Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland

Chair and Laboratory of Organic Chemistry, Faculty of Pharmacy, Medical University of Warsaw, 1 Banacha Street, PL 02-097 Warsaw, Poland
Molecules 2000, 5(12), 1331-1371; https://doi.org/10.3390/51201331
Submission received: 16 November 2000
/
Accepted: 16 November 2000
/
Published: 20 December 2000
Abstract
:This review reports some novel (or considerably improved) methods for the synthesis of aromatic iodides, (dichloroiodo)arenes, (diacetoxyiodo)arenes, iodylarenes and diaryliodonium salts, as well as some facile, oxidative anion metatheses in crude diaryliodonium halides and, for comparison, potassium halides. All these new results were obtained in our laboratory over the past decade (1990-2000). A full list of our papers dealing with the organic iodine(I, III and V) chemistry, covering exlusively the aromatic derivatives, is also provided.
Contents
- Introduction
- Syntheses of Aromatic Iodides
- Syntheses of (Dichloroiodo)arenes
- Syntheses od (Diacetoxyiodo)arenes
- Syntheses of Iodylarenes
- Syntheses of Diaryliodonium Salts
- Anion Metatheses in Diaryliodonium Halides
- Conclusions
1. Introduction
In the years 1980-1990 our research group had been mainly interested in the chemistry of aromatic, symmetric and unsymmetric organomercurials; see Refs. 22,23,24,25,26,27,28,29,30,31,32,33. In particular, we successfully synthesized a number of novel organomercurials derived from lactamic heterocycles as well as we discovered several novel cyano- and halo-demercuration reactions; this resulted in many effective syntheses of cyano or halo derivatives of those aromatics, whose corresponding organomercurials had been used in the said novel demercuration reactions. However, the quickly growing number of literature reports and reviews on the unique synthetic possibilities offered by organic hypervalent iodine reagents/compounds had been so promising and alluring that in 1990 we definitely decided that nearly all our future research work should be directed towards this area. Since all our previous research activity had been concerned with aromatic compounds, all our subsequent studies have also been limited to aromatic iodine(III or V) compounds. Additionally, we put special emphasis on the development of novel, easy and effective oxidative iodination procedures, applicable for numerous aromatics (both activated and deactivated), since the resulting iodo derivatives would then applied by us in our subsequent syntheses of the corresponding aromatic hypervalent iodine compounds. So far, mainly (dichloroiodo)arenes, (diacetoxyiodo)arenes, iodylarenes, and aromatic iodonium salts have been synthesized and studied by us, using novel (or considerably improved) methods. This short review presents mainly our own contributions to the chemistry of aromatic iodine(I, III and V) compounds over the past decade (1990-2000). At the end of this review a full list of our papers and Ph.D. theses dealing with this topic is provided (see Refs. 1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18).
2. Syntheses of Aromatic Iodides
Aromatic iodides, known for ca.150 years, are generally more reactive than the respective bromides and chlorides. There is a considerable number of different methods, direct or indirect, for their synthesis, and they are widely used in chemical laboratories and, sometimes, also in chemical industry and medicine [19]. Moreover, they are able to form a large variety of stable, aromatic polyvalent iodine compounds, which have found increasing applications in modern organic synthesis [21].
2.1. Our earlier results: aromatic iodides from aromatic organomercurials [22,23,24,25,26,27,28,29,30,31,32,33]
It is known [20] that aromatic iodides may be obtained indirectly from both symmetric and unsymmetric aromatic organomercurials, by reacting them with either hot aq. KI3 solutions or with hot solutions of diiodine in acetonitrile, DMF, etc. We had repeatedly applied the “classic” iododemercuration reactions to better elucidate the chemical structures of organomercurials prepared by us in various ways. Thus, we synthesized (the yields given in brackets) the following aromatic iodides:
i) 8-iodocaffeine (95%) from both 8-(acetoxymercurio)caffeine and 8,8’-mercuriobis(caffeine) [22];
ii) 5-iodouracil (60 or 93%, resp.) from either 1-acetyl-5-(trifluoroacetoxymercurio)uracil or 5,5’-mercuriobis(uracil) [23];
iii) 8-iodotheophylline (95%) from 8-(trifluoroacetoxymercurio)theophylline, and 8-iodotheobromine (95%) from 8-(trifluoroacetoxymercurio)theobromine [24];
iv) 2-iodofuran (60%) from 2,2’-difurylmercury, and 2-iodothiophene (65%) from 2,2’-dithienylmercury [25];
v) 5-iodo-1,3-dimethyluracil (87.5 or 82.5%, resp.) from either 5-(acetoxymercurio)-1,3-dimethyluracil or 5,5’-mercuriobis(1,3-dimethyluracil) [27];
vi) 5-iodo-2,4-dimethoxypyrimidine (58 or 52%, resp.) from either 5-(acetoxymercurio)-2,4-dimethoxypyrimidine or 5,5’-mercuriobis(2,4-dimethoxypyrimidine); the acid hydrolysis of pure 5-iodo-2,4-dimethoxypyrimidine afforded 5-iodouracil (69%) [28];
vii) 6-iodo-2,3-diphenyl-5-methoxybenzofuran (70%) from both 6-(acetoxymercurio)-2,3-diphenyl-5-methoxybenzofuran and 6,6’-mercuriobis(2,3-diphenyl-5-methoxybenzofuran) [29];
viii) iodobenzene (59%) from (chloromercurio)benzene, and 8-iodocaffeine (65%) from 8-(chloromercurio)caffeine [30];
ix) 8-iodotheobromine (97%) from 1,8-bis(acetoxydimercurio)theobromine; the latter compound was the first stable organic derivative of mercury(I) [32];
x) 5-iodouracil (61% crude yield) from 5-(chloromercurio)uracil, and 2-ethylthio-4-hydroksy-5-iodopyrimidine (64% crude yield) from 2-ethylthio-4-hydroxypyrimidine raw mercurial [33].
We also applied twice our novel iodo-demercuration method, by reacting two symmetric organomercurials with hot ethanolic solutions of cyanogen iodide, ICN. In this way we synthesized:
xi) 8-iodocaffeine (90%) from 8,8’-mercuriobis(caffeine) [22], and
xii) iodobenzene (72%) from diphenylmercury [26].
2.2. Aromatic Iodides from Some Highly Activated Aromatics with Lead(IV) Acetate as Oxidant [2]
In a very brief communication, without an experimental part, Soviet authors [34] have reported that on treating PhR (R = H, F, Cl, Br, I, Me, CF3) with diiodine and Pb(OAc)4 dissolved in costly CF3CO2H, the corresponding iodides are formed in 85-90% yields, as determined by GLC. Stoichiometric ratios of Pb4+/I2/Ar-H = 1:1:2 were mostly applied. They assumed that both iodine(I) and iodine(III) species would be the iodinating agents. A possible formation of intermediate ArI(OCOCF3)2 from Ar-H was claimed based only on the evidence of the reaction of PhCl (no yield given) with a Pb4+/I2 (3:1) system, in which the following stoichiometry was obeyed:
I2 + 3Pb4+ → 2I3+ + 3Pb2+
However, details of their iodination method have never been published.
In our first work [2] we carried out the oxidative aromatic iodination of eleven highly activated arenes and heterocycles in the presence of excess (10%) pure Pb(OAc)4 in glacial acetic acid, used as the inexpensive solvent of choice:
![Molecules 05 01331 i001]()

It is of interest that when using hot Pb(OAc)4 solutions prepared in situ from finely powdered, commercial minimum, Pb3O4, prior to the iodination, i.e.:
![Molecules 05 01331 i002]()

then the iodination yields were often somewhat better than those obtained with the pure oxidant. Also 4-bromo- and 2,4-dibromoanisoles were obtained from anisole by the respective oxidative brominations. We failed to iodinate benzene, toluene, 1,3-xylene and several less active arenes or heteroaromatics; for more information see Ref. 2.
2.3. Aromatic Iodides from Activated or Deactivated Arenes with Chromium(VI) Oxide as Oxidant [8]
Japanese chemists [35] iodinated benzene with diiodine, in pressurized air and oxygen gas and in acidic solvents using twenty four metal salts or oxides as catalysts; they mentioned also a chromate and MnO2 (see Section 2.5). The iodination rate was dependent on the acidity of the solvent applied: CF3SO3H > CH3SO3H > CF3COOH >> CH3COOH. PhI was obtained in excellent yield (91.9%) when NaVO3 and CF3SO3H were used. According to them, the combination of strong acidic solvent, oxygen gas, and oxidizing catalyst are indispensable in the iodination reaction, whereas the kind of metal counter ion used does not seem to be important. Later, Soviet chemists [36] used chromium (VI) salts for the oxidative aromatic iodination of benzene and toluene with diiodine in a mixture of CCl4 with acetic acid containing H2SO4, using four dichromates as the oxidants. They qualitatively arranged the oxidizing activity of the dichromates as follows: K2Cr2O7 < Na2Cr2O7 < Li2Cr2O7 < (NH4)2Cr2O7. We assume that their failure to iodinate PhCOOH and PhNO2 was mainly due to the presence of WATER in the reaction mixtures: they always used water acidified with H2SO4 for prior dissolution of the dichromates. It is known [37] that water, due to its high hydration power, diminishes the oxidizing activity of inorganic (and organic) oxidants. In contrast, this activity is increased when the overall acidity of the oxidizing mixtures rises to some optimum level. When this level is considerably exceeded, then the iodination yields may drop strikingly, due to a predominant protonation of the reacted aromatic molecules.
Note. Starting from our second paper in the series [8], in all our subsequent studies dealing with the oxidative aromatic iodination of various arenes/heteroarenes, the above general considerations have always been taken into account. The favorable anhydrous conditions were simply attained using mixtures of glacial acetic acid with acetic anhydride as the cheap solvents of choice. The optimum overall acidities of the iodinating mixtures, which are different for each arene/heteroarene iodinated, were attained by dropwise addition to the cooled reaction mixtures of widely varied quantities of conc. (98%) sulfuric acid, established experimentally in each case; see Experimental parts in our papers [8, 11, 15, 17, 18c,d] for more details.
For benzene, halobenzenes, and several activated arenes, the following reaction mechanism and general reaction stoichiometry were assumed and applied:
![Molecules 05 01331 i003]()

In spite of many attempts, working under widely varied reaction conditions, in practice we were unable to iodinate nitrobenzene. For a less strongly deactivated arene, benzoic acid, the final yield was low, i.e. 30%. Hence, for the first time in our research work, we deliberately changed the proportions of the reactants and, consequently, the reaction mechanism, by carrying out the oxidative aromatic iodination reactions as follows [the same general approach has later been used in all our subsequent iodination procedures applicable for deactivated aromatics, vide infra]:
![Molecules 05 01331 i004]()

After completing the above reactions, the resulting (final) reaction mixtures containing the assumed organic iodine(III) intermediates, ArISO4, were poured into excess aq. Na2SO3 solutions to give the corresponding crude iodoarenes, ArI, as follows:
2ArISO4 (not isolated) + 2Na2SO3 + 2H2O → 2ArI + 4NaHSO4
After the isolation and purification of crude ArI, we obtained pure 3-IC6H4COOH (86%), 3-IC6H4COOMe (66%), 3-IC6H4COOEt (64%), 3-IC6H4CONH2 (88%), 3-IC6H4SO2NH2 (74%), and 3-IC6H4NO2 (71%). Similarly, it was possible to obtain some pure diiodinated products, viz. 3,3’-diiodobenzophenone (57%), 3,3’-diiododibenzoyl (72%), 3,3’-diiododiphenyl sulfone (83%), and 3,5-diiodobenzoic acid (31%). These high iodination yields we achieved with deactivated arenes were due to the fact that the respective arenes had been reacted with some preponderant, more electrophilic transient iodine(III) species, I3+, formed momentarily in the reaction mixtures prepared in agreement with Eq. 3. Contrariwise, for benzene, halobenzenes, and activated arenes, the best iodination yields were attained with the reaction mixtures prepared in agreement with Eq. 2, where some less electrophilic, transient iodine(I) species, I+, had evidently been predominant.
When benzene was iodinated according to Eq. 3, the yield of PhI was low (10%). However, from the collected aqueous layers we precipitated out a yellow solid with excess aq. KBr. After recrystallization from MeOH is was identified as diphenyliodonium bromide (44%). We commented on this fact as follows [8]: “This novel “one-pot” method of preparing the symmetric diaryliodonium salts is presently being improved and extended [70], and will be published soon”. However, publication of this method has been delayed.
A more detailed discussion on the transient I+ or I3+ species that probably exist in the iodinating mixtures prepared in agreement either with Eq. 2 or with Eq. 3, was subsequently submitted in our last paper of the series [17]; see Section 2.6. The varied quantities of conc. H2SO4 (established experimentally for each of the arenes iodinated) added dropwise to the cooled reaction mixtures clearly depended on relative reactivities (and basicities) of the arenes investigated. The more deactivated an arene was, more conc. H2SO4 had to be added to catalyze better the iodination, and to possibly increase the concentration of still more reactive either iodine(I) or iodine(III) transient species:
![Molecules 05 01331 i005]()

