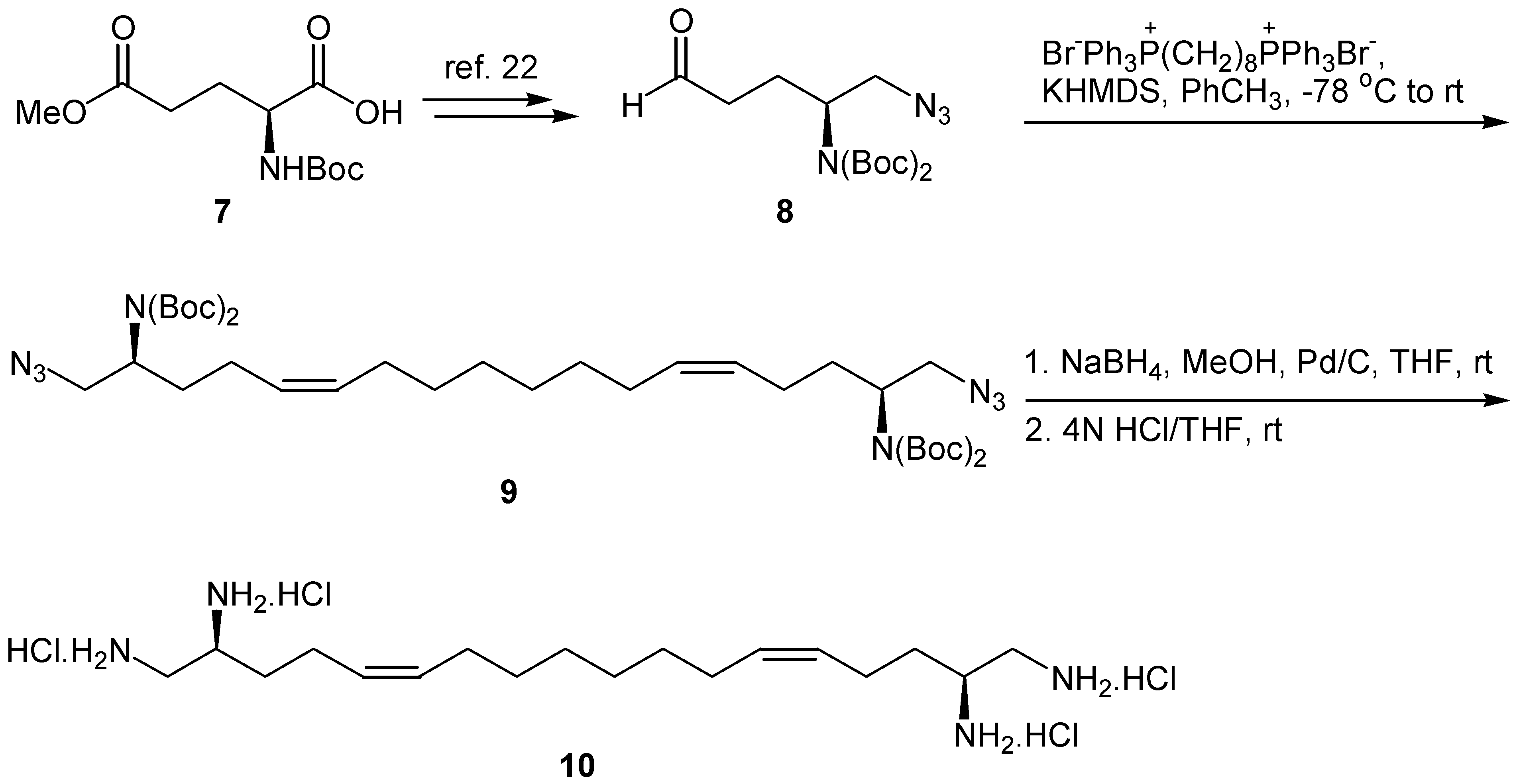
We have recently demonstrated that (2S)-1-azido-2-[bis(tert-butoxycarbonyl)amino]-5-oxopentane (8) is a useful synthon for the preparation of chiral 1,2-diamines [22]. This key-intermediate aldehyde is prepared from γ-methyl N-tert-butoxycarbonyl-L-glutamate in four steps and 40% overall yield. To prepare a chiral 1,2,17,18-tetramine, aldehyde 8 was submitted to Wittig olefination with Ph3P=CH(CH2)6CH=PPh3 under the conditions described for the synthesis of 2a,b, to produce chiral diamino diazide 9 (Scheme 3). The geometry of the double bonds was Z, based on NMR data. Free unsaturated tetramine 10 was obtained after selective reduction of the azide groups of 9 with NaBH4 in the presence of 10% Pd/C and subsequent treatment with HCl in THF. Under these conditions the double bonds remained unaffected.
Scheme 3.
Synthesis of tetramine 10
Conclusions
A route for the synthesis of chiral long chain diamines and tetramines starting from α-amino acids has been developed. The strengths of the method are in its: (i) simplicity and efficiency, (ii) flexibility with respect to the chain length, which depends on the chain length of the starting ylides used for the Wittig olefination reaction, (iii) applicability to the production of both enantiomers depending on the chirality of the starting α-amino acid.
Experimental
General
Melting points were determined on a Büchi 530 melting point apparatus and are uncorrected. Specific rotations were measured at 25 °C on a Perkin Elmer 141 polarimeter using a 10 cm cell. NMR spectra were recorded on a Varian Mercury (200 MHz) spectrometer. Where applicable, structural assignments were based on DEPT and COSY experiments. Analytical TLC plates (silica gel 60 F254) and silica gel 60 (70–230 or 230–400 mesh) for column chromatography were purchased from Merck. Visualization of spots was effected with UV light and/or staining with phosphomolybdic acid and/or ninhydrin, both in ethanol. Et2O was treated with calcium chloride and stored over Na. Toluene was distilled and stored over Na. All other solvents and chemicals were of reagent grade and used without further purification. The phosphonium salts were prepared [23] by refluxing PPh3 and the corresponding alkyl halide in MeCN and were used in the Wittig reactions without purification. The starting compounds 1a,b were prepared as described in the literature [16,17].
