Recent Developments in the Functionalization of Betulinic Acid and Its Natural Analogues: A Route to New Bioactive Compounds

 ,
, 
 and
and
Abstract
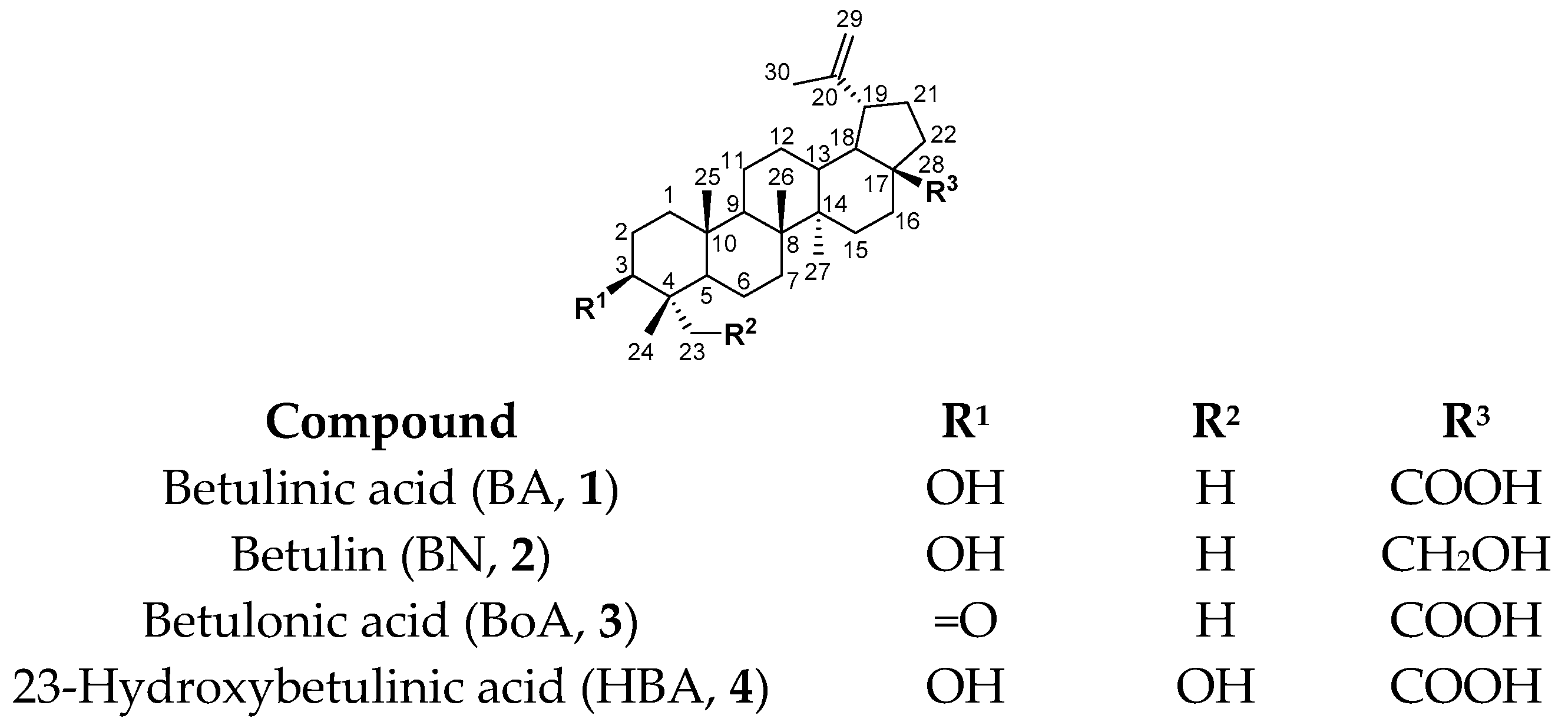
:1. Introduction
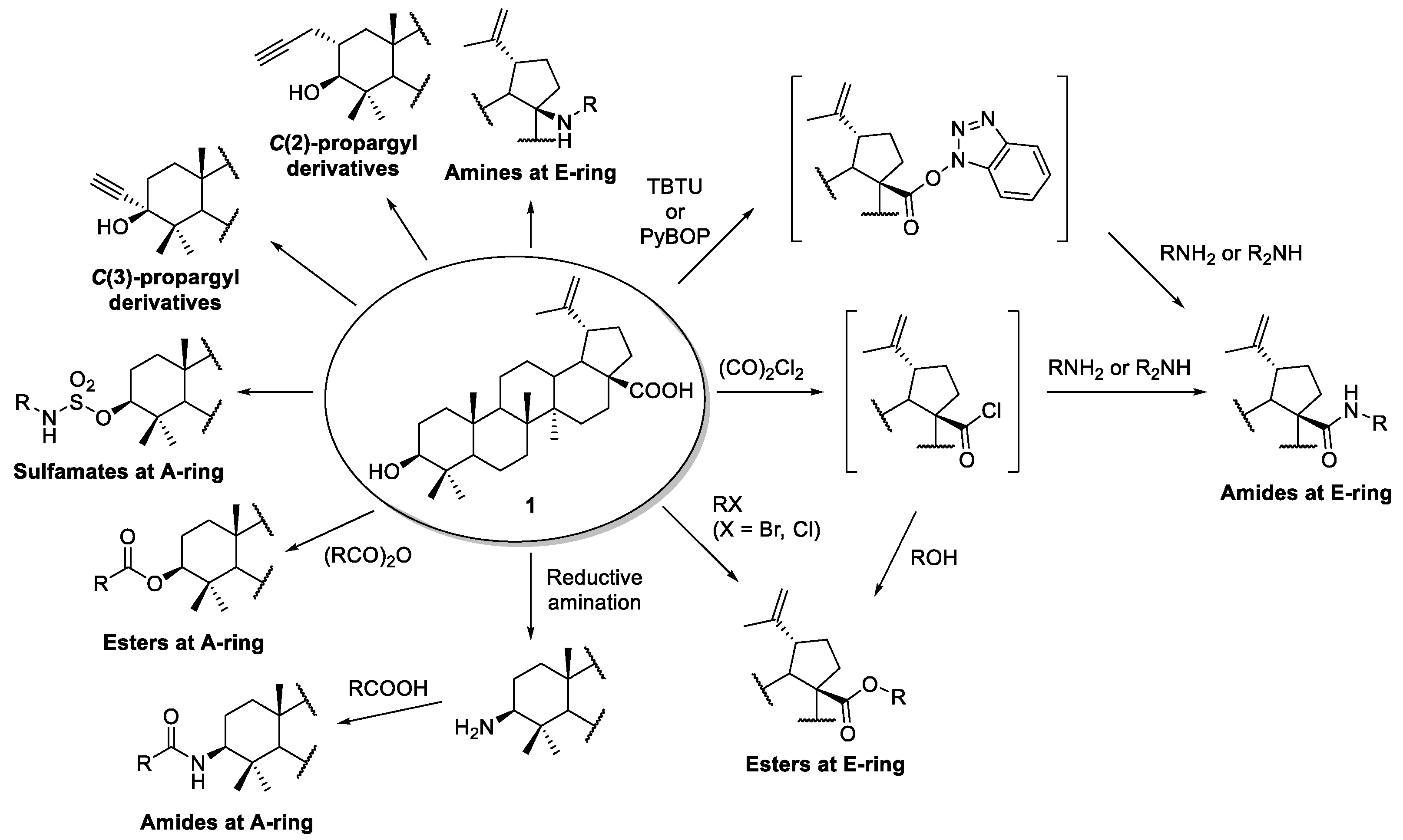
2. Simple Transformations
2.1. Amination
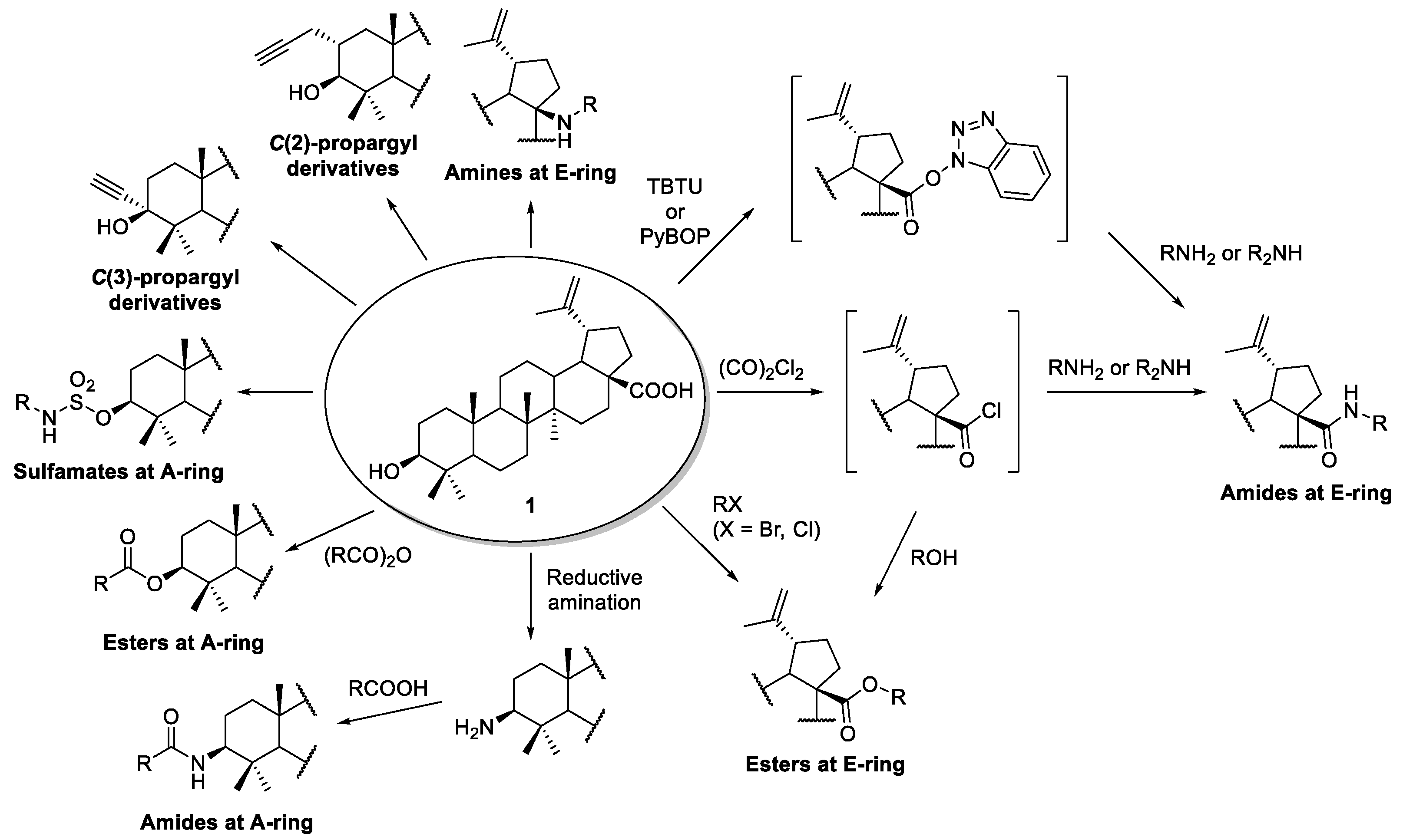
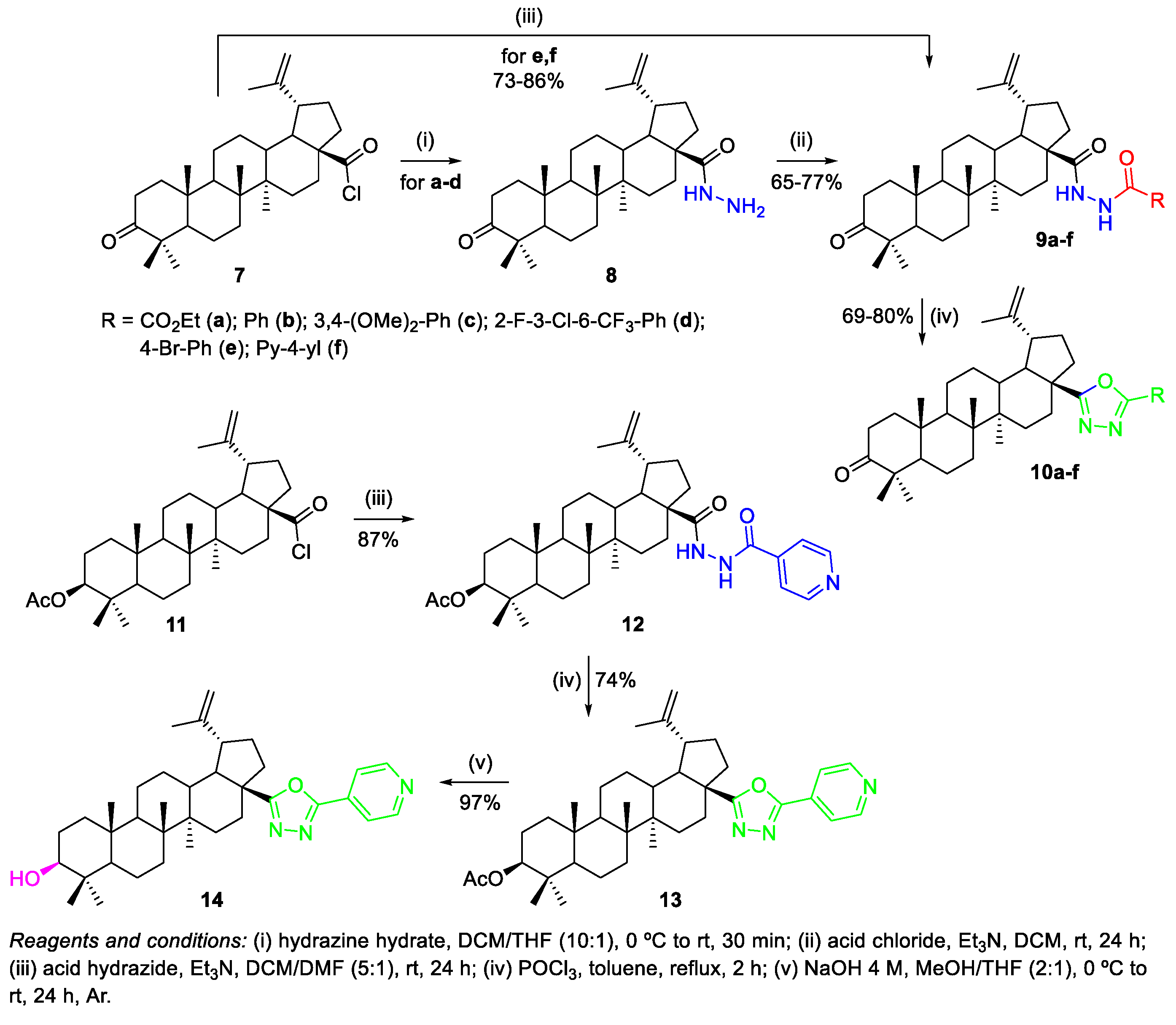
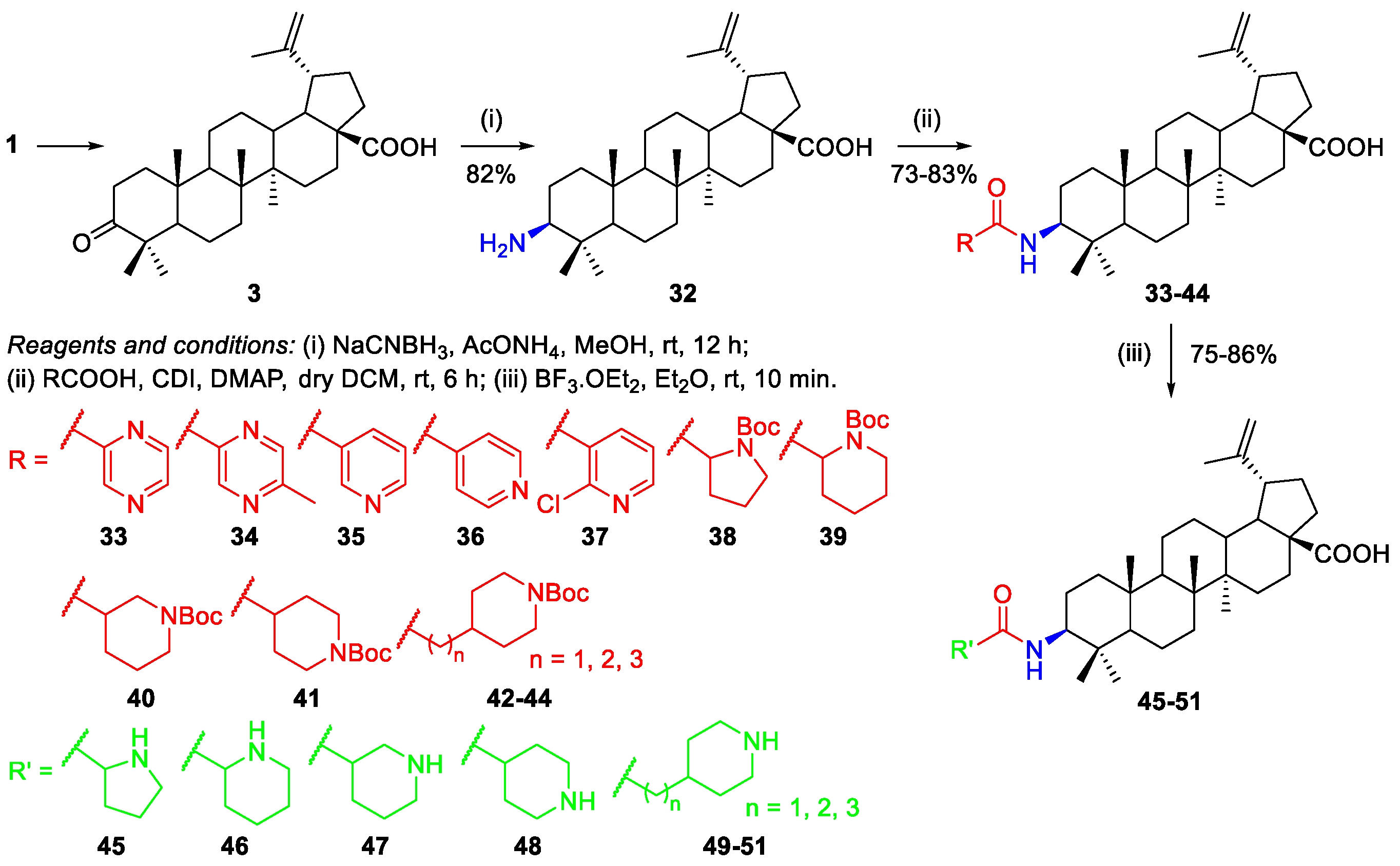
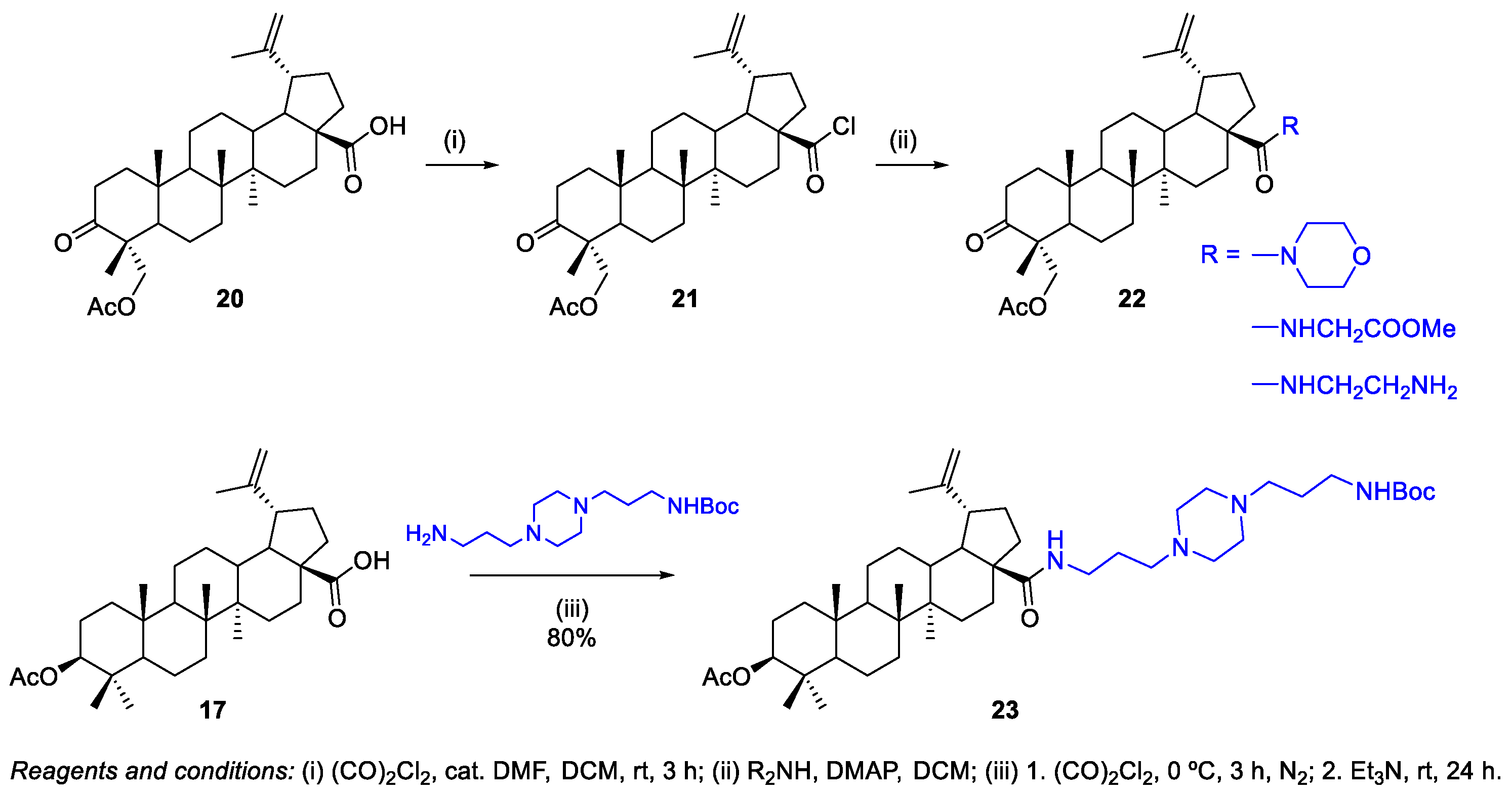
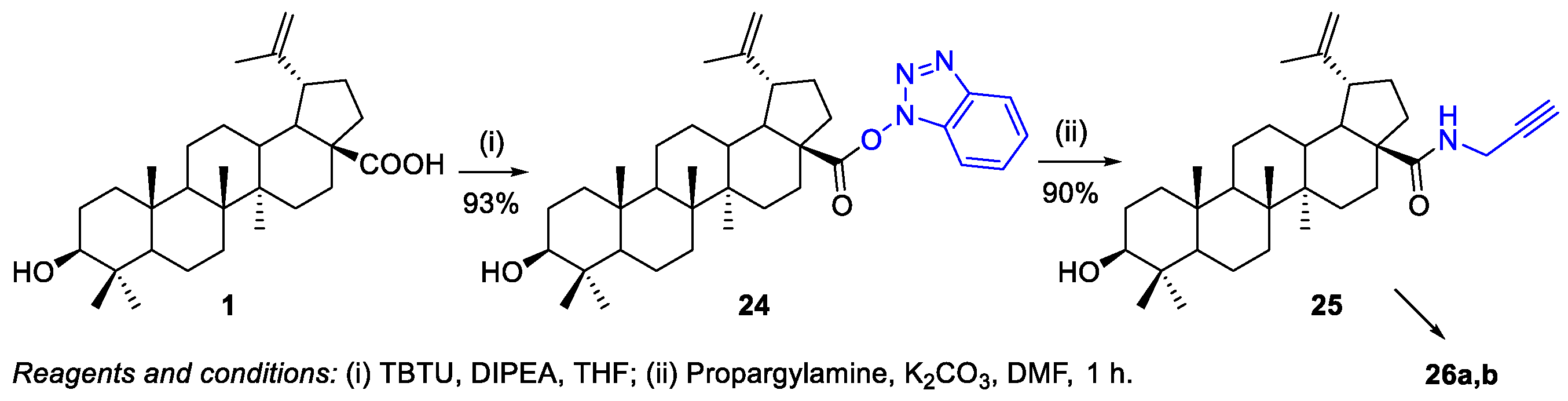
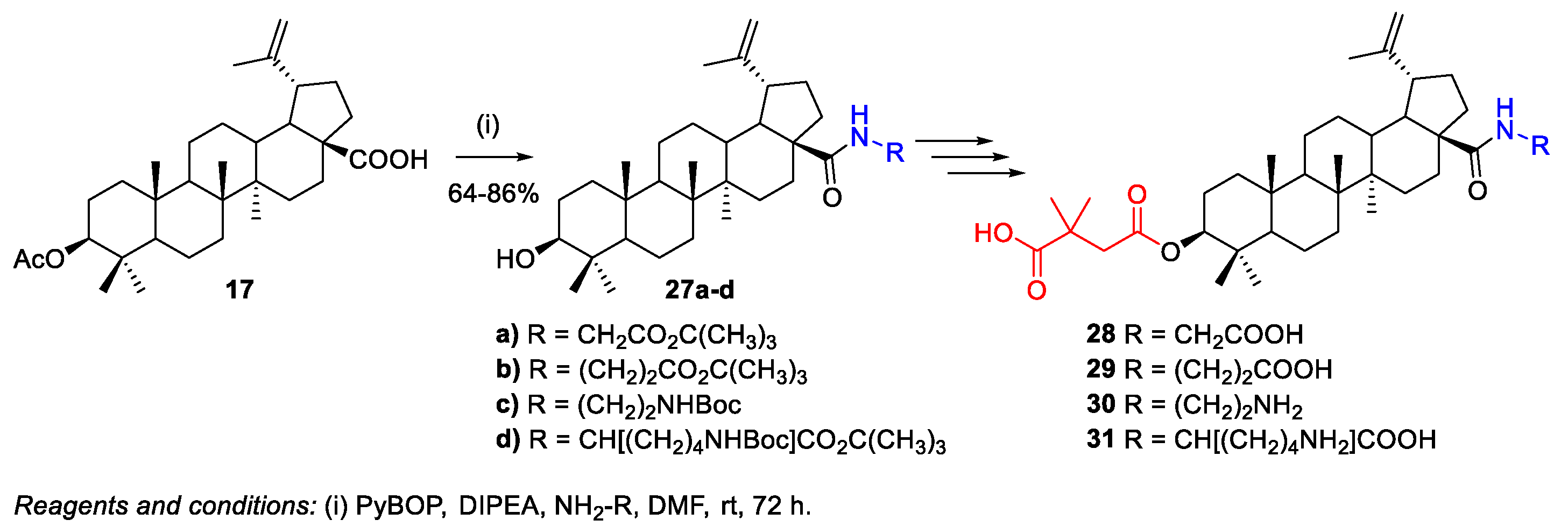
2.1.1. Synthesis of Amide Derivatives
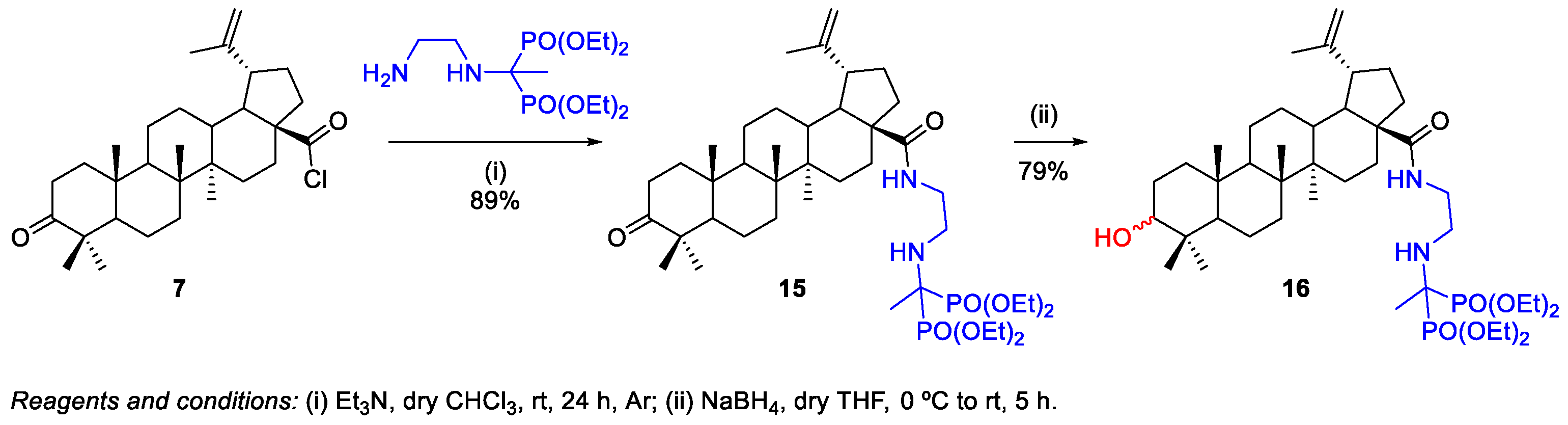
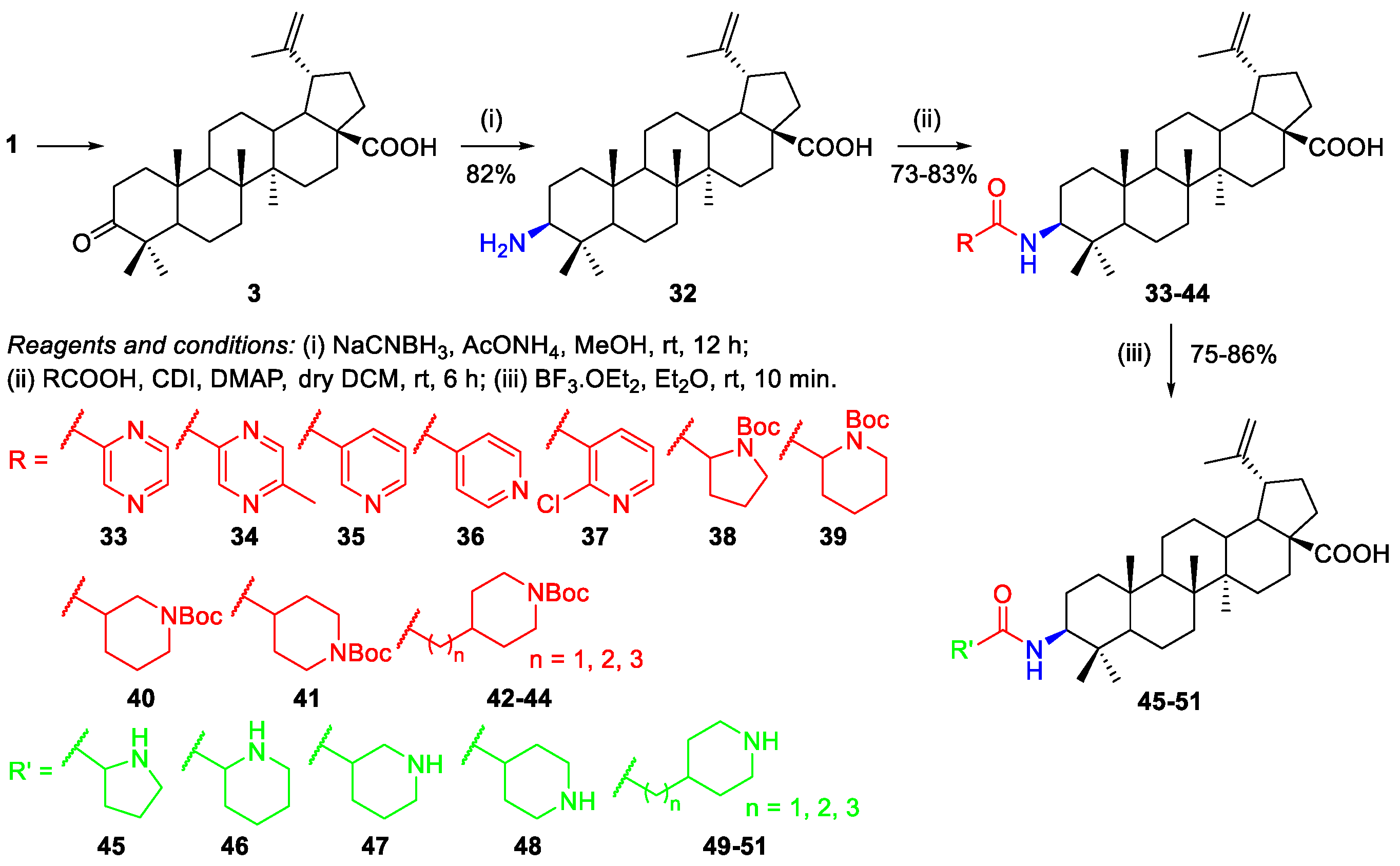
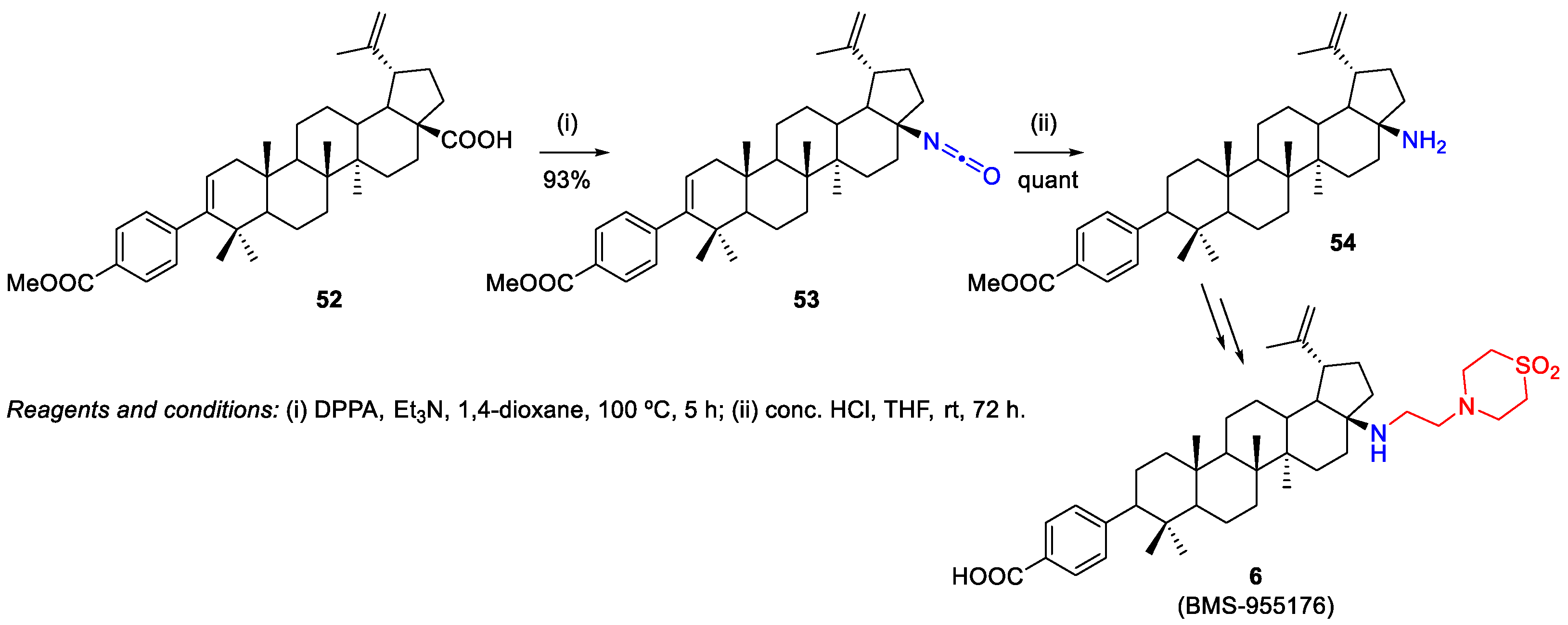
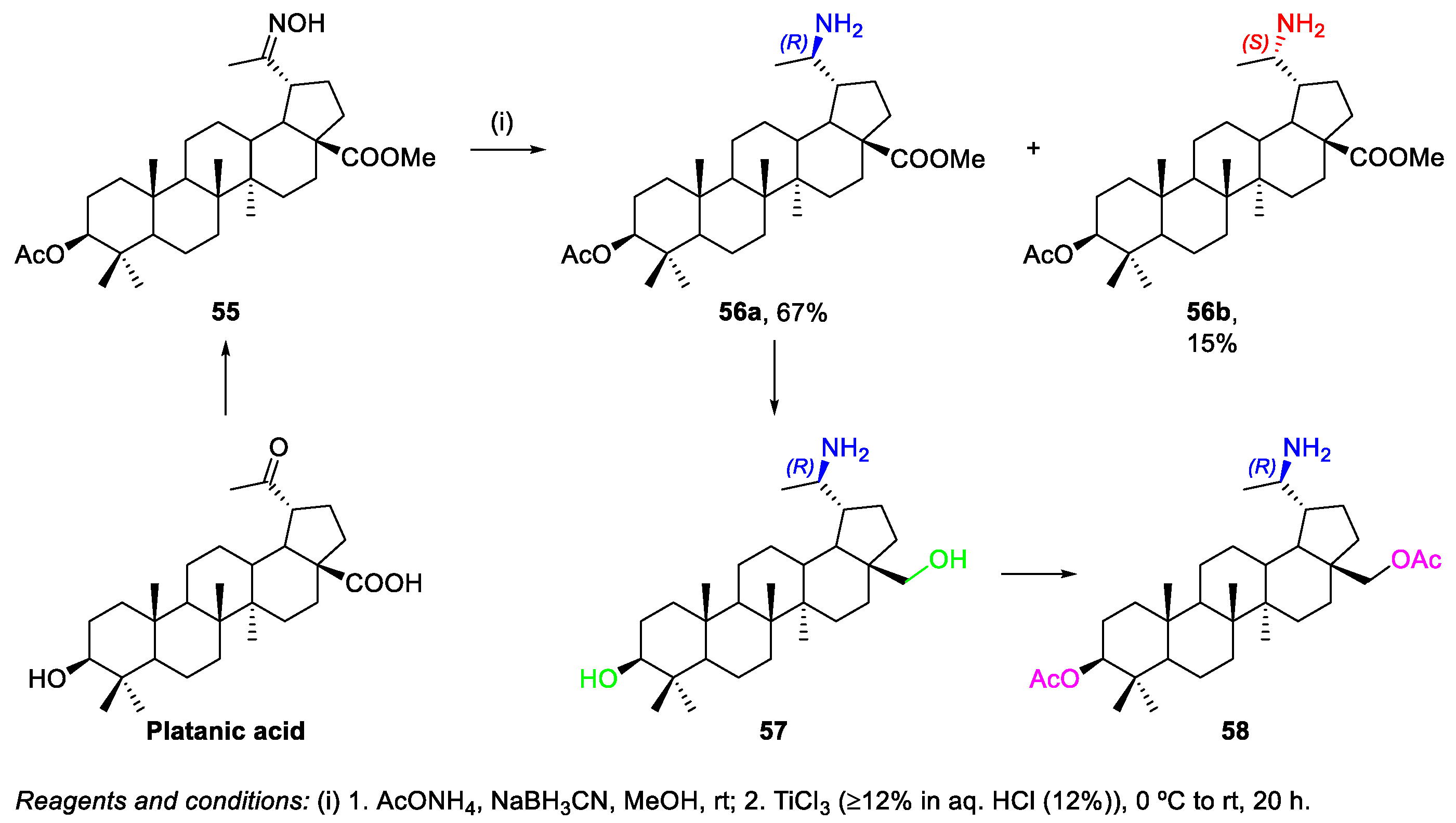
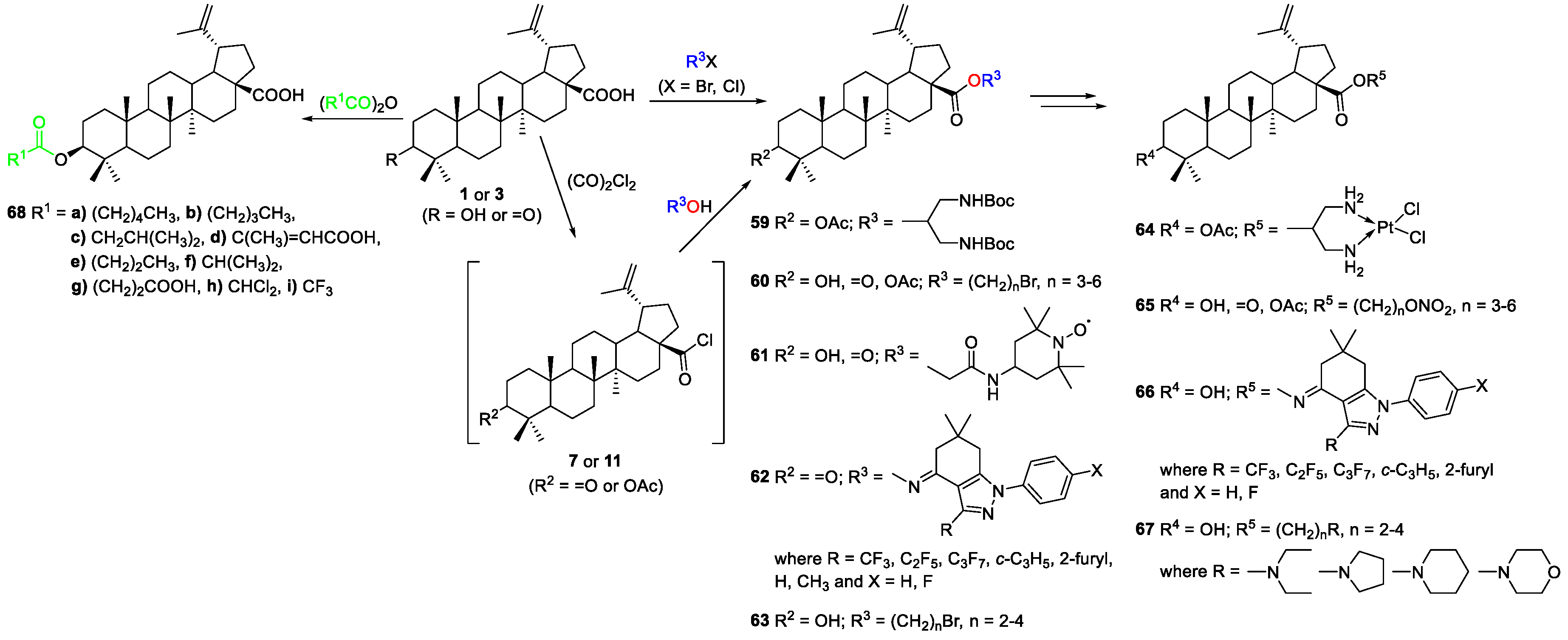
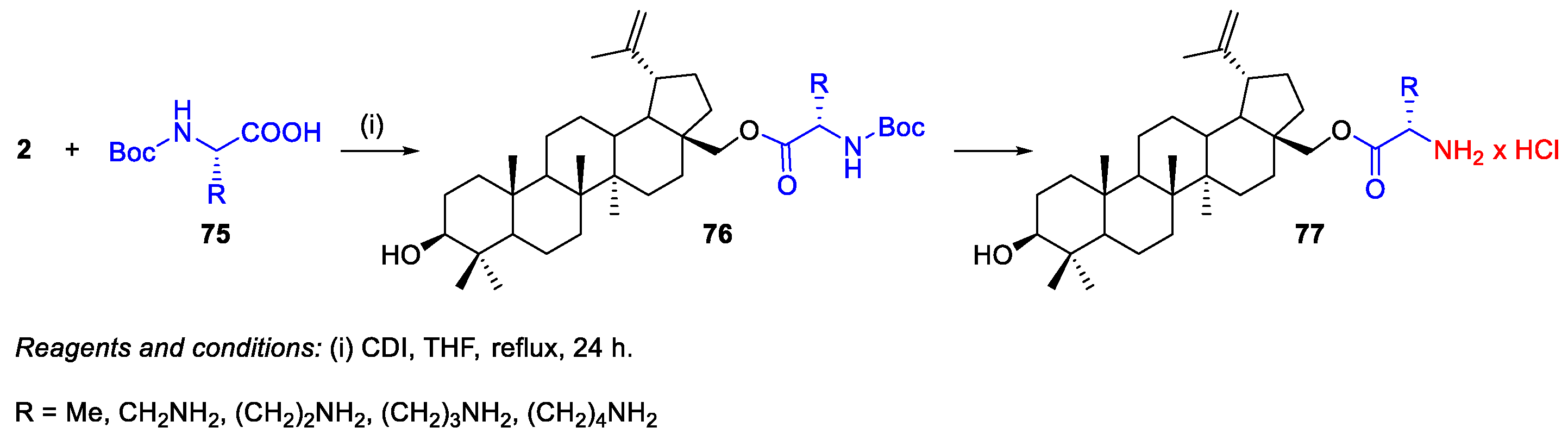
2.1.2. Synthesis of Amine Derivatives
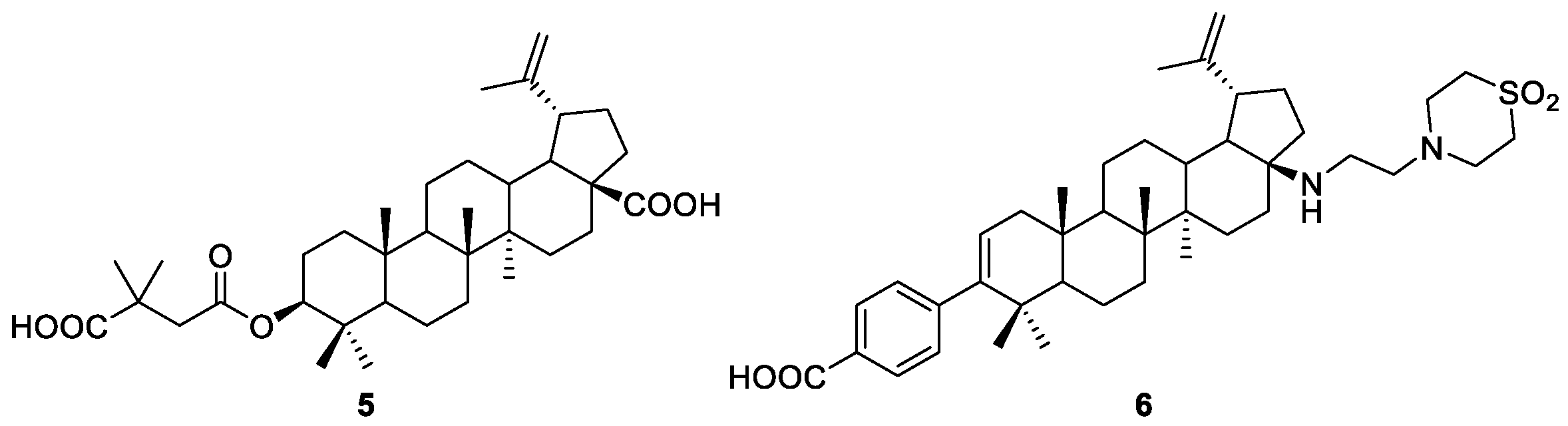
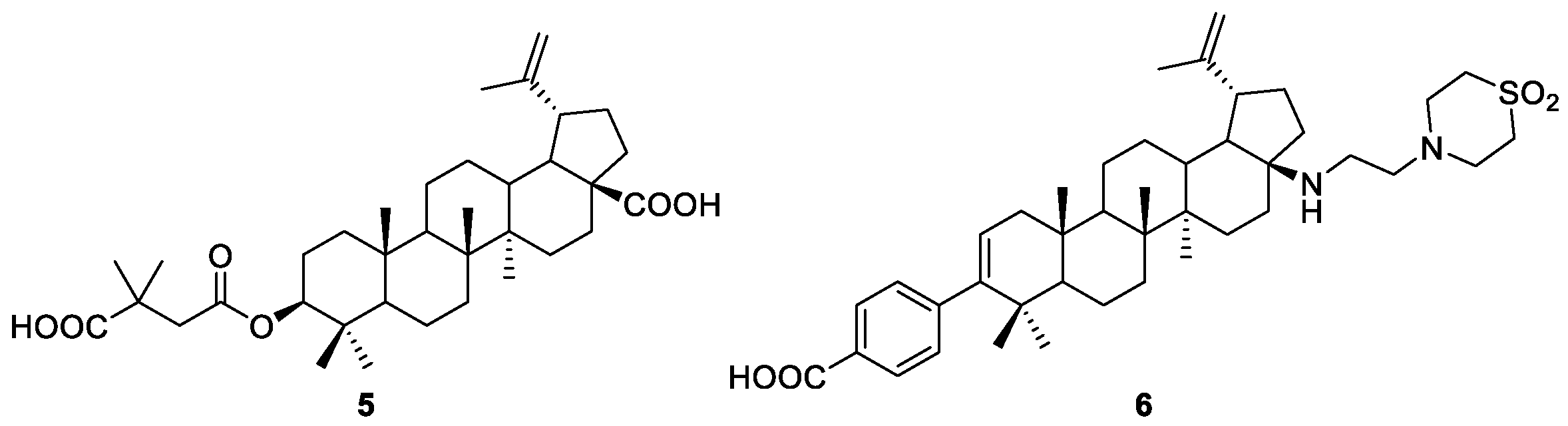
2.2. Esterification
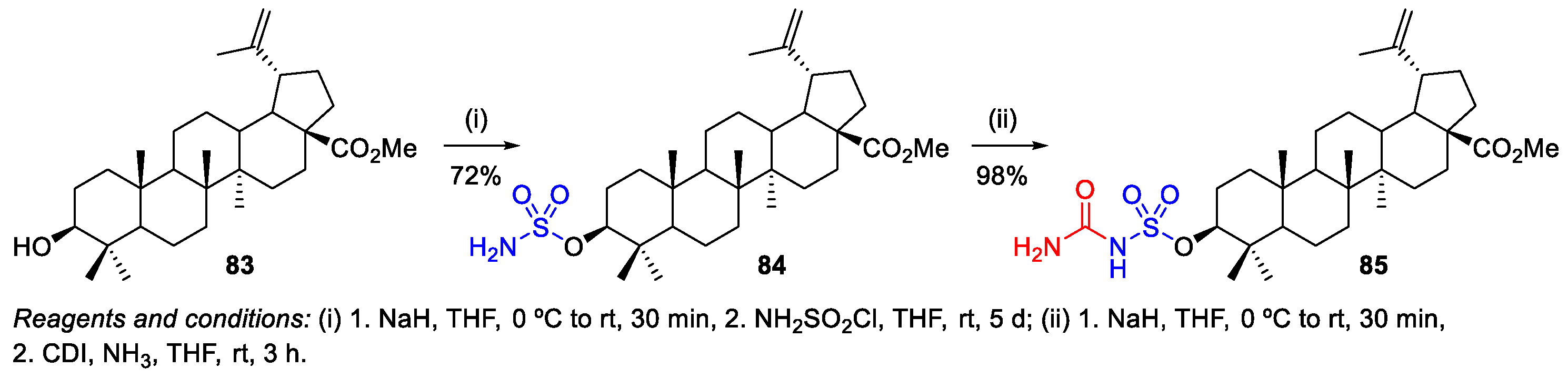
2.3. Sulfonation
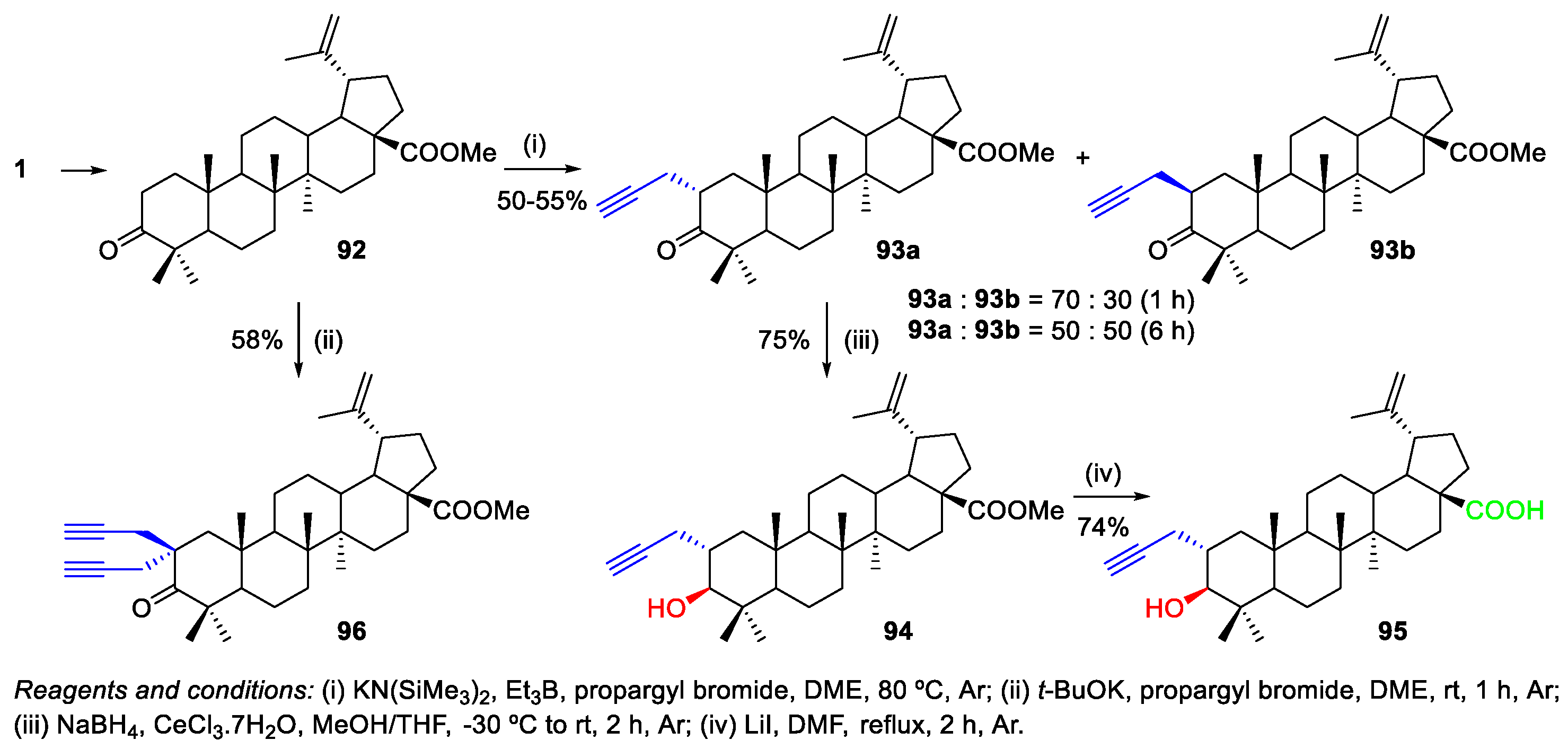
2.4. Alkylation
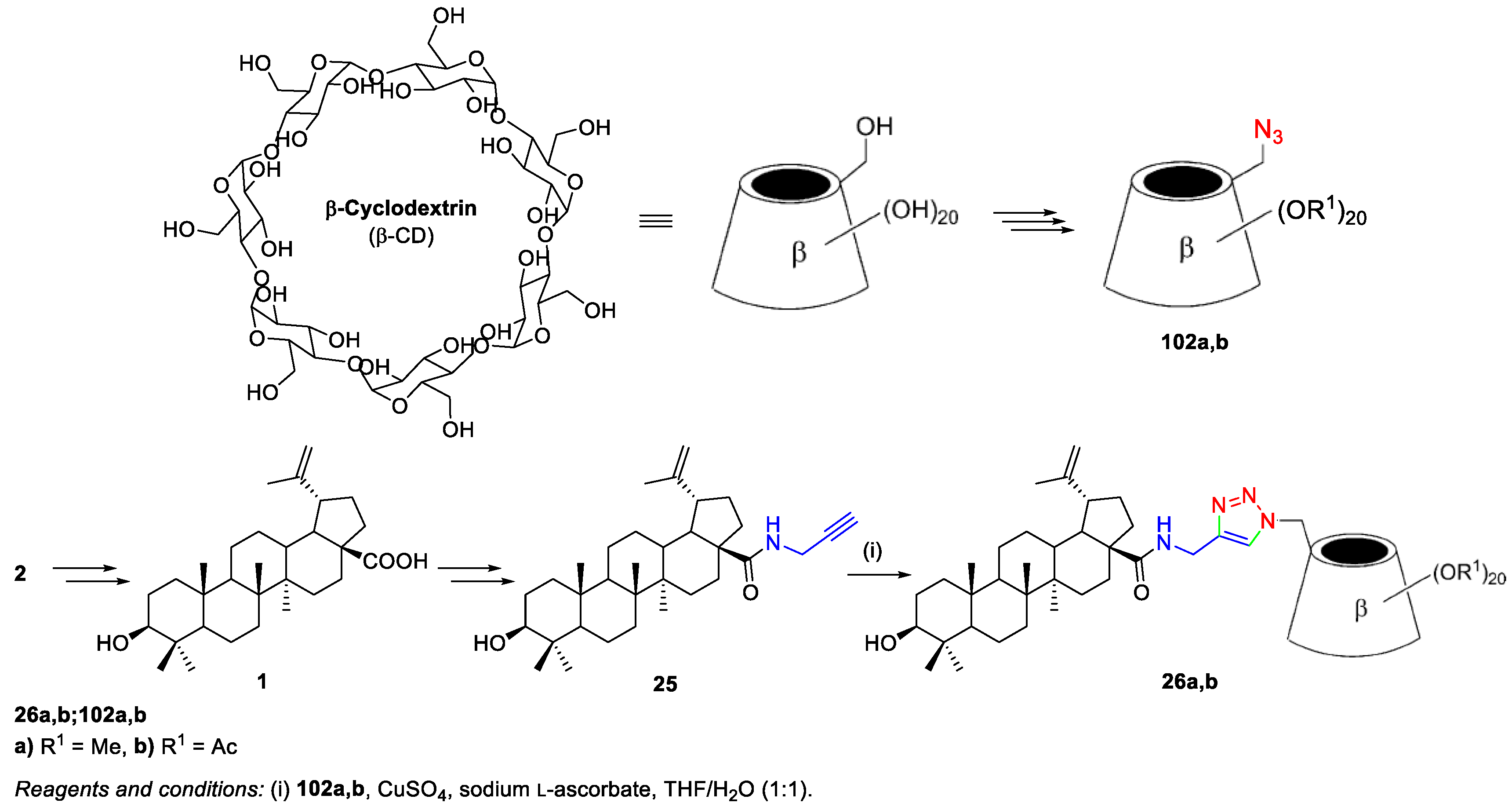
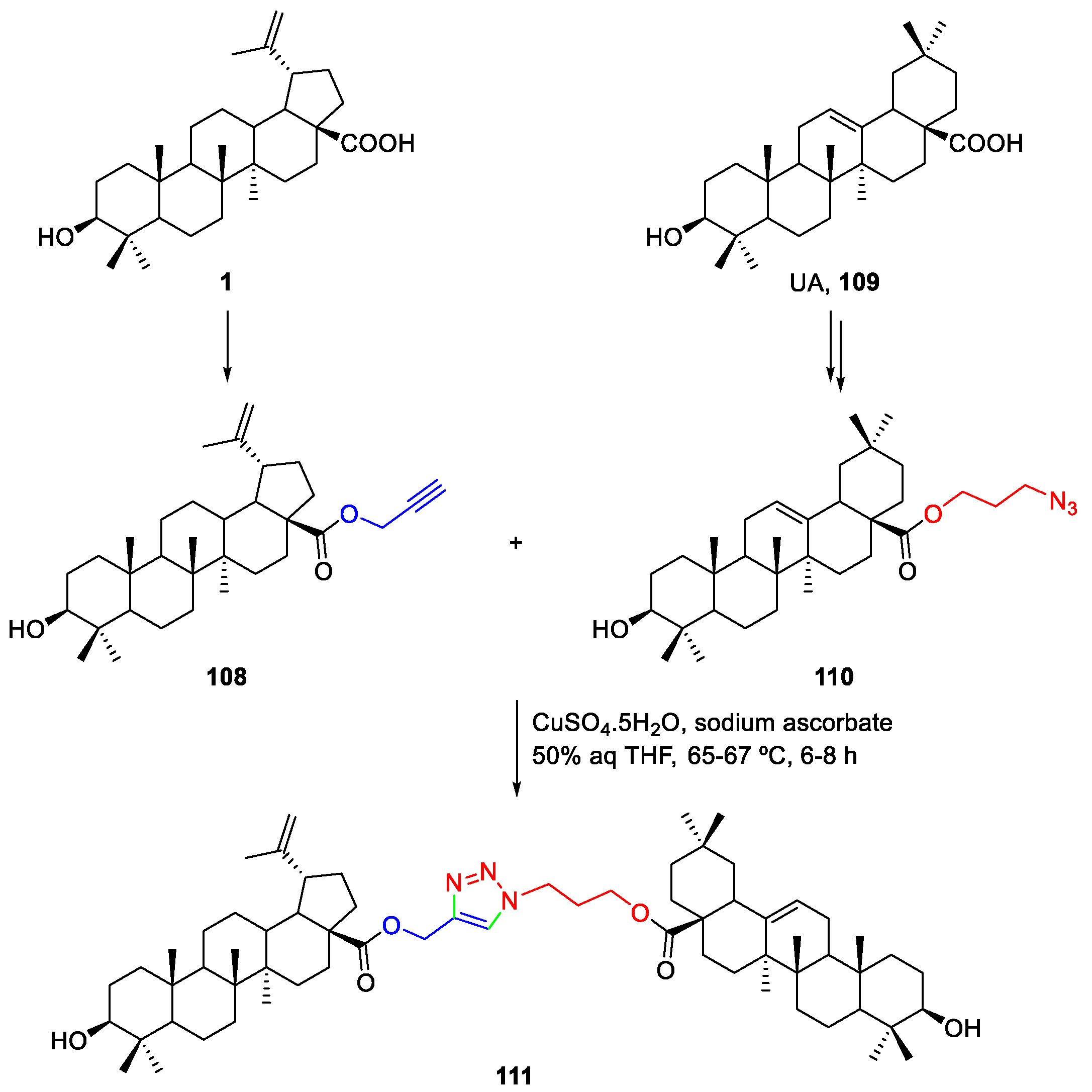
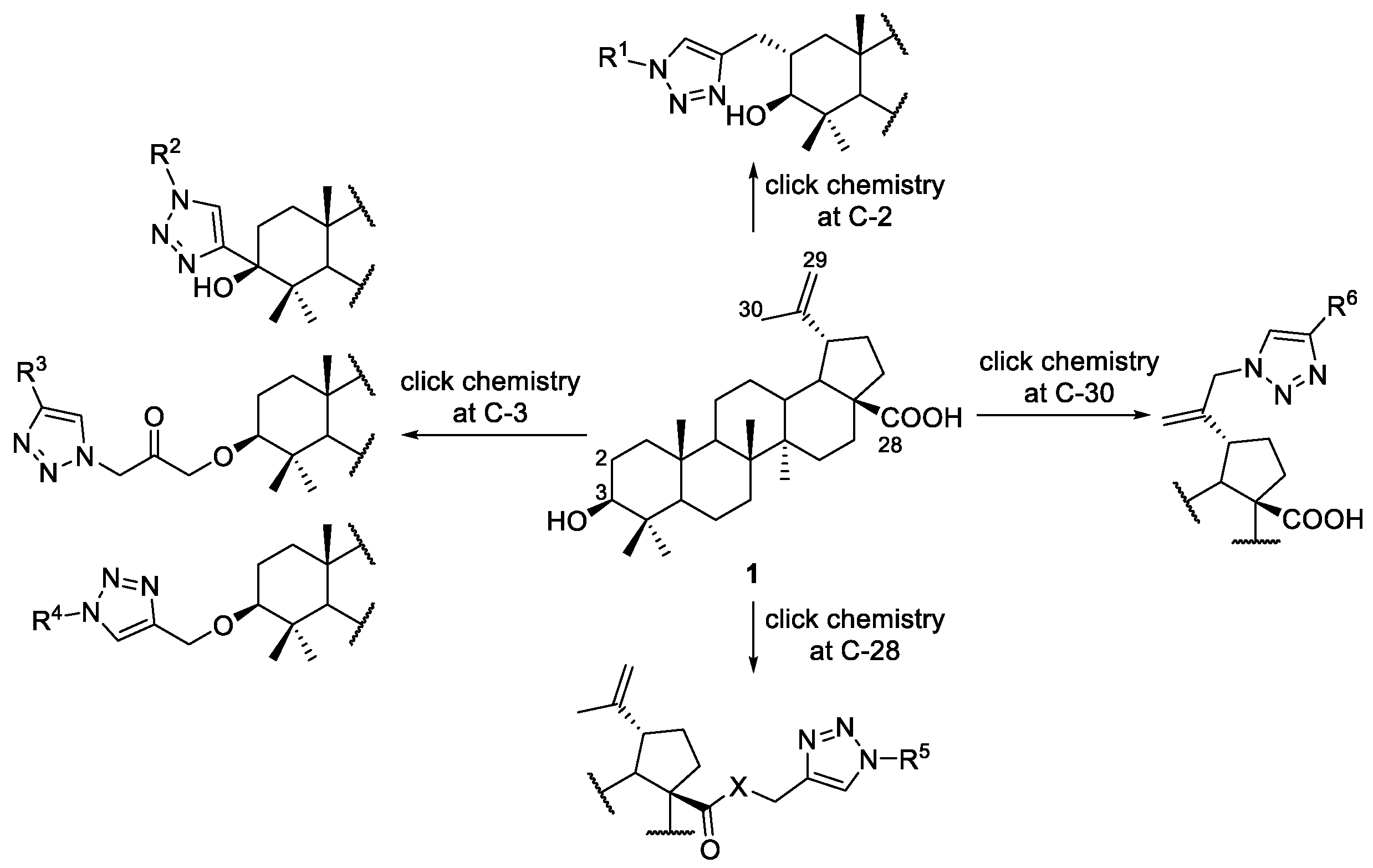
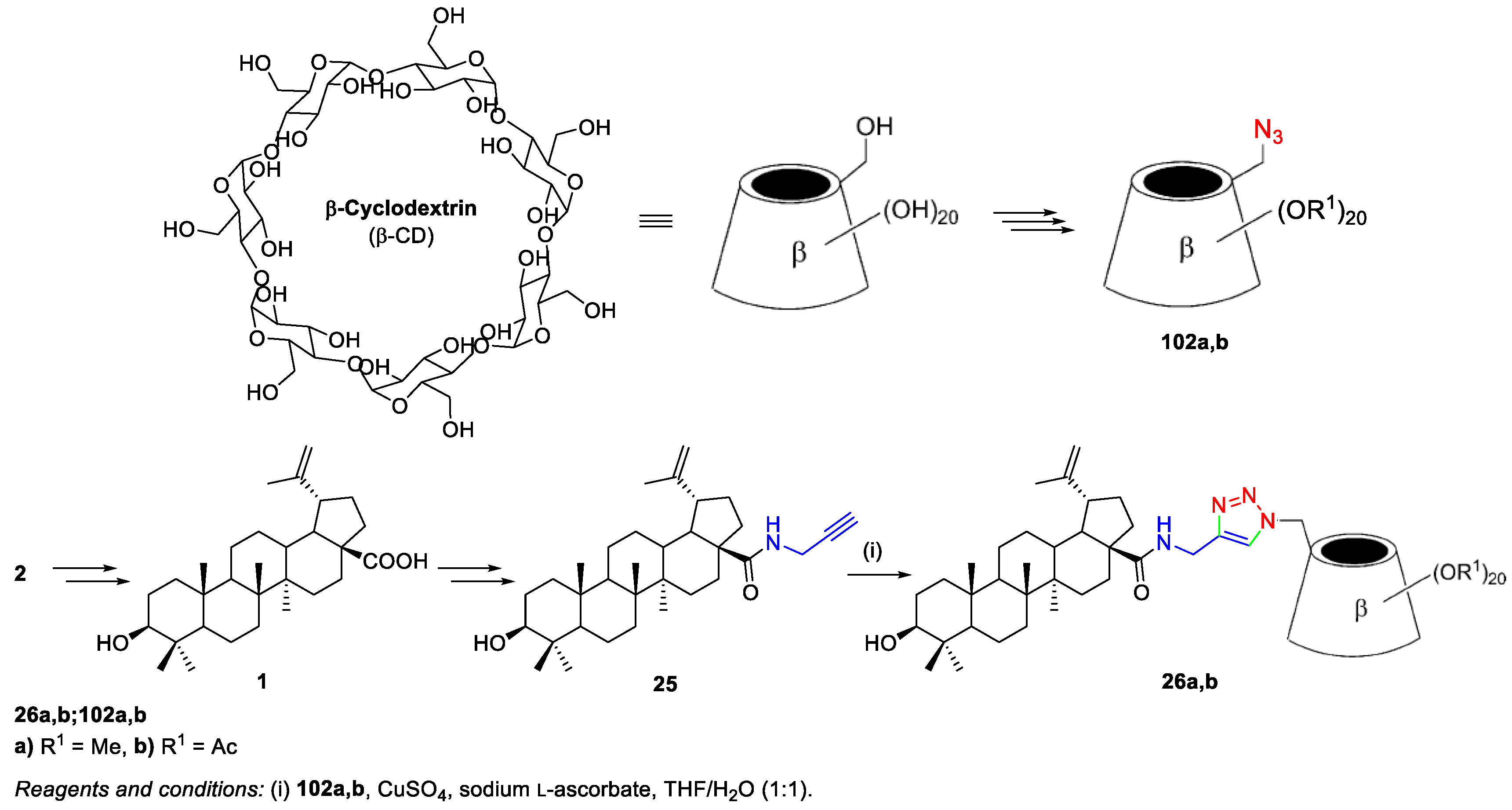
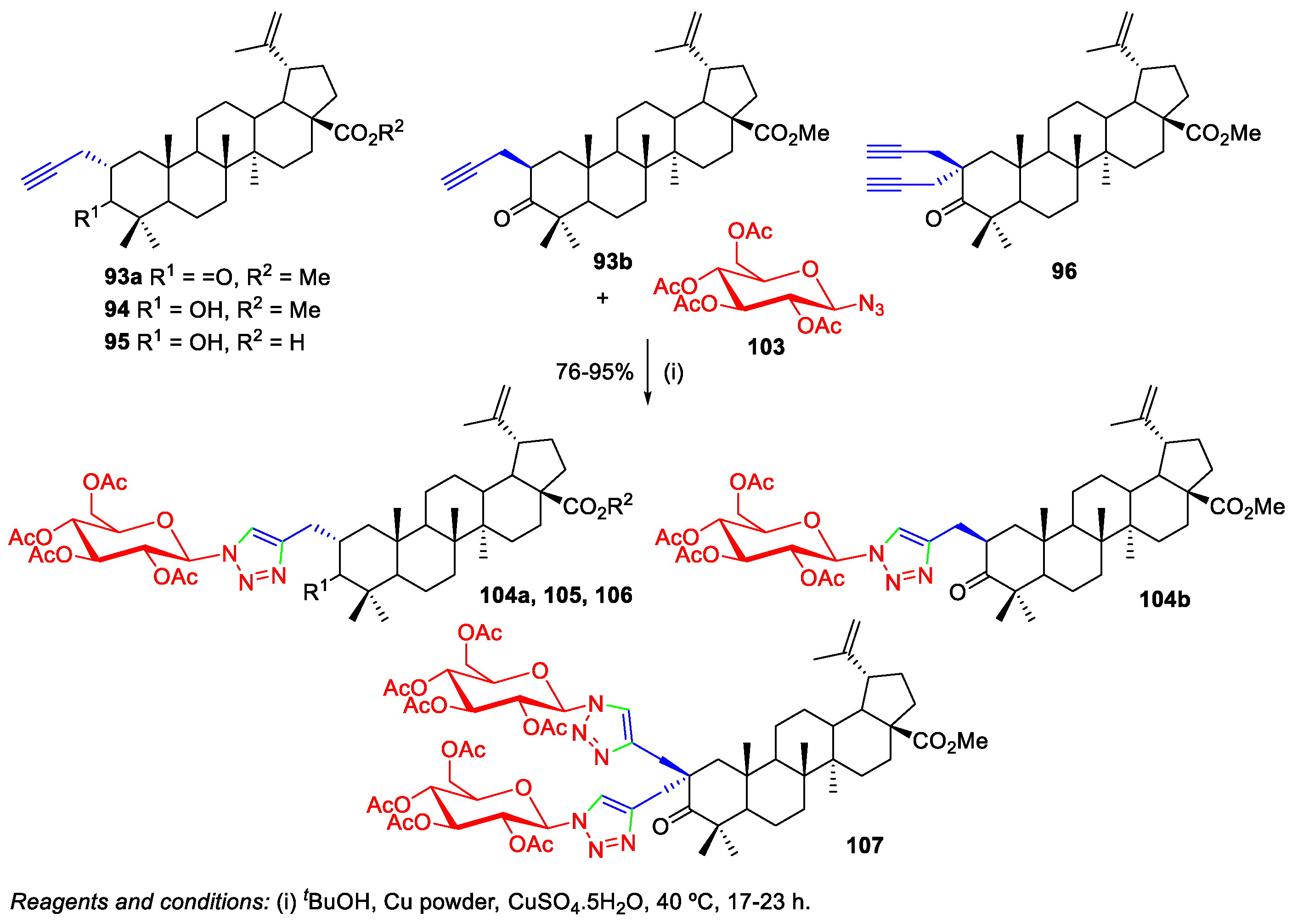
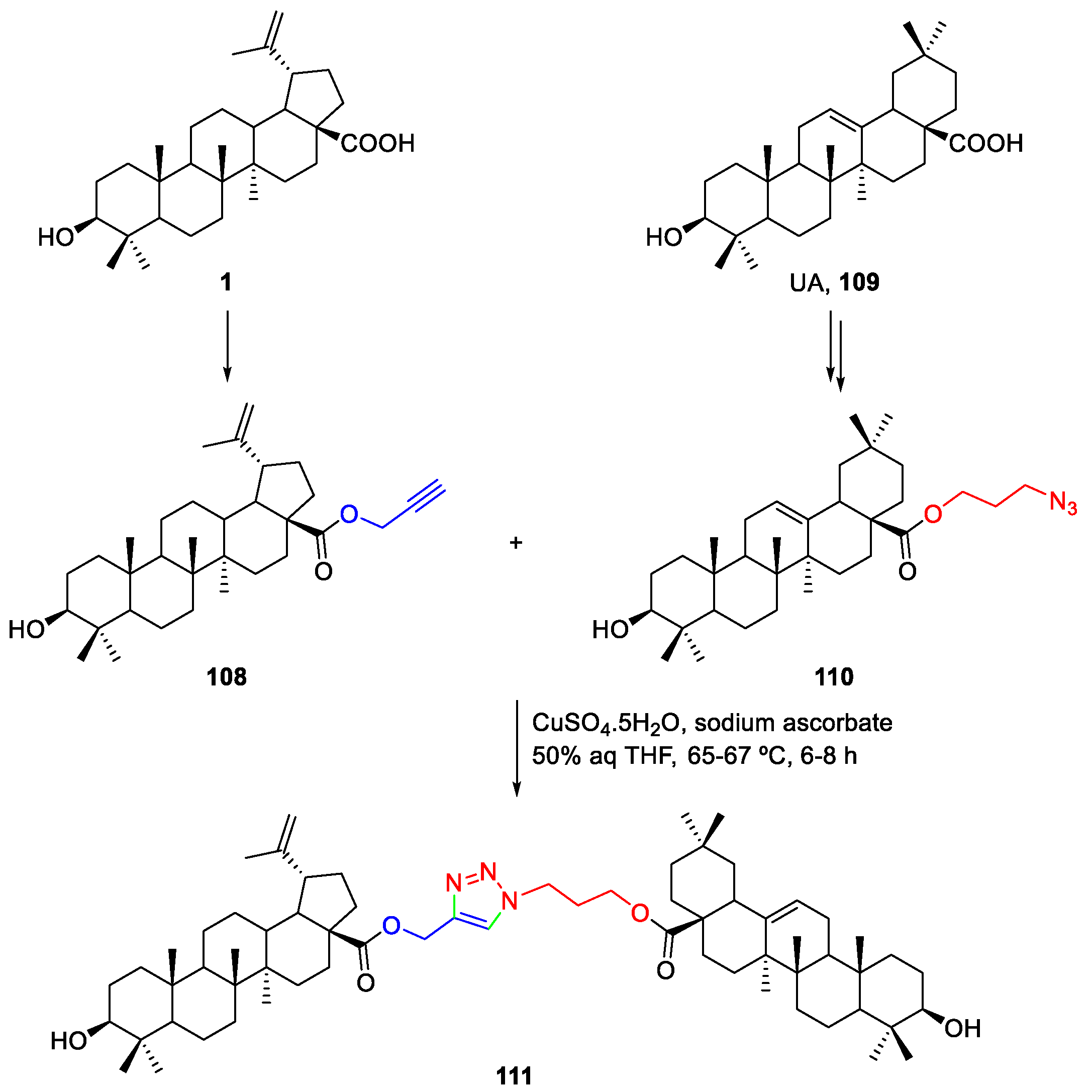
3. Click Chemistry—Copper-Catalyzed Azide-Alkyne Cycloaddition
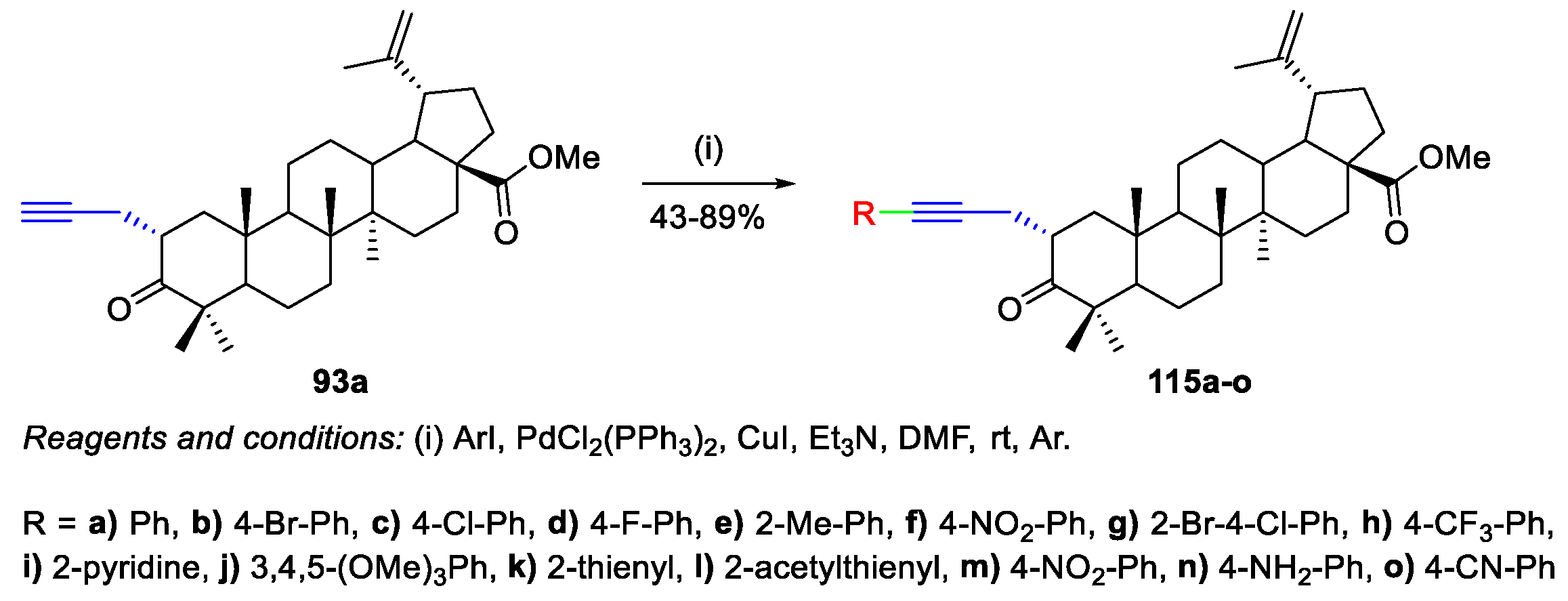
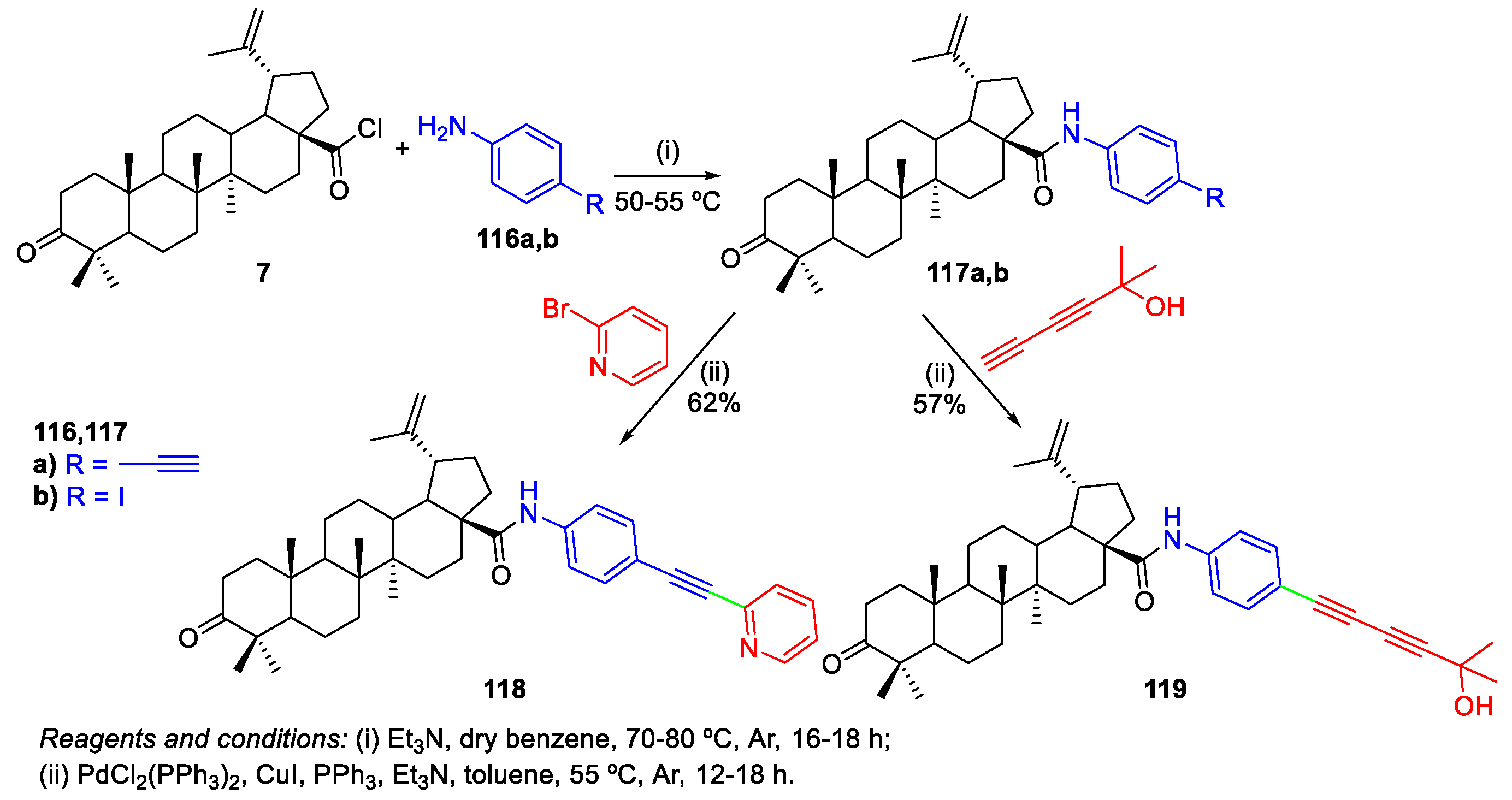
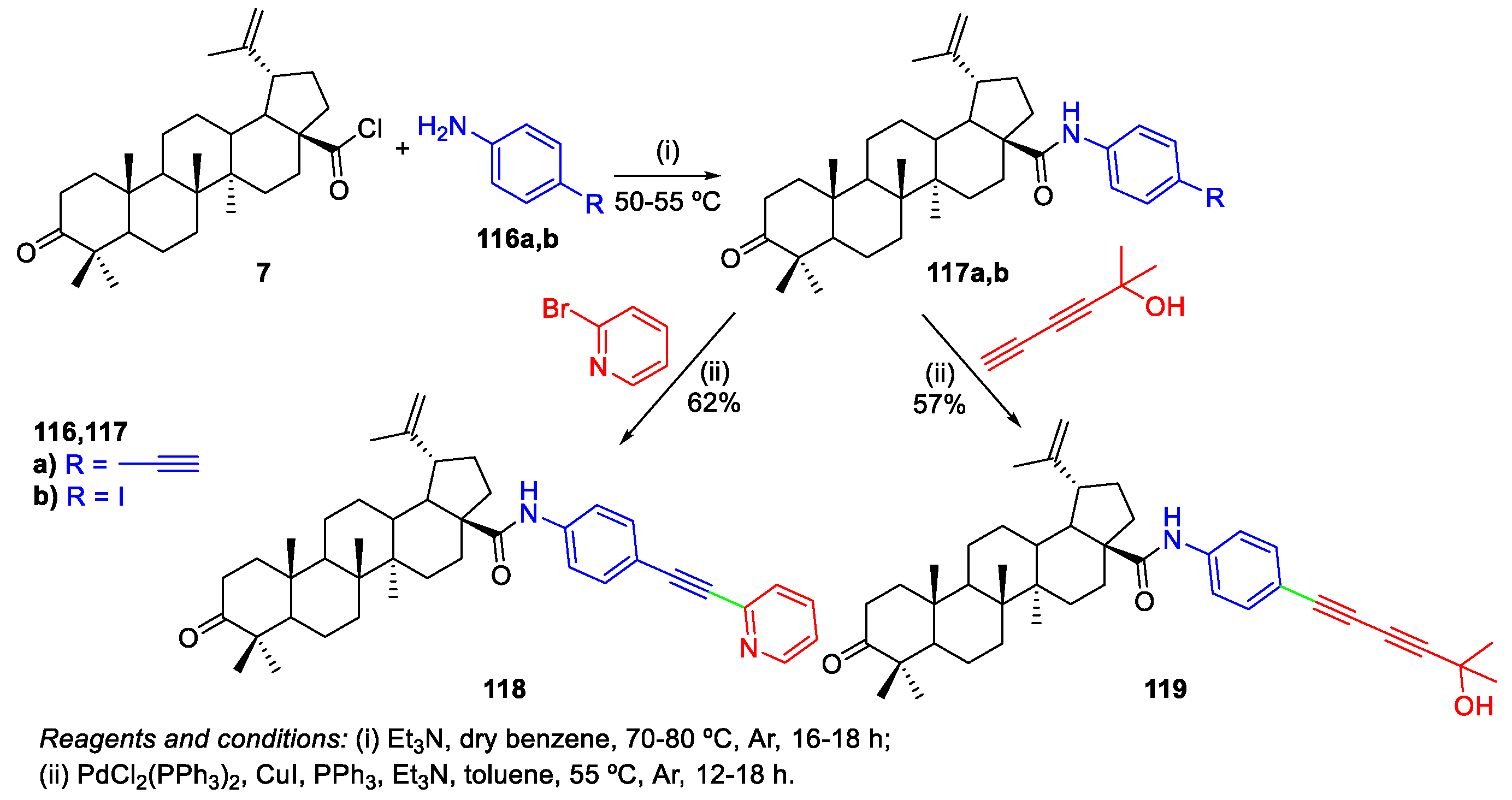
4. Palladium-Catalyzed Cross-Coupling Reactions
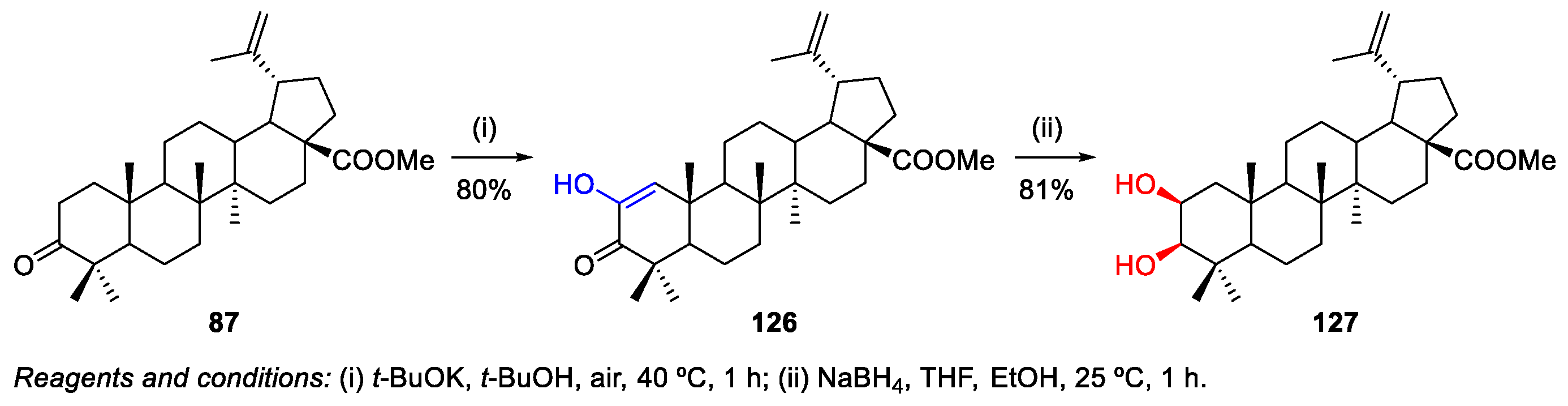
5. Hydroxylation
6. Aldol Condensation
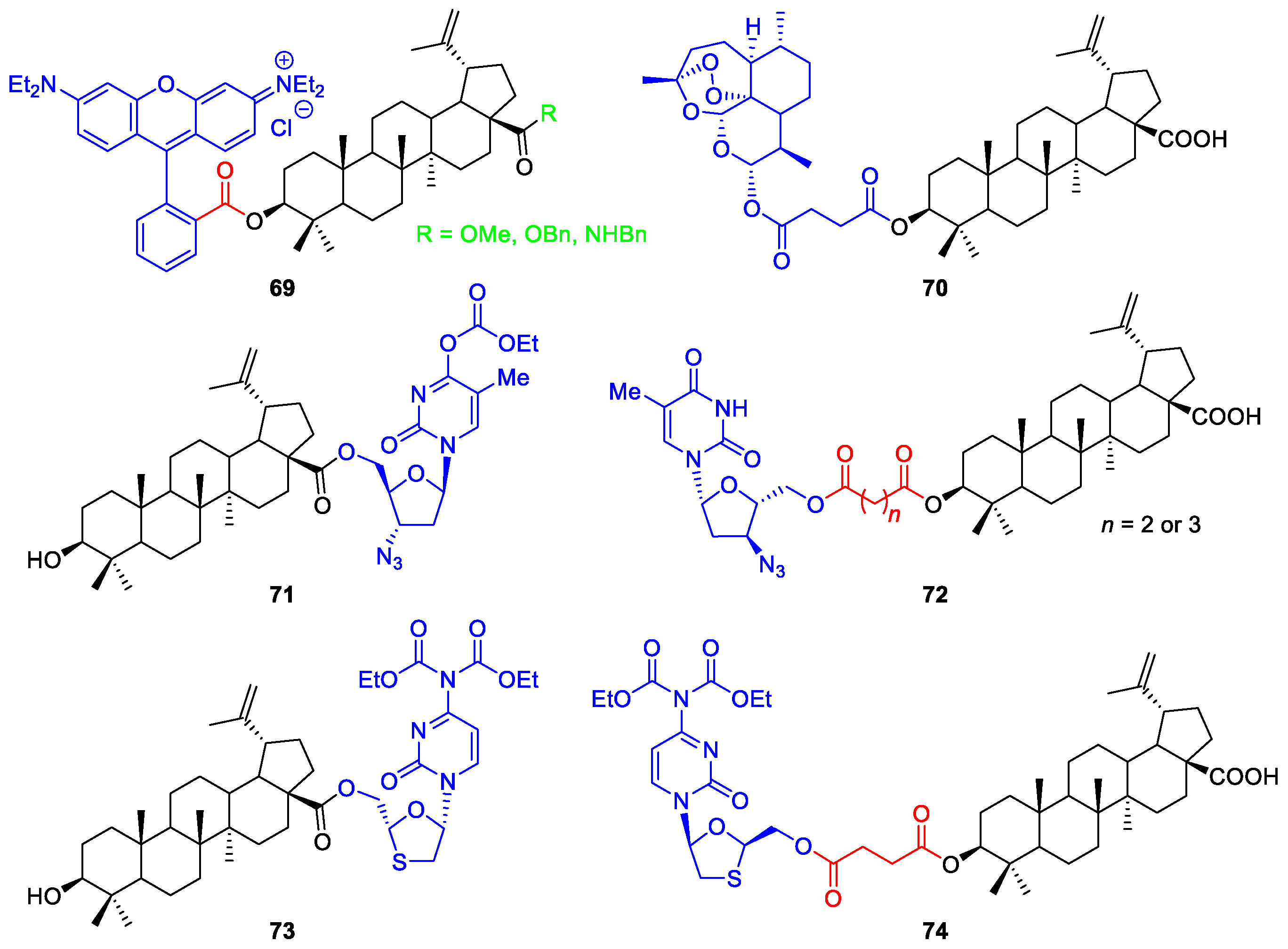
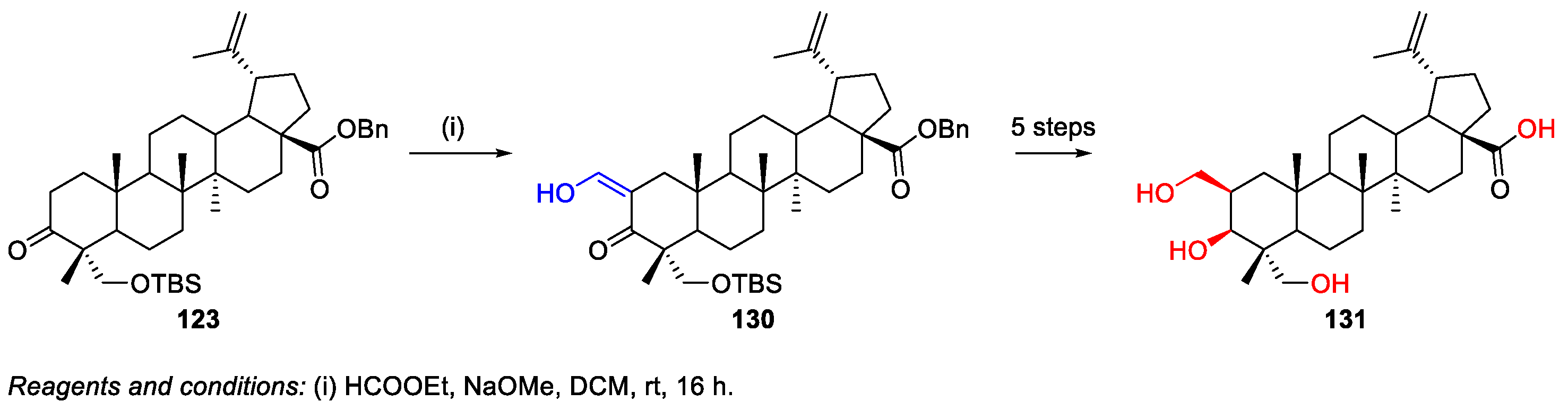
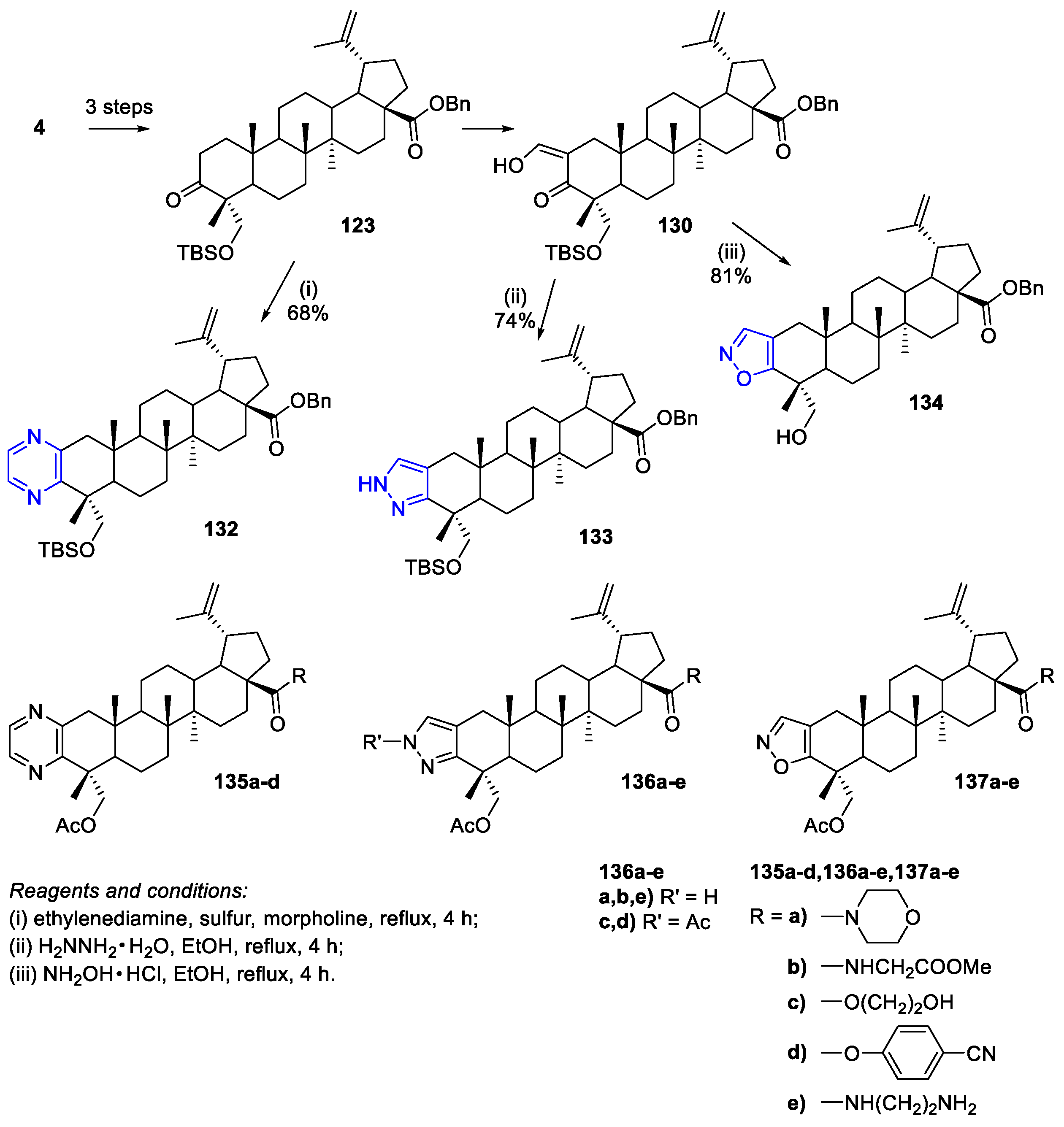
7. Synthesis of Heterocycle-Fused BA/HBA Derivatives
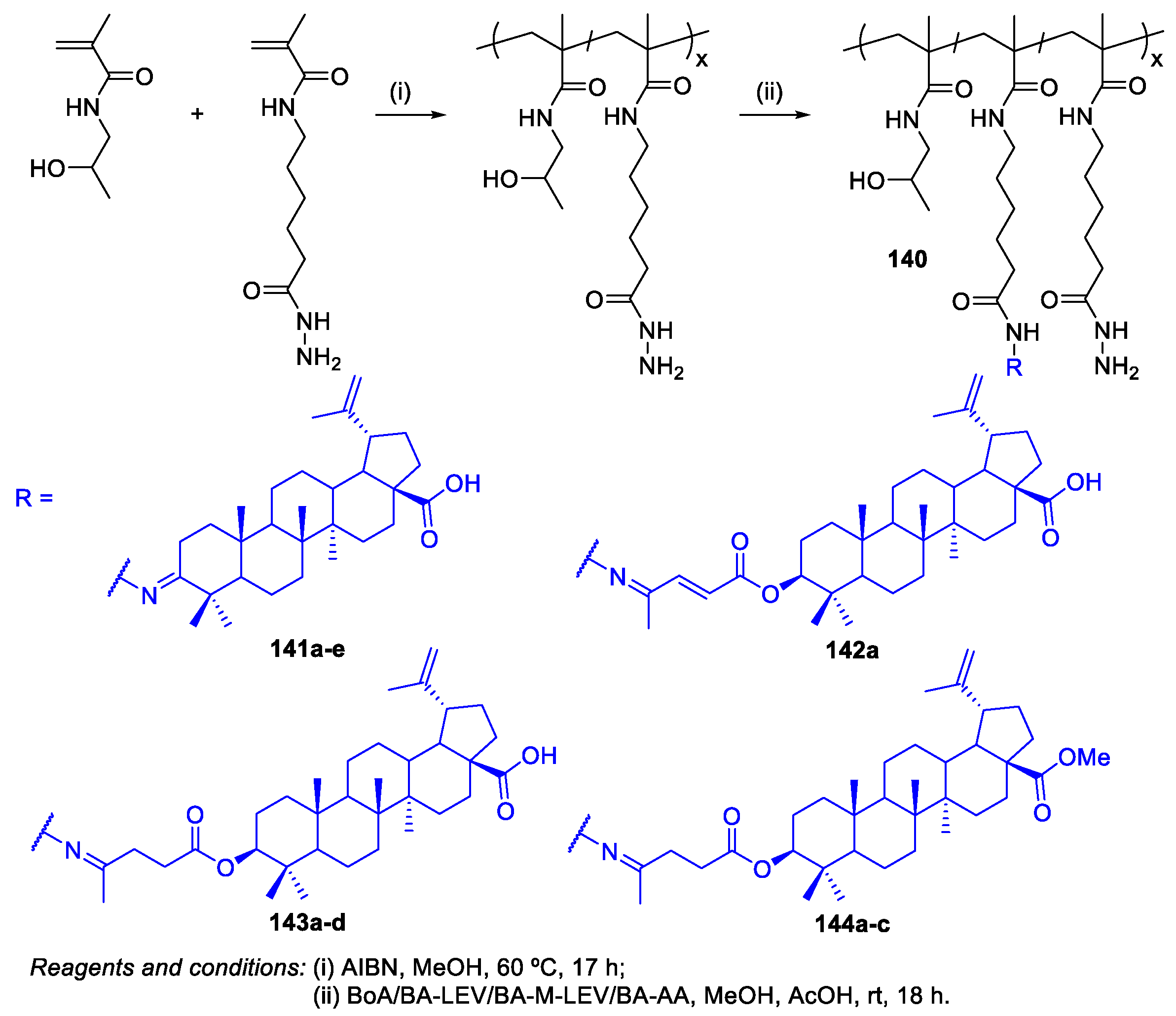
8. Synthesis of Polymer‒BA Conjugates
9. Miscellaneous Methodologies
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac | Acetyl |
| AIDS | Acquired immunodeficiency syndrome |
| Ala | Alanine |
| AZT | Azidothymidine |
| BA | Betulinic acid |
| BA-AA | Betulinic acid 3-acetyl acrylate |
| BA-LEV | Betulinic acid levulinate |
| BA-M-LEV | Methylated betulinic acid levulinate |
| BN | Betulin |
| Bn | Benzyl |
| BoA | Betulonic acid |
| Boc | tert-Butyloxycarbonyl |
| BODIPY | Boron-dipyrromethene |
| Bu | Butyl |
| cat | Catalyst |
| CCID50 | Cell culture infectious dose 50% |
| CD | Cyclodextrin |
| CDI | 1,1’-Carbonyl-diimidazole |
| CuAAC | Copper-catalyzed azide-alkyne cycloaddition |
| Dab | 2,4-Diaminobutyric acid |
| Dap | 2,3-Diaminopropionic acid |
| DCC | N,N’-Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DIC | N,N’-Diisopropylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DMA | N,N-Dimethylacetamide |
| DMAP | 4-(Dimethylamino)pyridine |
| DME | 1,2-Dimethoxyethane |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| DPPA | Diphenyl phosphoryl azide |
| EC50 | Half maximal effective concentration |
| EDC | N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide |
| equiv | Molar equivalents |
| Et | Ethyl |
| GI50 | Growth inhibition of 50% of cells |
| GSK | GlaxoSmithKline |
| HBA | 23-Hydroxybetulinic acid |
| HIV | Human Immunodeficiency Virus |
| HOBt | Hydroxybenzotriazole |
| HPMA | N-(2-Hydroxypropyl)methacrylamide |
| IC50 | Half maximal inhibitory concentration |
