A Deeper Investigation of Drug Degradation Mixtures Using a Combination of MS and NMR Data: Application to Indapamide

 , ,
, ,
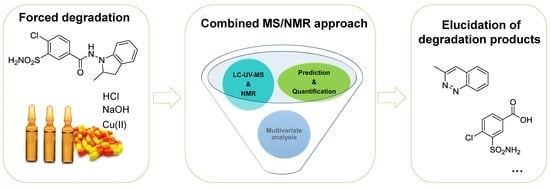
Abstract
:
1. Introduction
2. Results and Discussion
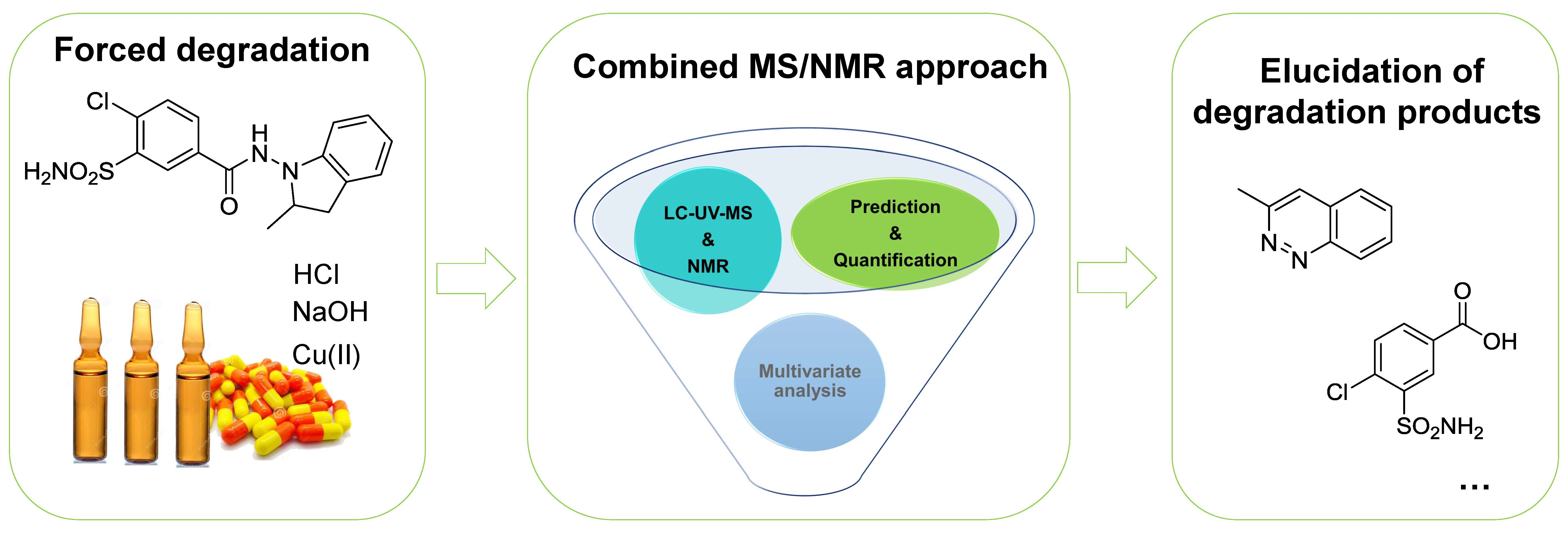
2.1. Methodology Development
2.2. Quantification of API in Degradation Mixtures
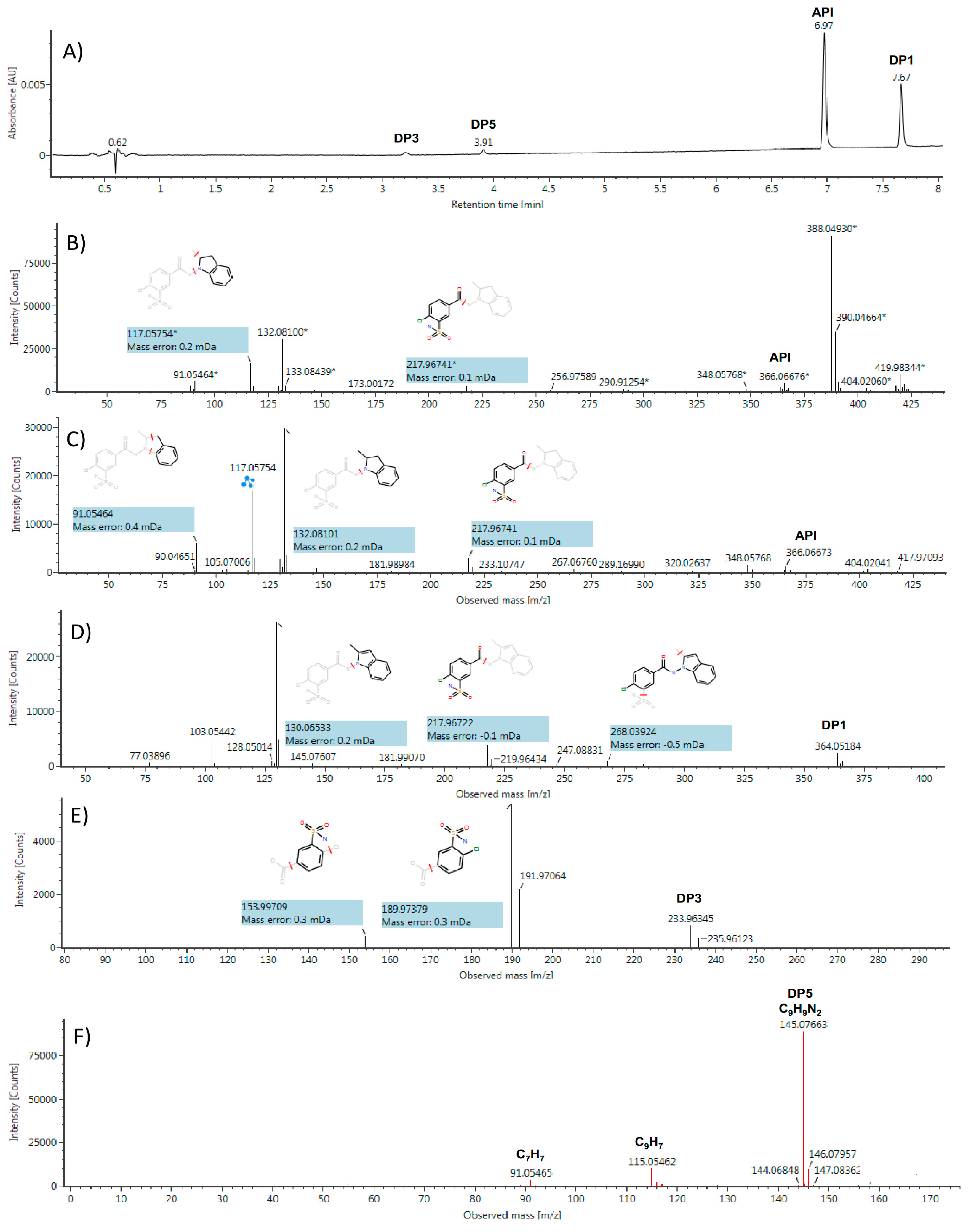
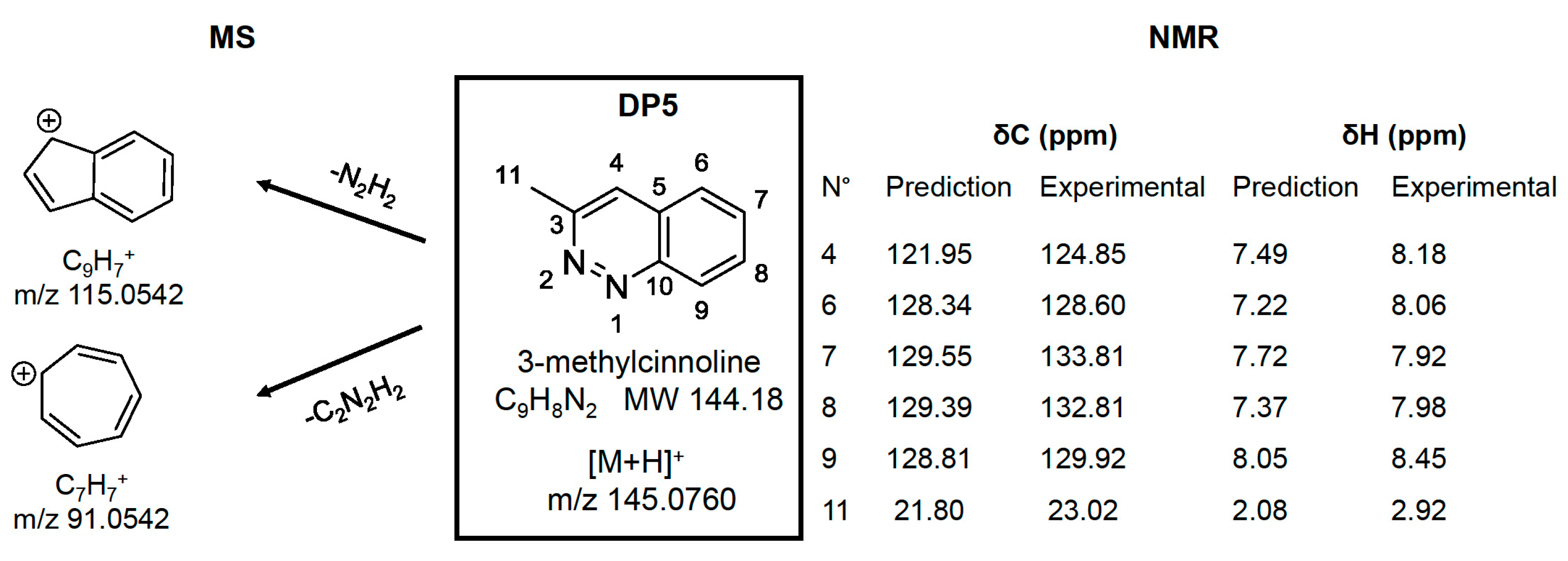
2.3. Identification/Prediction of DPs Using HDMSE Acquisitions and MNova Software
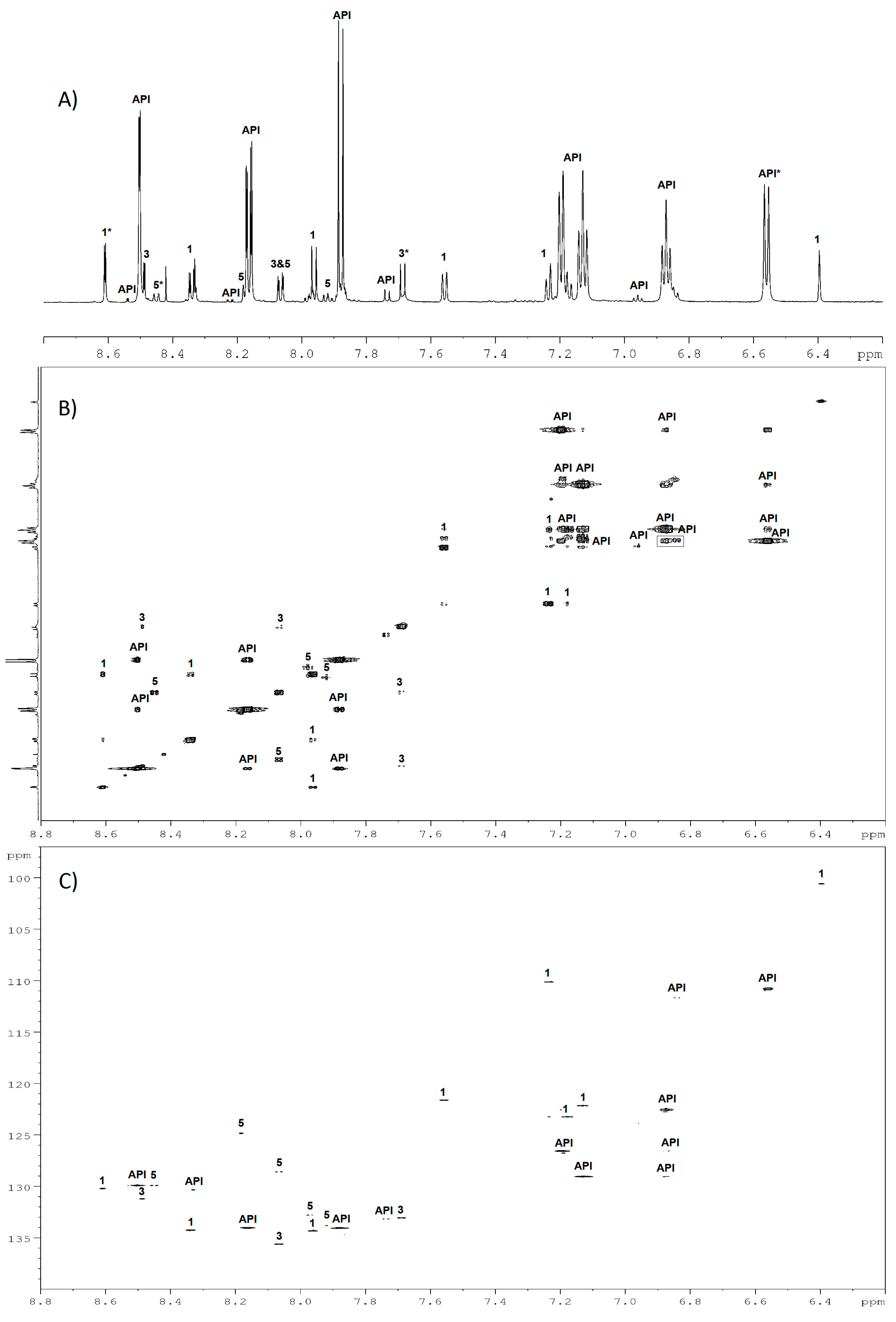
2.4. Structures Validation and Quantification of Degradation Products Using NMR
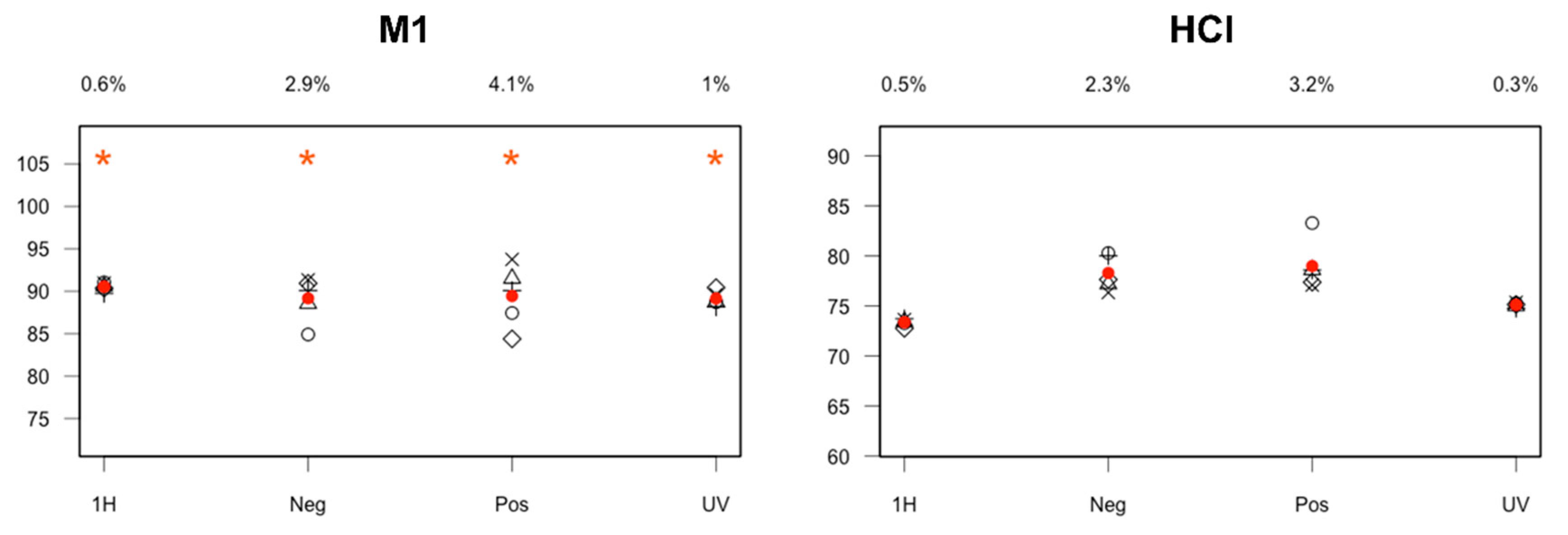
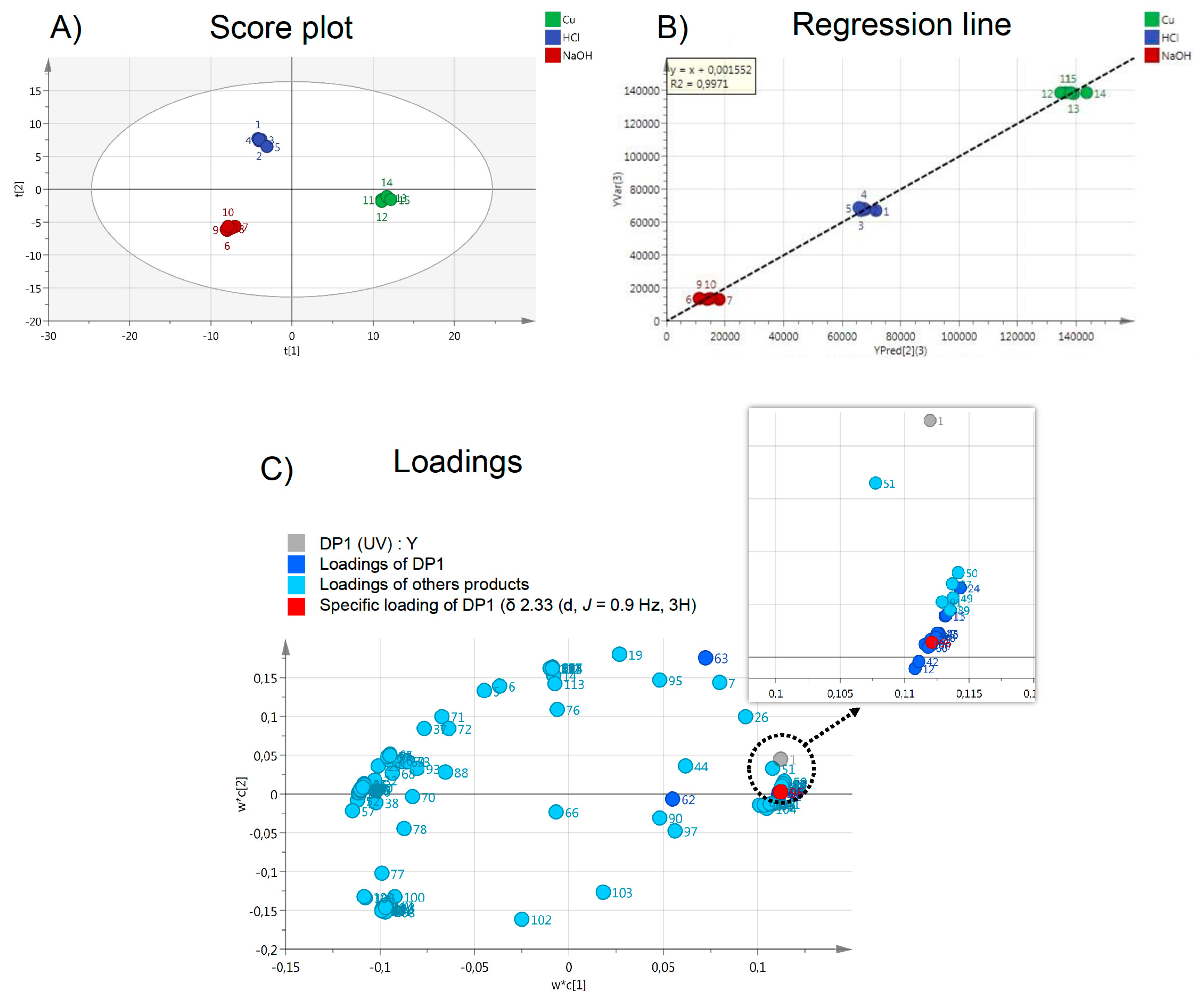
2.5. Contribution of Pre-Processing Filter Approach Using UV/MS and NMR Data for Multi-Degradation Studies
3. Materials and Methods
3.1. Chemicals
3.2. Forced Degradation Protocols and Sample Preparation
3.2.1. Forced Degradation Studies
3.2.2. Indapamide Calibration Curve and Model Mixtures
3.2.3. MS and NMR Samples
3.3. MS and NMR Analysis
3.3.1. LC-UV-MS Acquisition
3.3.2. LC-UV-MS Processing
3.3.3. NMR Acquisition