General Procedure for the Preparation of N-Protected Unsaturated Polyamines 2a,b and 4.
To a solution of N-protected 2-amino alcohol 1a,b (5.00 mmol) in a 1:1 mixture of toluene-EtOAc (30 mL) were added a solution of NaBr (540 mg, 5.25 mmol) in water (2.5 mL) and AcNH-TEMPO (11 mg, 0.050 mmol). The resulting biphasic system was cooled at –5 °C and an aqueous solution of 0.35 M NaOCl (15.7 mL, 5.50 mmol) containing NaHCO3 (1.26 g, 15 mmol) was added dropwise at ‑5°C over a period of 1 h under vigorous stirring. After stirring for an additional 15 min at 0 °C, EtOAc (30 mL) and water (10 mL) were added. The aqueous layer was separated and washed with EtOAc (10 ml). The combined organic layers were washed consecutively with 1% aqueous citric acid (30 mL) containing KI (0.18 g), 10% aqueous Na2S2O3 (30 mL), and brine and dried (Na2SO4). The solvents were evaporated under reduced pressure and the obtained crude aldehyde was immediately used for the Wittig reaction.
To a stirred suspension of the phosphonium salt Br-Ph3P+(CH2)6P+Ph3Br- (1.54 g, 2.00 mmol) or CH3(CH2)5P+Ph3Br- (2.14 g, 5.00 mmol) in dry toluene (27 mL) was added a 0.5 M solution of KHDMS (8.00 mL or 9.10 mL respectively) in toluene dropwise over a period of 5 min at 0 °C under N2. The bright red solution was stirred for another 15 min, cooled to –78 °C and a solution of the aldehyde in dry toluene (5 mL) was then added in one portion. The resulting light yellow mixture was stirred for 20 h at room temperature, then the reaction mixture was quenched with a saturated aqueous solution of NH4Cl (40 mL), and extracted with Et2O (3 × 10 mL). The combined organic layers were washed with brine and dried (Na2SO4). The solvents were removed and the residue was purified by column chromatography using a 3:7 mixture of EtOAc-petroleum ether as eluent.
(2S,11S,3Z,9Z)-2,11-Di[(tert-butoxycarbonyl)amino]-1,12-diphenyldodeca-3,9-diene (2a): yield 626 mg (57%); yellow solid; mp 91–93 oC; ![Molecules 07 00767 i001]() –4.7 (c 0.9, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.88–1.25 [m, 4H, innermost (CH2)2], 1.44 [br s, 18H, 2 × (CH3)3], 1.68–2.03 (m, 4H, 2 × CH2CH=CH), 2.69 (dd, 2H, J = 10.2, 12.8 Hz, 2 × CHHC6H5), 2.93 (dd, 2H, J = 4.4, 12.8 Hz, 2 × CHHC6H5), 4.34 (br, 2H, 2 × NH), 4.54 (m, 2H, 2 × CHN), 5.19 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CH=CHCH2), 5.36 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CH=CHCH2), 7.12–7.33 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CDCl3) δ 27.6, 28.4, 28.8, 42.1, 49.3, 79.2, 126.3, 128.1, 128.9, 129.7, 132.6, 137.7, 155.0; Anal. Calcd for C34H48N2O4 (548.77): C, 74.42; H, 8.82; N, 5.10. Found: C, 74.71; H, 8.91; N, 4.85.
–4.7 (c 0.9, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.88–1.25 [m, 4H, innermost (CH2)2], 1.44 [br s, 18H, 2 × (CH3)3], 1.68–2.03 (m, 4H, 2 × CH2CH=CH), 2.69 (dd, 2H, J = 10.2, 12.8 Hz, 2 × CHHC6H5), 2.93 (dd, 2H, J = 4.4, 12.8 Hz, 2 × CHHC6H5), 4.34 (br, 2H, 2 × NH), 4.54 (m, 2H, 2 × CHN), 5.19 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CH=CHCH2), 5.36 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CH=CHCH2), 7.12–7.33 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CDCl3) δ 27.6, 28.4, 28.8, 42.1, 49.3, 79.2, 126.3, 128.1, 128.9, 129.7, 132.6, 137.7, 155.0; Anal. Calcd for C34H48N2O4 (548.77): C, 74.42; H, 8.82; N, 5.10. Found: C, 74.71; H, 8.91; N, 4.85.
 –4.7 (c 0.9, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.88–1.25 [m, 4H, innermost (CH2)2], 1.44 [br s, 18H, 2 × (CH3)3], 1.68–2.03 (m, 4H, 2 × CH2CH=CH), 2.69 (dd, 2H, J = 10.2, 12.8 Hz, 2 × CHHC6H5), 2.93 (dd, 2H, J = 4.4, 12.8 Hz, 2 × CHHC6H5), 4.34 (br, 2H, 2 × NH), 4.54 (m, 2H, 2 × CHN), 5.19 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CH=CHCH2), 5.36 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CH=CHCH2), 7.12–7.33 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CDCl3) δ 27.6, 28.4, 28.8, 42.1, 49.3, 79.2, 126.3, 128.1, 128.9, 129.7, 132.6, 137.7, 155.0; Anal. Calcd for C34H48N2O4 (548.77): C, 74.42; H, 8.82; N, 5.10. Found: C, 74.71; H, 8.91; N, 4.85.