| i-Pr | Isopropyl |
| KHMDS | Potassium bis(trimethylsilyl)amide |
| LTA | Lead tetraacetate or lead(IV) acetate |
| Lys | Lysine |
| mCPBA | m-Chloroperoxybenzoic acid |
| Me | Methyl |
| Orn | Ornithine |
| PCC | Pyridinium chlorochromate |
| PEG | Polyethylene glycol |
| Ph | Phenyl |
| Py | Pyridine |
| PyBOP | Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate |
| quant | Quantitative (yield) |
| rt | Room temperature |
| SN2 | Bimolecular nucleophilic substitution |
| TBS | tert-Butyl(dimethyl)silyl |
| TBTU | N,N,N’,N’-Tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate |
| t-Bu | tert-Butyl |
| Tf | Triflate |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| UA | Ursolic acid |
| β-CD | β-Cyclodextrin |
References
- Moghaddam, M.G.; Ahmad, F.B.H.; Samzadeh-Kermani, A. Biological Activity of Betulinic Acid: A Review. Pharmacol. Pharm. 2012, 3, 119–123. [Google Scholar] [CrossRef]
- Ríos, J.L.; Máñez, S. New Pharmacological Opportunities for Betulinic Acid. Planta Med. 2018, 84, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Sami, A.; Taru, M.; Salme, K.; Jari, Y.-K. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar]
- Csuk, R. Betulinic acid and its derivatives: A patent review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-M.; Xu, H.-G.; Wang, L.; Li, Y.-J.; Sun, P.-H.; Wu, X.-M.; Wang, G.-J.; Chen, W.-M.; Ye, W.-C. Betulinic Acid and its Derivatives as Potential Antitumor Agents. Med. Res. Rev. 2015, 35, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Ali-Seyed, M.; Jantan, I.; Vijayaraghavan, K.; Bukhari, S.N.A. Betulinic Acid: Recent Advances in Chemical Modifications, Effective Delivery, and Molecular Mechanisms of a Promising Anticancer Therapy. Chem. Biol. Drug Des. 2016, 87, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.E.; Salzwedel, K.; Allaway, G.P. Bevirimat: A Novel Maturation Inhibitor for the Treatment of HIV-1 Infection. Antivir. Chem. Chemother. 2008, 19, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Regueiro-Ren, A.; Liu, Z.; Chen, Y.; Sin, N.; Sit, S.-Y.; Swidorski, J.J.; Chen, J.; Venables, B.L.; Zhu, J.; Nowicka-Sans, B.; et al. Discovery of BMS-955176, a Second Generation HIV-1 Maturation Inhibitor with Broad Spectrum Antiviral Activity. ACS Med. Chem. Lett. 2016, 7, 568–572. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Tang, N.; Yan, W. Research and Development in Betulin and Betulinic Acid Derived Triterpenoids. Mini-Rev. Org. Chem. 2014, 11, 343–354. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Zaprutko, L. Recent advances in synthesis and biological activity of triterpenic acylated oximes. Phytochem. Rev. 2015, 14, 203–231. [Google Scholar] [CrossRef]
- Kvasnica, M.; Urban, M.; Dickinson, N.J.; Sarek, J. Pentacyclic triterpenoids with nitrogen- and sulfur-containing heterocycles: Synthesis and medicinal significance. Nat. Prod. Rep. 2015, 32, 1303–1330. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, R.-H.; Wang, M.; Xu, G.-B.; Liao, S.-G. Prodrugs of triterpenoids and their derivatives. Eur. J. Med. Chem. 2017, 131, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Borkova, L.; Hodon, J.; Urban, M. Synthesis of Betulinic Acid Derivatives with Modified A-Rings and their Application as Potential Drug Candidates. Asian J. Org. Chem. 2018, 7, 1542–1560. [Google Scholar] [CrossRef]