In the strongly acidic reaction mixtures discussed, some fairly stable organic iodine(III) intermediates, ArISO4 (see Eq. 3), are, probably quickly equilibrating with some other intermediates, viz. ArI(OSO3H)2 (which, in fact, were solely shown in Ref. [8]) on adding more H2SO4. For the sake of simplicity, only one kind of them was shown and discussed in all our papers. Their real existence in the reaction mixtures has been experimentally demonstrated in our newest work [18c]; see Section 4.1.
2.4. Aromatic Iodides from Deactivated Arenes with Potassium Permanganate as Oxidant [11]
Chaikovski and Novikov [38] oxidatively iodinated benzene and various activated and deactivated derivatives, and some aromatic hydrocarbons, using stoichiometric quantities of KMnO4 in strongly acidic media. To explain the results, they proposed the following assumed reaction mechanism:
![Molecules 05 01331 i006]()

This shows, in their opinion, that a protonated form of hypoiodic acid, H2 +OI, generated from diiodine by the action of KMnO4 in strongly acidic solutions, is the main source of active, transient iodine(I) species, I+, which then react momentarily with the iodinated aromatics.
They worked out two simple procedures, carrying out the iodination reactions (at 100-115°C, for 1-5 h) using the following systems: Procedure 1: I2/KMnO4/glacial AcOH/conc. H2SO4 or Procedure 2: I2/KMnO4/80% AcOH/conc. H2SO4. Generally, the corresponding mono- or diiodinated products were obtained in 26-99% yields - hence, we made no attempt to improve their Procedure 2, suitable for aromatic hydrocarbons and activated arenes. But in the Procedure 1 they used, quite purposefully, an industrial grade glacial acetic acid - probably containing a considerable admixture of water which, due to its high hydration power, diminishes the oxidizing activity of inorganic oxidants [37]. Hence, on iodinating PhCOOH and 9,10-phenanthrenequinone they obtained the iodinated products in only moderate yields: 3-IC6H4COOH (49%) and 2,7-diiodo-9,10-phenanthrenequinone (35%). Taking into account our former experiments [8] explained in Section 2.3, we were able to improve considerably said Procedure 1, suitable for the iodination of deactivated arenes, by carrying out the iodination reactions under anhydrous conditions, and by applying the following reaction stoichiometry, which strongly favors the formation of more electrophilic, transient iodine(III) species, viz.
![Molecules 05 01331 i007]()

After pouring the final reaction mixtures containing fairly stable organic ArISO4 intermediates, into excess aq. Na2SO3 solutions (see Eq. 4 in Section 2.3), and the separation and purification of the crude products, we thus obtained 3-IC6H4COOH (78%), 3-IC6H4COOMe (83%) and 3-IC6H4COOEt (73%) as well as 2,7-diiodo-9,10-phenanthrenequinone (85%), by lowering the reaction temperature to 35°C, and shortening the reaction time to one hour.
It is necessary to add that Chinese chemists from Taiwan [39] iodinated various arylamines with a homogeneous mixture of hydroiodic acid and KMnO4 in acetonitrile, obtaining para-substituted products in high yields (71-78%) within twelve hours at room temperature. This reagent is highly selective for arylamines, but unsuitable for phenols, which undergo oxidative coupling easily in the presence of electron transfer agents.
2.5. Aromatic Iodides from Activated and Deactivated Arenes with Various Brands of Manganese(IV) Oxide as Oxidants [11, 18d]
Activated MnO2 is a common and easily handled oxidant, and its many applications in organic synthesis have been known for a long time [40]. Moreover, Mn(II) salts remaining in the residues after the oxidative iodination reactions are less toxic as compared with Pb(II) or Cr(III) salts, the byproducts in our former iodination experiments [2, 8]. Starting our iodination experiments with MnO2, we checked out various brands of this oxidant experimentally. Ordinary commercial MnO2 was not applicable. The best iodination yields were obtained with the activated MnO2 freshly prepared prior to use [41]. An activated commercial product (Aldrich AMD, suitable for organic reactions, ca. 85% MnO2) gave lower iodination yields by ca. 5-10%. The so-called Chemical Manganese Dioxide (CMD, suitable for use in batteries) has not been studied by us until recently [18d]; our newest results are briefly discussed below, at the end of this Section.
For benzene, toluene, and four halobenzenes, the iodination reactions were carried out according to the following stoichiometry, which favors the preponderant formation of transient iodine(I) species, viz.
![Molecules 05 01331 i008]()

Similarly, we obtained 1,4-diiodobenzene (66%) from benzene, when we halved the amount of Ar- H (i.e. benzene) in Eq. 6. For the oxidative iodination of deactivated arenes, the deliberately changed general reaction stoichiometry was applied, which favors the preponderant formation of transient iodine(III) species, viz.
![Molecules 05 01331 i009]()

After pouring the final reaction mixtures, containing the fairly stable organic ArISO4 intermediates, into excess aq. Na2SO3 solutions (see Eq. 4 in section 2.3), and purification of the crude products, we obtained: 3-IC6H4COOH (87%), 3-IC6H4COOMe (89%), 3-IC6H4COOEt (79%), 3-IC6H4CONH2 (63%), and 3-IC6H4NO2 (73%). As above, we also obtained the diiodinated products: 3,3’-diiodobenzophenone (78%) and 2,7-diiodo-9,10-phenanthrenequinone (72%).
When benzene was iodinated according to Eq. 7, the yield of PhI was negligible, but from the collected aqueous layers we precipitated a yellow solid with excess aq. KI. After its recrystallization from MeOH, it was identified as diphenyliodonium iodide (43%).
Japanese chemists [42] have used as an oxidant the so-called Chemical Manganese Dioxide (CMD, Wako product, min. 75% MnO2; its price in Poland: 378.00 DM/500 g + freight cost 60.00 DM) in numerous selective oxidations of various classes of organic compounds; these reactions often proceeded in nearly quantitative yields under relatively mild conditions. In some cases, the Wako product proved to be much superior to usual active MnO2 (AMD) commercially available from Aldrich, Fluka, Merck, Nakarei and Wako companies. Hence, we decided to check its application in the oxidative aromatic iodination reactions presented in Eqs. 6 and 7, respectively. With the Aldrich CMD (90+% MnO2; its price in Poland: 32.30 DM/500 g), the final yields for the purified ArI were practically the same as those obtained with the Aldrich AMD (its price in Poland: 107.30 DM/500 g). In contrast, with the said Wako CMD the crude iodinated products, isolated by more laborious methods, were dark-colored and heavily contaminated, which resulted in considerable losses during their troublesome purification. Though the self-prepared AMD (vide supra) is, in fact, somewhat more efficient, its tedious preparation is time-consuming and relatively expensive, so we would recommend the application of the Aldrich CMD as the oxidant of choice in oxidative aromatic iodination reactions, since is has satisfactory activity and is notably less costly.
2.6. Aromatic Iodides from Activated and Deactivated Arenes with Sodium Periodate or Sodium Iodate as Oxidants [17]
So far, a number of authors have used periodic acid or its salts, and iodic acid or its salts to oxidatively iodinate a variety of activated aromatics [19b, 43], including halobenzenes [44]. Only the most costly I2O5 has scarcely ever been used until recently [45, 46]. Suzuki [47] concluded his extensive studies by stating that the oxidative iodination with periodic acid is “the most convenient method for preparation of mono- or diiodo derivatives from various polyalkyl-benzenes in high yields”. Merkushev [19b] summarized the literature results as follows: “The iodination in the presence of iodic acid and periodic acid is accelerated considerably by the addition of sulfuric acid and, in some cases, of water. These mild oxidants are widely used in the iodination of aromatic compounds activated to electrophilic substitution reactions as well as in iodination reactions of polynuclear aromatic compounds”.
Periodic acid, NaIO4, iodic acid, and NaIO3 are commercially available, with the two sodium salts being notably cheaper [48]. These salts in solutions or suspensions acidified with excess conc. H2SO4 would momentarily form in situ either HIO4 or HIO3, respectively. Hence, they were used as such in all our oxidative iodination experiments [17] discussed below.
For benzene, toluene, four halobenzenes, 4-nitroanisole, and N,N-dimethylaniline their oxidative iodination reactions were carried out by us according to the following stoichiometry, which strongly favors the preponderant formation of less electrophilic, transient iodine(I) species, viz.
![Molecules 05 01331 i010]()

The least reactive 4-O2NC6H4OMe was at first stirred at room temperature for one hour, but its iodination was completed by a further stirring for one hour at 45°C. The most reactive PhNMe2 was iodinated in 65% yield by stirring at +5°C for only one hour; for some reasons, we failed to iodinate likewise PhNH2 and PhNEt2. When we halved the amount of Ar-H (i.e. benzene) added to the reaction mixture shown in Eq. 8, then we obtained the purified 1,4-C6H4I2 in 85% yield.
For nitrobenzene, 4-nitroanisole, (trifluoromethyl)benzene, benzoic acid and its methyl and ethyl esters, 4-toluic acid, 4-chlorobenzaldehyde [however, we failed to iodinate likewise benzaldehyde, benzonitrile, and benzamide] as well as (for comparison) iodobenzene, bromobenzene, and chlorobenzene, we deliberately applied the following general reaction stoichiometry, which strongly favors the preponderant formation of more electrophilic, transient iodine(III) species, viz.
![Molecules 05 01331 i011]()

After completing the above reactions, the resulting (final) reaction mixtures, containing the fairly stable organic iodine(III) intermediates, ArISO4, were poured into excess aq. Na2SO3 solutions to give the corresponding crude iodoarenes, ArI (see Eq. 4 in Section 2.3). After the purification of crude ArI, we obtained the eleven purified monoiodinated products in good or excellent yields (56-95%). However, when benzene was likewise monoiodinated, rather impure PhI was obtained in only 43% yield. This novel procedure is also suitable for the oxidative diiodination of deactivated arenes: the purified 3,3’-diiodobenzophenone was obtained in 51% yield.
Finally, using the appropriate I2/NaIO3/Ac2O/AcOH/conc. H2SO4 anhydrous system - equally suitable for the oxidative iodination of deactivated arenes - we monoiodinated nitrobenzene, benzoic acid, and 4-toluic acid, as well as (for comparison) bromobenzene, according to the general reaction stoichiometry, which strongly favors the preponderant formation of more electrophilic, transient iodine(III) species, viz.
![Molecules 05 01331 i012]()

After completing the main iodination reactions within 3-5 h at room temperature (only PhNO2 was stirred at r.t. for 4 h, and then at 65°C for 3 h), the strongly acidic reaction mixtures were poured into excess aq. Na2SO3 solutions (see Eq. 4 in Section 2.3). After recrystallization the crude aryl iodides, ArI, gave the four purified monoiodinated products in excellent yields (83-93%).
We have previously observed [8, 11] during our numerous experiments that in the presence of water (which always considerably diminishes the activity of all electrophilic iodinating species, owing to its high hydration power), the assumed transient iodine(III) species (briefly denoted above as I3+) are unstable and very quickly vanish to form some less reactive and more stable hydrated species, probably I-O+ H2. Presumably, these transformations would take place as follows [17]:
![Molecules 05 01331 i013]()

Hence, the anhydrous and strongly acidic conditions are indispensable to attain the possible highest yields of the assumed iodine(III) intermediates, ArISO4, derived from the reacted deactivated arenes by their electrophilic substitution with I3+; cf. Eqs. 3, 5, 7, 9, and 10.
The characteristic feature of all the above oxidative iodination reactions (undergoing in agreement with Eqs. 8-10) is that diiodine is oxidized there by HIO4 or HIO3 to form some reactive transient species, I+ or I3+, whereas the both oxidants applied are reduced by diiodine to form the same transient species, I+ or I3+ - which next react, more or less readily, with aromatic compounds, Ar-H, to form either ArI or ArISO4 intermediates, respectively. Hence, no strongly toxic residues remain after the oxidative iodination reactions using the inorganic iodine(VII) or iodine(V) oxidants. Such iodination reactions are, indeed, environmentally benign.
In our opinion, particularly interesting are our iodination results obtained with deactivated arenes (Eq. 9 and 10). It has been a generalized opinion [19b] that inorganic iodine(VII) and iodine(V) oxidants are mild - hence they can hardly be appropriate for the effective oxidative iodination of deactivated aromatic substrates. Our experimental results with deactivated arenes clearly show that this notion is incorrect provided that acidic and anhydrous conditions are maintained in the reactions as shown in Eqs. 9 and 10.
Note. Quite recently, Russian chemists [49] have developed easy and effective procedures for iodination of deactivated aromatics, without casual oxidation of CH3 or even CHO groups (e.g. they iodinated benzaldehyde in 61% yield). The reactions were carried out in conc. (90%) H2SO4 with some superactive iodine reagent “I+” prepared on a base of toxic iodine chloride with silver sulfate, at 0-20°C and within 15-150 min. Also, nearly all the former iodinating procedures suitable for deactivated aromatics are briefly reviewed therein - with excluding, however, our two former papers [8, 11].
2.7. Aromatic Iodides from Activated Aromatics Obtained by an Improved, Acid- catalyzed Iodinating Procedure with (Diacetoxyiodo)benzene as Oxidant [15]
In a full-text version of our paper [15] there is a comprehensive review on the aromatic halogenation reactions with organic trivalent iodine reagents as the oxidants. So far, nine organic iodine(III) compounds have been used as the oxidants for the oxidative halogenation of aromatic compounds:
![Molecules 05 01331 i014]()

