
The Drug Candidate BGP-15 Delays the Onset of Diastolic Dysfunction in the Goto-Kakizaki Rat Model of Diabetic Cardiomyopathy

 ,
,  ,
,  ,
,
Abstract
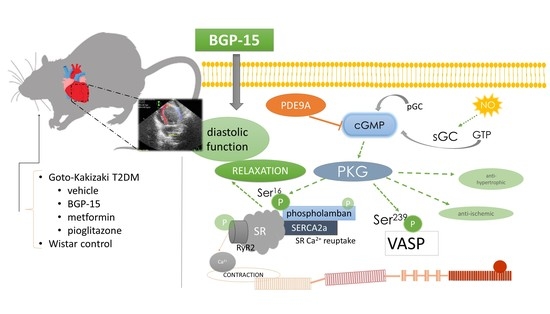
1. Introduction
2. Results
2.1. Metabolic Characterisation of Goto-Kakizaki and Wistar Rats
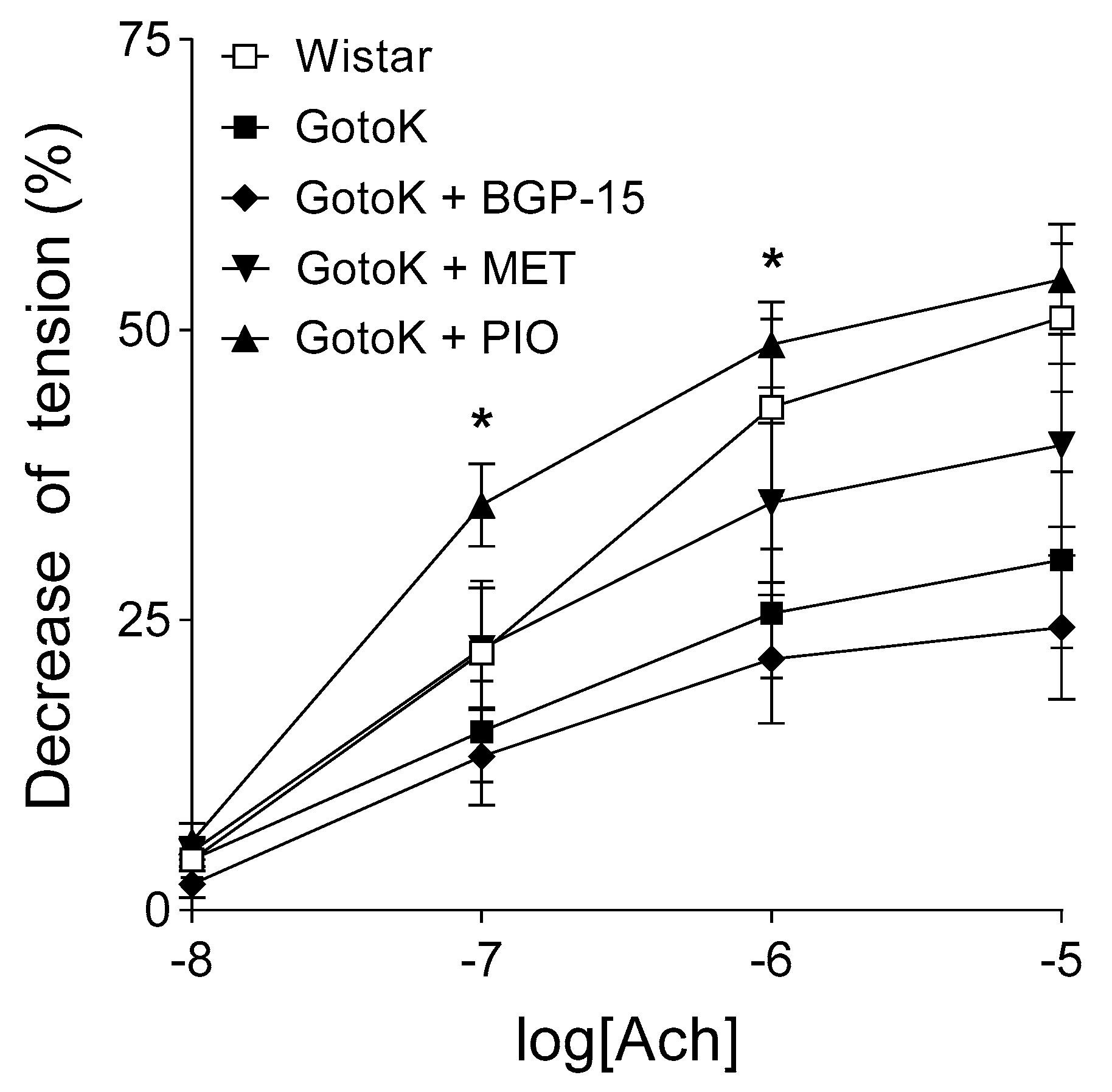
2.2. Endothelium-Dependent Vasorelaxation is Enhanced by Pioglitazone but not BGP-15
2.3. BGP-15 Enhances Diastolic Function Measured by Echocardiography
2.4. BGP-15 Elevates the Phosphorylation Level of Phospholamban and VASP Proteins
3. Discussion
4. Materials and Methods
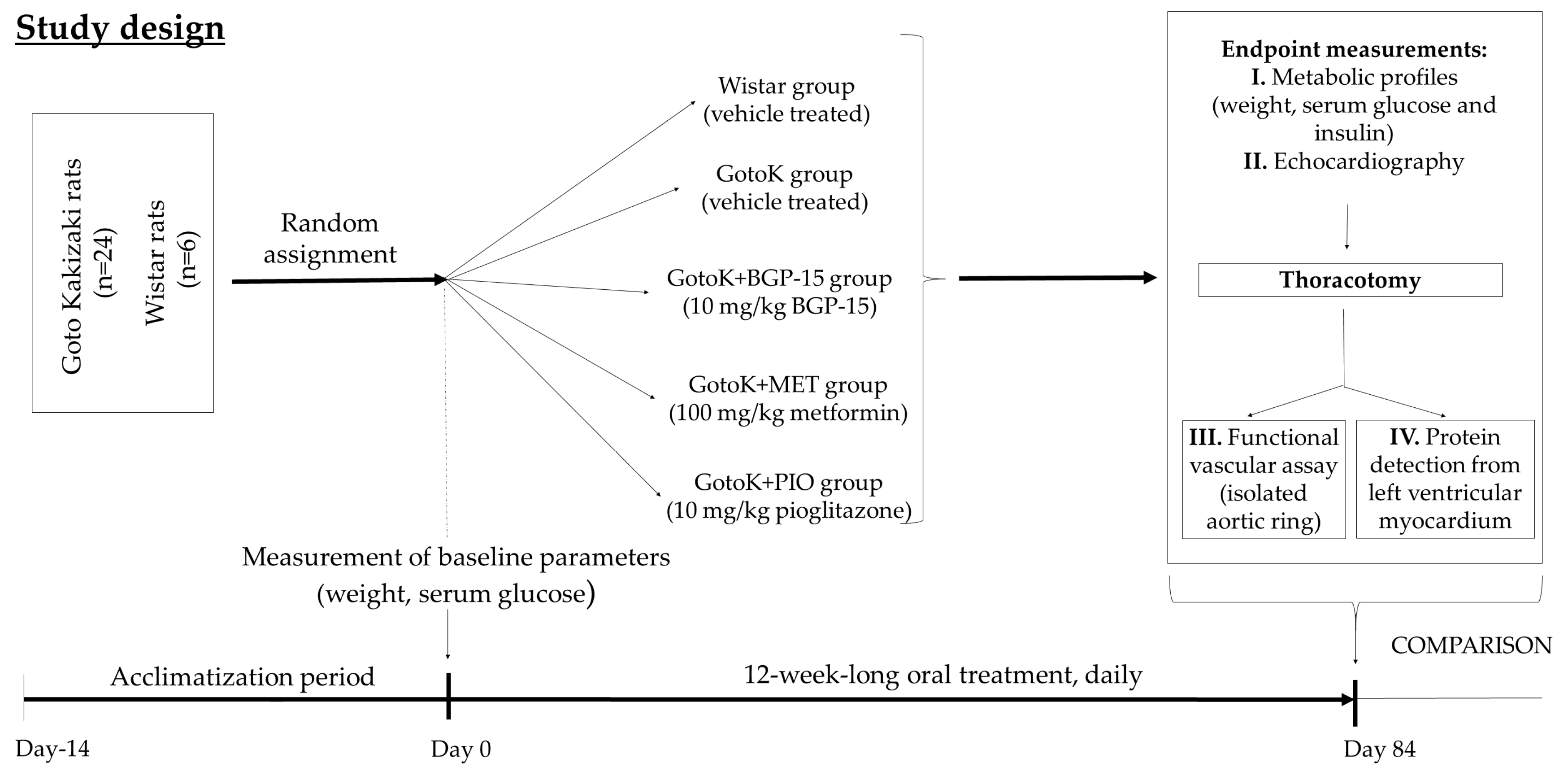
4.1. Animal Model and Study Design
4.2. Chemicals
4.3. Metabolic Profiles
4.4. Echocardiography
4.5. Functional Vascular Assay
4.6. Western blot
4.7. Statistical Procedures
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- De Ferranti, S.D.; de Boer, I.H.; Fonseca, V.; Fox, C.S.; Golden, S.H.; Lavie, C.J.; Magge, S.N.; Marx, N.; McGuire, D.K.; Orchard, T.J.; et al. Type 1 diabetes mellitus and cardiovascular disease: A scientific statement from the American Heart Association and American Diabetes Association. Circulation 2014, 130, 1110–1130. [Google Scholar] [CrossRef]
- Tate, M.; Grieve, D.J.; Ritchie, R.H. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin. Sci. 2017, 131, 897–915. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Gkogkolou, P.; Bohm, M. Advanced glycation end products: Key players in skin aging? Dermato-Endocrinology 2012, 4, 259–270. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef]
- King, A.J. The use of animal models in diabetes research. Brit. J. Pharmacol. 2012, 166, 877–894. [Google Scholar]
- D’Souza, A.; Howarth, F.C.; Yanni, J.; Dobryznski, H.; Boyett, M.R.; Adeghate, E.; Bidasee, K.R.; Singh, J. Left ventricle structural remodelling in the prediabetic Goto-Kakizaki rat. Expe. Physiol. 2011, 96, 875–888. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, M.M.; Yang, Z.K.; Phillips, A.O.; Shah, A.M. Cardiac dysfunction in the Goto-Kakizaki rat. A model of type II diabetes mellitus. Basic Res. Cardiol. 2004, 99, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Alonso, N.; Moliner, P.; Mauricio, D. Pathogenesis, Clinical Features and Treatment of Diabetic Cardiomyopathy. Adv. Exp. Med. Biol. 2018, 1067, 197–217. [Google Scholar]