- Pokorny, J.; Borkova, L.; Urban, M. Click Reactions in Chemistry of Triterpenes - Advances Towards Development of Potential Therapeutics. Curr. Med. Chem. 2018, 25, 636–658. [Google Scholar] [CrossRef]
- Innocente, A.; Casanova, B.B.; Klein, F.; Lana, A.D.; Pereira, D.; Muniz, M.N.; Sonnet, P.; Gosmann, G.; Fuentefria, A.M.; Gnoatto, S.C.B. Synthesis of Isosteric Triterpenoid Derivatives and Antifungal Activity. Chem. Biol. Drug Des. 2014, 83, 344–349. [Google Scholar] [CrossRef] [PubMed]
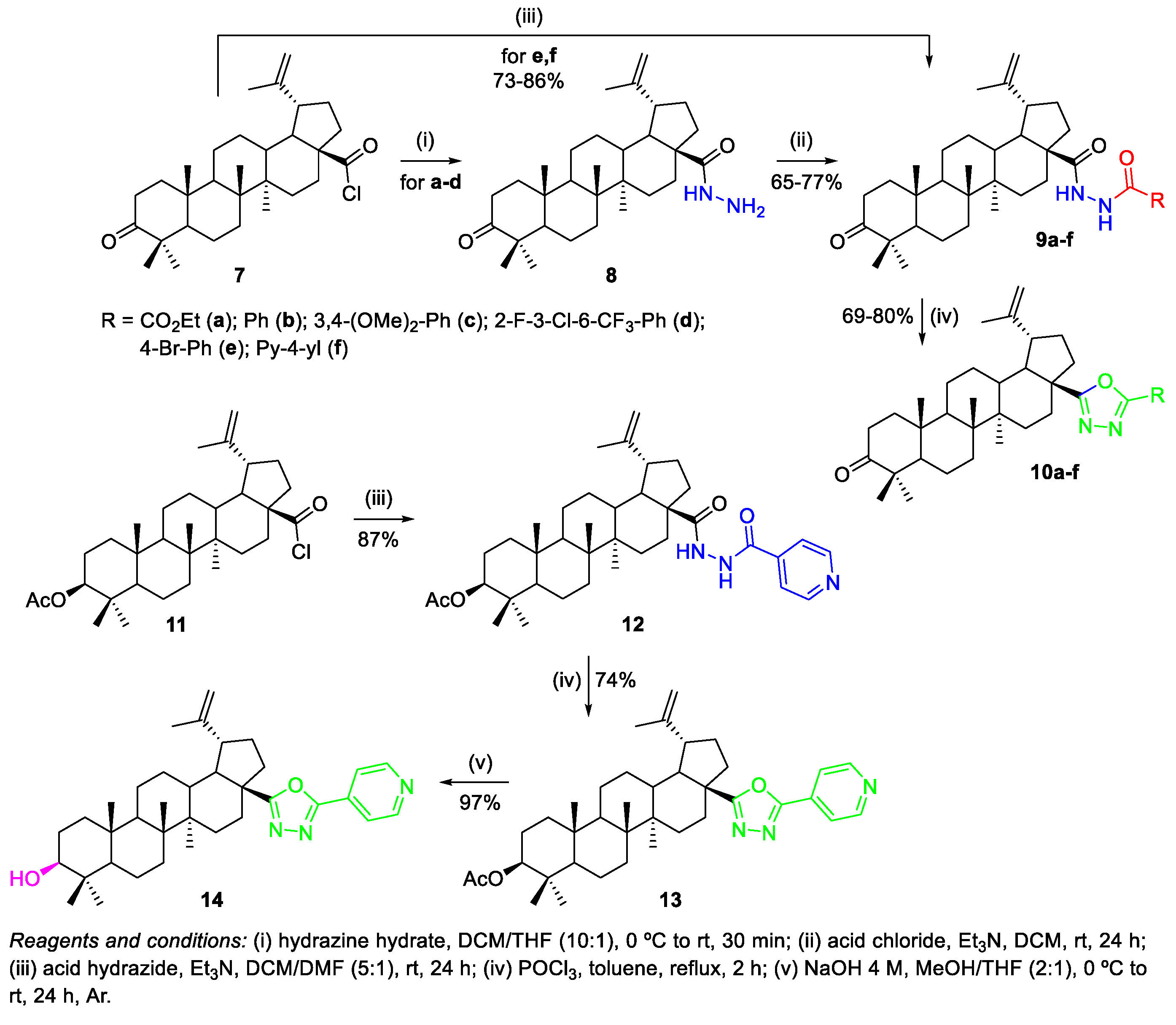
- Antimonova, A.N.; Petrenko, N.I.; Shakirov, M.M.; Pokrovskii, M.A.; Pokrovskii, A.G.; Shul’ts, E.E. Synthesis and Cytotoxic Activity of Lupane Triterpenoids Containing 1,3,4-Oxadiazoles. Chem. Nat. Compd. 2014, 50, 1016–1023. [Google Scholar] [CrossRef]
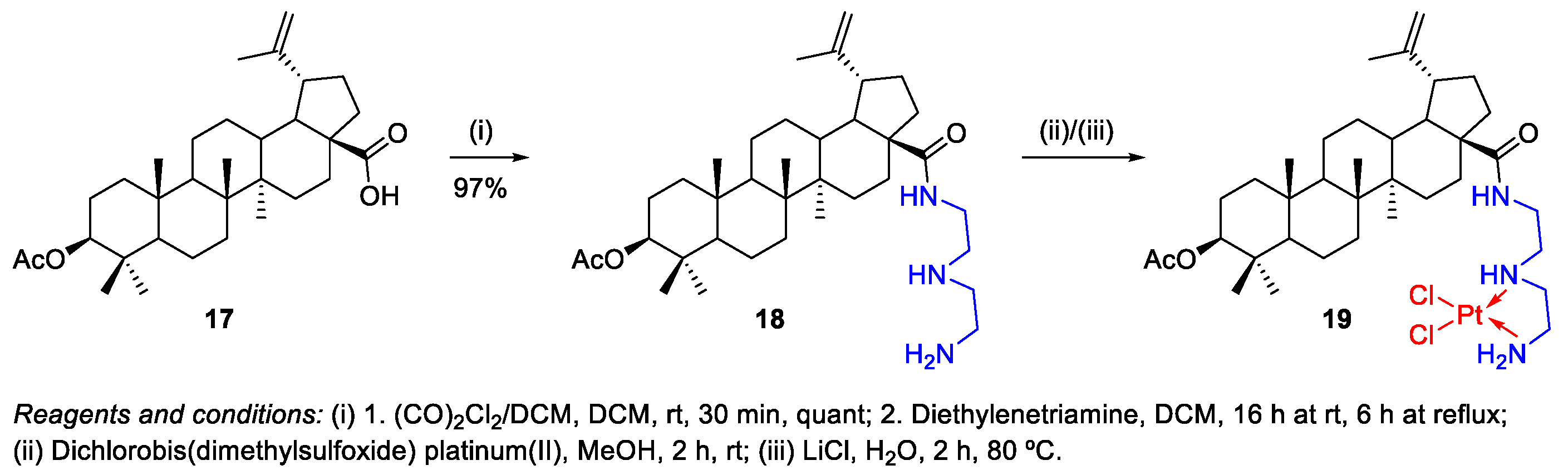
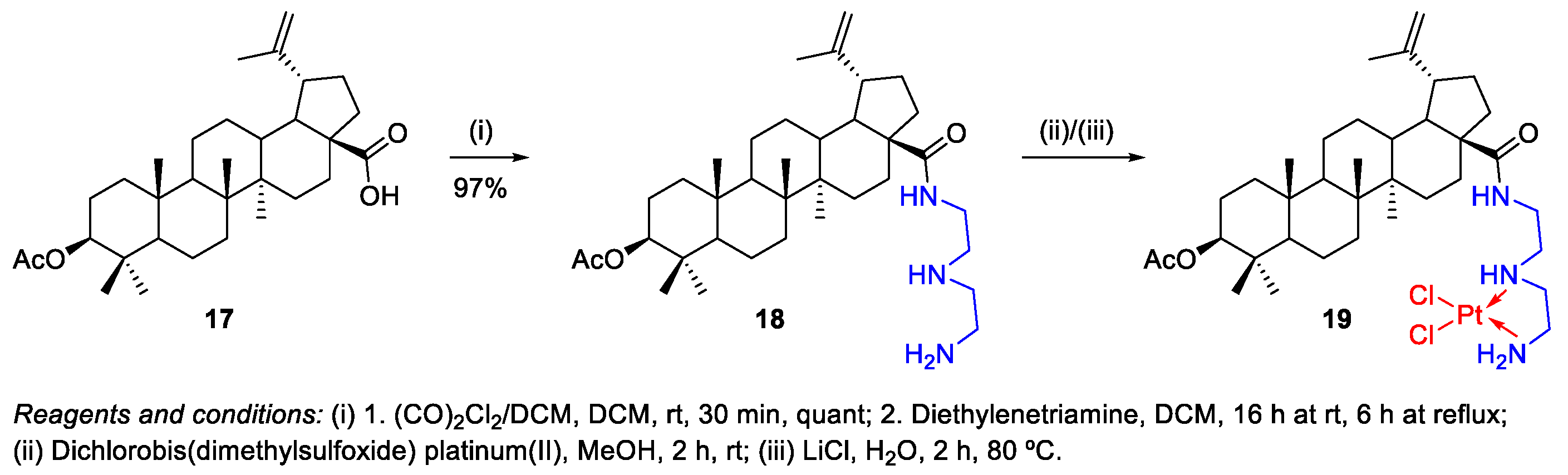
- Emmerich, D.; Vanchanagiri, K.; Baratto, L.C.; Schmidt, H.; Paschke, R. Synthesis and studies of anticancer properties of lupane-type triterpenoid derivatives containing a cisplatin fragment. Eur. J. Med. Chem. 2014, 75, 460–466. [Google Scholar] [CrossRef]
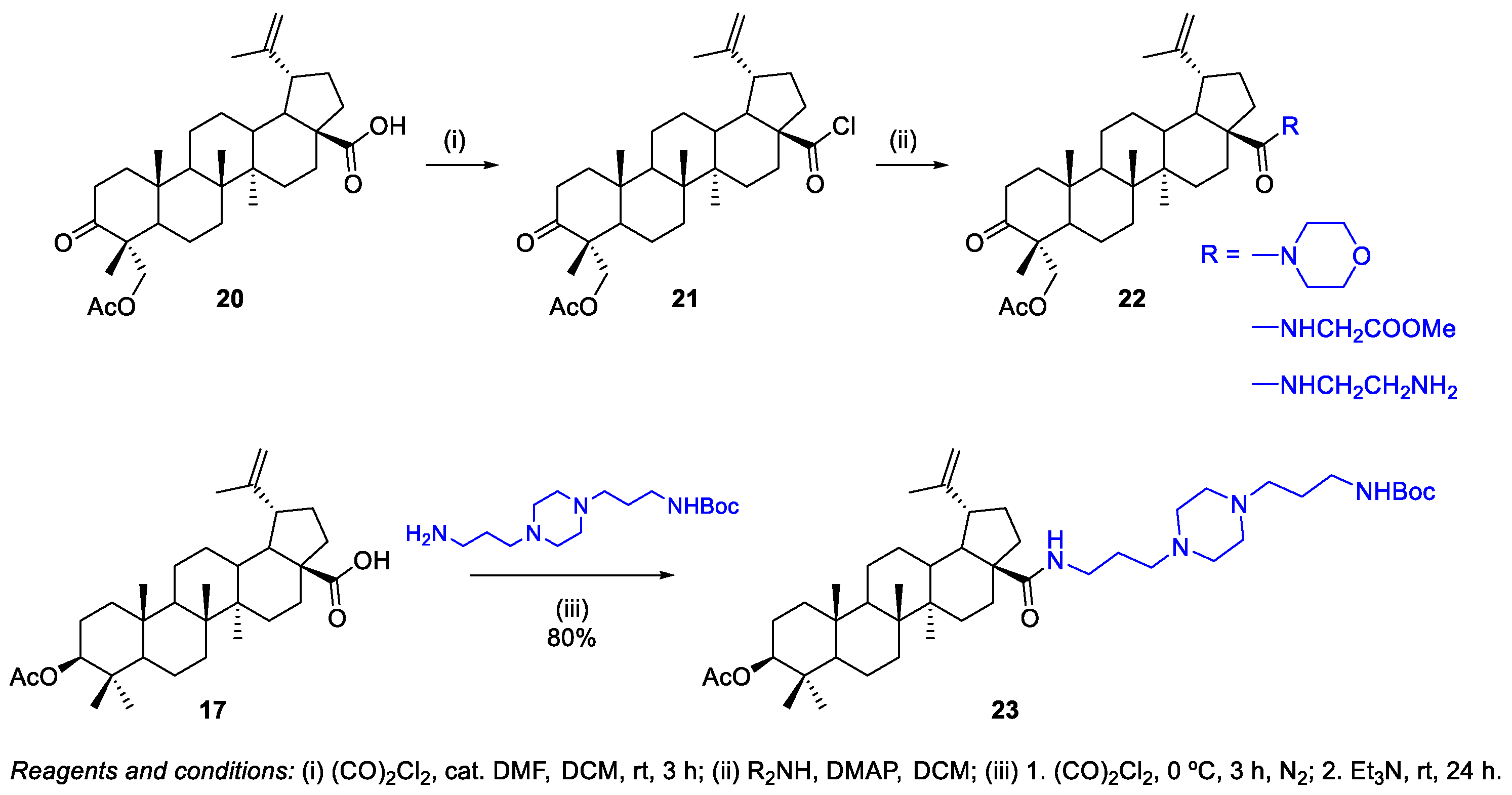
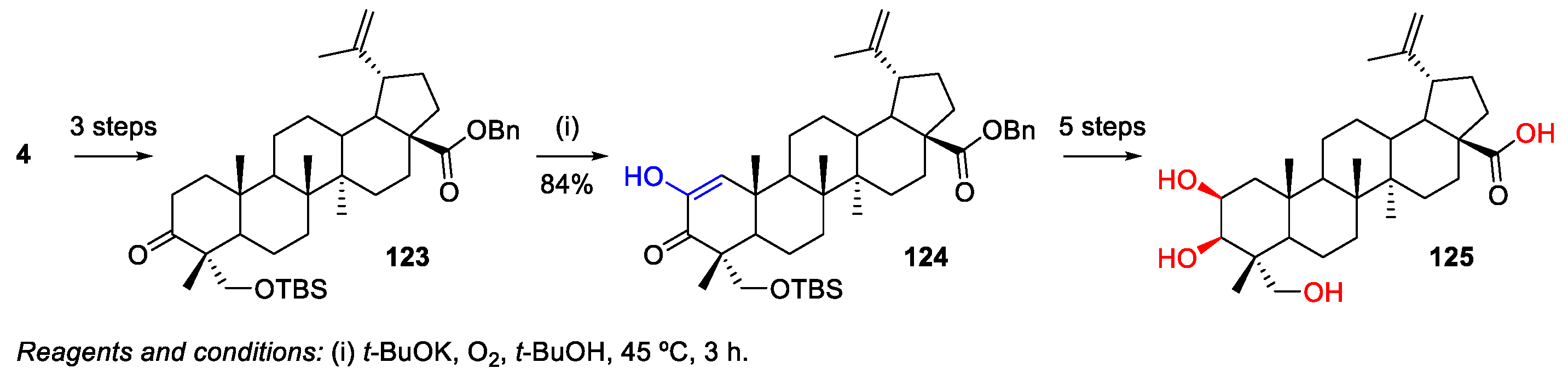
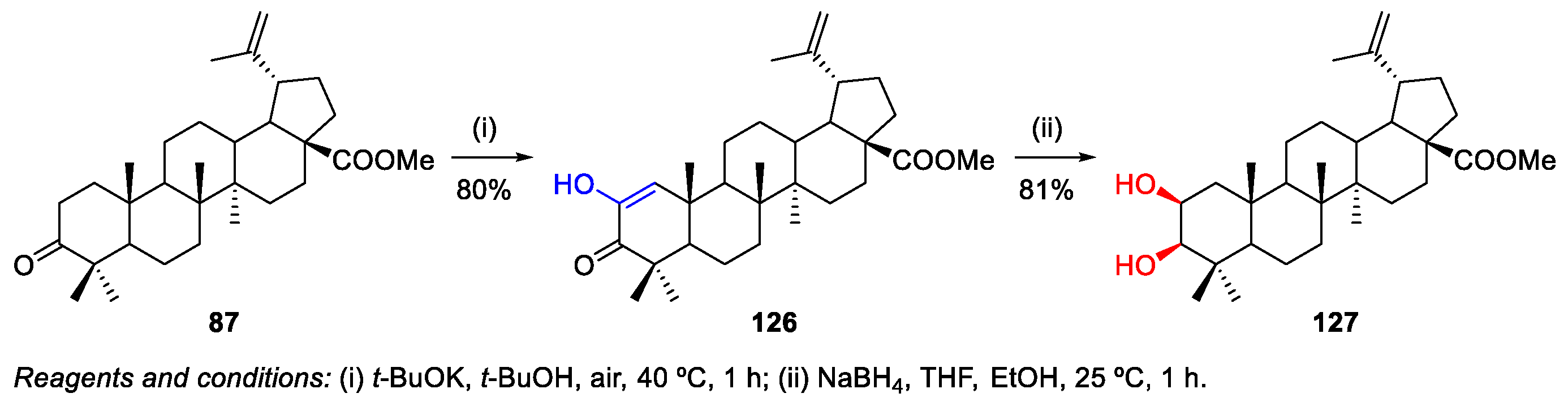
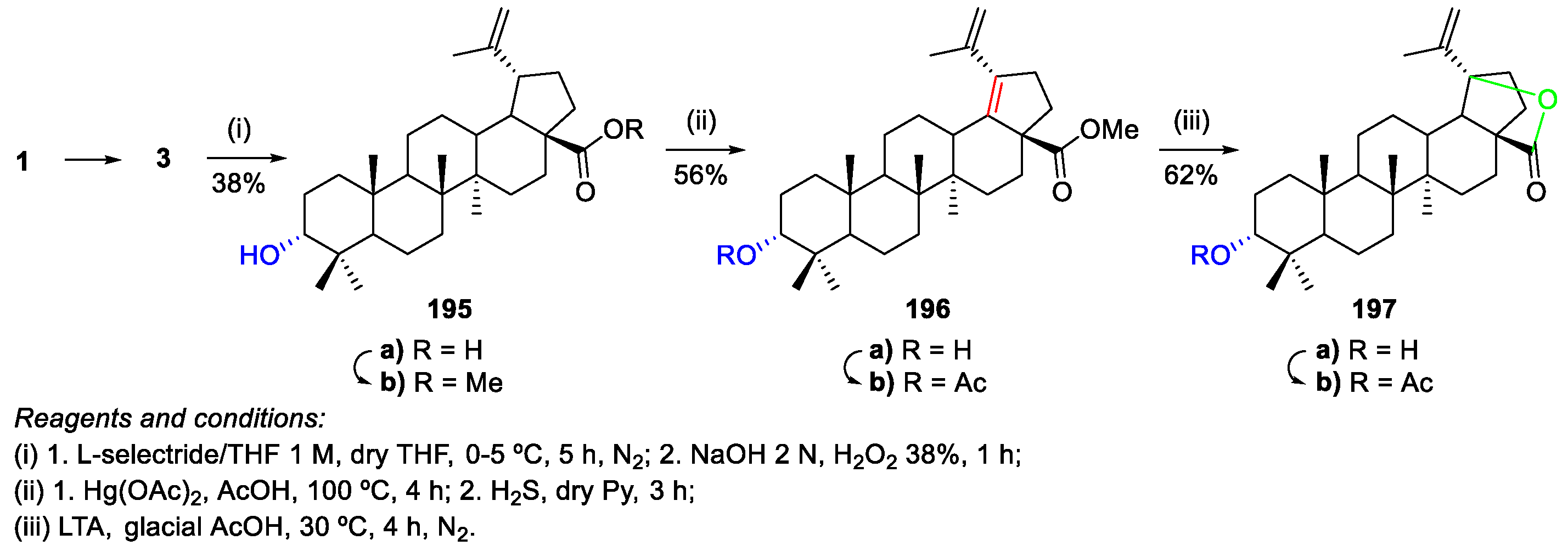
- Zhang, H.; Zhu, P.; Liu, J.; Yang, X.; Xu, S.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Synthesis and antitumor activity of novel 3-oxo-23-hydroxybetulinic acid derivatives. Eur. J. Med. Chem. 2014, 87, 159–167. [Google Scholar] [CrossRef]
- Innocente, A.; Silva, G.; Cruz, L.; Moraes, M.; Nakabashi, M.; Sonnet, P.; Gosmann, G.; Garcia, C.; Gnoatto, S. Synthesis and Antiplasmodial Activity of Betulinic Acid and Ursolic Acid Analogues. Molecules 2012, 17, 12003. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, Q.; Si, L.; Shi, Y.; Wang, H.; Yu, F.; Zhang, Y.; Li, Y.; Zheng, Y.; Zhang, C.; et al. Synthesis and Anti-HCV Entry Activity Studies of β-Cyclodextrin–Pentacyclic Triterpene Conjugates. ChemMedChem 2014, 9, 1060–1070. [Google Scholar] [CrossRef]
- Coric, P.; Turcaud, S.; Souquet, F.; Briant, L.; Gay, B.; Royer, J.; Chazal, N.; Bouaziz, S. Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. Eur. J. Med. Chem. 2013, 62, 453–465. [Google Scholar] [CrossRef]
- Xu, K.; Xu, X.; Chu, F.; Wang, M.; Wang, P.; Li, G.; Song, J.; Zhang, Y.; Lei, H. Synthesis and biological evaluation of T-OA analogues as the cytotoxic agents. Res. Chem. Intermed. 2015, 41, 6257–6269. [Google Scholar] [CrossRef]
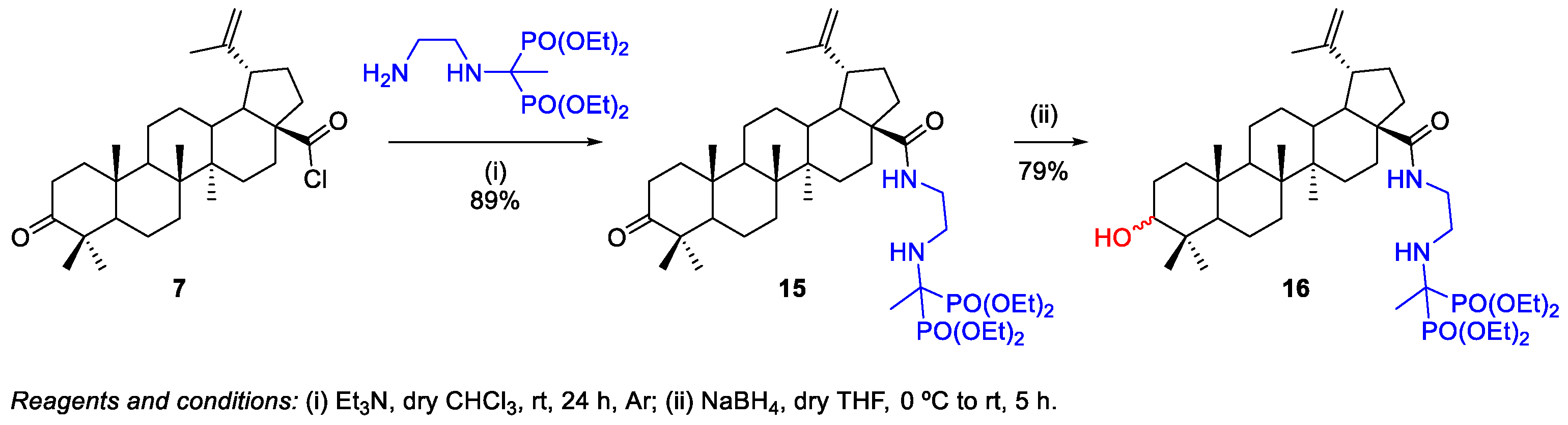
- Becker, C.S.; Chukanov, N.V.; Grigor’ev, I.A. New Amino-Bisphosphonate Building Blocks in the Synthesis of Bisphosphonic Derivatives Based on Lead Compounds. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1201–1212. [Google Scholar] [CrossRef]
- Wiemann, J.; Heller, L.; Perl, V.; Kluge, R.; Ströhl, D.; Csuk, R. Betulinic acid derived hydroxamates and betulin derived carbamates are interesting scaffolds for the synthesis of novel cytotoxic compounds. Eur. J. Med. Chem. 2015, 106, 194–210. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Govdi, A.I.; Shults, E.v.E.; Shakirov, M.M.; Sorokina, I.V.; Tolstikova, T.G.; Baev, D.S.; Tolstikov, G.A.; Alabugin, I.V. Efficient synthesis of the first betulonic acid–acetylene hybrids and their hepatoprotective and anti-inflammatory activity. Bioorg. Med. Chem. 2009, 17, 5164–5169. [Google Scholar] [CrossRef] [PubMed]