3.3.4. NMR Processing
3.4. Statistical Analysis
3.4.1. Quantification
3.4.2. UV/MS and NMR Data Correlation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Conference on Harmonization (ICH) Q1A(R2) Stability Testing of New Drug Substances and Products, 2003, CPMP/ICH/2736/99. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf (accessed on 10 January 2019).
- Görög, S. Critical review of reports on impurity and degradation product profiling in the last decade. TrAC Trends Anal. Chem. 2018, 101, 2–16. [Google Scholar] [CrossRef]
- De Vijlder, T.; Valkenborg, D.; Lemière, F.; Romijn, E.P.; Laukens, K.; Cuyckens, F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2018, 37, 607–629. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Maggio, R.M.; Calvo, N.L.; Vignaduzzo, S.E.; Kaufman, T.S. Pharmaceutical impurities and degradation products: Uses and applications of NMR techniques. J. Pharm. Biomed. Anal. 2014, 101, 102–122. [Google Scholar] [CrossRef]
- Webster, G.K.; Kumar, S. Expanding the Analytical Toolbox: Pharmaceutical Application of Quantitative NMR. Anal. Chem. 2014, 86, 11474–11480. [Google Scholar] [CrossRef]
- Thummar, M.; Kuswah, B.S.; Samanthula, G.; Bulbake, U.; Gour, J.; Khan, W.; Mohit, T.; Gananadhamu, S.; Upendra, B.; Jitendra, G.; et al. Validated stability indicating assay method of olaparib: LC-ESI-Q-TOF-MS/MS and NMR studies for characterization of its new hydrolytic and oxidative forced degradation products. J. Pharm. Biomed. Anal. 2018, 160, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Narayanam, M.; Sahu, A.; Singh, S. Use of LC–MS/TOF, LC–MSn, NMR and LC–NMR in characterization of stress degradation products: Application to cilazapril. J. Pharm. Biomed. Anal. 2015, 111, 190–203. [Google Scholar] [CrossRef]
- Bingol, K.; Brüschweiler, R. Knowns and unknowns in metabolomics identified by multidimensional NMR and hybrid MS/NMR methods. Curr. Opin. Biotechnol. 2017, 43, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boiteau, R.M.; Hoyt, D.W.; Nicora, C.D.; Kinmonth-Schultz, H.A.; Ward, J.K.; Bingol, K.; A Kinmonth-Schultz, H.; Kinmonth-Schultz, H. Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction. Metabolites 2018, 8, 8. [Google Scholar] [CrossRef]