- Sena, C.M.; Matafome, P.; Louro, T.; Nunes, E.; Fernandes, R.; Seica, R.M. Metformin restores endothelial function in aorta of diabetic rats. Brit. J. pharmacol. 2011, 163, 424–437. [Google Scholar] [CrossRef]
- Libby, P. Metformin and vascular protection: A cardiologist’s view. Diabetes Metab. 2003, 29, S117–S120. [Google Scholar] [CrossRef]
- Saremi, A.; Schwenke, D.C.; Buchanan, T.A.; Hodis, H.N.; Mack, W.J.; Banerji, M.; Bray, G.A.; Clement, S.C.; Henry, R.R.; Kitabchi, A.E.; et al. Pioglitazone slows progression of atherosclerosis in prediabetes independent of changes in cardiovascular risk factors. Arterioscl. Throm. Vas. 2013, 33, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Nowell, M.; Chima, R.; Zingarelli, B. Pioglitazone reduces inflammation through inhibition of NF-kappaB in polymicrobial sepsis. Innate immune. 2014, 20, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, S.M.; van der Poel, C.; Sayer, T.A.; Schertzer, J.D.; Henstridge, D.C.; Church, J.E.; Lamon, S.; Russell, A.P.; Davies, K.E.; Febbraio, M.A.; et al. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 2012, 484, 394–398. [Google Scholar] [CrossRef]
- Sapra, G.; Tham, Y.K.; Cemerlang, N.; Matsumoto, A.; Kiriazis, H.; Bernardo, B.C.; Henstridge, D.C.; Ooi, J.Y.; Pretorius, L.; Boey, E.J.; et al. The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat. commun. 2014, 5, 5705. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, T.L.; Silva, M.T.; Miyabara, E.H. BGP-15 improves contractile function of regenerating soleus muscle. J. Muscle Res. Cell M. 2018, 39, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, B.; Tory, K.; Peitl, B.; Bajza, A.; Koranyi, L.; Literati-Nagy, Z.; Hooper, P.L.; Vigh, L.; Szilvassy, Z. Improvement of insulin sensitivity by a novel drug candidate, BGP-15, in different animal studies. Metabo. Syndr. Relat. D. 2014, 12, 125–131. [Google Scholar] [CrossRef]