- Suman, P.; Patel, A.; Solano, L.; Jampana, G.; Gardner, Z.S.; Holt, C.M.; Jonnalagadda, S.C. Synthesis and cytotoxicity of Baylis-Hillman template derived betulinic acid-triazole conjugates. Tetrahedron 2017, 73, 4214–4226. [Google Scholar] [CrossRef]
- Bębenek, E.; Chrobak, E.; Wietrzyk, J.; Kadela, M.; Chrobak, A.; Kusz, J.; Książek, M.; Jastrzębska, M.; Boryczka, S. Synthesis, structure and cytotoxic activity of acetylenic derivatives of betulonic and betulinic acids. J. Mol. Struct. 2016, 1106, 210–219. [Google Scholar] [CrossRef]
- Khlebnikova, T.S.; Piven, Y.A.; Lakhvich, F.A.; Frolova, T.S.; Sorokina, I.V.; Tolstikova, T.G. Synthesis and Cytotoxicity of Conjugates of Betulinic Acid and F-Containing 2-Acylcycloalkane-1,3-Diones. Chem. Nat. Compd. 2018, 54, 1100–1105. [Google Scholar] [CrossRef]
- Krishna, C.; Bhargavi, M.V.; Krupadanam, G.L.D. Design, Synthesis, and Cytotoxicity of Semisynthetic Betulinic Acid-1,2,4-Oxadiazole Amide Derivatives. Russ. J. Gen. Chem. 2018, 88, 312–318. [Google Scholar] [CrossRef]
- Ali, M.T.M.; Zahari, H.; Aliasak, A.; Lim, S.M.; Ramasamy, K.; Macabeo, A.P.G. Synthesis, Characterization and Cytotoxic Activity of Betulinic Acid and Seco-Betulinic Acid Derivatives against Human Colorectal Carcinoma. Orient. J. Chem. 2017, 33, 242–248. [Google Scholar] [Green Version]
- Cui, H.-W.; He, Y.; Wang, J.; Gao, W.; Liu, T.; Qin, M.; Wang, X.; Gao, C.; Wang, Y.; Liu, M.-Y.; et al. Synthesis of heterocycle-modified betulinic acid derivatives as antitumor agents. Eur. J. Med. Chem. 2015, 95, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Kahnt, M.; Heller, L.; Al-Harrasi, A.; Schäfer, R.; Kluge, R.; Wagner, C.; Otgonbayar, C.; Csuk, R. Platanic acid-derived methyl 20-amino-30-norlupan-28-oates are potent cytotoxic agents acting by apoptosis. Med. Chem. Res. 2018, 27, 1757–1769. [Google Scholar] [CrossRef]
- Chen, Y.; Sit, S.-Y.; Chen, J.; Swidorski, J.J.; Liu, Z.; Sin, N.; Venables, B.L.; Parker, D.D.; Nowicka-Sans, B.; Lin, Z.; et al. The design, synthesis and structure-activity relationships associated with C28 amine-based betulinic acid derivatives as inhibitors of HIV-1 maturation. Bioorg. Med. Chem. Lett. 2018, 28, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-H.; Tang, J.; Zhu, Z.-F.; Chen, L. Design, synthesis, and anti-tumor activity of novel betulinic acid derivatives. J. Asian Nat. Prod. Res. 2014, 16, 34–42. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.N.; Maria, N.R.; Schuck, D.C.; Cruz, L.N.; de Moraes, M.S.; Nakabashi, M.; Graebin, C.; Gosmann, G.; Garcia, C.R.; Gnoatto, S.C. Two series of new semisynthetic triterpene derivatives: Differences in anti-malarial activity, cytotoxicity and mechanism of action. Malar. J. 2013, 12, 89. [Google Scholar] [CrossRef]
- Popov, S.A.; Shpatov, A.V.; Grigor’ev, I.A. Synthesis of Substituted Esters of Ursolic, Betulonic, and Betulinic Acids Containing the Nitroxyl Radical 4-Amino-2,2,6,6-Tetramethylpiperidine-1-Oxyl. Chem. Nat. Compd. 2015, 51, 87–90. [Google Scholar] [CrossRef]
- Khlebnicova, T.S.; Piven, Y.A.; Baranovsky, A.V.; Lakhvich, F.A.; Shishkina, S.V.; Zicāne, D.; Tetere, Z.; Rāviņa, I.; Kumpiņš, V.; Rijkure, I.; et al. Synthesis of novel lupane triterpenoid-indazolone hybrids with oxime ester linkage. Steroids 2017, 117, 77–89. [Google Scholar] [CrossRef]
