Applications of Microwave in Organic Synthesis: An Improved One-step Synthesis of Metallophthalocyanines and a New Modified Microwave Oven for Dry Reactions

Abstract
:Introduction
Results and Discussion
Synthesis of metallophthalocyanines from phthalodinitrile
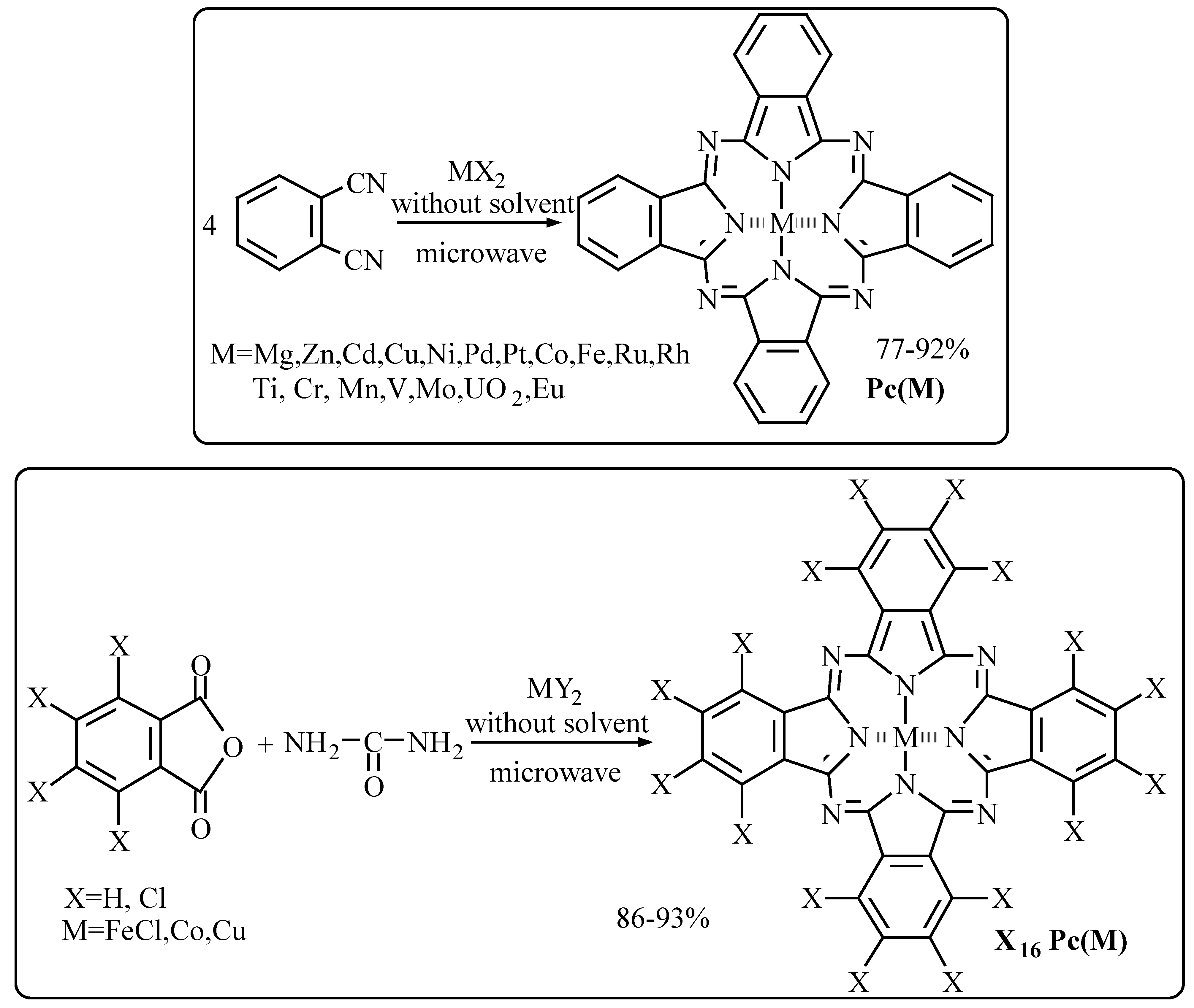

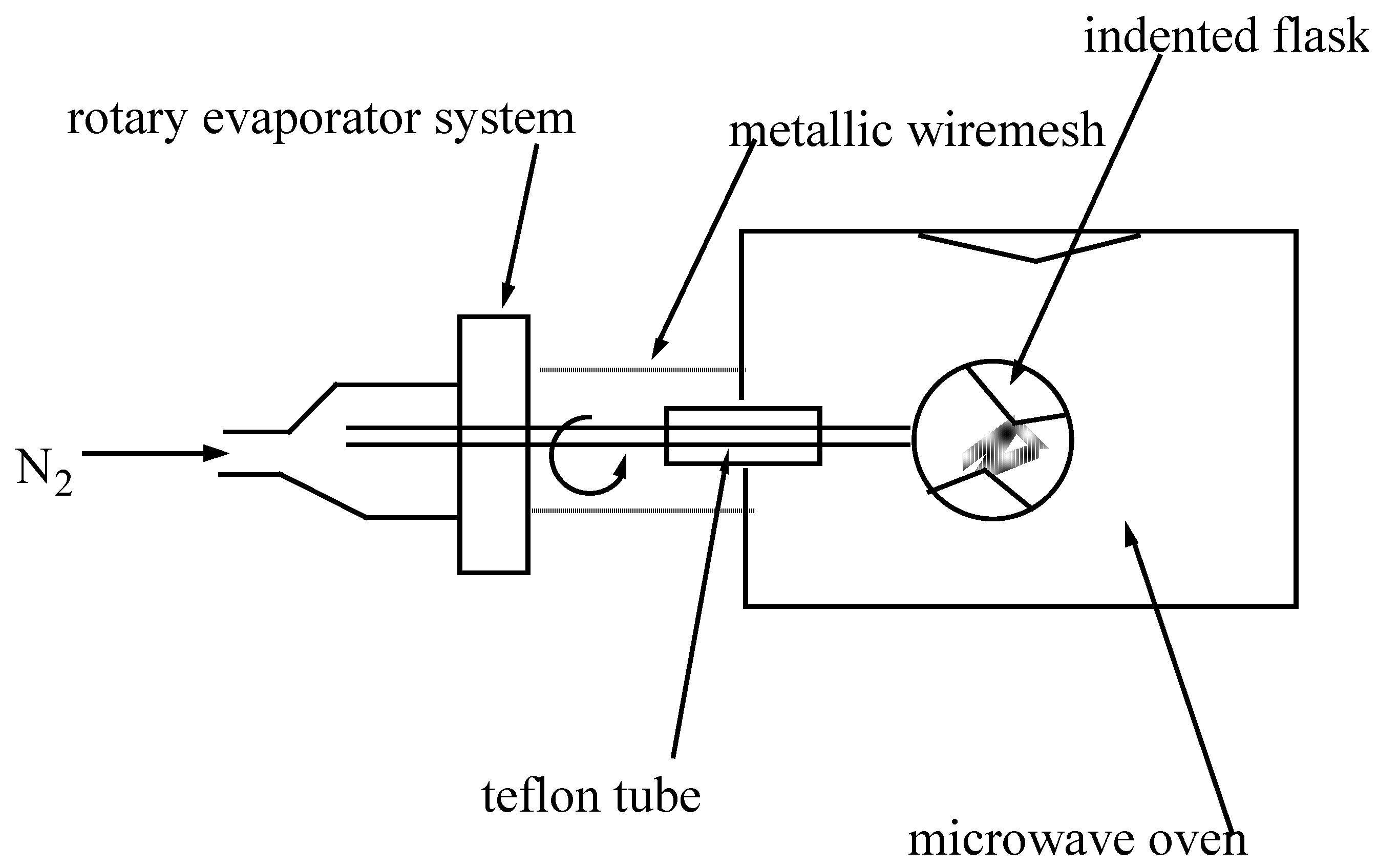
Choice of the mode of irradiation in the formation of Pc(Cu)

{kind=link}
{kind=link}
| Microwave Irradiation Sources | Time (min) | Power (W) | Yield (%) |
| microwave oven (commercial) | 10 | 560 | 92 |
| modified microwave oven | 10 | 480 | 98 |
| resonance cavity | 10 | 80 | 94 |
- -
- a better distribution and uniformity of the microwave irradiation
- -
- a mixing of solid reactants during the activation with microwave irradiation.