–4.7 (c 0.9, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.88–1.25 [m, 4H, innermost (CH2)2], 1.44 [br s, 18H, 2 × (CH3)3], 1.68–2.03 (m, 4H, 2 × CH2CH=CH), 2.69 (dd, 2H, J = 10.2, 12.8 Hz, 2 × CHHC6H5), 2.93 (dd, 2H, J = 4.4, 12.8 Hz, 2 × CHHC6H5), 4.34 (br, 2H, 2 × NH), 4.54 (m, 2H, 2 × CHN), 5.19 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CH=CHCH2), 5.36 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CH=CHCH2), 7.12–7.33 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CDCl3) δ 27.6, 28.4, 28.8, 42.1, 49.3, 79.2, 126.3, 128.1, 128.9, 129.7, 132.6, 137.7, 155.0; Anal. Calcd for C34H48N2O4 (548.77): C, 74.42; H, 8.82; N, 5.10. Found: C, 74.71; H, 8.91; N, 4.85.(5S,14S,6Z,12Z)-1,5,14,18-Tetra[(tert-butoxycarbonyl)amino]-octadeca-6,12-diene (2b): yield 611 mg (43%); white solid; mp 87–88 oC; ![Molecules 07 00767 i001]() +0.5 (c 1.0, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 1.05–1.68 [m, 52H, 4 × C(CH3)3, innermost (CH2)2, 2 × (CH2)3CHN], 1.89–2.17 (m, 4H, 2 × CH2CH=CH), 3.06 (dt, 4H, J = 6.4, 6.6 Hz, 2 × CH2N), 4.27 (m, 2H, 2 × CHN), 4.52 (br, 2H, 2 × CHNH), 4.67 (br, 2H, 2 × CH2NH), 5.13 (dd, 2H, J = 10.2, 10.6 Hz, 2 × CH=CHCH2), 5.40 (dt, 2H, J = 7.4, 10.2 Hz, 2 × CH=CHCH2); 13C-NMR (50 MHz, CDCl3) δ 22.8, 27.6, 28.3, 29.0, 29.7, 35.8, 40.3, 47.6, 79.0, 130.4, 132.2, 155.2, 156.0. Anal. Calcd for C38H70N4O8 (711.00): C, 64.19; H, 9.92; N, 7.88. Found: C, 64.38; H, 9.81; N, 7.98.
+0.5 (c 1.0, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 1.05–1.68 [m, 52H, 4 × C(CH3)3, innermost (CH2)2, 2 × (CH2)3CHN], 1.89–2.17 (m, 4H, 2 × CH2CH=CH), 3.06 (dt, 4H, J = 6.4, 6.6 Hz, 2 × CH2N), 4.27 (m, 2H, 2 × CHN), 4.52 (br, 2H, 2 × CHNH), 4.67 (br, 2H, 2 × CH2NH), 5.13 (dd, 2H, J = 10.2, 10.6 Hz, 2 × CH=CHCH2), 5.40 (dt, 2H, J = 7.4, 10.2 Hz, 2 × CH=CHCH2); 13C-NMR (50 MHz, CDCl3) δ 22.8, 27.6, 28.3, 29.0, 29.7, 35.8, 40.3, 47.6, 79.0, 130.4, 132.2, 155.2, 156.0. Anal. Calcd for C38H70N4O8 (711.00): C, 64.19; H, 9.92; N, 7.88. Found: C, 64.38; H, 9.81; N, 7.98.
+0.5 (c 1.0, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 1.05–1.68 [m, 52H, 4 × C(CH3)3, innermost (CH2)2, 2 × (CH2)3CHN], 1.89–2.17 (m, 4H, 2 × CH2CH=CH), 3.06 (dt, 4H, J = 6.4, 6.6 Hz, 2 × CH2N), 4.27 (m, 2H, 2 × CHN), 4.52 (br, 2H, 2 × CHNH), 4.67 (br, 2H, 2 × CH2NH), 5.13 (dd, 2H, J = 10.2, 10.6 Hz, 2 × CH=CHCH2), 5.40 (dt, 2H, J = 7.4, 10.2 Hz, 2 × CH=CHCH2); 13C-NMR (50 MHz, CDCl3) δ 22.8, 27.6, 28.3, 29.0, 29.7, 35.8, 40.3, 47.6, 79.0, 130.4, 132.2, 155.2, 156.0. Anal. Calcd for C38H70N4O8 (711.00): C, 64.19; H, 9.92; N, 7.88. Found: C, 64.38; H, 9.81; N, 7.98.(5S,6Z)-1,5-Di[(tert-butoxycarbonyl)amino]-dodec-6-ene (4): yield 1.14 g (57%); yellow oil; ![Molecules 07 00767 i001]() +4.3 (c 1.1, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.87 (t, 3H, J = 6.6 Hz, CH3), 1.18–1.62 [m, 30H, 2 × (CH3)3, (CH2)3CH3, (CH2)3CHN], 2.02–2.18 (m, 2H, CH2CH=CH), 3.08 (dt, 2H, J = 6.4, 6.6 Hz, CH2N), 4.22–4.47 (m, 2H, NH, CH), 4.55 (br, 1H, NH), 5.14 (dd, 1H, J = 10.0, 10.8 Hz, CHCH=CH), 5.44 (dt, J = 7.7, 10.0 Hz, CHCH=CH); 13C-NMR (50 MHz, CDCl3) δ 14.0, 22.3, 22.8, 27.8, 28.3, 28.4, 29.2, 29.7, 31.1, 35.9, 40.3, 47.6, 79.0, 130.2, 132.6, 155.2, 156.0. Anal. Calcd for C22H42N2O4 (398.59): C, 66.29; H, 10.62; N, 7.03. Found: C, 66.57; H, 10.73; N, 6.80.