- Palaric, C.; Pilard, S.; Fontaine, J.-X.; Cailleu, D.; Mesnard, F.; Renaud, T.; Petit, A.; Molinié, R. Combined MS-NMR approach for the quality control of food supplements containing amino acids. Food Control 2018, 88, 217–228. [Google Scholar] [CrossRef]
- European Pharmacopeia 9.0. Indapamide 01/2017:2214, 2953–2955. Available online: http://online6.edqm.eu/ep900/ (accessed on 10 January 2019).
- Attia, K.A.M.; Nassar, M.W.I.; El-Din, M.M.K.S.; Mohamad, A.A.A.; Kaddah, M.M.Y. A stability-indicating QTRAP LC-MS/MS method for identification and structural characterization of degradation products of indapamide. Anal. Methods 2016, 8, 1836–1851. [Google Scholar] [CrossRef]
- Otto, M. Chemometrics: Statistics and Computer Application in Analytical Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Monakhova, Y.B.; Holzgrabe, U.; Diehl, B.W. Current role and future perspectives of multivariate (chemometric) methods in NMR spectroscopic analysis of pharmaceutical products. J. Pharm. Biomed. Anal. 2018, 147, 580–589. [Google Scholar] [CrossRef]
- Fjeldsted, J. Advances in Time-of-Flight Mass Spectrometry. Compr. Anal. Chem. 2016, 71, 19–49. [Google Scholar]