- Yang, S.; Liang, N.; Li, H.; Xue, W.; Hu, D.; Jin, L.; Zhao, Q.; Yang, S. Design, synthesis and biological evaluation of novel betulinic acid derivatives. Chem. Cent. J. 2012, 6, 141. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Ghosh, M.; Dutta, S.K. A potent tumoricidal co-drug ‘Bet-CA’—An ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Sci. Rep. 2015, 5, 7762. [Google Scholar] [CrossRef]
- Ackermann, A.; Karagöz, A.Ç.; Ghoochani, A.; Buchfelder, M.; Eyüpoglu, I.; Tsogoeva, S.B.; Savaskan, N. Cytotoxic profiling of artesunic and betulinic acids and their synthetic hybrid compound on neurons and gliomas. Oncotarget 2017, 8, 61457–61474. [Google Scholar] [CrossRef]
- Xu, B.; Yan, W.-Q.; Xu, X.; Wu, G.-R.; Zhang, C.-Z.; Han, Y.-T.; Chu, F.-H.; Zhao, R.; Wang, P.-L.; Lei, H.-M. Combination of amino acid/dipeptide with ligustrazine-betulinic acid as antitumor agents. Eur. J. Med. Chem. 2017, 130, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chu, F.; Zhang, Y.; Wang, X.; Li, Q.; Liu, W.; Xu, X.; Xing, Y.; Chen, J.; Wang, P.; et al. A Series of New Ligustrazine-Triterpenes Derivatives as Anti-Tumor Agents: Design, Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2015, 16, 21035. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.T.T.; Pham, C.T.; Le, T.A.; Truong, H.H.; Vu, H.T.T.; Soldatenkov, A.T.; Van Nguyen, T. New hybrids between triterpenoid acids and nucleoside HIV-RT inhibitors. Mendeleev Commun. 2015, 25, 96–98. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Heller, L.; Csuk, R. Targeting mitochondria: Esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018, 152, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Eignerova, B.; Tichy, M.; Krasulova, J.; Kvasnica, M.; Rarova, L.; Christova, R.; Urban, M.; Bednarczyk-Cwynar, B.; Hajduch, M.; Sarek, J. Synthesis and antiproliferative properties of new hydrophilic esters of triterpenic acids. Eur. J. Med. Chem. 2017, 140, 403–420. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.N.S.; Atik, D.M.; Fernandes, J.L.A.; Nascimento, D.d.F.d.; Fazolo, T.; Souza, A.P.D.d.; Gnoatto, S.C.B. Synthesis of three triterpene series and their activity against respiratory syncytial virus. Arch. Pharm. 2018, 351, 1800108. [Google Scholar] [CrossRef] [PubMed]
- Komissarova, N.G.; Dubovitskii, S.N.; Orlov, A.V.; Shitikova, O.V.; Abdullin, M.F.; Spirikhin, L.V.; Yunusov, M.S. Synthesis of Sulfobetaines Based on Betulinic Acid and its Esters. Chem. Nat. Compd. 2015, 51, 746–751. [Google Scholar] [CrossRef]
- Drąg-Zalesińska, M.; Wysocka, T.; Borska, S.; Drąg, M.; Poręba, M.; Choromańska, A.; Kulbacka, J.; Saczko, J. The new esters derivatives of betulin and betulinic acid in epidermoid squamous carcinoma treatment–In vitro studies. Biomed. Pharmacother. 2015, 72, 91–97. [Google Scholar] [CrossRef]
- Komissarova, N.G.; Dubovitskii, S.N.; Shitikova, O.V.; Vyrypaev, E.M.; Spirikhin, L.V.; Eropkina, E.M.; Lobova, T.G.; Eropkin, M.Y.; Yunusov, M.S. Synthesis of Conjugates of Lupane-Type Pentacyclic Triterpenoids with 2-Aminoethane- and N-Methyl-2-Aminoethanesulfonic Acids. Assessment of in vitro Toxicity. Chem. Nat. Compd. 2017, 53, 907–914. [Google Scholar] [CrossRef]
- Bi, Y.; Xu, J.; Sun, F.; Wu, X.; Ye, W.; Sun, Y.; Huang, W. Synthesis and Biological Activity of 23-Hydroxybetulinic Acid C-28 Ester Derivatives as Antitumor Agent Candidates. Molecules 2012, 17, 8832. [Google Scholar] [CrossRef]