- Salah, H.; Li, M.; Cacciani, N.; Gastaldello, S.; Ogilvie, H.; Akkad, H.; Namuduri, A.V.; Morbidoni, V.; Artemenko, K.A.; Balogh, G.; et al. The chaperone co-inducer BGP-15 alleviates ventilation-induced diaphragm dysfunction. Sci. Transl. Med. 2016, 8, 350ra103. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Varughese, G.I.; Sriraman, R.; Arunagirinathan, G. Diabetic cardiomyopathy: Pathophysiology, diagnostic evaluation and management. World J. diabetes 2013, 4, 177–189. [Google Scholar] [CrossRef]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. Jama 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Radovits, T.; Korkmaz, S.; Matyas, C.; Olah, A.; Nemeth, B.T.; Pali, S.; Hirschberg, K.; Zubarevich, A.; Gwanmesia, P.N.; Li, S.; et al. An altered pattern of myocardial histopathological and molecular changes underlies the different characteristics of type-1 and type-2 diabetic cardiac dysfunction. J. Diabetes Res. 2015, 2015, 728741. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, W.M.T.; Panveloski-Costa, A.C.; Yokota, C.N.F.; Pereira, J.N.B.; Filho, J.M.; Torres, R.P.; Hirabara, S.M.; Curi, R.; Alba-Loureiro, T.C. Comparison of Goto-Kakizaki rats and high fat diet-induced obese rats: Are they reliable models to study Type 2 Diabetes mellitus? PLoS ONE 2017, 12, e0189622. [Google Scholar] [CrossRef] [PubMed]
- Sourij, H.; Zweiker, R.; Wascher, T.C. Effects of pioglitazone on endothelial function, insulin sensitivity, and glucose control in subjects with coronary artery disease and new-onset type 2 diabetes. Diabetes care 2006, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.; Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vascular health risk manage. 2007, 3, 853–876. [Google Scholar]
- Wen, Y.; Skidmore, J.C.; Porter-Turner, M.M.; Rea, C.A.; Khokher, M.A.; Singh, B.M. Relationship of glycation, antioxidant status and oxidative stress to vascular endothelial damage in diabetes. Diabetes Obes. Metab. 2002, 4, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.; Rehman, K.; Chen, S. Goto-Kakizaki rats: Its suitability as non-obese diabetic animal model for spontaneous type 2 diabetes mellitus. Curr. Diabetes Rev. 2013, 9, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G. Efficacy and tolerability of pioglitazone in patients with type 2 diabetes mellitus: Comparison with other oral antihyperglycaemic agents. Drugs 2010, 70, 1945–1961. [Google Scholar] [CrossRef]