- -
- possibility of working under controlled atmospheres (nitrogen) and avoiding hot points.
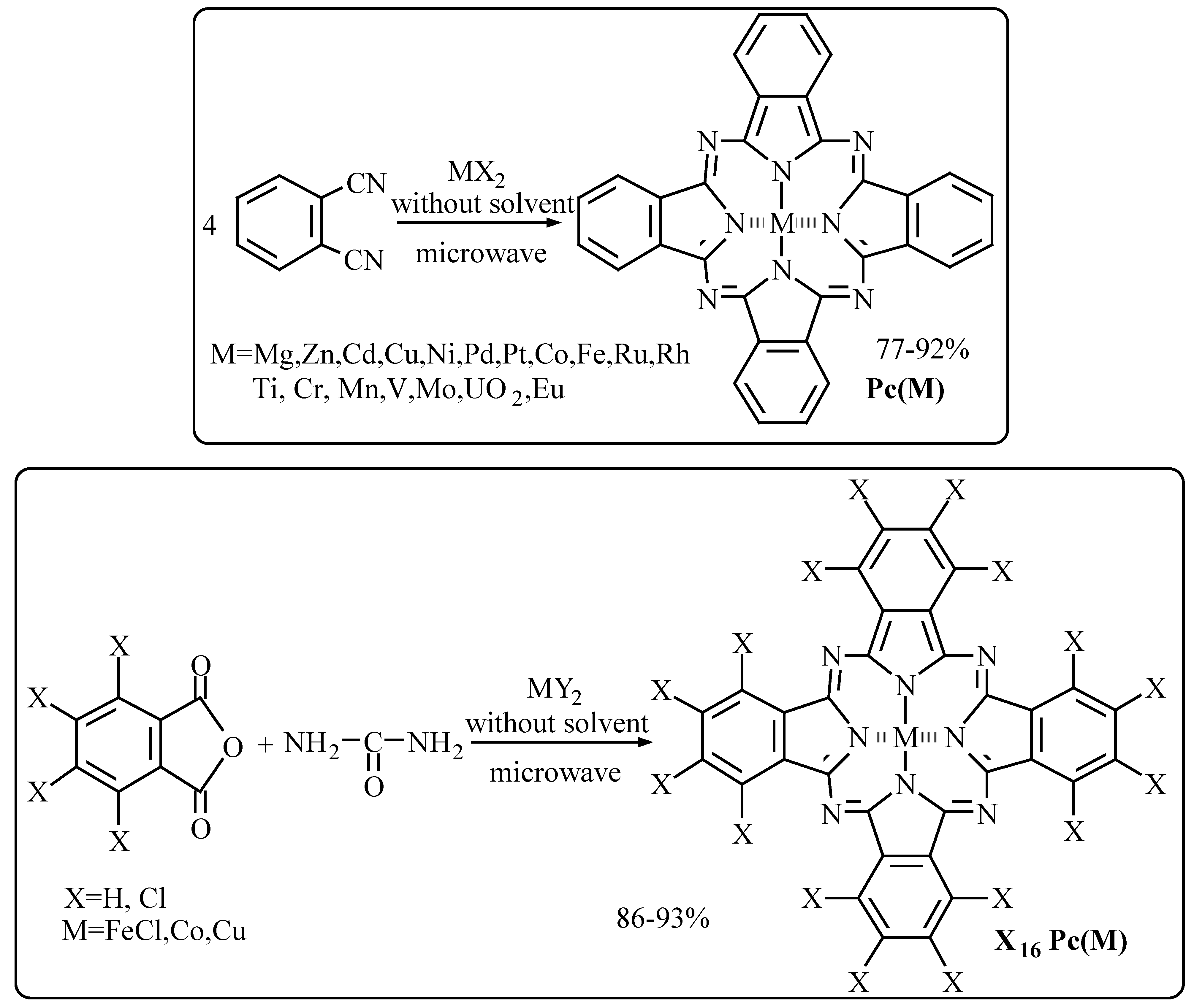
Synthesis of metallophthalocyanines from phthalic anhydrides
Conclusions
Experimental
General procedures
- 1)
- Irradiation with a commercial multimode microwave oven (Method A)
- 2)
- Irradiation with a modified multimode microwave oven (Method B)
- 3)
- Irradiation in a resonance cavity (focused irradiation) (Method C)
From phthalic anhydride: General procedure
From tetrachlorophthalic anhydride:
From tetrafluorophthalic anhydride:
References and Notes
- Moser, F.H.; Thomas, A.L. The Phthalocyanines; CRC Press: Boca Raton, FL, 1983. [Google Scholar] Bezerin, B.D. Coordination Compounds of Porphyrins and Phthalocyanines; J. Wiley and Sons: New York, NY, 1981. [Google Scholar] Bauman, F.; Bienert, B.; Rösch, G.; Vollman, H.; Wolf, W. Angew. Chem. 1956, 68, 133.