None of these reagents, however, permit aromatic fluorination. In view of electron density on the aromatic ring, it seems that halogenation of chlorobenzene represents the limit of the scope of the reactions with reagents 1-3 and 5-9; more deactivated aromatics cannot be halogenated with these reagents. But the evident advantages of these solid reagents are: 1) low toxity, 2) easy handling and simple experimental procedures, 3) usually high yields of halogenation products, 4) possibility (for reagents 1, 2, 5, 6 and 8) of recovery from the final reaction mixtures of iodobenzene which may be recycled after its oxidative conversion into the initial iodine(III) reagents, following known procedures [10, 21b]. Similarly, the polymeric reagent 3 can be recovered in good yield by filtration as poly(iodostyrene), and 3 can easily be regenerated and reused [50]. However, all the hypervalent iodine reagents are more or less light- and heat-sensitive, while 4 and 5 are also moisture-sensitive.
Reagents 2, 5 and 6 are commercially available with 2 being notably cheaper [48]. Moreover, pure 2 was synthesized by us in 79% yield with a novel procedure developed in our laboratory [10] (see Section 4.1); this procedure is ca. 5 times cheaper and 8-16 times faster than the former method [51]. Hence, in our work [15] we considerably improved the reported reaction conditions for the iodination of several activated arenes: benzene, iodobenzene, three xylenes, mesitylene, durene, di- and triphenylmethane, fluorene, fluoren-9-one, dibenzofuran, xanth-9-one, and uracil ― using just the reagent 2, which is more stable, preparatively more accessible, and notably cheaper than the moisture-sensitive reagent 5, strongly recommended by other authors [19b] in place of 2.
Ogata and Aoki [52] first iodinated 1,3-xylene to 4-iodo-1,3-xylene (96%) in warm (60°C) acetic acid with reagent 2 as the oxidant, with iodobenzene, produced as the side product, being recovered. Next [19b], several activated arenes were iodinated in warm (50°C) acetic acid with I2/2 to give monoiodinated products, within 1-5 hours, in 56-89% yields. Togo and co-workers [50] have compared the reagent 2 and the polymer-supported reagent 3 in the oxidative iodination of activated arenes. With I2/2 (in ethyl acetate, at room temperature or at 60°C) they monoiodinated nine activated arenes in 4-99% yields and within 4-16 hours; they also diiodinated mesitylene (96%), biphenyl (87%) and diphenyl ether (70%) at 60°C after 16 hours. After 1980, mono-, di-, and triiodinated activated arenes /hetero--arenes have instead been prepared with I2/5 (in chlorinated solvents) in good or excellent yields, mainly at room temperature and within 0.25-2 hours [19b]. The other hypervalent iodine reagents listed above have, so far, been used to a lesser extent.
Transient acetyl hypoiodite (CH3COI) was suggested as the iodinating species when reagent 2 is applied as the oxidant to iodinate aromatics [19b]. In the presence of strong acids, the reactivity of 2 is enhanced, because of its dissociation to more reactive cationic species [21b]:
![Molecules 05 01331 i015]()

As in our earlier works [8, 11], we deliberately applied anhydrous iodinating conditions, using AcOH/Ac2O mixtures as the cheap solvents of choice [15]. The addition of varying catalytic amounts of conc. (98%) H2SO4 (from one drop up to 1 ml, depending on the reactivity of iodinated aromatics) considerably accelerated the iodination reactions studied; only fluorene was effectively iodinated in absence of H2SO4 at room temperature and within 15 minutes. We put forward the following reaction mechanism and stoichiometry [15]:
![Molecules 05 01331 i016]()

The overall iodination rate is, in our opinion, mainly restricted by the rate of the first step (1), and is considerably accelerated by the anhydrous conditions applied as well as by catalytic amounts of conc. (98%) H2SO4 added to the iodinating (or brominating) reaction mixtures. The reactions proceeded quickly until the coloration of iodine faded, then the reaction mixtures were poured into excess aq. Na2SO3 solutions. The isolated crude products were purified by common methods. From the filtrates the iodobenzene was recovered in 40-75% yields.
Using our improved iodination procedure [15] it was possible to mono-, di-, or even triiodinate the studied aromatics, at or near room temperature and within at most 15 minutes, in 40-82% yields, except for uracil, which was also monoiodinated in 82% yield within 15 minutes, but at 40°C. For comparison, the oxidative bromination of mesitylene, durene and fluoren-9-one, carried out in a similar anhydrous system: arene/Br2/2/AcOH/Ac2O with only one drop of conc. H2SO4 added, resulted in tribromomesitylene (65%), dibromodurene (62%) and 2,7-dibromofluoren-9-one (73%). These bromination reactions even proceeded much faster than the corresponding iodination reactions. Hence, we recommend our improved method [15] as a very easy, fast, inexpensive and effective method to iodinate/brominate activated aromatic compounds.
Note. Dr. Maria Niemyjska [53] from our laboratory has carried out the oxidative iodination reactions using reagent 8 as the oxidant. The reactions took place in hot (95-100°C) AcOH/Ac2O mixtures acidified with conc. H2SO4, within several hours. She thus obtained 4-iodoanisole (68%, admixed with its ortho-isomer) and 1-iodo-2-methoxynaphthalene (10%, admixed with 6-iodo-2-methoxynaphthalene), but she failed to iodinate acetanilide, 4-methoxybenzophenone, methyl salicylate, and mesitylene. These reactions were undertaken only for the sake of comparing 8 with the other hypervalent iodine reagents, and were unworthy of further study.
2.8. Aromatic Iodides from Deactivated Aromatics with a Stable Urea•H2O2 Complex as Oxidant [98]
Recently [98], we succeeded in oxidatively iodinating several deactivated arenes (in 40-88% yields), with a stable, commercially available urea•H2O2 complex used as the oxidant, viz.
![Molecules 05 01331 i017]()

After pouring the resulting (final) reaction mixtures into excess aq. Na2SO3 solutions (see Eq. 4 in Section 2.3), the isolated crude ArI were purified. It should be emphasized that this effective iodination method is evidently environmentally benign.
3. Syntheses of (Dichloroiodo)arenes
(Dichloroiodo)arenes, ArICl2, have found growing importance in modern organic synthesis [21]. More stable, solid ArICl2, e. g. (dichloroiodo)benzene, are used as potent and fairly selective chlorinating and/or oxidizing agents. They have a practical advantage over elemental chlorine (dichlorine), due to their easy and safe handling. Moreover, they may be readily converted to other important organic hypervalent iodine reagents playing also an important role in organic synthesis, viz. iodosylarenes, iodylarenes, (diacyloxyiodo)arenes, (difluoroiodo)arenes [21], diaryliodonium salts [13], etc.
ArICl2, yellow crystalline compounds, are light- and heat-sensitive and often unstable to storage. They do not usually give satisfactory microanalyses and, due to their thermal lability, their melting points are rather uncertain, depending upon the purity of the crude products prepared, the time elapsed since their preparation, and the rate of heating. Keefer and Andrews [54] have observed that in the cases of 4-O2NC6H4ICl2 and 4-HOOCC6H4ICl2 all the dichlorine was expelled when their melting occured; the recorded melting points were the same as those of the initial iodoarenes, i. e. 4-O2NC6H4I and 4-HOOCC6H4I.
In 1885 Willgerodt [55] had prepared the first stable organic iodine(III) compound, (dichloroiodo)benzene: PhI + ICl3 → PhICl2 + ICl. ArICl2 can also be made as follows [20]: Ar2Hg + ICl3 → ArICl2 + ArHgCl; this reaction is still of preparative significance in the case of the vinyl-type organomercurials.
In 1886 Willgerodt [56] had developed the most common method up to now for preparing ArICl2, by passing the stream of Cl2 through solutions of ArI dissolved in CHCl3, at 0 °C. The yields are generally excellent when this method is applied; ArICl2 is not formed from ArI substituted with such groups as OH, NH2, ethylenic double bonds, etc. Anhydrous conditions previously demanded [57] are not necessary [58]. Quite recently [59], the repeated preparations of PhICl2 (in 94% crude yield) on a 20 kg scale have been conducted by the direct chlorination of PhI (dissolved in CH2Cl2), at -3 to +4 °C; next, it was possible to monochlorinate 4-aminoacetophenone with PhICl2 on 24.8 kg scale, in 87% yield.
The inconvenient use of gaseous Cl2 to afford ArICl2 from ArI may be avoided as follows:
(i) by the action of hydrochloric acid on either iodosylarenes, ArIO, or (diacyloxyiodo)arenes, ArI(OAc)2, previously otherwise obtained from ArI [21];
(ii) by using liquid SO2Cl2 added to a solution of ArI either in wet diethyl ether [60] or (much better) in 98% acetic acid [61]. Karele and Neiland [61] failed to chlorinate with SO2Cl2 three isomeric iodonitrobenzenes, 4-iodobenzoic acid, and 2-iodoacetophenone; for ten appropriate ArI the crude yields for ArICl2 were 73-96%;
(iii) by the action of a liquid mixture: cobalt(III) acetate - KCl - 67% aq. CF3COOH on PhI [62]; only iodobenzene was chlorinated there to give PhICl2 in 75% crude yield;
(iv) by the use of hydrochloric acid oxidized in situ with sodium perborate tetrahydrate in either acetonitrile or CCl4 containing a dissolved ArI [63]; when CCl4 was applied as co-solvent, then the chlorinations of three isomeric iodoanisoles were, in fact, biphasic ones. McKillop et al. [63] failed to chlorinate oxidatively 2-iodonitrobenzene; for fifteen appropriate iodobenzenes, including also 3- and 4-iodonitrobenzene, the obtained crude yields for ArICl2 were 60-98%.
3.1. Biphasic Chlorination of Iodoarenes to (Dichloroiodo)arenes [12,18a, 64]
In 1991 we already reported [64] a two-phase (CCl4/conc. hydrochloric acid) chlorination of iodobenzene and 4-iodoanisole to PhICl2 and 4-MeOC6H4ICl2, respectively, in yields exceeding 90%. The same biphasic procedure has later been applied in our laboratory [12] to chlorinate seventeen ArI compounds to the corresponding ArICl2 in 50-98% crude yields, including also three isomeric iodonitrobenzenes, 3-HOOCC6H4I, 3-MeOOCC6H4I, and 3-iodobenzophenone.
The essence of this method is as follows. A solution of ArI in CCl4 was vigorously stirred with excess conc. hydrochloric acid, at 0-5°C, and the powdered inorganic oxidant (KClO3 or chlorinated lime) was very slowly added portionwise. Dichlorine was produced in the aqueous phase as follows:
KClO3 (solid) + 6HCl (conc. aq.) → ↑3Cl2 + KCl + 3H2O, alternatively:
CaCl(OCl) + 2HCl (conc. aq.) → ↑Cl2 + CaCl2 + H2O (less effective)
CaCl(OCl) + 2HCl (conc. aq.) → ↑Cl2 + CaCl2 + H2O (less effective)
Most of the dichlorine evolved was absorbed and consumed in the organic phase to chlorinate effectively a iodoarene dissolved therein. After 3 hours of stirring at 0-5°C, the yellow precipitates, ArICl2, were washed on the filter with dilute hydrochloric acid, water and CCl4, and air-dried in the dark.
In our next work [18a] we used sodium peroxodisulfate (sodium persulfate), Na2S2O8, a very potent, inexpensive [48] and often applied oxidant [40] to oxidize in situ conc. hydrochloric acid to chlorinate ArI to the corresponding ArICl2, using either the liquid-phase (see Section 3.2) or biphasic chlorination procedures. Dichlorine was produced there as follows:
Na2S2O8 + 2HCl (conc. aq.) → ↑Cl2 + 2NaHSO4
However, a short preheating period (at 45-50°C) was necessary to start the above oxidative reaction. Only PhI, 4-MeCONHC6H4I, 4-FC6H4I, 4-MeOC6H4I, and 2-IC6H4COOH were chlorinated by the biphasic method to the corresponding ArICl2 obtained in 89-100% crude yields. 4-Acetamidoiodobenzene gave 4-acetamido(dichloroiodo)benzene in only 60% crude yield when it was chlorinated with the liquid-phase method, but its chlorination with the biphasic method afforded the same product in 90% crude yield. This example shows that for iodoarenes substituted with hydrolyzable groups, the biphasic chlorination is evidently preferable.
In comparison with the classic Willgerodt method [56, 57, 58, 59], the biphasic chlorination method avoids the inconvenient use of gaseous Cl2, hence it is relatively safe. If an oxidant, e.g. KClO3 [12], is very slowly added to the vigorously stirred biphasic reaction mixture, then the escape of Cl2 to the outer atmosphere is considerably limited. The limits of the scope of the two methods are the same or nearly so.
3.2. Oxidative Liquid-Phase Chlorination of Iodoarenes to (Dichloroiodo)arenes [14,18a,b]
In 1999 we reported [14] a novel, very simple, and efficient laboratory method for the conversion of seventeen iodoarenes to the corresponding (dichloroiodo)arenes, with including 2- and 3-iodonitro-benzene and 3-iodobenzoic acid; although we failed to chlorinate 4-iodonitrobenzene, 4-iodobenzonitrile, and 1, 3, 5-trichloro-2-iodobenzene. We used conc. hydrochloric acid oxidized in situ with varied amounts of chromium(VI) oxide, a common and easily handled oxidant [37, 40], dissolved in aq. acetic acid, and acting upon a dissolved (or suspended) iodoarene. The reactions proceeded according to the following stoichiometry:
![Molecules 05 01331 i018]()