- Hallakou, S.; Doare, L.; Foufelle, F.; Kergoat, M.; Guerre-Millo, M.; Berthault, M.F.; Dugail, I.; Morin, J.; Auwerx, J.; Ferre, P. Pioglitazone induces in vivo adipocyte differentiation in the obese Zucker fa/fa rat. Diabetes 1997, 46, 1393–1399. [Google Scholar] [CrossRef]
- Fontaine, K.R.; Redden, D.T.; Wang, C.; Westfall, A.O.; Allison, D.B. Years of life lost due to obesity. Jama 2003, 289, 187–193. [Google Scholar] [CrossRef]
- Chawla, S.; Kaushik, N.; Singh, N.P.; Ghosh, R.K.; Saxena, A. Effect of addition of either sitagliptin or pioglitazone in patients with uncontrolled type 2 diabetes mellitus on metformin: A randomized controlled trial. J. Pharmacol. Pharmacot. 2013, 4, 27–32. [Google Scholar]
- Liao, H.W.; Saver, J.L.; Wu, Y.L.; Chen, T.H.; Lee, M.; Ovbiagele, B. Pioglitazone and cardiovascular outcomes in patients with insulin resistance, pre-diabetes and type 2 diabetes: A systematic review and meta-analysis. BMJ Open 2017, 7, e013927. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F.; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.C.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiog. 2016, 29, 277–314. [Google Scholar] [CrossRef]
- Oh, T.J.; Shin, J.Y.; Kang, G.H.; Park, K.S.; Cho, Y.M. Effect of the combination of metformin and fenofibrate on glucose homeostasis in diabetic Goto-Kakizaki rats. Exp. Mol. Med. 2013, 45, e30. [Google Scholar] [CrossRef] [PubMed]
- Portha, B.; Lacraz, G.; Kergoat, M.; Homo-Delarche, F.; Giroix, M.H.; Bailbe, D.; Gangnerau, M.N.; Dolz, M.; Tourrel-Cuzin, C.; Movassat, J. The GK rat beta-cell: A prototype for the diseased human beta-cell in type 2 diabetes? Mol. Cell. Endocrinol. 