- Leznoff, C.C.; Lever, A.B.P. Phthalocyanines, Properties and Applications; VCH Publishers: New-York, 1989-1993; pp. 1–3. [Google Scholar] Hanack, M.; Lang, M. Adv. Mater. 1994, 6, 819.
- Borsenger, P.M.; Weiss, D.S. Organic Photoreceptors for Imaging System; Marcel Dekker, Inc.: New York, 1993. [Google Scholar] Law, K.Y. Chem. Rev. 1993, 93, 499.
- Casstevens, M.; Samok, M.; Pfleger, J.; Prasard, P.N. Chem. Phys. 1990, 92, 2019. Simon, J.; Bassoul, P.; Norvez, S. New J. Chem. 1994, 13, 13.
- Van Der Pol, J. F.; Neeleman, E.; Zwikker, J.W.; Nolte, R.J.M.; Drenth, W.; Aerts, J.; Visser, R.; Picken, J. J. Liq. Cryst. 1989, 6, 577. [CrossRef] Simon, J.; Sirlin, C. Pure Appl. Chem. 1989, 61, 1625.
- Kato, M.; Nishioka, Y.; Kaifu, K.; Kawamura, K.; Ohno, S. Appl. Chem. Lett. 1985, 86, 196.
- Kuder, J. E. J. Imag. Sci. 1988, 32, 51.
- Temofonte, T. A.; Schoch, K.F. J. Appl. Phys. 1989, 65, 1350. [CrossRef] Sadaoka, Y.; Jones, T.A.; Göpel, W. Sensors Actuators B. 1990, I, 148.
- Riou, M.T.; Clarisse, C.J. Electroanal. Chem. 1988, 249, 181.
- Law, K.Y. Chem. Rev. 1993, 93, 449.
- Darwent, J.R.; Douglas, P.; Harriman, A.; Porter, G.; Richoux, M.C. Coord. Chem. Rev. 1982, 44, 83.
- Wöhrle, D.; Gitzel, J.; Krawczyk, G.; Tsuchida, E.; Ohno, H.; Nishisaka, T.J. Macromol. Sci. C. A. 1988, 25, 1227.
- Henderson, B.A.; Dougherty, T.J. Photochem. Photobiol. 1992, 55, 145. [CrossRef]
- Wöhrle, D.; Ardeschirpur, A.; Heuermann, A.; Muller, S.; Graschew, G.; Rinneberg, H.; Kohl, M.; Neukammer. J. Makromol. Chem. Makromol. Symp. 1992, 59, 17. [CrossRef]
- Our work on synthesis of metallophthalocyanines was partially described in a monograph and in a communication: Bram, G.; Loupy, A.; Villemin, D. Solid Supports and Catalysts in Organic Synthesis; Smith, K., Ed.; Ellis Horwood and Prentice Hall: Chichester, U.K., 1992; Volume 12, pp. 302–326. [Google Scholar] Villemin, D; Hammadi, M. Actes of 3 ème Colloque Franco-Maghrebin de Catalyse, CFMC 1996, 622.