+4.3 (c 1.1, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.87 (t, 3H, J = 6.6 Hz, CH3), 1.18–1.62 [m, 30H, 2 × (CH3)3, (CH2)3CH3, (CH2)3CHN], 2.02–2.18 (m, 2H, CH2CH=CH), 3.08 (dt, 2H, J = 6.4, 6.6 Hz, CH2N), 4.22–4.47 (m, 2H, NH, CH), 4.55 (br, 1H, NH), 5.14 (dd, 1H, J = 10.0, 10.8 Hz, CHCH=CH), 5.44 (dt, J = 7.7, 10.0 Hz, CHCH=CH); 13C-NMR (50 MHz, CDCl3) δ 14.0, 22.3, 22.8, 27.8, 28.3, 28.4, 29.2, 29.7, 31.1, 35.9, 40.3, 47.6, 79.0, 130.2, 132.6, 155.2, 156.0. Anal. Calcd for C22H42N2O4 (398.59): C, 66.29; H, 10.62; N, 7.03. Found: C, 66.57; H, 10.73; N, 6.80.
+4.3 (c 1.1, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.87 (t, 3H, J = 6.6 Hz, CH3), 1.18–1.62 [m, 30H, 2 × (CH3)3, (CH2)3CH3, (CH2)3CHN], 2.02–2.18 (m, 2H, CH2CH=CH), 3.08 (dt, 2H, J = 6.4, 6.6 Hz, CH2N), 4.22–4.47 (m, 2H, NH, CH), 4.55 (br, 1H, NH), 5.14 (dd, 1H, J = 10.0, 10.8 Hz, CHCH=CH), 5.44 (dt, J = 7.7, 10.0 Hz, CHCH=CH); 13C-NMR (50 MHz, CDCl3) δ 14.0, 22.3, 22.8, 27.8, 28.3, 28.4, 29.2, 29.7, 31.1, 35.9, 40.3, 47.6, 79.0, 130.2, 132.6, 155.2, 156.0. Anal. Calcd for C22H42N2O4 (398.59): C, 66.29; H, 10.62; N, 7.03. Found: C, 66.57; H, 10.73; N, 6.80.General Procedure for the Preparation of Free Unsaturated Polyamines 3a,b and 6.
A solution of N-protected polyamine (1.00 mmol) in 4 N HCl in MeOH (25 mL) was stirred for 30 min at room temperature. After evaporation, dry Et2O was added and the product was filtered and recrystallized from MeOH/Et2O.
(2S,11S,3Z,9Z)-1,12-Diphenyl-dodeca-3,9-diene-2,11-diamine dihydrochloride (3a): yield 329 mg (78%); white solid; ![Molecules 07 00767 i001]() +1.9 (c 0.6, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.76–1.08 [m, 4H, innermost (CH2)2], 1.50–1.92 (m, 4H, 2 × CH2CH=CH), 2.76 (dd, 2H, J = 10.2, 12.9 Hz, 2 × CHHPh), 3.14 (dd, 2H, J = 4.4, 12.9 Hz, 2 × CHHPh), 4.20 (m, 2H, 2 × CHN), 5.32 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CHCH=CH), 5.56 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CHCH=CH), 7.13–7.47 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CD3OD) δ 28.4, 29.4, 40.8, 51.4, 125.2, 128.4, 130.0, 131.0, 137.1, 138.6; MS (FAB) m/z (%): 349 (M++1, 100), 332 (28), 240 (12). Anal. Calcd for C24Cl2H34N2 (421.46): C, 68.40; H, 8.13; N, 6.65. Found: C, 68.19; H, 8.25; N, 6.43.
+1.9 (c 0.6, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.76–1.08 [m, 4H, innermost (CH2)2], 1.50–1.92 (m, 4H, 2 × CH2CH=CH), 2.76 (dd, 2H, J = 10.2, 12.9 Hz, 2 × CHHPh), 3.14 (dd, 2H, J = 4.4, 12.9 Hz, 2 × CHHPh), 4.20 (m, 2H, 2 × CHN), 5.32 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CHCH=CH), 5.56 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CHCH=CH), 7.13–7.47 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CD3OD) δ 28.4, 29.4, 40.8, 51.4, 125.2, 128.4, 130.0, 131.0, 137.1, 138.6; MS (FAB) m/z (%): 349 (M++1, 100), 332 (28), 240 (12). Anal. Calcd for C24Cl2H34N2 (421.46): C, 68.40; H, 8.13; N, 6.65. Found: C, 68.19; H, 8.25; N, 6.43.