The crude yellow products precipitated by water were washed well on the filter with ice-cold water to remove CrCl3, HCl, and AcOH, then with CCl4 to remove most of the unreacted ArI, and air-dried in the dark; we observed that by drying the crude ArICl2 in a vacuum desiccator, the chlorine percentage was lowered: ArICl2 → ArI + ↑ Cl2.
Note: easily oxidizable ArI, e.g. 4-iodoanisole and iodotoluenes, should be added to the reaction mixture after the addition of the whole portion of conc. HCl. Otherwise, the final yields of the corresponding ArICl2 were very low.
The same procedure is effective for the aromatic chlorination of activated arenes. Exemplarily, we obtained the purified chlorinated products: 2-chloro-4-nitroanisole (64%), 2-chloro-4-nitroacetanilide (58%), and 3,5-dichloro-4-methoxybenzoic acid (42%) from the respective arenes.
Later, we reported [18a] the preparation of (dichloroiodo)arenes, ArICl2, from the respective iodoarenes, ArI, by the use of hydrochloric acid oxidized in situ with sodium peroxodisulfate (sodium persulfate), Na2S2O8, in acetic acid containing a dissolved ArI; it was necessary to warm up preliminarily the stirred reaction mixture to 45-50°C to initiate the oxidative chlorination of ArI. We failed to chlorinate 2-iodo-1,4-xylene, iodomesitylene, and 1,3,5-trichloro-2-iodobenzene. For sixteen appropriate ArI, with including all the isomeric iodonitrobenzenes and iodobenzoic acids, the crude yields for ArICl2 were 60-100%. The chlorination reactions underwent according to the following stoichiometry:
![Molecules 05 01331 i019]()

Finally, we presented [18b] one improved (cf. Ref. 63) and eight novel oxidative, liquid-phase chlorination procedures for the preparation of (dichloroiodo)arenes, ArICl2, from always the same nine exemplary iodoarenes, ArI, using KMnO4, activated (85%) MnO2, KClO3, NaIO4, NaIO3•H2O, conc. nitric acid, NaBO3•H2O, 2Na2CO3•3H2O2, and a stable urea•H2O2 complex, used as the oxidants, which oxidized hydrochloric acid to produce in situ an active chlorine (in statu nascendi). The crude yields for the nine ArICl2 obtained were good or excellent (63-99%).
The following general reaction stoichiometries (Eqs. 14-22) may be deduced for the oxidative, liquid-phase chlorination procedures applied in our work [18b], viz.
ArI + NaBO3•H2O + 2HCl → ArICl2 + [NaBO2] + H2O
McKillop et al. [63] applied NaBO3•4H2O as the oxidant used in the 400% excess, using acetonitrile (or, only for iodoanisoles, carbon tetrachloride) as co-solvent. They stirred the reaction mixtures, at room temperature, mostly for 2 hours, but for 1,4-diiodobenzene and 4-iodonitrobenzene the stirring had to be prolonged to 3 days; no chlorination of 2-iodonitrobenzene was attained even after 3 days. In our present work [18b], Na2BO3•H2O was used in only the 200% excess, the reaction times did not exceed 2 hours, but we unexpectedly failed to chlorinate 4-iodonitrobenzene, in spite of several attempts (we also met with the same failure using the other oxidants in our work [18b]).
ArI + HNO3 + 3HCl → ArICl2 + [NOCl] + 2H2O
We excluded the possibility of chlorination of ArI with NOCl (formed there as a side product [65]), carrying out the separate, biphasic experiments. ArI were dissolved in CHCl3 or CCl4, excess conc. hydrochloric acid was added, then solid NaNO2 was slowly added portionwise with vigorous stirring. In the aqueous phase the following reaction took place [66]: NaNO2 + 2HCl → NOCl + NaCl + H2O. The NOCl formed was surely present in excess in the organic phase, since it is readily soluble in chlorinated aliphatic hydrocarbons. However, no trace of any yellow coloration was developed, characteristic of ArICl2.
5ArI + 2KMnO4 + 16HCl → 5ArICl2 + 2MnCl2 + 2KCl + 8H2O
ArI + activated MnO2 + 4HCl → ArICl2 + MnCl2 + 2H2O
We observed that ArICl2, produced according to Eqs. 16 and 17, were more contaminated than those obtained with the other procedures.
3ArI + NaIO4 + 8HCl → 3ArICl2 + [ICl] + NaCl + 4H2O
2ArI +NaIO3•H2O + 6HCl → 2ArICl2 + [ICl] + NaCl + 4H2O
So far, we have had no experimental evidence suggesting that iodine chloride was formed as a side product in the reaction mixtures studied. Nevertheless, high crude yields of ArICl2 formed from ArI in the chlorination reactions undergoing in agreement with such the supposed stoichiometries would suggest that Eqs. 18 and 19 are plausible.
3ArI + KClO3 + 6HCl → 3ArICl2 + KCl + 3H2O
Knowing the explosive properties of KClO3, it must not be pulverized in a mortar prior to its use in the reactions.
3ArI + 2Na2CO3•3H2O2 + 10HCl → 3ArICl2 + 4NaCl + 2CO2 + 8H2O
ArI + urea•H2O2 + 2HCl → ArICl2 + [urea] + 2H2O
Sodium percarbonate and sodium perborate are both available at a low price, and are widely used in chemical laboratories or in industry [67]. They may be considered as “dry carries” of the hazardous and unstable hydrogen peroxide, are easy to handle, safe and stable at room temperature; the same is true in respect to a commercially available, stable complex of urea with hydrogen peroxide [68]. Their ability to release oxidative species in organic media has made them useful reagents in organic synthesis. Hence, they were also checked in our work [18b].
When the oxidative chlorination reactions are carried out in a liquid phase containing ArI, an appropriate strong oxidant, and conc. hydrochloric acid mixed with an inert solvent, then a very active chlorine, in statu nascendi, is instantly produced there, which is apt to react not only with the iodine atoms of ArI, but also with aromatic rings of highly activated ArI, e. g. iodoanisoles, which were formerly chlorinated by McKillop with co-workers [63]. There underwent rapid aromatic chlorinations when acetonitrile was used as co-solvent (a liquid-phase protocol). 3-Iodoanisole gave 2,4,6-trichloro-3-methoxyiodobenzene, whereas 4-iodoanisole gave 3-chloro-4-methoxy(dichloroiodo)benzene initially, but this rapidly decomposed to give 3,5-dichloro-4-methoxyiodobenzene. However, the same iodoanisoles could be satisfactorily chlorinated to the corresponding ArICl2 using CCl4 as co-solvent (a biphasic protocol) - it means that the two-phase systems allow milder chlorination conditions. The latter may be explained as follows. An active chlorine in statu nascendi, formed in the aqueous phase of the biphasic system, is very quickly deactivated there to form the “ordinary” dichlorine, Cl2, which next is readily absorbed and consumed in the organic phase containing ArI to produce there ArICl2. Similarly, in the Willgerodt method [56, 57, 58, 59], the stream of this “ordinary” dichlorine, Cl2, is passed through solutions of ArI in inert solvents to form there ArICl2. Thus, we previously expressed the opinion [12] that “the limits of the scope of the two methods are the same or nearly so.” Later [18a], we concluded our chlorination experiments as follows: “... it is preferable to obtain ArICl2 from iodoanisoles and highly activated ArI with using either the biphasic method or the classic Willgerodt method, where such ArI and a less active molecular chlorine Cl2, are both dissolved and reacting in an inert solvent. Contrariwise, more suitable for deactivated ArI seem to be appropriate liquid-phase chlorinating protocols - because such ArI are reacted upon with some more active, though short-living, chlorine species, formed there in statu nascendi.”
3.3. A One-Pot Method for Preparing (Dichloroiodo)arenes from Arenes [18c]
Almost all former methods of preparing ArICl2 demanded the use of costly iodoarenes, ArI, as the starting substrates to be then chlorinated at their iodine atoms. In our newest paper [18c] we have presented a quite novel, one-pot (two-stage) method for preparing eleven exemplary ArICl2 from the respective arenes, Ar-H, used as the starting substrates to afford ArICl2, isolated in 46-88% crude yields, which were possibly optimized. Ar-H were, at first, oxidatively substituted in appropriate, anhydrous I2/NaIO4 or NaIO3/AcOH/Ac2O/conc. H2SO4 mixtures (discussed previously in Section 2.6), with some transient iodine(III) species, denoted as I3+, to form in situ the respective organic iodine(III) intermediates, denoted as ArISO4. Formerly [17], the resulting reaction mixtures, containing ArISO4 intermediates, were poured into excess aq. Na2SO3 solutions to afford iodoarenes, ArI (see Eq. 4) In our work [18c], ArICl2 were precipitated from the same, resulting (final) reaction mixtures, containing ArISO4 intermediates, by adding to them a large excess of conc. hydrochloric acid. Thus, the essence of this quite novel method is following:
![Molecules 05 01331 i020]()

The yellow precipitates were collected by filtration, washed well with ice-cold water, a little CCl4, and air-dried in the dark.
When NaIO4 was used as the oxidant, the following RC6H4ICl2 were obtained: R = H (88%), 4-F (70%), 4-Cl (70%), 4-Br (75%), 4-I (81%), 3-COOH (86%), 3-COOMe (69%), 3-COOEt (74%), 3-CF3 (57%), 4-OMe (57%); also 2,4-Cl2C6H3ICl2 (46%) was obtained from 1,3-C6H4Cl2. When NaIO3 was used as the oxidant, the following RC6H4ICl2 were obtained: R = H (87%), 4-Br (63%), 3-COOH (80%), 3-COOMe (64%). Of course, only those isomeric RC6H4ICl2 may predominantly be obtained from the monosubstituted benzenes, RC6H5, which are formed in agreement with common orientation rules in the electrophilic substitutions of RC6H5 with strongly electrophilic I3+ transient species. We failed to synthesize ArICl2 from toluene, phenetole, 4-nitroanisole, 4-nitrotoluene, 2-methylbenzaldehyde, 2-methoxybenzaldehyde, 4-chlorobenzaldehyde, 4-toluic acid, and 4-bromobenzoic acid. Further studies are necessary to establish the limits of the scope of this novel method, which is evidently environmentally benign - hence it would be particularly suitable for large-scale preparations of ArICl2, e.g. PhICl2 (cf. Ref. 59).
4. Syntheses of (Diacetoxyiodo)arenes
(Diacetoxyiodo)arenes, ArI(OAc)2, and particularly the parent (diacetoxyiodo)benzene, PhI(OAc)2, have been known for a long time [21, 56]. They are potent, often fairly selective, oxidizing agents, hence the interest in ArI(OAc)2 and PhI(OAc)2 is growing rapidly, as demonstrated by a number of recent books [21] and reviews [69]. They are also used for the facile syntheses of, for example, [bis(trifluoroacetoxy)iodo]arenes, [hydroxy(tosyloxy)iodo]arenes (selective oxidants), aromatic iodonium salts (arylating reagents), etc. [21, 69].
There are several preparative methods for these compounds. So far the substrates have generally been [21, 56, 69]:
(i) iodosylarenes dissolved in glacial acetic acid;
(ii) (dichloroiodo)arenes in which the chlorine atoms are exchanged by acetoxy groups coming either from silver, lead(II) or sodium acetate, or from acetic acid in the presence of mercury(II) oxide in chlorinated solvents [61];
(iii) iodoarenes are oxidized in warm glacial acetic acid by either peracetic acid, or sodium perborate [51], or electrolytically.
The standard, and most general, method for the synthesis of ArI(OAc)2 (oxidative diacetoxylation of ArI by warm peracetic acid) is, in fact, a very prolonged reaction (12-16 hours), and the utmost care should be taken to maintain the exact temperature, 40°C [21b]. ArI(OAc)2 are generally crystalline compounds, fairly stable in the air, which may be stored for long periods by avoidance of light, and preferably in a cooler.
4.1. A Two-Step Conversion of Iodoarenes to (Diacetoxyiodo)arenes with Chromium(VI) Oxide as Oxidant [10]
In our earlier papers [3, 4, 5, 6b, 9] many short-cut syntheses of diaryliodonium salts were reported (for more details see Section 6). We oxidized various ArI (excluding those substituted solely with stronger electron-donating groups, e.g. OMe, NHAc) with appropriate mixtures of anhydrous CrO3/ AcOH/Ac2O/conc. H2SO4, immediately followed by the acidic coupling of the in situ formed iodine (III) intermediates, ArISO4 and/or ArI(OSO3H)2, with many activated arenes, Ar’-H. The soluble diaryliodonium hydrogensulfates thus obtained, Ar(Ar’)I+HSO4 -, were next precipitated out in the form of insoluble diaryliodonium bromides, iodides, or perchlorates. Alternatively, by pouring the deep-green solutions containing Cr(III) salts and ArISO4 and/or ArI(OSO3H)2 into excess aq. Na2SO3 solutions, the respective ArI were obtained in high yields; see Eqs. 3 and 4 in Section 2.3.
In our work [10] we applied the same method to the oxidation of seventeen ArI with the appropriate CrO3/AcOH/Ac2O/conc. H2SO4 liquid systems, followed by mixing the resulting deep-green reaction mixtures with excess 20% aq. ammonium acetate solutions. Crude crystalline ArI(OAc)2, obtained according to Eqs. 24 and 25, viz.
![Molecules 05 01331 i021]()
![Molecules 05 01331 i022]()