- Villemin, D.; Hammadi, M.; Hachemi, M. Synth. Commun. 2001, in press.
- Kingston, H. M.; Haswell, S. J. (Eds.) Microwave-Enhanced Chemistry. Fundamentals, Sample Preparation, and Applications; American Chemical Society: Washington, D.C., 1997. Varma, R.S. Green Chem. 1999, 43.
- Shaabani, A. J. Chem. Res 1998, 672.
- Marvel, C.S.; Rassweiler, J. J. Amer. Chem. Soc. 1958, 80, 1197. [CrossRef]
- Linstead, R.P.; Robertson, J.M. J. Chem. Soc. 1936, 173. Lexas, D.; Reix, M. J. Chim. Phys. Physicochim. Biol. 1974, 71, 517. Lhost, J.M; Grivet, J.Ph. Adv. Radiat. Res. Phys. Chem. 1973, 327–1. Shablya, A.V.; Terenin, A.N. Optika Spectroskopiya. 1961, 9, 533, [C. A. 1961, 55, 12028i]. Seybold, P.G.; Gouterman, M. J. Mol. Spectrosc. 1939, 31, 1. [CrossRef]
- Linstead, R.P. J. Mol. Spectrosc. 1934, 1010. Dent, C.E.; Linstead, R.P. J. Chem. Soc. 1934, 1027. Linstead, R.P.; Lowe, A.R. J. Chem. Soc. 1934, 1031. Linstead, R.P. J. Chem. Soc. 1934, 1016. Barrett, P.A; Dent, C.E.; Linstead, R.P. J. Chem. Soc. 1936, 1720. Linstead, R.P.; Robertson, J.M. J. Chem. Soc. 1936, 1736. Berezin, B.D. Takashi, O.; Michiakazu, N.; Atsushi, K.; Shuji, F.; Japan Catalytic Chemical Industry Co., Ltd. Japan Pat. 70, 07,659; [C.A. 1970, 73, 16287], 1970.
- Seybold, P.G.; Gouterman, M. J. Mol. Spectrosc 1969, 31, 1. [CrossRef] Barrett, P.A.; Dent, C.E.; Linstead, R.P. J. Chem. Soc. 1936, 1719. Vacus, J.; Memetzidis, G.; Doppelt, P.; Simmon, J. J. Chem. Soc. Chem. Commun. 1994, 697.
- Lexas, D.; Reix, M. J. Chim. Phys. Physicochim. Biol. 1974, 71, 517.
- Block, B.P.; Meloni, E.G. Inorg. Chem. 1965, 4, 111. Taube, R. Z. Chem. 1963, 3, 194.
- Plyushchev, V.E.; Shkolner, L.P.; Rodin, I.A. Zh. Neorgan. Khim. 1967, 9, 125. Mühler, P. Z. Chem. 1967, 1, 352.
- Mitsuo, S.; Takao, K. Chem. Pharm. Bull. 1968, 16, 2517. Berezin, B.D. Izv. Vyssh. Ucheb. Zaved., Khim. Khim. Tekhnol. 1968, 11, 537–541, [C.A. 1968, 69, 72393c].
- James, S.A.; Ray, A.K; Silver, J. Phys. Stat. Sol. 1992, 129, 435. [CrossRef]
- Lever, A.B.P. J. Chem. Soc. 1965, 1821. Barrett, P.A.; Dent, C.E.; Linstead, R.P. J. Chem. Soc. 1936, 1719. Rutter, H.A.; Mc Queen, J.D. J. Inorg. Nucl. Chem. 1960, 12, 361. [CrossRef] Mason, R.; Willems, G.A.; Fielding, P.E. J. Chem. Soc. Dalton Trans 1991, 676.