+1.9 (c 0.6, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.76–1.08 [m, 4H, innermost (CH2)2], 1.50–1.92 (m, 4H, 2 × CH2CH=CH), 2.76 (dd, 2H, J = 10.2, 12.9 Hz, 2 × CHHPh), 3.14 (dd, 2H, J = 4.4, 12.9 Hz, 2 × CHHPh), 4.20 (m, 2H, 2 × CHN), 5.32 (dd, 2H, J = 10.0, 10.8 Hz, 2 × CHCH=CH), 5.56 (dt, 2H, J = 7.7, 10.0 Hz, 2 × CHCH=CH), 7.13–7.47 (m, 10H, 2 × C6H5); 13C‑NMR (50 MHz, CD3OD) δ 28.4, 29.4, 40.8, 51.4, 125.2, 128.4, 130.0, 131.0, 137.1, 138.6; MS (FAB) m/z (%): 349 (M++1, 100), 332 (28), 240 (12). Anal. Calcd for C24Cl2H34N2 (421.46): C, 68.40; H, 8.13; N, 6.65. Found: C, 68.19; H, 8.25; N, 6.43.(5S,14S,6Z,12Z)-Οctadeca-6,12-diene-1,5,14,18-tetramine tetrahydrochloride (3b): yield 374 mg (82%); white solid; ![Molecules 07 00767 i001]() +2.4 (c 2.5, EtOH); 1H-NMR (200 MHz, CD3OD) δ 1.47 [br, 8H, innermost (CH2)2, 2 × CH2CH2CHN], 1.55–1.97 (m, 8H, 2 × CH2CHN, 2 × CH2CH2N), 2.19 (br, 4Η, 2 × CH2CH=CH), 2.94 (t, 4H, J = 7.2 Hz, 2 × CH2N), 4.10 (br, 2H, 2 × CHN), 5.35 (dd, 2H, J = 10.2, 10.6 Hz, 2 × CHCH=CH), 5.82 (dt, 2H, J = 7.4, 10.2 Hz, 2 × CHCH=CH); 13C-NMR (50 MHz, CD3OD) δ 22.4, 27.0, 27.6, 29.0, 32.9, 39.3, 53.7, 124.9, 137.5; MS (FAB) m/z (%): 311 (M++1, 100), 277 (18). Anal. Calcd for C18Cl4H42N4.H2O (474.39): C, 45.57; H, 9.35; N, 11.81. Found: C, 45.31; H, 9.54; N, 11.70.
+2.4 (c 2.5, EtOH); 1H-NMR (200 MHz, CD3OD) δ 1.47 [br, 8H, innermost (CH2)2, 2 × CH2CH2CHN], 1.55–1.97 (m, 8H, 2 × CH2CHN, 2 × CH2CH2N), 2.19 (br, 4Η, 2 × CH2CH=CH), 2.94 (t, 4H, J = 7.2 Hz, 2 × CH2N), 4.10 (br, 2H, 2 × CHN), 5.35 (dd, 2H, J = 10.2, 10.6 Hz, 2 × CHCH=CH), 5.82 (dt, 2H, J = 7.4, 10.2 Hz, 2 × CHCH=CH); 13C-NMR (50 MHz, CD3OD) δ 22.4, 27.0, 27.6, 29.0, 32.9, 39.3, 53.7, 124.9, 137.5; MS (FAB) m/z (%): 311 (M++1, 100), 277 (18). Anal. Calcd for C18Cl4H42N4.H2O (474.39): C, 45.57; H, 9.35; N, 11.81. Found: C, 45.31; H, 9.54; N, 11.70.
+2.4 (c 2.5, EtOH); 1H-NMR (200 MHz, CD3OD) δ 1.47 [br, 8H, innermost (CH2)2, 2 × CH2CH2CHN], 1.55–1.97 (m, 8H, 2 × CH2CHN, 2 × CH2CH2N), 2.19 (br, 4Η, 2 × CH2CH=CH), 2.94 (t, 4H, J = 7.2 Hz, 2 × CH2N), 4.10 (br, 2H, 2 × CHN), 5.35 (dd, 2H, J = 10.2, 10.6 Hz, 2 × CHCH=CH), 5.82 (dt, 2H, J = 7.4, 10.2 Hz, 2 × CHCH=CH); 13C-NMR (50 MHz, CD3OD) δ 22.4, 27.0, 27.6, 29.0, 32.9, 39.3, 53.7, 124.9, 137.5; MS (FAB) m/z (%): 311 (M++1, 100), 277 (18). Anal. Calcd for C18Cl4H42N4.H2O (474.39): C, 45.57; H, 9.35; N, 11.81. Found: C, 45.31; H, 9.54; N, 11.70.(5R)-Dodecane-1,5-diamine dihydrochloride (6) [24]: yield 183 mg (67%); pale yellow solid; ![Molecules 07 00767 i002]() ‑1.3 (c 0.8, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.90 (t, 3H, J = 6.4 Hz, CH3), 1.19–1.82 [m, 18H, (CH2)6, (CH2)3CH], 2.96 (t, 2H, J = 7.6 Hz, CH2N), 3.18 (m, 1H, CH); 13C-NMR (50 MHz, CD3OD) δ 14.4, 23.2, 23.7, 26.1, 28.3, 30.2, 30.5, 32.9, 33.2, 33.6, 40.4, 52.9. Anal. Calcd for C12Cl2H30N2 (273.29): C, 52.74; H, 11.07; N; 10.25. Found: C, 52.51; H, 11.33; N, 10.09.
‑1.3 (c 0.8, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.90 (t, 3H, J = 6.4 Hz, CH3), 1.19–1.82 [m, 18H, (CH2)6, (CH2)3CH], 2.96 (t, 2H, J = 7.6 Hz, CH2N), 3.18 (m, 1H, CH); 13C-NMR (50 MHz, CD3OD) δ 14.4, 23.2, 23.7, 26.1, 28.3, 30.2, 30.5, 32.9, 33.2, 33.6, 40.4, 52.9. Anal. Calcd for C12Cl2H30N2 (273.29): C, 52.74; H, 11.07; N; 10.25. Found: C, 52.51; H, 11.33; N, 10.09.
 ‑1.3 (c 0.8, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.90 (t, 3H, J = 6.4 Hz, CH3), 1.19–1.82 [m, 18H, (CH2)6, (CH2)3CH], 2.96 (t, 2H, J = 7.6 Hz, CH2N), 3.18 (m, 1H, CH); 13C-NMR (50 MHz, CD3OD) δ 14.4, 23.2, 23.7, 26.1, 28.3, 30.2, 30.5, 32.9, 33.2, 33.6, 40.4, 52.9. Anal. Calcd for C12Cl2H30N2 (273.29): C, 52.74; H, 11.07; N; 10.25. Found: C, 52.51; H, 11.33; N, 10.09.