2009, 297, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Okuno, A.; Tanaka, J.; Takahashi, K.; Nakashima, R.; Kanda, S.; Ogawa, J.; Hagisawa, Y.; Fujiwara, T. Metformin primarily decreases plasma glucose not by gluconeogenesis suppression but by activating glucose utilization in a non-obese type 2 diabetes Goto-Kakizaki rats. Eur. J. Pharmacol. 2009, 623, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Yuda, S.; Kouzu, H.; Miura, T. Diabetic cardiomyopathy: Pathophysiology and clinical features. Heart Fail. Rev. 2013, 18, 149–166. [Google Scholar] [CrossRef]
- Park, J.H.; Marwick, T.H. Use and Limitations of E/e’ to Assess Left Ventricular Filling Pressure by Echocardiography. J. Card. 2011, 19, 169–173. [Google Scholar] [CrossRef]
- Colyer, J. Phosphorylation states of phospholamban. Ann. N.Y. Acad. Sci. 1998, 853, 79–91. [Google Scholar] [CrossRef]
- Ramirez-Correa, G.A.; Murphy, A.M. Is phospholamban or troponin I the "prima donna" in beta-adrenergic induced lusitropy? Circ. Res. 2007, 101, 326–327. [Google Scholar] [CrossRef]
- Frantz, S.; Klaiber, M.; Baba, H.A.; Oberwinkler, H.; Volker, K.; Gabetaner, B.; Bayer, B.; Abebetaer, M.; Schuh, K.; Feil, R.; et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I. Eur. Heart J. 2013, 34, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Gorbe, A.; Giricz, Z.; Szunyog, A.; Csont, T.; Burley, D.S.; Baxter, G.F.; Ferdinandy, P. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Rese. Cardiol. 2010, 105, 643–650. [Google Scholar] [CrossRef]
- Smolenski, A.; Bachmann, C.; Reinhard, K.; Honig-Liedl, P.; Jarchau, T.; Hoschuetzky, H.; Walter, U. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J. Biol. Chem. 1998, 273, 20029–20035. [Google Scholar] [CrossRef]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Priksz, D.; Bombicz, M.; Varga, B.; Kurucz, A.; Gesztelyi, R.; Balla, J.; Toth, A.; Papp, Z.; Szilvassy, Z.; Juhasz, B. Upregulation of Myocardial and Vascular Phosphodiesterase 9A in A Model of Atherosclerotic Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 2882. [Google Scholar] [CrossRef]