- Regueiro, J.; Negreira, N.; Hannisdal, R.; Berntssen, M.H. Targeted approach for qualitative screening of pesticides in salmon feed by liquid chromatography coupled to traveling-wave ion mobility/quadrupole time-of-flight mass spectrometry. Food Control 2017, 78, 116–125. [Google Scholar] [CrossRef]
- Campuzano, I.D.; Lippens, J.L. Ion mobility in the pharmaceutical industry: An established biophysical technique or still niche? Curr. Opin. Chem. Biol. 2018, 42, 147–159. [Google Scholar] [CrossRef]
- Blessy, M.; Patel, R.D.; Prajapati, P.N.; Agrawal, Y. Development of forced degradation and stability indicating studies of drugs—A review. J. Pharm. Anal. 2014, 4, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Masanori, S.; Kazuko, U. Ring enlargement reaction of 1-aminoindoles to cinnoline derivatives. Chem. Lett. 1978, 7, 707–708. [Google Scholar]
- Peyrot, L.; Elkhatib, M.; Vignalou, J.R.; Metz, R.; Elomar, F.; Delalu, H. Synthesis and characterization of new heterocyclic derivatives by oxidation of 1-amino-2-methylindoline. J. Heterocycl. Chem. 2001, 38, 885–893. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Kim, H.K.; Verpoorte, R. Sample preparation for plant metabolomics. Phytochem. Anal. 2010, 21, 4–13. [Google Scholar] [CrossRef]
- Swann, J.; Garcia-Perez, I.; Braniste, V.; Wilson, I.; Sidaway, J.; Nicholson, J.; Pettersson, S.; Holmes, E. Application of 1H-NMR spectroscopy to the metabolic phenotyping of rodent brain extracts: A metabonomic study of gut microbial influence on host brain metabolism. J. Pharm. Biomed. Anal. 2017, 143, 141–146. [Google Scholar] [CrossRef]
- Mukerjee, P. Effects of added dimetylsulfoxide on pKa values of uncharged organic acids and pH values of aqueous buffers. Tetrahedron Lett. 1998, 39, 423–426. [Google Scholar] [CrossRef]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef]
- Thrippleton, M.J.; Keeler, J. Elimination of Zero-Quantum Interference in Two-Dimensional NMR Spectra. Angew. Chem. 2003, 115, 4068–4071. [Google Scholar] [CrossRef]
- Le Guennec, A.; Dumez, J.-N.; Giraudeau, P.; Caldarelli, S. Resolution-enhanced 2D-NMR of complex mixtures by non-uniform sampling: Resolution-enhanced 2D-NMR of complex mixtures by non-uniform sampling. Magn. Reson. Chem. 2015, 53, 913–920. [Google Scholar] [CrossRef]