‑1.3 (c 0.8, EtOH); 1H-NMR (200 MHz, CD3OD) δ 0.90 (t, 3H, J = 6.4 Hz, CH3), 1.19–1.82 [m, 18H, (CH2)6, (CH2)3CH], 2.96 (t, 2H, J = 7.6 Hz, CH2N), 3.18 (m, 1H, CH); 13C-NMR (50 MHz, CD3OD) δ 14.4, 23.2, 23.7, 26.1, 28.3, 30.2, 30.5, 32.9, 33.2, 33.6, 40.4, 52.9. Anal. Calcd for C12Cl2H30N2 (273.29): C, 52.74; H, 11.07; N; 10.25. Found: C, 52.51; H, 11.33; N, 10.09.(5R)-1,5-Di[(tert-butoxycarbonyl)amino]-dodecane (5)
To a solution of 4 (399 mg, 1.00 mmol) in MeOH (10 mL) was added 10% Pd/C (40 mg). The reaction mixture was stirred under H2 for 18 h at room temperature. After filtration through a pad of Celite®, the solvent was evaporated under reduced pressure. Yield 365 mg (91%); white solid; mp 63–64 oC, ![Molecules 07 00767 i001]() –2.3 (c 1.0, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.85 (t, 3H, J = 6.4 Hz, CH3), 1.15–1.62 [m, 18H, 2 × (CH3)3], 3.08 (dt, 2H, J = 6.4, 6.6 Hz, CH2N), 3.37–3.61 (m, 1H, CH2NH), 4.28 (d, 1H, J = 9.2 Hz, CHNH), 4.64 (br, 1H, CH); 13C-NMR (50 MHz, CDCl3) δ 14.0, 22.6, 23.0, 25.8, 28.3, 28.4, 29.2, 29.5, 29.7, 31.7, 35.2, 35.6, 40.3, 50.3, 78.8, 155.8, 156.0; Anal. Calcd for C22H44N2O4 (400.60): C, 65.96; H, 11.07; N; 6.99. Found: C, 65.89; H, 11.01; N, 7.03.
–2.3 (c 1.0, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.85 (t, 3H, J = 6.4 Hz, CH3), 1.15–1.62 [m, 18H, 2 × (CH3)3], 3.08 (dt, 2H, J = 6.4, 6.6 Hz, CH2N), 3.37–3.61 (m, 1H, CH2NH), 4.28 (d, 1H, J = 9.2 Hz, CHNH), 4.64 (br, 1H, CH); 13C-NMR (50 MHz, CDCl3) δ 14.0, 22.6, 23.0, 25.8, 28.3, 28.4, 29.2, 29.5, 29.7, 31.7, 35.2, 35.6, 40.3, 50.3, 78.8, 155.8, 156.0; Anal. Calcd for C22H44N2O4 (400.60): C, 65.96; H, 11.07; N; 6.99. Found: C, 65.89; H, 11.01; N, 7.03.
–2.3 (c 1.0, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 0.85 (t, 3H, J = 6.4 Hz, CH3), 1.15–1.62 [m, 18H, 2 × (CH3)3], 3.08 (dt, 2H, J = 6.4, 6.6 Hz, CH2N), 3.37–3.61 (m, 1H, CH2NH), 4.28 (d, 1H, J = 9.2 Hz, CHNH), 4.64 (br, 1H, CH); 13C-NMR (50 MHz, CDCl3) δ 14.0, 22.6, 23.0, 25.8, 28.3, 28.4, 29.2, 29.5, 29.7, 31.7, 35.2, 35.6, 40.3, 50.3, 78.8, 155.8, 156.0; Anal. Calcd for C22H44N2O4 (400.60): C, 65.96; H, 11.07; N; 6.99. Found: C, 65.89; H, 11.01; N, 7.03.(2S,17S,5Z,13Z))-1,18-Diazido-2,17-di[bis(tert-butoxycarbonyl)amino]-octadeca-5,13-diene (9)
To a stirred suspension of phosphonium salt Br-Ph3P+(CH2)8PPh3+Br- (4.00 mmol, 3.19 g) in dry toluene (54 mL) was added a 0.5 M solution of KHMDS (16.0 mL) in toluene dropwise over a period of 5 min at 0 °C under N2. The bright red solution was stirred for another 15 min and cooled to –78 °C, and a solution of the aldehyde 8 (8.00 mmol, 2.76 g) in dry toluene (8 mL) was instantly added. The light yellow mixture was stirred at room temperature for 20 h. Then, the reaction mixture was quenched with a saturated aqueous solution of NH4Cl (70 mL) and extracted with Et2O (3 × 16 mL). The combined organic phases were washed with brine and dried (Na2SO4). The solvent was removed, and the residue was purified by column chromatography using a 1:9 mixture of EtOAc-petroleum ether as eluent. Yield 1.31 g (43%); colorless oil; ![Molecules 07 00767 i001]() = –3.5 (c 1.2, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 1.28 [br s, 8H, (CH2)4], 1.43–1.65 [m, 38H, 4 × C(CH3)3, 2 × CHHCHN], 1.74–1.90 (m, 2H, 2 × CHHCHN), 1.91–2.13 (m, 8H, 2 × CH2CH=CHCH2), 3.29 (dd, 2H, J = 12.2, 5.6 Hz, 2 × CHHN3), 3.76 (dd, 2H, J = 12.2, 9.4, 2 × CHHN3), 4.35 (m, 2H, 2 × CHN), 5.23–5.46 (m, 4H, 2 × CH=CH); 13C‑NMR (50 MHz, CDCl3) δ 24.0, 27.2, 28.0, 29.3, 29.7, 30.2, 53.6, 56.6, 82.5, 128.0, 131.1, 153.2; MS (FAB) m/z (%): 785 (M++Na, 100), 736 (8). Anal. Calcd for C38H66N8O8 (762.99): C, 59.82; H, 8.72; N; 14.69. Found: C, 59.56; H, 8.99; N, 14.63.