- Kovacs, D.; Simon, Z.; Hari, P.; Malnasi-Csizmadia, A.; Hegedus, C.; Drimba, L.; Nemeth, J.; Sari, R.; Szilvassy, Z.; Peitl, B. Identification of PPARgamma ligands with One-dimensional Drug Profile Matching. Drug Des, Dev. Ther. 2013, 7, 917–928. [Google Scholar]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Szabo, K.; Gesztelyi, R.; Lampe, N.; Kiss, R.; Remenyik, J.; Pesti-Asboth, G.; Priksz, D.; Szilvassy, Z.; Juhasz, B. Fenugreek (Trigonella Foenum-Graecum) Seed Flour and Diosgenin Preserve Endothelium-Dependent Arterial Relaxation in a Rat Model of Early-Stage Metabolic Syndrome. Int. J. Mol. Sci. 2018, 19, 798. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Fasting Glucose (mmol/L) | Fasting Insulin (μIU/mL) | Baseline BODYWEIGHT (g) | Endpoint Bodyweight (g) |
|---|---|---|---|---|
| Wistar | 6.280 ± 0.428 | 5.850 ± 0.132 | 407.8 ± 3.798 | 539.8 ± 22.45 |
| GotoK | 13.62 ± 0.626 * | 12.53 ± 3.292 | 313.8 ± 4.070 * | 374.0 ± 6.266 * |
| GotoK+BGP15 | 10.92 ± 0.561 | 12.30 ± 3.793 | 318.3 ± 5.566 | 383.8 ± 9.024 |
| GotoKMET | 11.20 ± 1.002 | 12.82 ± 4.104 | 312.7 ± 5.655 * | 361.7 ± 12.27 * |
| GotoK+PIO | 7.880 ± 0.312 # | 14.83 ± 2.768 | 316.3 ± 3.658 | 411.3 ± 10.40 |
| Parameter | Wistar | GotoK | GotoK + BGP-15 | GotoK + MET | GotoK + PIO |
|---|---|---|---|---|---|
| LVIDd (mm) | 7.267 ± 0.278 | 6.918 ± 0.167 | 7.243 ± 0.183 | 6.483 ± 0.425 | 7.323 ± 0.400 |
| LVIDs (mm) | 4.365 ± 0.201 | 4.515 ± 0.095 | 4.69 ± 0.193 | 3.940 ± 0.200 | 4.478 ± 0.345 |
| FS (%) | 39.94 ± 1.474 | 33.27 ± 1.667 | 37.62 ± 0.727 | 38.85 ± 1.715 | 39.21 ± 1.608 |
| EF (%) | 68.92 ± 1.813 | 60.36 ± 2.343 | 66.13 ± 0.891 | 67.94 ± 2.068 | 67.98 ± 2.110 |
| MV E vel. (m/s) | 0.612 ± 0.047 | 0.883 ± 0.024 ** | 0.648 ± 0.021 ## | 0.763 ± 0.025 | 0.810 ± 0.033 |
| MV A vel. (m/s) | 0.402 ± 0.048 | 0.507 ± 0.023 | 0.372 ± 0.022 # | 0.457 ± 0.021 | 0.480 ± 0.032 |
| E/A ratio | 1.554 ± 0.070 | 1.735 ± 0.117 | 1.762 ± 0.066 | 1.698 ± 0.0840 | 1.645 ± 0.089 |
| MV DecT (ms) | 53.83 ± 3.692 | 62.33 ± 2.883 | 55.00 ± 3.759 | 58.00 ± 3.425 | 53.00 ± 3.162 |
| e′/a′ ratio | 1.284 ± 0.158 | 0.744 ± 0.056 * | 1.458 ± 0.155 # | 1.249 ± 0.153 | 0.968 ± 0.032 |
| E/e′ | 16.48 ± 1.512 | 24.92 ± 1.114 ** | 16.31 ± 0.918 ## | 20.58 ± 0.766 | 19.74 ± 1.156 |
| LAP * (mmHg) | 22.34 ± 1.875 | 32.79 ± 1.381 ** | 22.12 ± 1.138 ## | 27.42 ± 0.949 | 26.38 ± 1.433 |