were admixtured with a little of the hydrolyzed yellowish side products, viz. ArI(OH)OAc, ArIO, and [ArI(OAc)]2O. Crude yellowish ArI(OAc)2 were washed on the filter with a cold 10% aq. AcOH (in which they are less soluble than in water), air-dried in the dark, and recrystallized from either AcOH or AcOMe mixed with Ac2O (9:1) to acetylate the yellowish side products back to ArI(OAc)2. Then, either hexane or Et2O were added in excess to the cooled solutions to improve the crystallization yields. After washing on the filter with hexane or Et2O, the final yields of pure ArI(OAc) were 58-82%. This method is unsuitable for iodoanisoles and iodoacetanilides. 4-Iodotoluene gave 4-(diacetoxyiodo)toluene in only 20% yield; as a side product we identified 4-iodobenzoic acid (40%), the unreacted 4-IC6H4Me as well as 4-tolyl acetate by TLC. Pure Ph(OAc)2 was obtained from PhI in 79% yield. Taking into account the reaction times as well as total amounts, and the respective costs, of all the reagents and solvents necessary to prepare pure ArI(OAc)2, our method [10] is 8-16 times faster (30 min as compared with 4-8 h) and ca. 5 times less expensive than the method of McKillop and Kemp [51].
4.2. (Diacetoxyiodo)arenes from Iodoarenes with Sodium Perborate Monohydrate as Oxidant [71]
McKillop and Kemp [51] oxidized thirteen ArI to the corresponding ArI(OAc)2 with NaBO3•4H2O (900% excess) in a large volume of glacial AcOH within 4-8 hours, at 40-45°C. After recrystallizations from AcOH/hexane or cyclohexane, they obtained pure ArI(OAc)2 in 66-80% yields; no attempt was made to optimize yields.
In our unpublished work [71] we used acetic anhydride as the solvent of choice and NaBO3•H2O as the oxidant, which allowed to lower considerably an excess of the oxidant applied to only 200%. The reactions underwent as follows:
![Molecules 05 01331 i023]()

This work is still in progress and will be published soon.
4.3. (Diacetoxyiodo)arenes from Iodoarenes with Sodium Periodate as Oxidant [18e]
The essence of this novel method is following:
![Molecules 05 01331 i024]()

The parent pure PhI(OAc)2 was obtained from PhI in 73% yield; this method is not applicable for ArI substituted with strong electron-withdrawing groups. The presence of sodium acetate (in stochiometric quantities) in the reaction mixtures is indispensable - without its addition the oxidation reactions did not proceed. When sodium acetate was replaced for pyridine, then the final yields of ArI(OAc)2 were lowered by ca. 10-20%. The purities and homogeneities of the purified ArI(OAc)2 were checked by TLC, mixed melting points with authentic specimens as well as with 1H-NMR spectra and elemental analyses. This novel method nicely complements our former method [10], which was hardly applicable for iodotoluenes and quite inappropriate for ArI substituted with stronger electron-donating groups, e. g. iodoanisoles and iodoacetanilides.
5. Syntheses of Iodylarenes
Rather few iodylarenes, ArIO2, have been used as useful oxidants in organic synthesis. These include iodylbenzene, PhIO2, and some of its substituted derivatives, e.g. 3-iodylbenzoic acid or 4-tert-butyliodylbenzene. In some cases, the reactivity of PhIO2 was increased in the presence of CF3CO2H. But their former limited applications were partly due to their polymeric nature which makes them insoluble in most ordinary solvents, excepting water.Their synthetic utility is at present quickly growing and is more appreciated. The most applied reagents of iodine(V) are: cyclic “2-iodylbenzoic acid” (1-oxido-1-hydroxy-1,2-benziodoxol-3(1H)-one) and particularly the Dess-Martin reagent (1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one), which synthetic applications, as versatile selective oxidants, are growing nearly exponentially; for more details see Refs. [21, 69a]. ArIO2 are also useful in the preparation of iodonium salts, Ar2I+X- or Ar(Ar’)I+X- [56, 72, 73].
ArIO2 are polymeric and they not dissolve in ordinary organic solvents. They are rather stable thermally, but their melting points are, in fact, their decomposition points, accompanied by explosion. A violent decomposition of PhIO2 (dry sample) has been induced by scraping with a spatula [21b].
Sharefkin and Saltzman [74] briefly summarized the former methods of preparing PhIO2 as follows (see their Refs. 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14): “PhIO2 has been prepared by the disproportionation of PhIO, by oxidation of PhIO with hypochlorous acid or bleaching powder, and by oxidation of PhI with hypochlorous acid or with sodium hydroxide and bromine.Other oxidizing agents used with PhI include air, chlorine in pyridine, Caro’s acid, conc. chloric acid, and peracetic acid solution. Hypochloric oxidation of PhICl2 has also been employed”.Their one-step method of preparing PhIO2 (which seems general for ArIO2, at least those with electron-releasing substituents) depended on the oxidation of PhI with commercial 40% peracetic acid, which resulted in pure PhIO2 (72-80%). Indian chemists [75] also briefly reviewed the former methods of preparing ArIO2 (see their Refs. 1, 2, 3, 4, 5, 6, 7, 8), and made the following comments: “The reported methods of Lucas and Kennedy [76] for the preparation of PhIO2 from PhI proved to be lenghty and irksome in our hands while that of Sharefkin and Saltzman [74] was neither economical nor free of danger for the use of peracetic acid, though the yield of the desired product in this single step reaction was excellent. Other effective oxidizing agents employed are oxygen, perbenzoic acid, potassium peroxymonosulfate,...and dibenzoyl peroxide and iodine;...also the preparation of ArIO2 from ArI was reported using nitric acid and trifluoroacetic anhydride followed by hydrolysis of the intermediate PhI(OCOCF3)2”, and next followed by steam-distillation. They developed the preparation of ArIO2 by the oxidation of six ArI using KBrO3 under acidic conditions, which afforded the following ArIO2: PhIO2 (45%), 4-BrC6H4IO2(98%), 3-O2NC6H4IO2 (46%), 4-O2NC6H4IO2 (88%), 2-HOOCC6H4IO2 (95%), and 3-MeC6H4IO2 (poor yield). KBrO3 and dilute H2SO4 produced PhIO2 from PhIO in excellent yield.
The treatment of halobenzenes, benzoic acid, or nitrobenzene, dissolved in unspecified solvents, with HIO3 and conc. H2SO4 possibly resulted in the formation of the respective iodylarenes, ArIO2; next, they may be reduced to the corresponding iodides [77]. This short communication, with no experimental and physico-chemical support, was never followed up either then or after 1969 by the more detailed experimental paper. This should be checked and, possibly, extended, because this direct method seems to be promising.
A considerable progress in the synthesis of ArIO2 is greatly due to a quickly growing importance of “2-iodylbenzoic acid” and its triacetoxy derivative, i.e. the Dess-Martin reagent (vide supra). From the last short review [78] it is seen that they were obtained, most satisfactorily, in the following ways [79, 80]:
![Molecules 05 01331 i025]()

The monoacetylated form of the above “2-iodylbenzoic acid”, i.e. 1-acetoxy-1,2-benziodoxol- 3(1H)-one-1-oxide, is probably the actual oxidizing species [81]. Note. “2-Iodylbenzoic acid” is explosive on heating above 200°C and also upon impact; the Dess-Martin reagent explodes violently on heating under confinement, at 130°C [21b].
5.1. Early Results: Biphasic Oxidation of Iodobenzene to Iodylbenzene [64]
In 1990/1991 a biphasic oxidation of PhI to PhIO2 (77%), in the presence of a phase transfer catalyst, Bu4N+Br-, was performed as follows [64]. PhI was dissolved in CCl4, while excess NaOCl and a catalytic amount of Bu4N+Br- were dissolved or suspended in water. The both phases mixed together in the reaction vessel were vigorously stirred and warmed up to boiling under a reflux condenser. 800% excess conc. hydrochloric acid was slowly added dropwise to the boiling mixture within a few hours. After cooling, crude PhIO2 was collected, washed with water, and recrystallized from boiling water. KMnO4 used as the oxidant was somewhat less effective, and yielded crude PhIO2 contaminated with MnO2. Since Dr. J. Golinski left our laboratory, this work has not been continued.
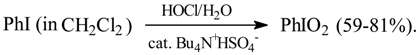
5.2. Iodylarenes from Iodoarenes with Sodium Periodate as Oxidant [18e]
The heterophasic reactions between various iodoarenes suspended in boiling aq. NaIO4 solutions proceeded smoothly within 8-16 hours to give the corresponding, colorless iodylarenes in 58-91% crude yields, viz.
![Molecules 05 01331 i027]()

When the above reactions were completed, the cooled reaction mixtures were diluted with cold water. The collected precipitates were washed on the filter with cold water, next with CHCl3 or acetone (to remove the unreacted ArI), and air-dried in the dark. The following crude RC6H4IO2 were obtained (yields, %): R = H (86), 4-OMe (85), 2-Me (61), 3-Me (77), 4-Me (80), 4-F (91), 3-Cl (75), 4-Cl (80), 4-Br (73), 3-NO2 (85), 4-NO2 (58), and 3-COOH (89). Iodometric titrations [56, 97] showed that the contents of ArIO2 in the crude products varied between 98.5 and 99.2%. Small samples of the crude products were recrystallized from boiling water to give the analytically pure specimens, which melting/detonation points were close to those reported in the literature. Their IR spectra displayed the characteristic frequencies for iodylarenes [21a].
The reaction mixtures discussed above were nearly neutral. When they were made alkaline, the unreactive Na3H2IO6 precipitated out, and the reactions did not proceed. The same negative results were obtained, when NaIO4 was replaced with NaIO3 used as the oxidant under the same reaction conditions. When the reaction mixtures discussed above were acidified with either acetic acid or aq. H2SO4, then the yellow colored iodosylarenes, ArIO, were the main products of the reactions; they were not studied further.
Exceptionally, when 2-iodobenzoic acid was oxidized as above with boiling aq. NaIO4 solution, then “2-iodosylbenzoic acid” (i.e. 1-hydroxy-1,2-benziodoxol-3(1H)-one) was isolated in 76% crude yield. After recrystallization from boiling water, its structure was confirmed as above. Contrariwise, when sodium salt of 2-iodobenzoic acid was reacted as above with boiling aq. NaIO4 solution, then this resulted in the formation of “2-iodylbenzoic acid” (i.e. 1-hydroxy-1,2-benziodoxol-3(1H)-one-1-oxide), isolated in 71% crude yield. After recrystallization from boiling water, its chemical structure was supported analytically, spectrally (IR), and with its characteristic melting/detonation point. Further informations will be given in our just completed paper [18e].
6. Syntheses of Diaryliodonium Salts
Symmetric and unsymmetric diaryliodonium salts, Ar2I+X- and Ar(Ar’)I+X-, represent a very important class of aromatic iodine(III) compounds. They are widely used in organic synthesis as arylating reagents for organic and inorganic nucleophiles, some of them display a biological activity and photochemical properties, hence they are often applied as efficient catalysts for radiation initiated polymerization [21a,b]. Generally, their reactivity is less pronounced than that of other hypervalent iodine compounds. In several of their reactions relatively drastic conditions may be necessary, especially for the least reactive heterocyclic iodonium salts. The search for optimum conditions is often desirable even for well-established reactions, by applying new findings concerning the use of specific solvents, catalysts or radical traps [21b].
Diaryliodonium salts are generally solid compounds, mostly stable towards heat, oxygen and humidity; they are mildly light-sensitive and should be stored in the dark, without refrigeration [21b].
A large number of methods are available for the preparation of diaryliodonium salts. A brief account of the most often used methods is given by Varvoglis in his latest book [21b]; for more details see Refs. 21a, 69a, 83, 84.
6.1. Short-Cut Syntheses of Diaryliodonium Salts with Chromium(VI) Oxide as Oxidant [3, 4, 5, 6b, 9, 13]
In 1995 we reported [3] a one-pot (short-cut) synthesis of sparingly soluble diaryliodonium bromides from various ArI oxidized with anhydrous CrO3/AcOH/Ac2O/conc. H2SO4 mixtures, then coupled in situ with various arenes and, finally, precipitated with excess aq. KBr solutions. The essence of this novel method is shown below (for experimental details see [3]).
![Molecules 05 01331 i028]()