= –3.5 (c 1.2, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 1.28 [br s, 8H, (CH2)4], 1.43–1.65 [m, 38H, 4 × C(CH3)3, 2 × CHHCHN], 1.74–1.90 (m, 2H, 2 × CHHCHN), 1.91–2.13 (m, 8H, 2 × CH2CH=CHCH2), 3.29 (dd, 2H, J = 12.2, 5.6 Hz, 2 × CHHN3), 3.76 (dd, 2H, J = 12.2, 9.4, 2 × CHHN3), 4.35 (m, 2H, 2 × CHN), 5.23–5.46 (m, 4H, 2 × CH=CH); 13C‑NMR (50 MHz, CDCl3) δ 24.0, 27.2, 28.0, 29.3, 29.7, 30.2, 53.6, 56.6, 82.5, 128.0, 131.1, 153.2; MS (FAB) m/z (%): 785 (M++Na, 100), 736 (8). Anal. Calcd for C38H66N8O8 (762.99): C, 59.82; H, 8.72; N; 14.69. Found: C, 59.56; H, 8.99; N, 14.63.
= –3.5 (c 1.2, CHCl3); 1H-NMR (200 MHz, CDCl3) δ 1.28 [br s, 8H, (CH2)4], 1.43–1.65 [m, 38H, 4 × C(CH3)3, 2 × CHHCHN], 1.74–1.90 (m, 2H, 2 × CHHCHN), 1.91–2.13 (m, 8H, 2 × CH2CH=CHCH2), 3.29 (dd, 2H, J = 12.2, 5.6 Hz, 2 × CHHN3), 3.76 (dd, 2H, J = 12.2, 9.4, 2 × CHHN3), 4.35 (m, 2H, 2 × CHN), 5.23–5.46 (m, 4H, 2 × CH=CH); 13C‑NMR (50 MHz, CDCl3) δ 24.0, 27.2, 28.0, 29.3, 29.7, 30.2, 53.6, 56.6, 82.5, 128.0, 131.1, 153.2; MS (FAB) m/z (%): 785 (M++Na, 100), 736 (8). Anal. Calcd for C38H66N8O8 (762.99): C, 59.82; H, 8.72; N; 14.69. Found: C, 59.56; H, 8.99; N, 14.63.(2S,17S,5Z,13Z)-Octadeca-5,13-diene-1,2,17,18-tetramine tetrahydrochloride (10)
To a stirred mixture of the diazide 9 (1.53 g, 2.00 mmol) and 10% Pd/C (100 mg) in THF (15 mL), through which N2 had been passed for 5 min, were added NaBH4 (454 mg, 12.00 mmol) and MeOH (40 mL) dropwise. After stirring for 20 min, the catalyst was filtered, the solution was neutralized with 1 M KHSO4 and the organic solvents were removed. The aqueous phase was extracted with EtOAc (2 × 30 mL), the combined organic phases were dried (Na2SO4) and the solvent was removed. The tert-butoxycarbonyl group was removed by treatment with 4 N HCl in THF (30 mL) for 30 min at room temperature. After evaporation, Et2O was added and the product was filtered, and recrystallized from MeOH-Et2O. Yield 612 mg (67%); yellow solid; 1H-NMR (200 MHz, CDCl3) δ 1.24 [br s, 8H, innermost (CH2)4], 1.58–1.71 (m, 4H, 2 × CH2CHCH2N), 1.89–2.19 (m, 8H, 2 × CH2CH=CHCH2), 3.00–3.17 (m, 4H, 2 × CH2N), 3.30–3.56 (m, 2H, 2 × CHN), 5.21–5.45 (m, 4H, 2 × CH=CH); MS (FAB) m/z (%): 311 (Μ++1 – 4HCl, 100). Anal. Calcd for C18Cl4H42N4·H2O (474.39): C, 45.57; H, 9.35; N; 11.81. Found: C, 45.35; H, 9.49; N, 11.69.
Acknowledgements
V.L. thanks the Greek Government and the European Commission for a fellowship (E.P.E.A.E.K.). This work was supported in part by the University of Athens (Special Account for Research Grants).