- Yao, H.; Wei, G.; Liu, Y.; Yao, H.; Zhu, Z.; Ye, W.; Wu, X.; Xu, J.; Xu, S. Synthesis, Biological Evaluation of Fluorescent 23-Hydroxybetulinic Acid Probes, and Their Cellular Localization Studies. ACS Med. Chem. Lett. 2018, 9, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Csuk, R. Synthesis and Cytotoxic Activity of Pentacyclic Triterpenoid Sulfamates. Arch. Pharm. 2015, 348, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Vanchanagiri, K.; Emmerich, D.; Bruschke, M.; Bache, M.; Seifert, F.; Csuk, R.; Vordermark, D.; Paschke, R. Synthesis and biological investigation of new carbonic anhydrase IX (CAIX) inhibitors. Chem.-Biol. Interact. 2018, 284, 12–23. [Google Scholar] [CrossRef] [PubMed]
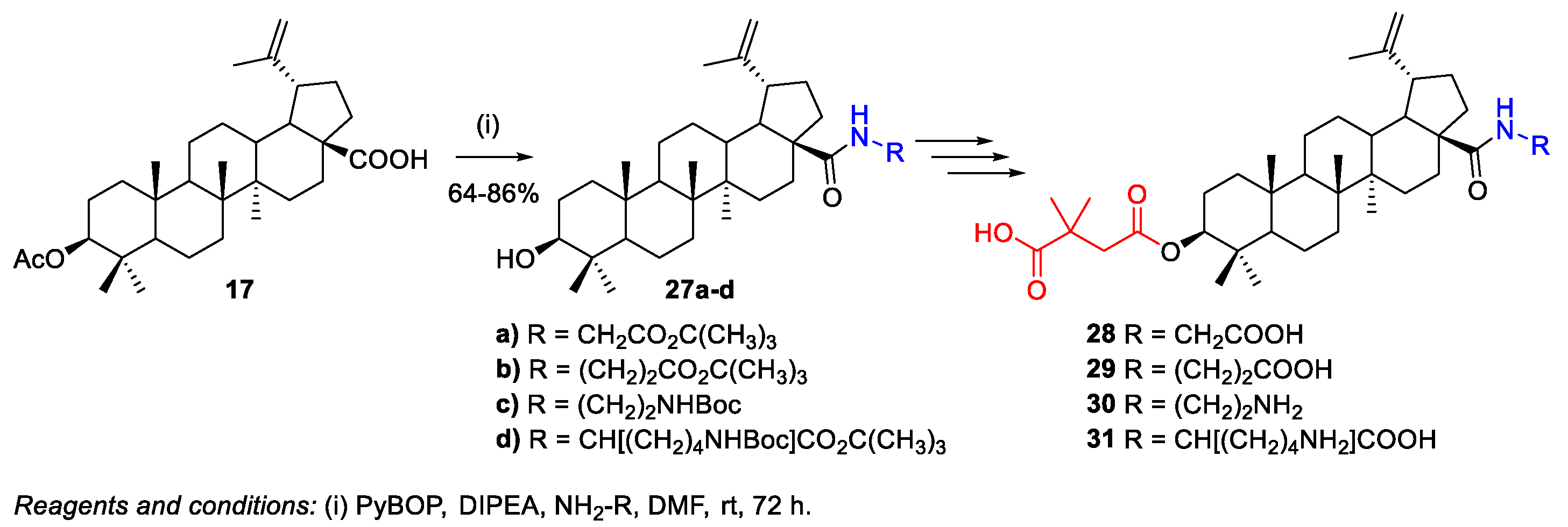
- Govdi, A.I.; Sokolova, N.V.; Sorokina, I.V.; Baev, D.S.; Tolstikova, T.G.; Mamatyuk, V.I.; Fadeev, D.S.; Vasilevsky, S.F.; Nenajdenko, V.G. Synthesis of new betulinic acid-peptide conjugates and in vivo and in silico studies of the influence of peptide moieties on the triterpenoid core activity. MedChemComm 2015, 6, 230–238. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Gubaidullin, R.R.; Galimshina, Z.R.; Nedopekina, D.A.; Odinokov, V.N. Effective synthesis of novel C(2)-propargyl derivatives of betulinic and ursolic acids and their conjugation with β-d-glucopyranoside azides via click chemistry. Tetrahedron 2016, 72, 1249–1256. [Google Scholar] [CrossRef]
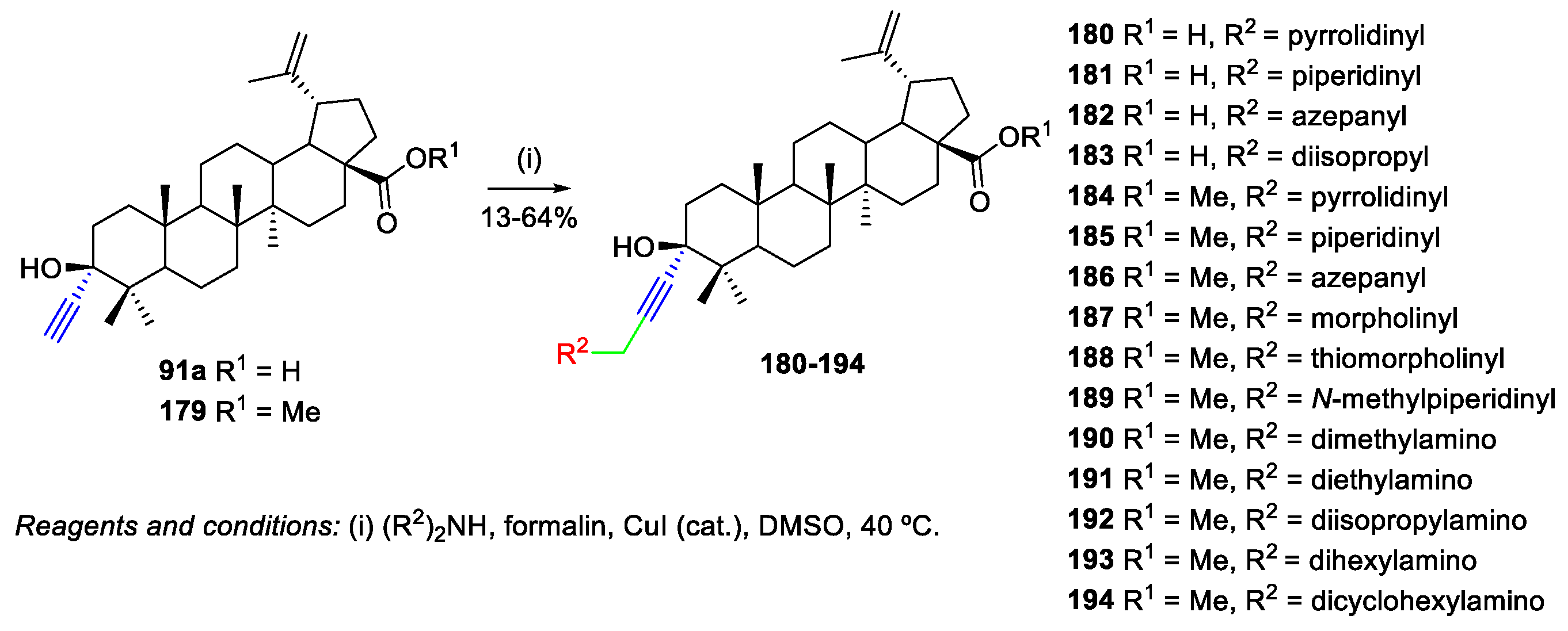
- Csuk, R.; Nitsche, C.; Sczepek, R.; Schwarz, S.; Siewert, B. Synthesis of Antitumor-Active Betulinic Acid-Derived Hydroxypropargylamines by Copper-Catalyzend Mannich Reactions. Arch. Pharm. 2013, 346, 232–246. [Google Scholar] [CrossRef] [PubMed]
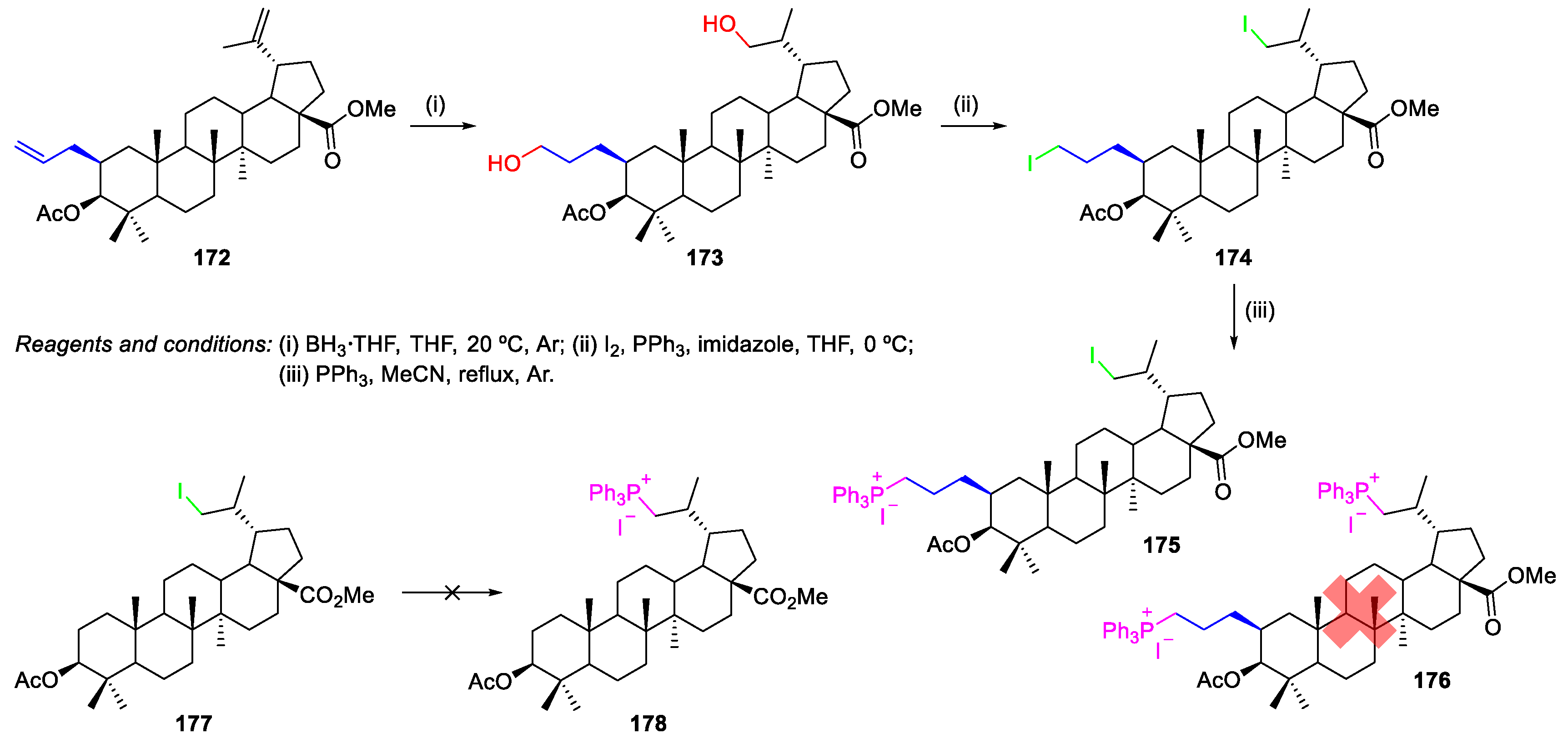
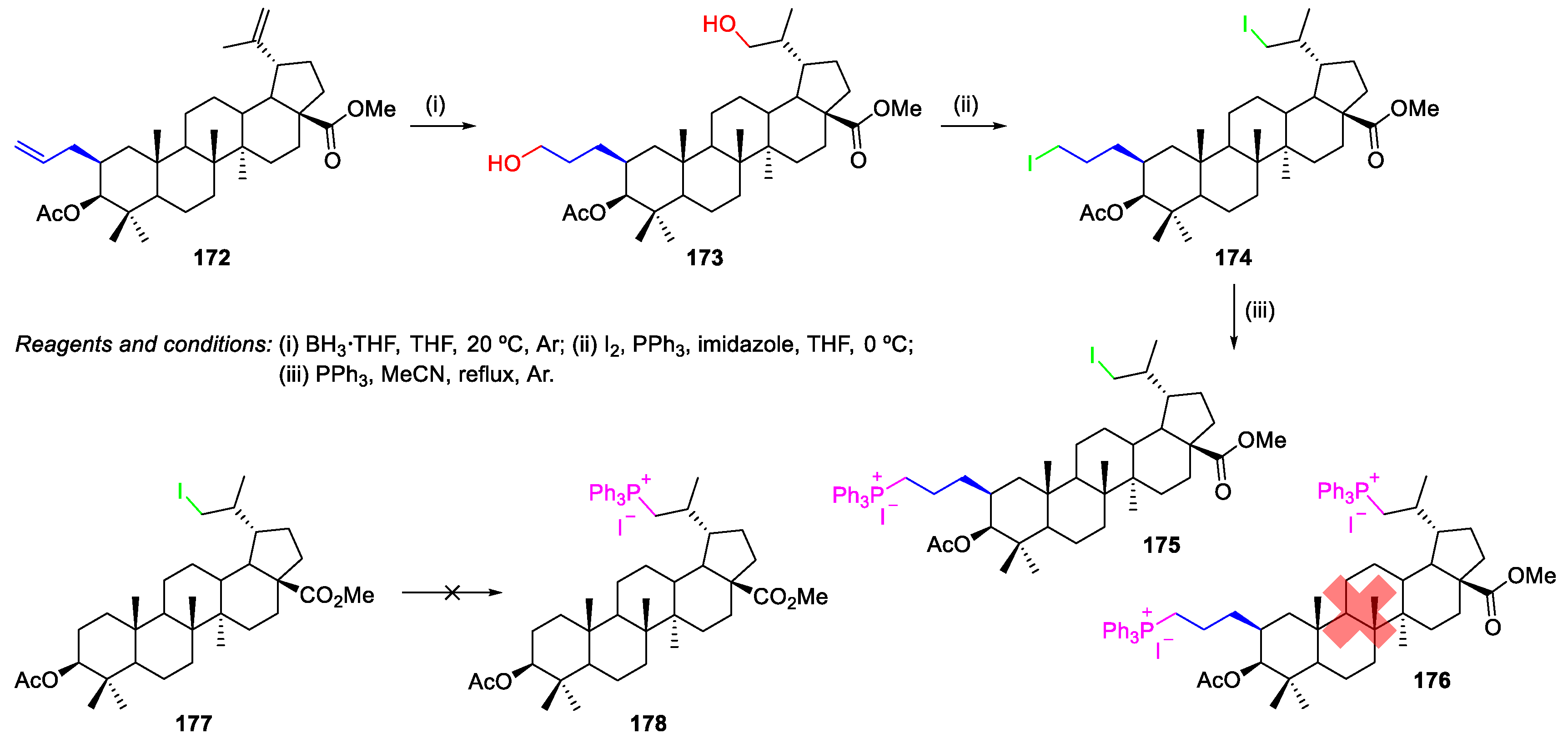
- Spivak, A.Y.; Shakurova, E.R.; Nedopekina, D.A.; Khalitova, R.R.; Khalilov, L.M.; Odinokov, V.N.; Bel’skii, Y.P.; Ivanova, A.N.; Bel’skaya, N.V.; Danilets, M.G.; Ligacheva, A.A. Novel lupane triterpenoids containing allyl substituents in ring A: Synthesis and in vitro study of antiinflammatory and cytotoxic properties. Russ. Chem. Bull. 2011, 60, 694. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Galimshina, Z.R.; Nedopekina, D.A.; Odinokov, V.N. Synthesis of New C-2 Triazole-Linked Analogs of Triterpenoid Pentacyclic Saponins. Chem. Nat. Compd. 2018, 54, 315–323. [Google Scholar] [CrossRef]
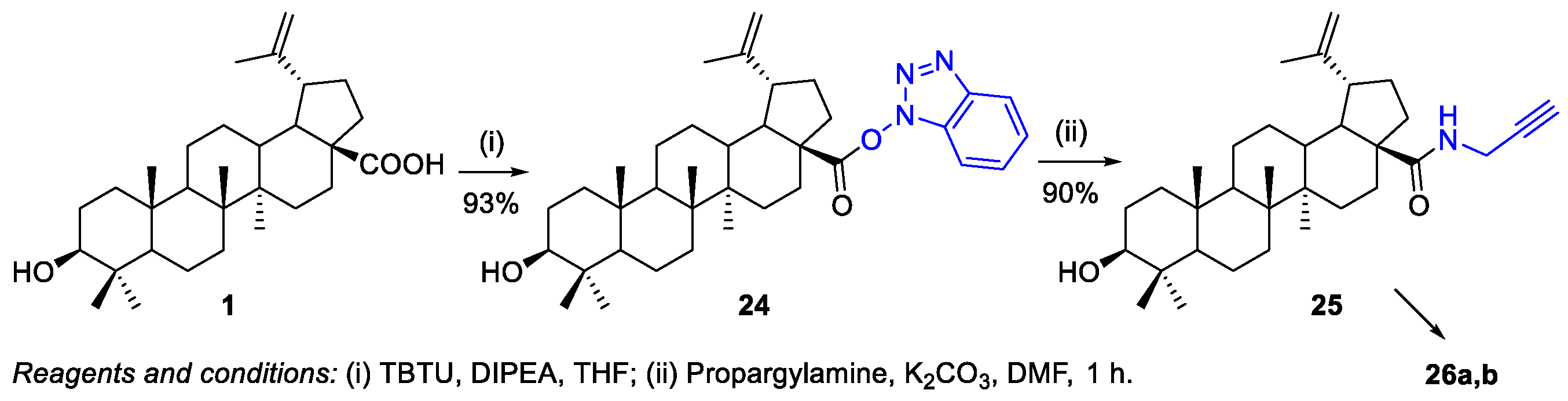
- Majeed, R.; Sangwan, P.L.; Chinthakindi, P.K.; Khan, I.; Dangroo, N.A.; Thota, N.; Hamid, A.; Sharma, P.R.; Saxena, A.K.; Koul, S. Synthesis of 3-O-propargylated betulinic acid and its 1,2,3-triazoles as potential apoptotic agents. Eur. J. Med. Chem. 2013, 63, 782–792. [Google Scholar] [CrossRef]
- Chakraborty, B.; Dutta, D.; Mukherjee, S.; Das, S.; Maiti, N.C.; Das, P.; Chowdhury, C. Synthesis and biological evaluation of a novel betulinic acid derivative as an inducer of apoptosis in human colon carcinoma cells (HT-29). Eur. J. Med. Chem. 2015, 102, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lu, L.; Na, H.; Li, X.; Wang, Q.; Jiang, X.; Xu, X.; Yu, F.; Zhang, T.; Li, J.; et al. Conjugation of a Nonspecific Antiviral Sapogenin with a Specific HIV Fusion Inhibitor: A Promising Strategy for Discovering New Antiviral Therapeutics. J. Med. Chem. 2014, 57, 7342–7354. [Google Scholar] [CrossRef] [PubMed]