For X = H, CH3, Cl, NO2; if X = OCH3 or NHCOCH3, effective oxidation was not achieved.
Ar-H = benzene, toluene, mesitylene, anisole, chlorobenzene, acetanilide, anisic acid, and thiophene.
We have established that our one-pot method is unsuitable to couple the oxidized iodoarenes with nitrobenzene, benzoic acid and its methyl and ethyl esters, benzonitrile, acetophenone, benzophenone, cinnamic acid and its ethyl ester, beta-nitrostyrene, and chalcone under reaction conditions given in our paper [3].
The soluble diaryliodonium hydrogensulfates, Ar(Ar’)I+HSO4 -, formed in the last stage were metathesized into the insoluble bromides by adding excess aq. KBr to the final, deep-green reaction mixtures, also containing the soluble chromium(III) salts. The chromium salts were completely washed off with water on the filter, followed with acetone (which removed organic impurities), leaving the nearly pure diaryliodonium bromides in 20 - 88% crude yields. The sparingly soluble diaryliodonium iodides or perchlorates may also be obtained by the same one-pot method by adding excess aq. KI or HClO4 to the final, deep-green reaction mixtures; they were obtained in 40 - 89% crude yields.
Note. Mgr Anna Kryska from our laboratory has recently established that CrO3 (Eq. 28) may be replaced by finely powdered K2Cr2O7, with preserving the same crude yields of the iodonium bromides/iodides.
There are repeated reports in the literature [84, 85] that diaryliodonium halides are thermally unstable on prolonged heating, hence their recrystallizations should be carried out as quickly as possible from boiling solvents. Analytical purity of the iodonium bromides and iodides obtained by us was achieved by quick dissolving small crude samples in boiling MeOH, followed by the immediate hot filtration neglecting the losses, and rapid cooling. More stable thermally iodonium perchlorates were recrystallized in usual way; after drying they should be handled with care.
The aforementioned new method [3] is easier and shorter than many earlier methods [21, 69a, 72, 83, 84], hence this method was also used in our subsequent work [4, 5, 6b, 9, 13] (see Section 6.2 and Section 6.3).
In the same paper [3] and in the following paper [6b] we reported the oxidative metatheses in the crude iodonium bromides, iodides and chlorides into the corresponding pure tetrafluoroborates, tosylates, trifluoroacetates, etc. This will be dealt with in Section 7.1.
6.2. Iodonium/Diiodonium Salts Derived from Various Tricyclic Carbo- and Heteroaromatic Systems [4, 5, 9]
A considerable number of iodonium/diiodonium salts, mostly isolated from the final reaction mixtures in the form of sparingly soluble bromides/dibromides were synthesized, and their physical characterics, after the purification, as well as the IR and 1H NMR spectra were reported. The following tricyclic aromatic systems were covered:
Nearly all the iodonium salts synthesized were new. The aim of this work was to obtain several tens novel iodine(III) derivatives of the aforementioned tricyclic aromatic systems, which previously had scarcely ever been covered by the organic chemistry of polyvalent iodine. The crude iodonium bromides / dibromides obtained, after their effective oxidative metatheses into e.g. pure tetrafluoroborates (see Section 7.1), would next be reacted with various nucleophiles (inorganic and organic) to produce the corresponding mono- or disubstituted derivatives of the above tricyclic aromatic systems.
6.3. Improved Syntheses of Some Diaryliodonium Salts from Symmetric Diarylmercurials and (Dichloroiodo)arenes (Willgerodt Method) [13]
Willgerodt [56, 86] had reacted cold (or hot [86a]) aqueous suspensions of equal masses [it practically means that the mercurials were used in a deficit] of powdered PhICl2 with powdered Ar2Hg (where Ar = phenyl, 2- and 4-tolyl, and 2-naphthyl) to afford the respective diaryliodonium chlorides (yields were not reported); sparingly soluble ArHgCl and other admixtures, e.g. PhIO [87], were hot-filtered off and discarded, viz.
![Molecules 05 01331 i029]()

Beringer and Lillien [88] applied the Willgerodt method to obtain three unsymmetric diaryliodonium chlorides. They obtained only 4-acetamidophenyl(phenyl)iodonium chloride (which was precipitated out as its sparingly soluble iodide, isolated in 10% crude yield) by reacting equimolar amounts of Ph2Hg with 4-AcNHC6H4ICl2 in hot water (40-50°C) for 12 hours. We obtained [13] the same iodide, but in 80% crude yield, by reacting equal masses of PhICl2 with symmetric 4,4’-mercuriobis(acetanilide) suspended in stirred hot water (40-50°C) for 12 hours. We explained this evident yield increase as follows:
![Molecules 05 01331 i030]()

Consequently, by reacting equal masses of PhICl2 with symmetric 4,4’-mercuriobis(N,N-dimethylaniline), suspended in stirred hot water (40-50°C) for 12 hours, we isolated from the hot filtrate, after its cooling, 4-dimethylaminophenyl(phenyl)iodonium chloride in 60% crude yield. Previously, Beringer and Lillien [88] failed to obtain this iodonium salts, para-substituted with only one NMe2 group; the same failure was also reported by Neiland [89]. A similar iodonium salt bearing the two p,p’-substituted NMe2 groups was synthesized quite otherwise [90]; this synthesis is shown (Scheme 7) in our paper [13].
We also attempted, without effect, to synthesize various 8-(aryliodonio)caffeine halides with using the Willgerodt method. Hence, we used our short-cut, oxidative method [3] (see Section 6.1) to obtain 8-(4-methoxyphenyliodonio)caffeine bromide (49% crude yield) by acidic coupling of the oxidized 8-iodocaffeine with anisole. It is, in fact, the first iodine(III) derivative of caffeine, which may open novel routes for preparing 8-substituted caffeines by its reactions with various nucleophiles [21, 69, 84].
6.4. Short-cut Syntheses of Diaryliodonium Bromides with Sodium Perborate as Oxidant [96]
This novel method is just completed and will be published soon. Its essence is as follows:
- (1)
- ArI + NaBO3•H2O + 2Ac2O + 2H2SO4 → ArI(OSO3H)2 + [NaBO2] + 4AcOH
- (2)
- ArI(OSO3H)2 (not isolated) + Ar’-H (in excess) → Ar(Ar’)I+HSO4 - + H2SO4
- (3)
- Ar(Ar’)I+HSO4 - (not isolated) + aq. KBr (in excess) → ↓ Ar(Ar’)I+Br- + KHSO4 (Eq. 30)
Powdered NaBO3•H2O (used in the 200% excess) was suspended in Ac2O and this suspension was stirred for 30 min, at 30°C. Appropriate ArI (0% excess) followed by Ar’-H (used in the 400% excess) were slowly added at 40°C with stirring, and next the whole was cooled to 10°C. Varied amounts of conc. H2SO4 were slowly added dropwise with stirring while keeping the temperature below 15°C. The stirring was continued for a further 5-7 h while keeping the temperature at 15°C; some acidic couplings were carried out at 25-30°C. The mixtures were poured into ice water, followed by the extraction with Et2O to remove the unreacted Ar’-H and other organic impurities. Excess aq. KBr was added to the aqueous layers to precipitate the respective bromides, Ar(Ar’)I+Br-. They were collected by filtration, washed well on the filter with water, then with acetone and Et2O, and air-dried in the dark; the crude bromides were obtained in 45-91% yields. Small crude samples were quickly recrystallized from boiling MeOH to give the analytically pure specimens.
7. Anion Metatheses in Diaryliodonium Halides
One of the most important applications of diaryliodonium salts is their use in the arylation of organic and inorganic nucleophiles [21, 69a, 84, 91], including the fluoride anion, which results in the formation of aromatic fluorides [92]. It has been observed [21, 84, 91] that the best arylation yields of the nucleophilic bases are achieved by applying the diaryliodonium salts with substantially non-nucleophilic counterions, viz. tosylates, triflates, hydrogensulfates, perchlorates, etc.; the direct synthesis of diaryliodonium triflates has been published by Japanese workers [93]. Beringer and co-workers [91] have observed the superior yields with diphenyliodonium tetrafluoroborate in the phenylation of organic and inorganic bases; several diaryliodonium tetrafluoroborates were directly synthesized by Neiland [89]. The sparingly soluble perchlorates as well as bromides, iodides, picrates, tetraphenylborates, and 2,4,6-tribromobenzenesulfonates [72] were often nearly quantitatively precipitated out from hot solutions or suspensions of more soluble diaryliodonium chlorides or hydrogensulfates by the solutions of cheap sodium, potassium, ammonium or calcium salts with an appropriate counterion. However, a considerable number of necessary diaryliodonium salts were only attainable from the corresponding halides or hydrogensulfates by using costly and toxic lead(II), silver and barium salts, e.g. silver tetrafluoroborate, trifluoroacetate and nitrate, or lead(II), silver and barium organosulfates or organocarboxylates, or otherwise [21, 72, 84]. These approaches have been briefly reviewed [ 21b, 84].
Ionic inorganic and organic halides, except of fluorides, in hot aqueous or boiling alcoholic solutions (acidified with e.g. H2SO4) were more or less readily oxidized (I- > Br- > Cl-, fluorides do not react) by an excess of 30% aq. H2O2, with evolution of the respective halogens [94]. We have applied this information to effect a variety of the oxidative anion metatheses in a large number of crude diaryliodonium bromides and iodides, which were formerly precipitated out from the final reaction mixtures with excess aq. KBr or KI solutions [3, 4, 5, 6b, 9, 13] (see Section 6.1, Section 6.2 and Section 6.3). This is explained below in Section 7.1 and Section 7.2.
7.1. Oxidative Anion Metatheses in Crude Diaryliodonium Bromides [3]
In our paper [3] we have reported various oxidative anion metatheses in the crude diaryliodonium bromides, previously obtained by us with the one-pot protocols discussed in Section 6.1, which produced the corresponding pure diaryliodonium tetrafluoroborates, tosylates, trifluoroacetates, hydrogensulfates, nitrates, and chlorides in 57-80% yields. These new preparative procedures are easier and shorter, and also less expensive, than many earlier methods.
The essence of our oxidative anion metatheses is given below:
![Molecules 05 01331 i031]()

Eleven crude diaryliodonium bromides were suspended with stirring in pure MeOH (unless otherwise stated) mixed with the 100% excess of cyclohexene (acting there as a “halogen scavenger”), then an appropriate strong acid, HX (see above), was added with stirring, followed by 30% aq. H2O2 used in the 196% excess. The mixtures were stirred and boiled under a reflux condenser until complete dissolution of all the reaction components. The reflux was prolonged for a further 15 min. The solvent was distilled off under vacuum, and the crude products left in the flask were triturated with either Et2O or acetone to remove any organic impurities. The following individual workups and recrystallizations resulted in the analytically pure iodonium salts, Ar(Ar’)I+X-. For the metathesis into trifluoroacetates, a catalytic amount of ammonium molybdate was added to the reaction mixtures. For the metathesis into nitrates, 50% aq. AcOH at 70°C was used as the solvent, instead of boiling MeOH. For the metathesis into chlorides, a much slower competitive reaction also occurs [2HCl + H2O2 → 2H2O + Cl2 (captured by cyclohexene)], hence somewhat increased amounts (by ca. 10%) of H2O2, HCl, and cyclohexene were used.
Note. Dr. Pawel Kazmierczak has recently established that dry acetone used as the solvent of choice in the oxidative anion metatheses of crude diaryliodonium bromides and chlorides (see Section 7.2) may act itself as a very efficient “halogen scavanger”, hence the addition of cyclohexene to the respective reaction mixtures is not necessary.
7.2. Oxidative Anion Metatheses in Crude Diaryliodonium Iodides and Chlorides [6b]
The oxidative anion metatheses, similar to those reported in Section 7.1., in six crude diaryliodonium iodides and two chlorides produced the corresponding pure hydrogensulfates, nitrates, tetrafluoroborates, triflates, tosylates, as well as bromides and chlorides (only from the iodides) in 54-86% yields. By using the modified oxidative metatheses in the iodides (in the presence of HBr or HCl, but in the absence of cyclohexene) it was possible either to isolate, or to detect only, the intermediate dihaloiodates(I), [Ar2I]+[IX2]- (X = Br or Cl). A complex Ph2I+Cl-•1/2I2 was also obtained in 56% yield. Tetraethylammonium iodide, Et4N+I-, was similarly converted into pure tetrafluoroborate or dibromoiodate(I) in 75 and 76% yields, respectively.
The essence of our oxidative anion metatheses is as follows:
![Molecules 05 01331 i032]()