- Linstead, R.P. Chem. Soc. 1934, 1016. Linstead, R.P.; Robertson, J.M. J. Chem. Soc. 1936, 1736. Barrett, P.A.; Dent, C.E.; Linstead, R.P. J. Chem. Soc. 1936, 1719. Barrett, P.A.; Frye, D. A.; Linstead, R.P. J . Chem. Soc. 1938, 1157. Metz, J.; Schneider, O.; Hanack, M. Inorg. Chem. 1984, 23, 1065. Rudenko, A.P.; Dobrosel'skaya, N.P. Zh. Obshch. Khim. 1961, 31, 3667, [C. A. 1962, 57, 9856b]. Taube, R.; Drevs, H.; Fluck, E.; Kuhn, P.; Brauch, K.F. Z. Anorg. Allg. Chem. 1969, 364, 297. [CrossRef]
- Krueger, P.C.; Kenney, M.E. J. Inorg. Nucl. Chem. 1963, 25, 303. [CrossRef]
- Enokida, T.; Hirohashi, R. Chem. Letters. 1991, 2155. Berezin, B.D. Izv. Vyssh. Ucheb. Zaved. Khim. Khim. Tekhnol. 1968, 11, 537, [C. A. 1968, 69, 72393c].
- Keen, I. M.; Malerbi, B. W. J. Inorg. Nucl. Chem 1965, 27, 1311. [CrossRef]
- Vacus, J.; Memetzidis, G.; Doppelt, P.; Simmon, J. J. Chem. Soc. Chem. Commun. 1994, 697. Keen, I. M.; Malerbi, B.W. J. Inorg. Nucl. Chem. 1965, 27, 1311. [CrossRef] Barrett, P.A.; Frye, D. A.; Linstead, R. P. J. Chem. Soc. 1938, 1157.
- Bansho, Y.; Suzuki, S.; Sekiguchi, T.; Saito, I. Kogyo Kagaku Zasshi 1962, 65, 2005. Metz, J.; Schneider, O.; Hanack, M. Inorg. Chem. 1984, 23, 1065.
- Berezin, B.D. Izv. Vyssh. Ucheb. Zaved., Khim. Khim. Tekhnol. 1968, 11, 537, [C. A. 1968, 69, 72393c]. Keen, I.M.; Malerbi, B.W. J. Inorg. Nucl. Chem. 1965, 27, 1311. [CrossRef] Menzel, E.R.; Rieckhoff, K.E.; Voigt, E.M. Chem. Phys. Lett. 1972, 13, 604.
- Gurevich, M.G.; Solov'ev, K.N. Dokl. Akad. Nauk. Beloruss. SSR. 1961, 5, 291, [C. A. 1962, 57, 15948].
- Edward, A.; Cuellar, G.; Marks, J.M. Inorg. Chem. 1981, 20, 3766.
- Zhenxiang, L.; Jiazuan, N.; Guangxian, N.; Lemin, L. Wuji. Huaxue. 1986, 2, 8, [C. A. 1987, 106, 202111e].
- Elvidge, A.; Lever, A.B.P. J. Chem. Soc. 1961, 1257. Meloni, E.G.; Ocone, L.R.; Block, B.P. Inorg. Chem. 1967, 6, 424.
- Barmasov, A.V.; Korotkov, V.I.; Kholmogorov, V.E. Khim. Fiz. 1986, 5, 414, [C. A. 1992, 116, 197588r].
- Sample Availability: Available from MDPI.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Villemin, D.; Hammadi, M.; Hachemi, M.; Bar, N. Applications of Microwave in Organic Synthesis: An Improved One-step Synthesis of Metallophthalocyanines and a New Modified Microwave Oven for Dry Reactions. Molecules 2001, 6, 831-844. https://doi.org/10.3390/61000831
Villemin D, Hammadi M, Hachemi M, Bar N. Applications of Microwave in Organic Synthesis: An Improved One-step Synthesis of Metallophthalocyanines and a New Modified Microwave Oven for Dry Reactions. Molecules. 2001; 6(10):831-844. https://doi.org/10.3390/61000831
Chicago/Turabian StyleVillemin, Didier, Mohamed Hammadi, Messaoud Hachemi, and Nathalie Bar. 2001. "Applications of Microwave in Organic Synthesis: An Improved One-step Synthesis of Metallophthalocyanines and a New Modified Microwave Oven for Dry Reactions" Molecules 6, no. 10: 831-844. https://doi.org/10.3390/61000831