- Thi, T.A.D.; Tuyet, N.T.K.; The, C.P.; Nguyen, H.T.; Thi, C.B.; Duy, T.D.; D’hooghe, M.; Nguyen, T.V. Synthesis and cytotoxic evaluation of novel ester-triazole-linked triterpenoid–AZT conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 5190–5194. [Google Scholar]
- Khan, I.; Guru, S.K.; Rath, S.K.; Chinthakindi, P.K.; Singh, B.; Koul, S.; Bhushan, S.; Sangwan, P.L. A novel triazole derivative of betulinic acid induces extrinsic and intrinsic apoptosis in human leukemia HL-60 cells. Eur. J. Med. Chem. 2016, 108, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, B.; Lakshmi, J.K.; Kavitha, R.; Jagadeesh, B.; Bhattacharjee, D.; Jain, N.; Mallavadhani, U.V. Synthesis, structural studies, and cytotoxic evaluation of novel ursolic acid hybrids with capabilities to arrest breast cancer cells in mitosis. J. Asian Nat. Prod. Res. 2017, 19, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Thi, T.A.D.; Tuyet, N.T.K.; The, C.P.; Nguyen, H.T.; Thi, C.B.; Phuong, H.T.; Boi, L.V.; Nguyen, T.V.; D’hooghe, M. Synthesis and cytotoxic evaluation of novel amide–triazole-linked triterpenoid–AZT conjugates. Tetrahedron Lett. 2015, 56, 218–224. [Google Scholar]
- Bębenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Latocha, M.; Boryczka, S. Novel triazoles of 3-acetylbetulin and betulone as anticancer agents. Med. Chem. Res. 2018, 27, 2051–2061. [Google Scholar] [CrossRef]
- Wang, H.; Xu, R.; Shi, Y.; Si, L.; Jiao, P.; Fan, Z.; Han, X.; Wu, X.; Zhou, X.; Yu, F.; et al. Design, synthesis and biological evaluation of novel l-ascorbic acid-conjugated pentacyclic triterpene derivatives as potential influenza virus entry inhibitors. Eur. J. Med. Chem. 2016, 110, 376–388. [Google Scholar] [CrossRef]
- Dangroo, N.A.; Singh, J.; Rath, S.K.; Gupta, N.; Qayum, A.; Singh, S.; Sangwan, P.L. A convergent synthesis of novel alkyne–azide cycloaddition congeners of betulinic acid as potent cytotoxic agent. Steroids 2017, 123, 1–12. [Google Scholar] [CrossRef]
- Han, X.; Shi, Y.; Si, L.; Fan, Z.; Wang, H.; Xu, R.; Jiao, P.; Meng, K.; Tian, Z.; Zhou, X.; et al. Design, synthesis and biological activity evaluation of novel conjugated sialic acid and pentacyclic triterpene derivatives as anti-influenza entry inhibitors. MedChemComm 2016, 7, 1932–1945. [Google Scholar] [CrossRef]
- Antimonova, A.N.; Petrenko, N.I.; Shakirov, M.M.; Rybalova, T.V.; Frolova, T.S.; Shul’ts, E.E.; Kukina, T.P.; Sinitsyna, O.I.; Tolstikov, G.A. Synthesis and study of mutagenic properties of lupane triterpenoids containing 1,2,3-triazole fragments in the C-30 position. Chem. Nat. Compd. 2013, 49, 657–664. [Google Scholar] [CrossRef]
- Shi, W.; Tang, N.; Yan, W.-D. Synthesis and cytotoxicity of triterpenoids derived from betulin and betulinic acid via click chemistry. J. Asian Nat. Prod. Res. 2015, 17, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Sidova, V.; Zoufaly, P.; Pokorny, J.; Dzubak, P.; Hajduch, M.; Popa, I.; Urban, M. Cytotoxic conjugates of betulinic acid and substituted triazoles prepared by Huisgen Cycloaddition from 30-azidoderivatives. PLoS ONE 2017, 12, e0171621. [Google Scholar] [CrossRef] [PubMed]
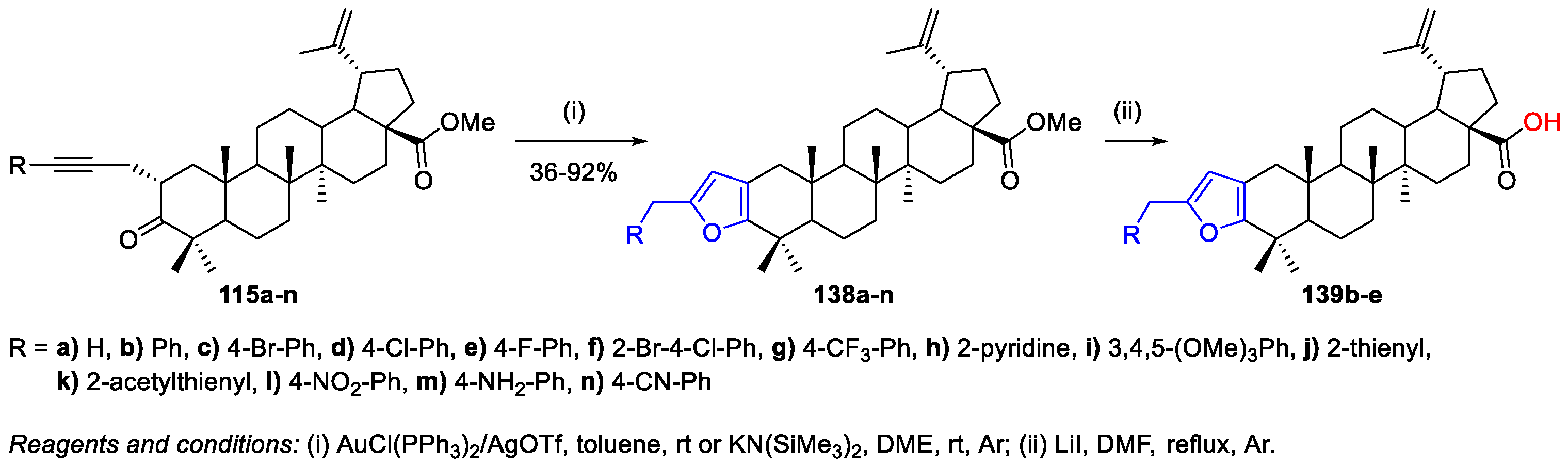
- Gubaidullin, R.R.; Yarmukhametova, D.S.; Nedopekina, D.A.; Khalitova, R.R.; Spivak, A.Y. Effective synthesis of novel furan-fused pentacyclic triterpenoids via anionic 5-exo dig cyclization of 2-alkynyl-3-oxotriterpene acids. Arkivoc 2017, v, 100–116. [Google Scholar] [CrossRef]