Y = I or Cl; X = HSO4, NO3, BF4, CF3SO3, TsO, as well as Cl, Br (only when Y = I).
As previously Br2 [3], also molecular chlorine, Y2 = Cl2, evolved in the oxidative reactions, was fully captured by the 100% excess of cyclohexene added on purpose as a “halogen scavanger”; 1,2-dichlorocyclohexane as well as the other possible cyclohexene adducts with reactive agents (H2O2, ClO+H2, etc.) were next removed from the crude metathesized products by their prior trituration with anhydrous Et2O, followed by washing on the filter with the same solvent. For the diaryliodonium iodides, the diiodine, Y2 = I2, evolved was simply washed off from the crude metathesized products with anhydrous Et2O. The following recrystallizations (see [6b] for details) gave analytically pure metathesized products.
Diaryliodonium chlorides were oxidized by H2O2 in acidified, boiling alcoholic solutions much less readily (3 h; 1076% excess of H2O2 added portionwise) than the respective bromides [3] (15 min; 196% excess of H2O2) and iodides (10 min; 36% excess of H2O2); the final yields of the respective pure metathesized products were 54-76, 57-80 [3], and 54-86%, respectively.
In our earlier work [3] we metathesized oxidatively two aryliodonium bromides into the respective chlorides with no comments. However, in our second work [6b] we found that such oxidative reactions are more complex than expected. This is likely to be due to a series of fast consecutive reactions taking place in the investigated solutions of diaryliodonium iodides containing H2O2 and acidified with excess HBr or HCl, but in the absence of cyclohexene, (a)-(d):
![Molecules 05 01331 i033]()

It is known that both diaryliodonium iodides and tetraalkylammonium iodides are easily converted into the respective dihaloiodates(I) either by the reaction of Cl2 or Br2 with the iodides, or by the reaction of IBr or ICl with the corresponding bromides or chlorides. For more details see Ref. 21a, Chap. 7.
By carrying out the oxidative anion metatheses in either diphenyliodonium iodide or tetraethylammonium iodide with excess HBr, we succeeded in the isolation of the intermediate, deep-orange colored, dibromoiodates(I) in 56 and 76% yields, respectively. The other diaryliodonium iodides gave only oily or semisolid, deep-yellow or deep-orange mixtures of dihaloiodates(I) with the respective diaryliodonium bromides or chlorides, when their oxidative metatheses were performed in the presence of excess HBr or HCl, but in the absence of cyclohexene. These mixtures may be easily converted into the unmixed bromides or chlorides in 86-92% crude yields by short (5 min) refluxing in anhydrous MeOH admixtured with excess cyclohexene (see Eq. 33 below). This procedure for converting the dihaloiodates(I) into the corresponding diaryliodonium bromides or chlorides (X = Br or Cl) is more effective than that offered in the literature [91, 95]. This involved chlorination of the diaryliodonium iodides to the dichloroiodates(I) or tetrachloroiodates(III), which when dissolved in hot acetone reacted to give the iodonium chlorides.
![Molecules 05 01331 i034]()