- Jacob, D.; Deborde, C.; Lefebvre, M.; Maucourt, M.; Moing, A. NMRProcFlow: a graphical and interactive tool dedicated to 1D spectra processing for NMR-based metabolomics. Metabolomics 2017, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- The R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing 2017. Available online: https://www.r-project.org/foundation/ (accessed on 10 January 2019).
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Structure | Chemical Formula | [M + H]+ m/z | [M − H]− m/z |
|---|---|---|---|---|
| Indapamide (API) |  | C16H16ClN3O3S | 366.0674 | 364.0528 |
| DP1 |  | C16H14ClN3O3S | 364.0517 | 362.0372 |
| DP2 |  | C9H10N2 | 147.0917 | - |
| DP3 |  | C7H6ClNO4S | - | 233.9633 |
| DP4 |  | C9H12N2 | 149.1073 * | - |
| Stress Factor | Exposure | % of Remaining PA ± 2% (Mean of UV and 1H-NMR) | % of Individual DPs ± 5% (Based on qNMR) |
|---|---|---|---|
| Acid | 0.1 N HCl at 70 °C for 24 h | 74% | DP1: 14%; DP3: 8%; DP5: 5% |
| Base | 0.1 N NaOH at 70 °C for 20 min | 94% | DP1: 3% |