| LVOT Vmax (m/s) | 0.768 ± 0.037 | 0.998 ± 0.026 ** | 0.780 ± 0.029 # | 0.768 ± 0.008 # | 0.812 ± 0.039 |
| IVCT (ms) | 15.67 ± 2.906 | 18.00 ± 3.777 | 18,83 ± 3.516 | 22.83 ± 3.167 | 16.83 ± 2.272 |
| IVRT (ms) | 32.50 ± 1.258 | 30.33 ± 3.007 | 32.50 ± 1.057 | 35.17 ± 1.515 | 34.00 ± 1.438 |
| LV ET (ms) | 87.67 ± 3.116 | 63.67 ± 1.961 * | 101.7 ± 3.703 ### | 87.83 ± 5.282 | 80.67 ± 1.745 |
| Tei-index (MPI) | 0.558 ± 0.058 | 0.724 ± 0.051 | 0.471 ± 0.027 # | 0.669 ± 0.055 | 0.632 ± 0.039 |
| RV E vel. (m/s) | 0.332 ± 0.026 | 0.497 ± 0.044 | 0.368 ± 0.034 | 0.383 ± 0.048 | 0.364 ± 0.027 |
| RV A vel. (m/s) | 0.498 ± 0.041 | 0.628 ± 0.073 | 0.578 ± 0.033 | 0.235 ± 0.003 # | 0.356 ± 0.074 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bombicz, M.; Priksz, D.; Gesztelyi, R.; Kiss, R.; Hollos, N.; Varga, B.; Nemeth, J.; Toth, A.; Papp, Z.; Szilvassy, Z.; et al. The Drug Candidate BGP-15 Delays the Onset of Diastolic Dysfunction in the Goto-Kakizaki Rat Model of Diabetic Cardiomyopathy. Molecules 2019, 24, 586. https://doi.org/10.3390/molecules24030586
Bombicz M, Priksz D, Gesztelyi R, Kiss R, Hollos N, Varga B, Nemeth J, Toth A, Papp Z, Szilvassy Z, et al. The Drug Candidate BGP-15 Delays the Onset of Diastolic Dysfunction in the Goto-Kakizaki Rat Model of Diabetic Cardiomyopathy. Molecules. 2019; 24(3):586. https://doi.org/10.3390/molecules24030586
Chicago/Turabian StyleBombicz, Mariann, Daniel Priksz, Rudolf Gesztelyi, Rita Kiss, Nora Hollos, Balazs Varga, Jozsef Nemeth, Attila Toth, Zoltan Papp, Zoltan Szilvassy, and et al. 2019. "The Drug Candidate BGP-15 Delays the Onset of Diastolic Dysfunction in the Goto-Kakizaki Rat Model of Diabetic Cardiomyopathy" Molecules 24, no. 3: 586. https://doi.org/10.3390/molecules24030586
APA StyleBombicz, M., Priksz, D., Gesztelyi, R., Kiss, R., Hollos, N., Varga, B., Nemeth, J., Toth, A., Papp, Z., Szilvassy, Z., & Juhasz, B. (2019). The Drug Candidate BGP-15 Delays the Onset of Diastolic Dysfunction in the Goto-Kakizaki Rat Model of Diabetic Cardiomyopathy. Molecules, 24(3), 586. https://doi.org/10.3390/molecules24030586