where: HX = HBr or HCl; only diphenyliodonium dibromoiodate(I) was isolated in 56% yield as the intermediate. 1-X(Cl or Br)-2-iodocyclohexanes were washed off in full from the crude products, Ar(Ar’)I+X-, with anhydrous diethyl ether.
Finally, we also performed some “model” study [6a] on quite similar oxidative anion metatheses in boiling methanolic solutions of KI, KBr, and KCl, acidified with H2SO4, HBF4 or p-toluenesulfonic acid. Each of the halides was effectively converted into pure potassium hydrogensulfate, tetrafluoroborate, and tosylate. This also confirms the usefulness of the presented method in inorganic chemistry.
8. Conclusions
This review shows our small research group’s main interests in developing novel (or considerably improved) preparative procedures, which are suitable for easy, quick, cheap, effective, and possibly environmentally benign preparations of iodoarenes and some basic organic hypervalent iodine reagents: ArICl2, ArI(OAc)2, ArIO2, and diaryliodonium salts with substantially non-nucleophilic counterions (suitable for the arylation of organic and inorganic nucleophiles). We hope that some of our methods will be applied as such in other organic laboratories and, possibly, in the pharmaceutical industry, but after their enlargements worked up in the experienced and well-equipped industrial laboratories; see e.g. Ref. 59.
References
- A list of our papers and Ph.D. theses on Organic Iodine(I, III and V) Chemistry:
- Krowczynski, A.; Skulski, L. p-Iodo-N-methylacetanilide and its Polyvalent Iodine Derivatives. Polish J. Chem. 1993, 67, 67–70. [Google Scholar]
- Krassowska-Swiebocka, B.; Lulinski, P.; Skulski, L. Aromatic Iodination of Activated Arenes With Lead Tetraacetate as the Oxidant. Synthesis 1995, 926–929. [Google Scholar] [CrossRef]
- Kazmierczak, P.; Skulski, L. A Short-Cut Synthesis of Diaryliodonium Bromides Followed by Oxidative Anion Metatheses. Synthesis 1995, 1027–1032. [Google Scholar] [CrossRef]
- Farhan, A.N.; Skulski, L. 2- and 2,8-Substituted Iodine(III) Derivatives of Dibenzofuran. Polish J. Chem. 1995, 63, 1663–1668. [Google Scholar]
- Farhan, A.N.; Skulski, L. Synthesis of N-acyl-3-(Br, Cl or NO2)-6-(aryliodonio)carbazole Bromides. Polish J. Chem. 1996, 70, 211–216. [Google Scholar]
- Two complementary papers: Farhan, A.N.; Kryska, A.; Skulski, L. Exemplary Oxidative and Evaporative Anion Metatheses in Potassium Halides. Bull. Polish Acad. Sci., Chem. 1996, 44, 201–203. [Google Scholar] Kazmierczak, P.; Skulski, L. Oxidative Anion Metatheses in Diaryliodonium Iodides and Chlorides. Bull. Chem. Soc. Jpn. 1997, 70, 219–224. [Google Scholar] [CrossRef]
- Abdo Nagi Farhan (Yemen), Ph.D. thesis entitled: “Syntheses of novel aromatic iodonium salts substituted with various carbo- and heterocyclic groups” [in Polish]; Fac. of Pharm., Med. Acad., Warsaw 1996, 146 pp. + Addenda, 173 refs. Defended on March 5, 1997.
- Lulinski, P; Skulski, L. The Direct Iodination of Arenes with Chromium(VI) Oxide as the Oxidant. Bull. Chem. Soc. Jpn. 1997, 70, 1665–1669, Erratum. See Ref. 11, p. 118. [Google Scholar]
- Farhan, A.N.; Skulski, L.; Kryska, A. 2- and 2,7-Substituted Iodonium/Diiodonium Salts Derived from Xanthene, Xanth-9-one, Fluorene, and Fluoren-9-one. Polish J. Chem. 1997, 71, 1236–1245. [Google Scholar]
- Kazmierczak, P.; Skulski, L. A Simple, Two-Step Conversion of Various Iodoarenes to (Diacetoxyiodo)arenes with Chromium(VI) Oxide as the Oxidant. Synthesis 1998, 1721–1723. [Google Scholar] [CrossRef]
- Lulinski, P; Skulski, L. Oxidative Iodination of Arenes with Manganese(IV) Oxide or Potassium Permanganate as the Oxidants. Bull. Chem. Soc. Jpn. 1999, 115–120. [Google Scholar]
- Krassowska-Swiebocka, B.; Prokopienko, G.; Skulski, L. Biphasic Chlorination of Iodoarenes to (Dichloroiodo)arenes. Synlett 1999, 1409–1410. [Google Scholar] [CrossRef]
- Skulski, L.; Wroczynski, P. Improved Syntheses of Some Diaryliodonium Salts from Symmetric Diarylmercurials and (Dichloroiodo)arenes (Willgerodt Method). Bull. Polish Acad. Sci., Chem. 1999, 47, 231–238. [Google Scholar]
- Kazmierczak, P.; Skulski, L.; Obeid, N. Oxidative Chlorination of Various Iodoarenes to (Dichloro-iodo)arenes with Chromium(VI) Oxide as the Oxidant. J. Chem. Res., Synop. 1999, 64–65. [Google Scholar] [CrossRef]
- Kryska, A.; Skulski, L. Improved, Acid-catalyzed Iodinating Procedures for Activated Aromatics with (Diacetoxyiodo)benzene as the Oxidant. J. Chem. Res., Synop. 1999, 590–591. [Google Scholar] and (M), 2501-2517. In its full-text version there is a review on the aromatic halogenation reactions with organic trivalent iodine reagents as the oxidants.
- Pawel Kazmierczak, Ph. D. thesis entitled: “New synthetic methods for preparing diaryliodonium salts, (diacetoxyiodo)arenes, and (dichloroiodo)arenes” [in Polish], Fac. of Pharm., Med. Acad., Warsaw 1999., 186 pp. + Addenda, 314 refs. Defended on December 8, 1999.
- Lulinski, P.; Skulski, L. Iodination of Both Deactivated and Activated Arenes with Sodium Periodate or Sodium Iodate as the Oxidants. Bull. Chem. Soc. Jpn. 2000, 73, 951–956. [Google Scholar] [CrossRef]
- Papers in press or in preparation for publication: Baranowski, A.; Plachta, D.; Skulski, L.; Klimaszewska, M. Liquid-Phase and Biphasic Chlorination of Some Iodoarenes to (Dichloroiodo)arenes with Sodium Peroxodisulfate as the Oxidant. J. Chem. Res., Synop. 2000, 435–437. [Google Scholar] [CrossRef] Obeid, N.; Skulski, L. Novel Oxidatixe, Liquid-Phase Chlorination Procedures for the Preparation of (Dichloroiodo)arenes from Iodoarenes. Polish J. Chem. 2000, 74, 1609–1615. [Google Scholar] (c)Lulinski, P.; Obeid, N.; Skulski, L. A One-Pot Method for Preparing (Dichloroiodo)arenes from Arenes. (d)Krassowska-Swiebocka, B.; Lulinski, P.; Skulski, L. Chemical Manganese Dioxide (CMD): Its Application to Oxidative Iodination of Benzene, Halobenzenes and Some Deactivated Arenes. (e)Kazmierczak, P.; Skulski, L.; Kraszkiewicz, L. A Facile Synthesis of (Diacetoxyiodo)arenes and Iodylarenes from Iodoarenes with Sodium Periodate as the Oxidant.
- Reviews on Aromatic Iodination: Roedig, A. Houben-Weyl, 4th ed.; Vol. V/4a, Mueller, E., Ed.; Stuttgart, 1960; pp. 517–678. [Google Scholar] Merkushev, E.B. Synthesis 1988, 923. March, J. Advanced Organic Chemistry, 4th ed.; New York, 1992; pp. 531–534. [Google Scholar] Grimmet, M.R. Adv. Heterocyclic Chem. 1993, 57, 291, ibid., 58, 271; ibid., 59, 245. Sainsburry, M. Halobenzenes. In Rodd’s Chem. Carbon Compounds, 2nd ed.; Sainsburry, M. Amsterdam, 1996; Vol. 3, Part 1; pp. 178–224. [Google Scholar] The Merck Index, 12th ed.; CD-ROM, Merck Co., 1998.
- Nesmeyanov, A.N.; Makarova, L.G. Organic Compounds of Mercury; Amsterdam, 1967. [Google Scholar] Makarova, L.G. Organometallic Reactions; Becker, E.I., Tsutsui, M., Eds.; Vol. 1, New York, 1970; pp. 119–348, Vol. 2, New York 1971, pp. 355-423. [Google Scholar] Mueller, E. (Ed.) Methoden der organischen Chemie (Houben-Weyl); Vol. 13(2b), Stuttgart, 1974.
- Varvoglis, A. The Organic Chemistry of Polycoordinated Iodine; Weinheim, 1992. [Google Scholar] Varvoglis, A. Hypervalent Iodine in Organic Synthesis; San Diego, 1997. [Google Scholar] Chemistry of Hypervalent Compounds; Akiba, K. (Ed.) New York, 1999; Chap. 11 and 12.
- Skulski, L.; Wroczynski, P. Polish J. Chem. 1982, 56, 975.
- Skulski, L.; Wroczynski, P. Khim. Geterotsikl. Soed. 1983, 543.
- Wroczynski, P.; Kujawa, A.; Skulski, L. Khim. Geterotsikl. Soed. 1985, 386.
- Khodeir, N.M.; Skulski, L.; Wroczynski, P. Bull. Polish Acad. Sci., Chem. 1986, 34, 443.
- Khodeir, N.M.; Skulski, L.; Wroczynski, P. Bull. Polish Acad. Sci., Chem. 1987, 35, 9.
- Skulski, L.; Kujawa, A.; Kujawa, T.M. Bull. Polish Acad. Sci., Chem. 1987, 35, 499.
- Skulski, L.; Kujawa, A.; Wroczynski, P. Khim. Geterotsikl. Soed. 1989, 249.
- Kandeel, E.; Skulski, L.; Wroczynski, P. Bull. Polish Acad. Sci., Chem. 1990, 38, 37.
- Niemyjska, M.; Skulski, L.; Zdrojewska, H. Bull. Polish Acad. Sci., Chem. 1990, 38, 41.
- Skulski, L.; Kujawa, A.; Wroczynski, P. Bull. Polish Acad. Sci., Chem. 1991, 39, 23.
- Baranowski, A.; Skulski, L. Bull. Polish Acad. Sci., Chem. 1991, 39, 29.
- Skulski, L.; Baranowski, A.; Lempke, T. Bull. Polish. Acad. Sci., Chem. 1991, 39, 459.
- Serguchev, Yu. A.; Davydowa, V.G.; Makhon’kov, D.I.; Cheprakov, A.V.; Beletskaya, I.P. Zh. Org. Khim. 1985, 21, 2010, J. Org. Chem. USSR 1985, 21, 1841.
- Shimizu, A.; Yamataka, K.; Isoya, T. Bull. Chem. Soc. Jpn. 1985, 58, 1611. [CrossRef]
- Grigor’ev, M.G.; Buketova, I. A.; Polevshchikov, P.F. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol. 1987, 30, 22, Chem. Abstr. 1998, 109, 6142.
- Cainelli, G.; Cardillo, G. Chromium Oxidations in Organic Chemistry; Berlin, 1984. [Google Scholar]
- Chaikovskii, V.K.; Novikov, A.N. Zh. Prikl. Khim. 1984, 57, 134, Chem. Abstr. 1984, 100, 191452.
- Chen, J.-A.; . Lin, C.-S.; Liu, L.K. J. Chin. Chem. Soc. (Taiwan) 1996, 43, 95.
- Fieser, L.F.; Fieser, M. Reagents in Organic Synthesis; Vols. 1-18. Oxidation in Organic Chemistry; Wiberg, K.W. (Ed.) New York, 1965. Oxidation. Techniques and Application in Organic Synthesis; Augustine, R.L. New York, 1969. [Google Scholar]
- Galecki, J. Preparatyka Nieorganiczna; Warsaw, 1964; p. 439. [Google Scholar] Karyakin, Yu.V.; Angelov, I.I. Chistye Khimicheskie Reaktivy; Moscow, 1955; p. 333. [Google Scholar]
- Aoyama, T.; Sonoda, N.; Yamaguchi, M.; Toriyama, K.; Anzai, M.; Ando, A.; Schioiri, T. Synlett 1998, 35. see also Refs. 4, 5, 6, 7, 8 therein for their former works in which the CMD (Wako product) was used as the oxidant. Hirano, M.; Yakabe, S.; Hikamori, H.; Clark, J.H.; Morimoto, T. J. Chem. Res. Synop. 1998, (i) Part 1, p. 308; (ii) Part 2, p. 310; (iii) Part 3. 771.
- Fatiadi, A.J. Synthesis 1974, 229. see particularly Table 4 therein, p. 243. Pizey, J.S. Synthetic Reagents; Pizey, J.S. New York, 1981; Vol. 4, pp. 184–187, A brief review on the I2/H5IO6 reagent. [Google Scholar]
- Wirth, H.O.; Koenigstein, O.; Kern, W. Liebigs Ann. Chem. 1960, 634, 84.
- Keys, A.; Johnson, D. R.; Soulen, R. L. CHED Newsletter Fall 1994, Abstr. No.72. In Paper presented at a 208th National Meeting of the American Chemical Society, Washington, D.C., August 21-25, 1994. Brazdil, L.C.; Cutler, C.J. J. Org. Chem. 1996, 61, 9621. Brazdil, L.C.; Fitch, J.L.; Cutler, C.J.; Haynik, D.M.; Ace, E.R. J. Chem. Soc.,Perkin Trans. 2 1998, 933.
- Also polystyrene was previously iodinated with I2/I2O5 reagent, see: Yamada, Y.; Okawara, M. Macromol. Chem. 1972, 152, 153. Togo, H.; Abe, S.; Nogami, G.; Yokoyama, M. Bull. Chem. Soc. Jpn. 1999, 72, 2351 and 2354.
- Suzuki, H. Org. Synth. 1971, 51, 94, or Coll. Vol. VI, p.700.
- Aldrich Catalogue Handbook of Fine Chemicals 1999-2000.
- Chaikovskii, V.K.; Kharlova, T.S.; Filimonov, V.D.; Saryucheva, T.A. Synthesis 1999, 748.
- Togo, H.; Nogami, G.; Yokoyama, M. Synlett 1998, 534.
- McKillop, A.; Kemp, D. Tetrahedron 1989, 45, 3299.
- Ogata, Y.; Aoki, K. J. Am. Chem. Soc. 1968, 90, 6187.
- Niemyjska, M. unpublished results.
- Keefer, R.M.; Andrews, L.J. J. Am. Chem. Soc. 1958, 80, 277.
- Willgerodt, C. Tageblatt der 58. Vers. deutscher Naturforscher u. Aertzte; Strassburg, 1885. [Google Scholar]
- Willgerodt, C. Die organischen Verbindungen mit mehrwertigem Jod. Stuttgart, 1914. [Google Scholar]
- Lucas, H.J.; Kennedy, E.R. Org. Synth.; Coll. Vol. III, New York, 1955; p. 482. [Google Scholar]
- Taylor, R.T.; Stevenson, T.A. Tetrahedron Lett. 1988, 29, 2033.
- Zanka, A.; Takeuchi, H.; Kubota, A. Org. Process Res. Dev. 1998, 2, 270. [CrossRef]
- Toehl, A. Ber. dtsch. chem. Ges. 1893, 26, 2949.
- Karele, B. Ya.; Neiland, O. Ya. Latv. PSR Zinat. Acad. Vestis, Kim. Ser. 1970, 587. Chem. Abstr.1971, 74, 42033.
- Makhon’kov, D.I.; Cheprakov, A.V.; Beletskaya, I.P. Zh. Org. Khim. 1986, 22, 681.
- Koyuncu, D.; McKillop, A.; McLaren, L. J. Chem. Res., Synop. 1990, 21.
- Golinski, J.; Kazmierczak, P.; Skulski, L. communicated at a Meeting of the Polish Chemical Society, Krakow, September 4-7, 1991; Abstracts of Papers, (1). p. 25.
- Nekrasov, B. Textbook of General Chemistry; Moscow, 1969; p. 258. [Google Scholar]
- Morton, J.R.; Wilcox, H.W. Inorg. Synth.; Vol. 4, New York, 1953; p. 48. [Google Scholar]
- Muzart, J. Synthesis 1995, 1325. McKillop, A.; Sanderson, W.R. Tetrahedron 1995, 51, 6145. McKillop, A.; Sanderson, W. R. J. Chem. Soc., Perkin Trans. 1 2000, 471.
- Lu, C.-S.; Hughes, E.W.; Giguere, P.A. J. Am. Chem. Soc. 1941, 53, 1507.
- Stang, P.J.; Zhdankin, V.V. Chem. Rev. 1996, 96, 1123. Kitamura, T.; Fujiwara, Y. Org. Prep. Proced. Int. 1997, 29, 409. Varvoglis, A. Tetrahedron 1997, 53, 1179. Muraki, T.; Togo, H.; Yokoyama, M. Rev. Heteroatom Chem. 1997, 17, 213. Moriarty, R.M.; Prakash, O. Adv. Heterocycl. Chem. 1998, 69, 1. Varvoglis, A.; Spyroudis, S. Synlett. 1998, 221. [Google Scholar] Kirschning, A. J. Prakt. Chem./Chem.-Ztg. 1998, 340, 184. [CrossRef] Ochiai, M. Kikan Kagaku Sosetsu 1998, 34, 181. (in Japanese). Hypervalent Organic Compounds (book in Japanese); Anon.; Tokyo, 1998. Muraki, T.; Togo, H.; Yokoyama, M. J. Org. Chem. 1999, 64, 2883. Wirth, T.; Hirt, V.H. Synthesis 1999, 1271. 2nd Symposium on Iodine Utilization, Chiba, Japan, Oct. 16, 1999. Proceedings (FIU Rep. No. 2); see pp. 5-8 and 116-136..
- Kazmierczak, P.; Skulski, L. communicated at a Meeting of the Polish Chemical Society, Poznan, Sept. 23-26, 1996.
- Kryska, A.; Lechnio, J.; Skulski, L. communicated at an All-Polish Meeting of Medicine Students, Warsaw, April 15-17, 1999.
- Beringer, F.M.; Falk, R.A.; Karniol, M.; Lillien, J.; Masullo, G.; Mausner, M.; Sommer, E. J. Am. Chem. Soc. 1959, 81, 342.
- Hartmann, C.; Meyer, V. Ber. dtsch. chem. Ges. 1894, 27, 426. [CrossRef]
- Sharefkin, J.G.; Saltzman, H. Org. Synth. 1963, 43, 65.
- Banarjee, A.; Banarjee, G.C.; Bhattacharia, S.; Banarjee, S.; Sammadar, H. J. Indian Chem. Soc. 1981, 58, 605.
- Lucas, H.J.; Kennedy, E.R. Org. Synth. 1942, 22, 72.
- Ciustea, G.; Panescu, A. Rev. Chim. (Bucharest) 1969, 20, 216.
- Chaundhari, S.S. Synlett 2000, 278.
- Ireland, R.E.; Liu, L. J. Org. Chem. 1993, 58, 2899.
- Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537.
- Meyer, S.D.; Schreiber, S.L. J. Org. Chem. 1994, 59, 7549.
- Bayraktaroglou, T.O.; Gooding, M.A.; Khatib, S.F.; Lee, M.; Kourouma, M.; Landot, R.G. J. Org. Chem. 1993, 58, 1264.
- Olah, G.A. Halonium Ions; New York, 1975. [Google Scholar]
- Koser, G.F. Halonium Ions. In The Chemistry of Functional Groups, Supplement D; Patai, S., Rappoport, Z., Eds.; New York, 1983; Chap. 25. [Google Scholar]
- Sandin, R.B. Chem .Rev. 1943, 32, pp. 249, pp. 266.
- Willgerodt, C. Ber. dtsch. chem. Ges. (a) 1897, 30, 56; (b) 1898, 31, 915.
- Freidlina, R. Kh.; Nesmeyanov, A.N. Compt .Rend. [Doklady] Acad. Sci. U.R.S.S. 1940, 29, 567, Chem. Abstr. 1941, 35, 3614.
- Beringer, F.M.; Lillien, I. J. Am. Chem. Soc. 1960, 82, 725.
- Neiland, O. Ya. Latv. PSR Zinat. Akad. Vestis, Kim. Ser. 1964, pp. 589., pp. 591.
- Beringer, F.M.; Chang, L.L. J. Org. Chem. 1972, 37, 1516.
- Beringer, F.M.; Briedley, A.; Drexler, M.; Gindler, E.M.; Lumpkin, C.C. J. Am. Chem. Soc. 1953, 75, 2708.
- van der Puy, M. J. Fluorine Chem. 1982, 21, 385.
- Kitamura, T.; Matsuyuki, J.; Taniguchi, H. Synthesis 1994, 147.
- Jurd, L. Austral. J. Sci. Res. 1949, 2[A], 595. Seikel, M.K. Org .Synth. 1944, 24, 47. Gaca, J. personal communication.
- Forster, M.O.; Schaeppi, J.H. J. Chem. Soc. 1912, 101, 382.
- Kryska, A.; Skulski, L. communicated at an Annual Meeting of the Polish Chemical Society, Lodz, Sept. 10-15, 2000.
- Lucas, H. J.; Kennedy, E. R.; Formo, M. W. Org. Synth. 1942, 22, 70.
- Kryska, A.; Lulinski, P.; Skulski, L. communicated at an Annual Meeting of the Polish Chemical Society, Lodz, Sept. 10-15, 2000.
© 2000 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Skulski, L. Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland. Molecules 2000, 5, 1331-1371. https://doi.org/10.3390/51201331
AMA Style
Skulski L. Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland. Molecules. 2000; 5(12):1331-1371. https://doi.org/10.3390/51201331
Chicago/Turabian StyleSkulski, Lech. 2000. "Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland" Molecules 5, no. 12: 1331-1371. https://doi.org/10.3390/51201331