| Metal ion | 0.05 M CuCl2, immediate | 0% | DP1: 100% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palaric, C.; Molinié, R.; Cailleu, D.; Fontaine, J.-X.; Mathiron, D.; Mesnard, F.; Gut, Y.; Renaud, T.; Petit, A.; Pilard, S. A Deeper Investigation of Drug Degradation Mixtures Using a Combination of MS and NMR Data: Application to Indapamide. Molecules 2019, 24, 1764. https://doi.org/10.3390/molecules24091764
Palaric C, Molinié R, Cailleu D, Fontaine J-X, Mathiron D, Mesnard F, Gut Y, Renaud T, Petit A, Pilard S. A Deeper Investigation of Drug Degradation Mixtures Using a Combination of MS and NMR Data: Application to Indapamide. Molecules. 2019; 24(9):1764. https://doi.org/10.3390/molecules24091764
Chicago/Turabian StylePalaric, Cécile, Roland Molinié, Dominique Cailleu, Jean-Xavier Fontaine, David Mathiron, François Mesnard, Yoann Gut, Tristan Renaud, Alain Petit, and Serge Pilard. 2019. "A Deeper Investigation of Drug Degradation Mixtures Using a Combination of MS and NMR Data: Application to Indapamide" Molecules 24, no. 9: 1764. https://doi.org/10.3390/molecules24091764