References
- For reviews see: (a) Kuksa, V.; Buchan, R.; Lin, P. K. T. Synthesis of polyamines, their derivatives, analogues and conjugates. Synthesis 2000, 1189–1207. [Google Scholar] (b) Karigiannis, G.; Papaioannou, D. Structure, biological activity and synthesis of polyamine analogues and conjugates. Eur. J. Org. Chem. 2000, 1841–1863. [Google Scholar]
- Lucet, D.; Le Gall, T.; Mioskowski, C. The chemistry of vicinal diamines. Angew. Chem., Int. Ed. 1998, 37, 2580–2627. [Google Scholar]
- Donner, B. G. Conversion of chiral amino acids to enantiomerically pure α-methylamines. Tetrahedron Lett. 1995, 36, 1223–1226. [Google Scholar]
- Kokotos, G.; Markidis, T.; Constantinou-Kokotou, V. Synthesis of chiral triamines and diamines from amino acids. Synthesis 1996, 1223–1226. [Google Scholar]
- Gurjar, M. K.; Pal, S.; Rao, A. V. R. Synthesis of novel C2-symmetric and pseudo C2-symmetric based diols, epoxides and dideoxy derivatives of HIV protease inhibitors. Tetrahedron 1997, 53, 4769–4778. [Google Scholar]
- Yamauchi, T.; Higashiyama, K.; Kubo, H.; Ohmiya, S. Synthesis of C2-symmetrical chiral diamines: diastereoselective addition to bis(1,3-oxazolidinyl)alkanes with Grignard reagents. Tetrahedron: Asymmetry 2000, 11, 3003–3015. [Google Scholar]
- Mamos, P.; Karigiannis, G.; Athanassopoulos, C.; Bichta, S.; Kalpaxis, D.; Papaioannou, D.; Sindona, G. Simple total syntheses of N-substituted polyamine derivatives using N-tritylamino acids. Tetrahedron Lett. 1995, 36, 5187–5190. [Google Scholar]
- Kokotos, G.; Constantinou-Kokotou, V.; Noula, C.; Hadjipavlou-Litina, D. Synthetic routes to lipidic diamines and amino alcohols: a class of potential antiinflammatory agents. Lipids 1999, 34, 307–311. [Google Scholar]
- Kokotos, G.; Theodorou, V.; Constantinou-Kokotou, V.; Gibbons, W. A.; Roussakis, C. Synthesis and in vitro cytotoxicity of lipophilic platinum(II) complexes. Bioorg. Med. Chem. Lett. 1998, 8, 1525–1530. [Google Scholar]
- Constantinou-Kokotou, V.; Kokotos, G.; Roussakis, C. Synthesis and in vitro cytotoxicity of lipidic alcohols and amines. Anticancer Res. 1998, 18, 3439–3442. [Google Scholar]
- Markidis, T.; Padrón, J. M.; Martín, V. S.; Peters, G. J.; Kokotos, G. Synthesis and in vitro cytotoxicity of long chain 2-amino alcohols and 1,2-diamines. Anticancer Res. 2001, 21, 2835–2840. [Google Scholar]
- Karikas, G.; Constantinou-Kokotou, V.; Kokotos, G. An HPLC method for the measurement of polyamines and lipidic amines binding to DNA. J. Liq. Chrom. & Rel. Technol. 1997, 20, 1789–1796. [Google Scholar]
- Constantinou-Kokotou, V.; Karikas, G.; Kokotos, G. Study of aminoglycoside-nucleic acid interactions by an HPLC method. Bioorg. Med. Chem. Lett. 2001, 11, 1015–1018. [Google Scholar]
- For reviews see: (a) Jurczak, J.; Golebiowski, A. Optically active N-protected α-amino aldehydes in organic synthesis. Chem. Rev. 1989, 89, 149–164. [Google Scholar] (b) Reetz, M. T. Synthesis and diastereoselective reactions of N,N-dibenzylamino aldehydes and related compounds. Chem. Rev. 1999, 99, 1121–1162. [Google Scholar]
- Jurczak, J.; Gryko, D.; Kobrzycka, E.; Gruza, H.; Prokopowicz, P. Effective and mild method for preparation of optically active α-amino aldehydes via TEMPO oxidation. Tetrahedron 1998, 54, 6051–6064. [Google Scholar]
- Kokotos, G. A convenient one-pot conversion of N-protected amino acids and peptides into alcohols. Synthesis 1990, 299–301. [Google Scholar]
- Kokotos, G.; Noula, C. Selective one-pot conversion of carboxylic acids into alcohols. J. Org. Chem. 1996, 61, 6994–6996. [Google Scholar]
- Ma, Z.; Bobbitt, J. M. Organic oxoammonium salts. 3. A new convenient method for the oxidation of alcohols to aldehydes and ketones. J. Org. Chem. 1991, 56, 6110–6114. [Google Scholar]
- Leanna, M. R.; Sowin, T. J.; Morton, H. E. Synthesis of α-amino and α-alkoxy aldehydes via oxoammonium oxidation. Tetrahedron Lett. 1992, 33, 5029–5032. [Google Scholar]
- Schlosser, M.; Schaub, B.; de Oliveira-Neto, J.; Jeganathan, S. Practical guidance for obtaining optimum cis-selectivities in Wittig reactions with triphenylphosphonio-alkanides. Chimia 1986, 40, 244–245. [Google Scholar]
- Maryanoff, B. E.; Reitz, A. B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar]
- Markidis, T.; Kokotos, G. A novel approach to the synthesis of chiral terminal 1,2-diamines. J. Org. Chem. 2001, 66, 1919–1923. [Google Scholar]
- Dawson, M. I.; Vasser, M. Synthesis of prostaglandin synthetase substrate analogues. 1. (Z)-14-Hydroxy-12,13-methano-8-nonadecenoic acid. J. Org. Chem. 1977, 42, 2783–2785. [Google Scholar]
- For synthesis of racemic 1,5-bis-p-tolouenesulfonylamino-dodecane see: Wiesner, K.; Valenda, Z.; Orr, D. E.; Liede, V.; Kohan, G. Stucture of pithecolobine. III. The synthesis of the 1,5- and 1,3-deoxypithecolobines. Can. J. Chem. 1968, 46, 3617–3624. [Google Scholar]
- Sample Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Loukas, V.; Markidis, T.; Kokotos, G. Amino Acid Based Synthesis of Chiral Long Chain Diamines and Tetramines. Molecules 2002, 7, 767-776. https://doi.org/10.3390/71000767
AMA Style
Loukas V, Markidis T, Kokotos G. Amino Acid Based Synthesis of Chiral Long Chain Diamines and Tetramines. Molecules. 2002; 7(10):767-776. https://doi.org/10.3390/71000767
Chicago/Turabian StyleLoukas, Vassilios, Theodoros Markidis, and George Kokotos. 2002. "Amino Acid Based Synthesis of Chiral Long Chain Diamines and Tetramines" Molecules 7, no. 10: 767-776. https://doi.org/10.3390/71000767