- Gubaidullin, R.R.; Khalitova, R.R.; Galimshina, Z.R.; Spivak, A.Y. Synthesis of novel [3,2-b]furan-fused pentacyclic triterpenoids via gold-catalyzed intramolecular heterocyclization of 2-alkynyl-3-oxotriterpene acids. Tetrahedron 2018, 74, 1888–1899. [Google Scholar] [CrossRef]
- Patrick, G.L. An Introduction to Medicinal Chemistry; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
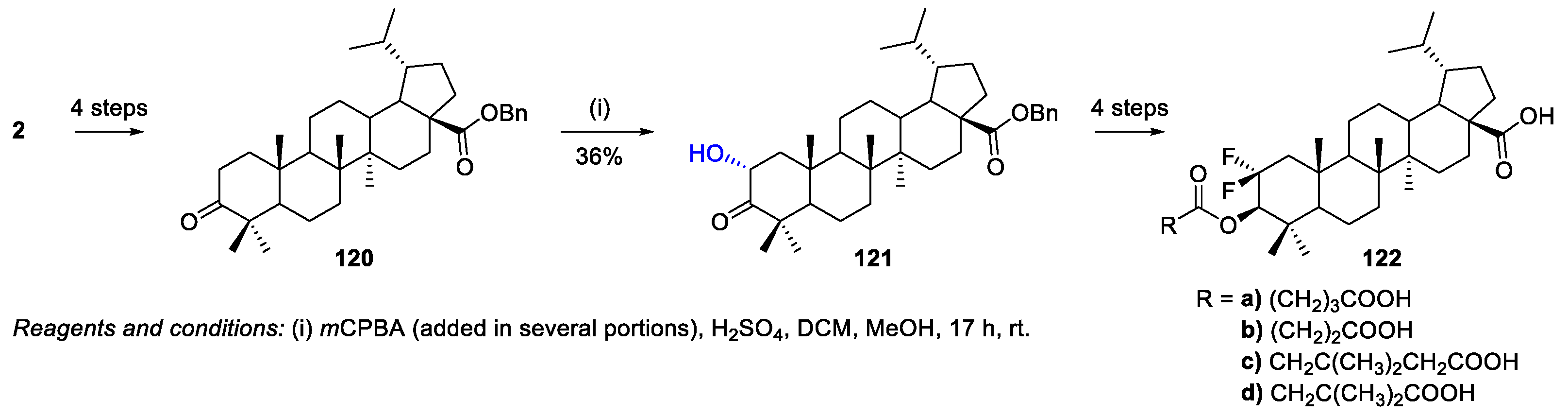
- Borkova, L.; Jasikova, L.; Rehulka, J.; Frisonsova, K.; Urban, M.; Frydrych, I.; Popa, I.; Hajduch, M.; Dickinson, N.J.; Vlk, M.; et al. Synthesis of cytotoxic 2,2-difluoroderivatives of dihydrobetulinic acid and allobetulin and study of their impact on cancer cells. Eur. J. Med. Chem. 2015, 96, 482–490. [Google Scholar] [CrossRef] [PubMed]
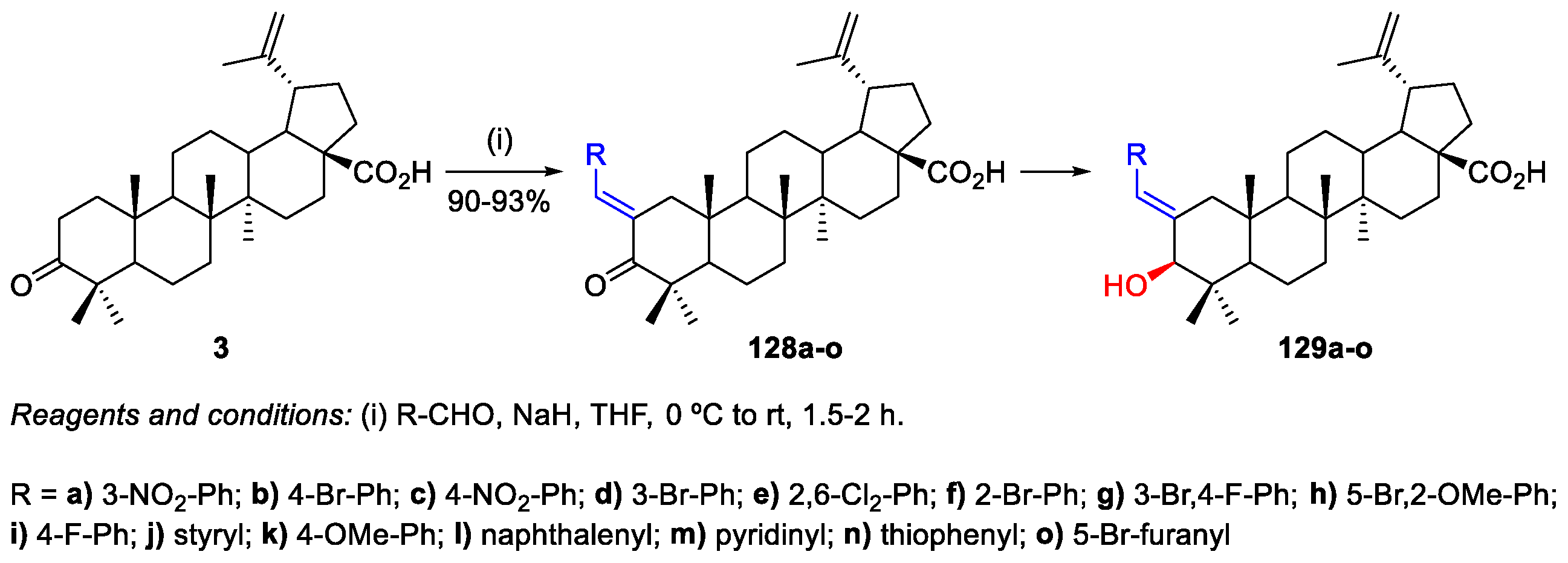
- Gupta, N.; Rath, S.K.; Singh, J.; Qayum, A.; Singh, S.; Sangwan, P.L. Synthesis of novel benzylidene analogues of betulinic acid as potent cytotoxic agents. Eur. J. Med. Chem. 2017, 135, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Zhu, P.; Liu, J.; Xu, S.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Design, synthesis and antitumor activity of triterpenoid pyrazine derivatives from 23-hydroxybetulinic acid. Eur. J. Med. Chem. 2015, 97, 235–244. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, P.; Liu, J.; Lin, Y.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Synthesis, in vitro and in vivo antitumor activity of pyrazole-fused 23-hydroxybetulinic acid derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 728–732. [Google Scholar] [CrossRef]
- Zhang, H.; Li, F.; Zhu, P.; Liu, J.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Synthesis and Biological Evaluation of Oxygen-containing Heterocyclic Ring-fused 23-Hydroxybetulinic Acid Derivatives as Antitumor Agents. Chem. Biol. Drug Des. 2015, 86, 424–431. [Google Scholar] [CrossRef]
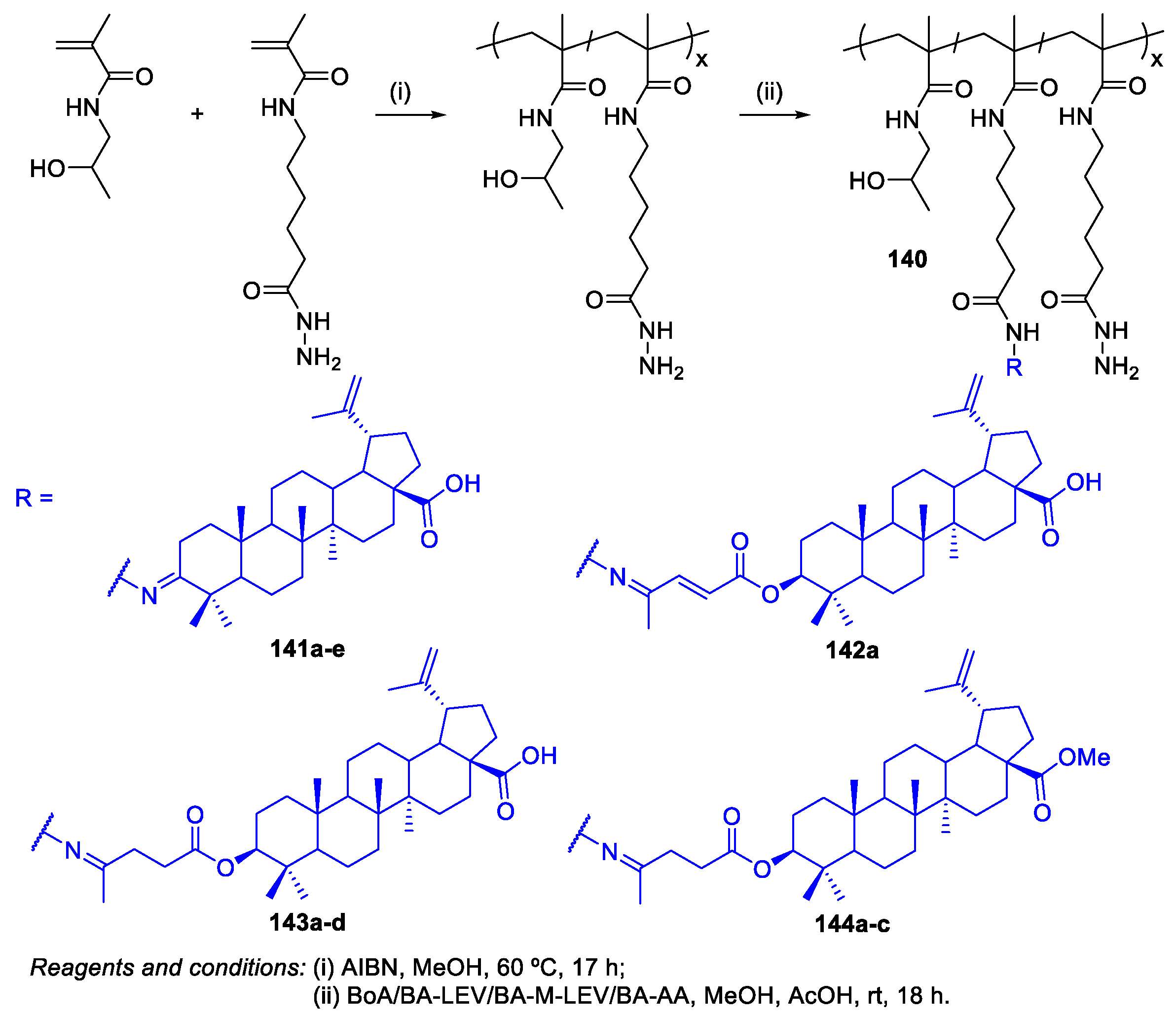
- Lomkova, E.A.; Chytil, P.; Janoušková, O.; Mueller, T.; Lucas, H.; Filippov, S.K.; Trhlíková, O.; Aleshunin, P.A.; Skorik, Y.A.; Ulbrich, K.; Etrych, T. Biodegradable Micellar HPMA-Based Polymer–Drug Conjugates with Betulinic Acid for Passive Tumor Targeting. Biomacromolecules 2016, 17, 3493–3507. [Google Scholar] [CrossRef] [PubMed]
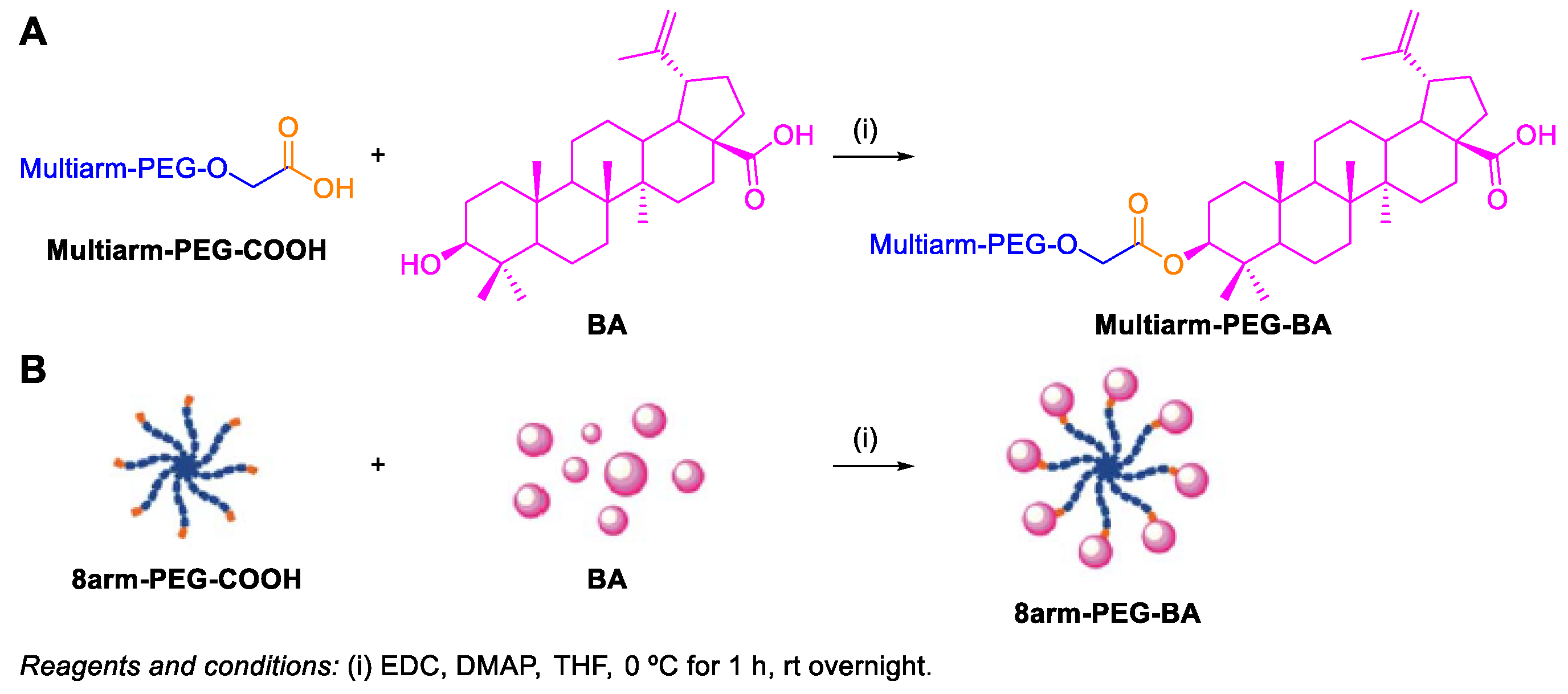
- Dai, L.; Li, D.; Cheng, J.; Liu, J.; Deng, L.-H.; Wang, L.-Y.; Lei, J.-D.; He, J. Water soluble multiarm-polyethylene glycol–betulinic acid prodrugs: Design, synthesis, and in vivo effectiveness. Polym. Chem. 2014, 5, 5775–5783. [Google Scholar] [CrossRef]
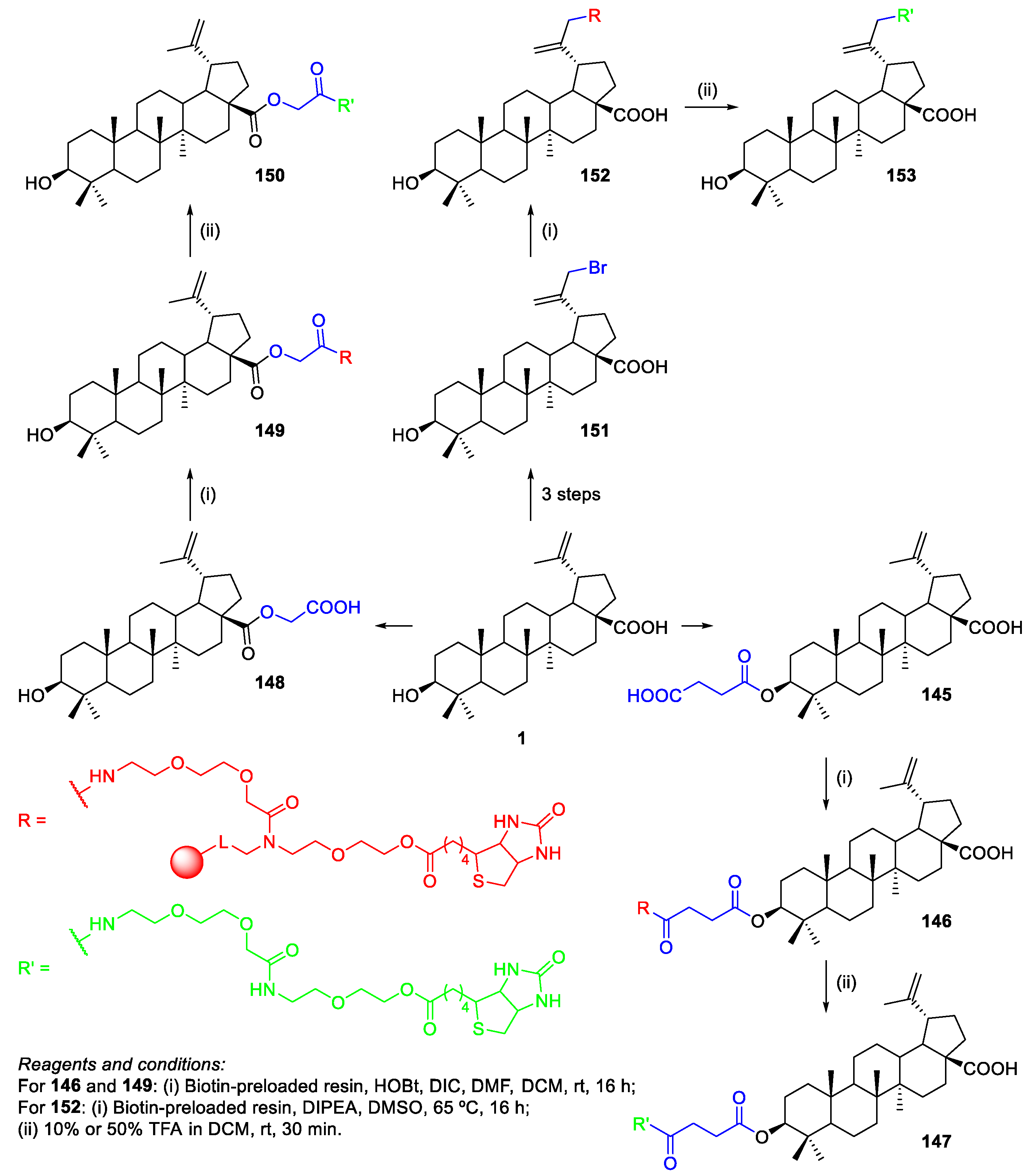
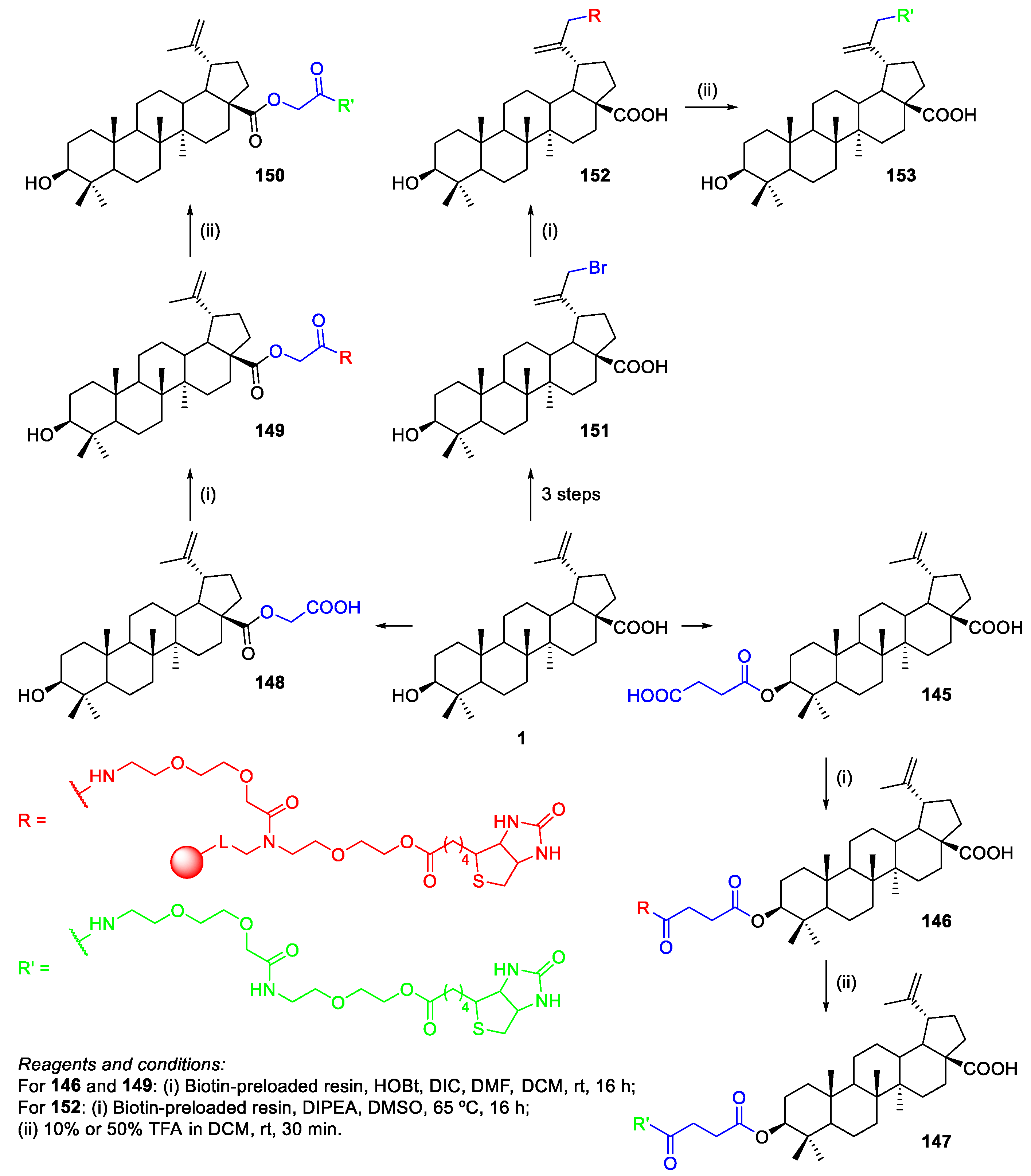
- Soural, M.; Hodon, J.; Dickinson, N.J.; Sidova, V.; Gurska, S.; Dzubak, P.; Hajduch, M.; Sarek, J.; Urban, M. Preparation of Conjugates of Cytotoxic Lupane Triterpenes with Biotin. Bioconjugate Chem. 2015, 26, 2563–2570. [Google Scholar] [CrossRef] [PubMed]
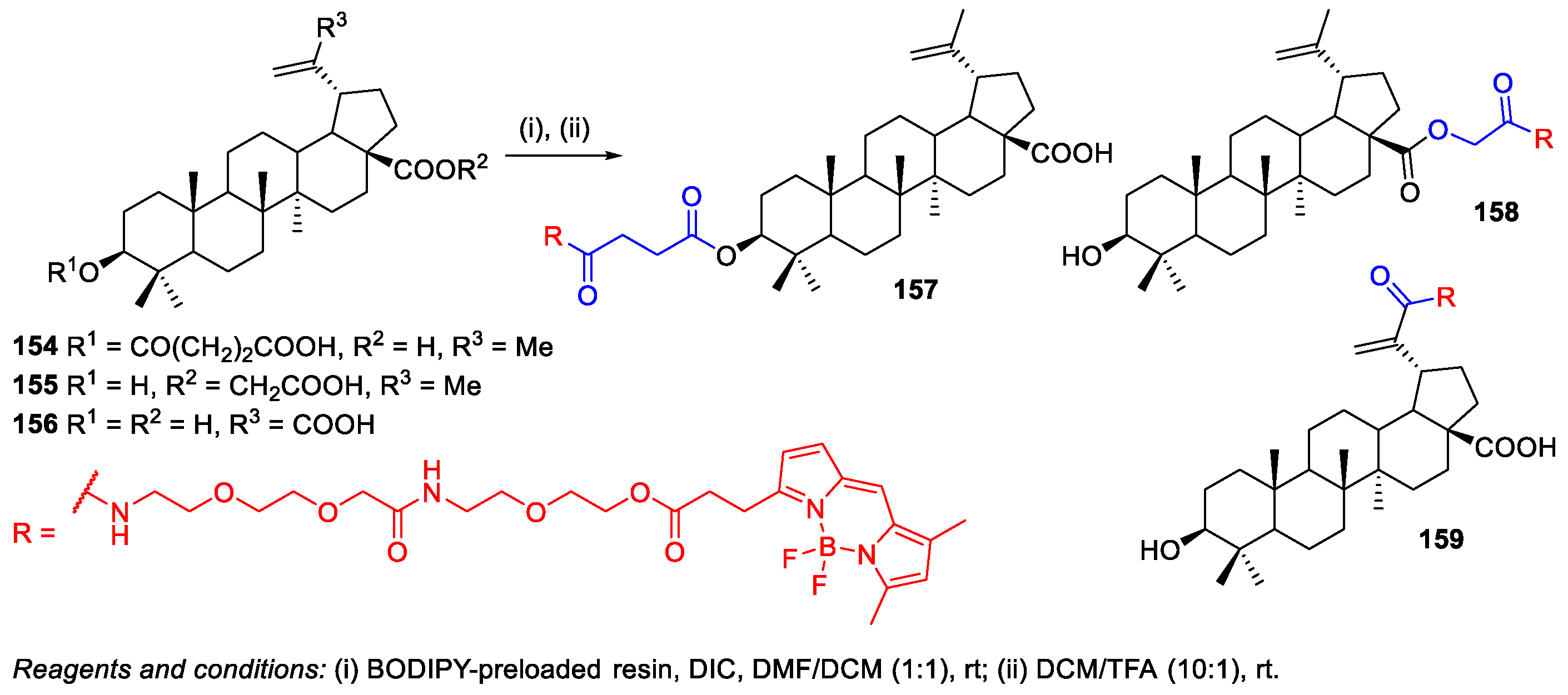
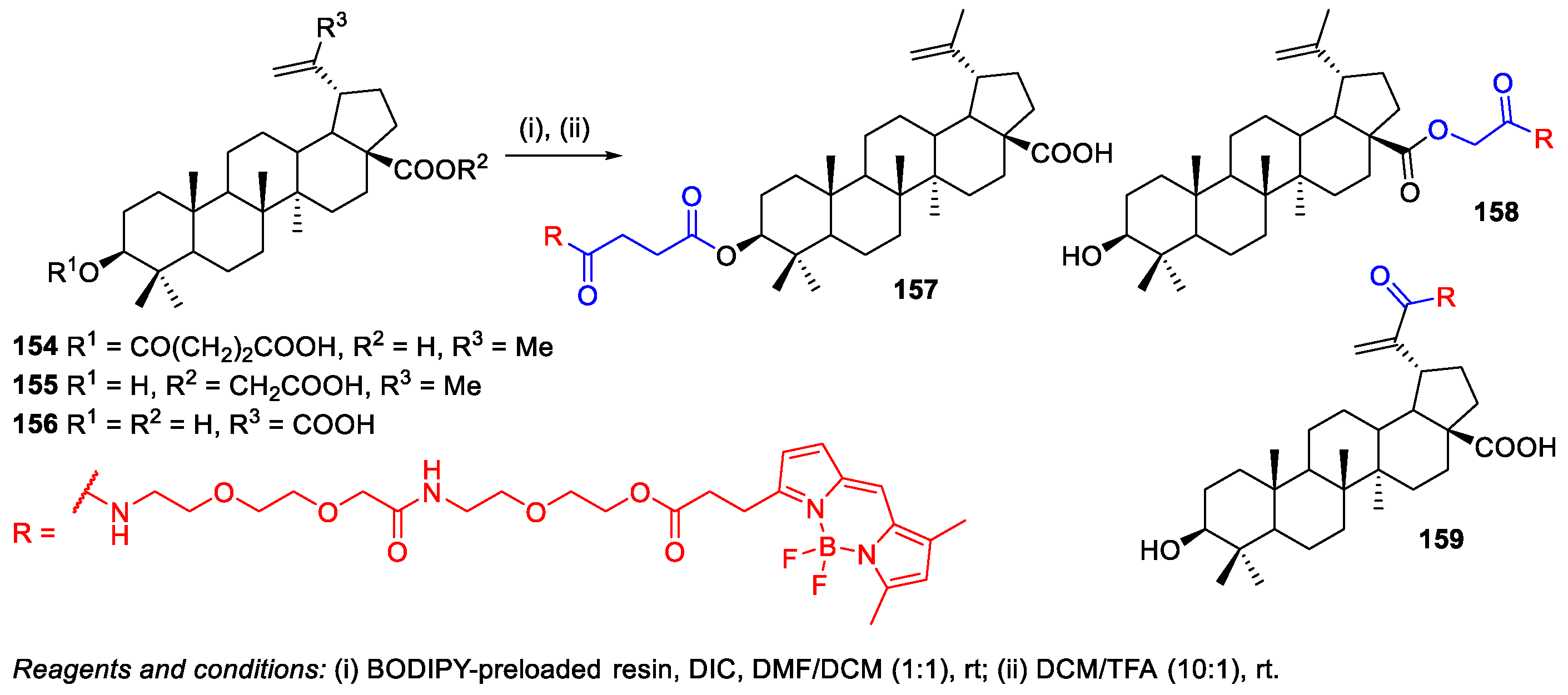
- Krajcovicova, S.; Stankova, J.; Dzubak, P.; Hajduch, M.; Soural, M.; Urban, M. A Synthetic Approach for the Rapid Preparation of BODIPY Conjugates and their use in Imaging of Cellular Drug Uptake and Distribution. Chem. Eur. J. 2018, 24, 4957–4966. [Google Scholar] [CrossRef]
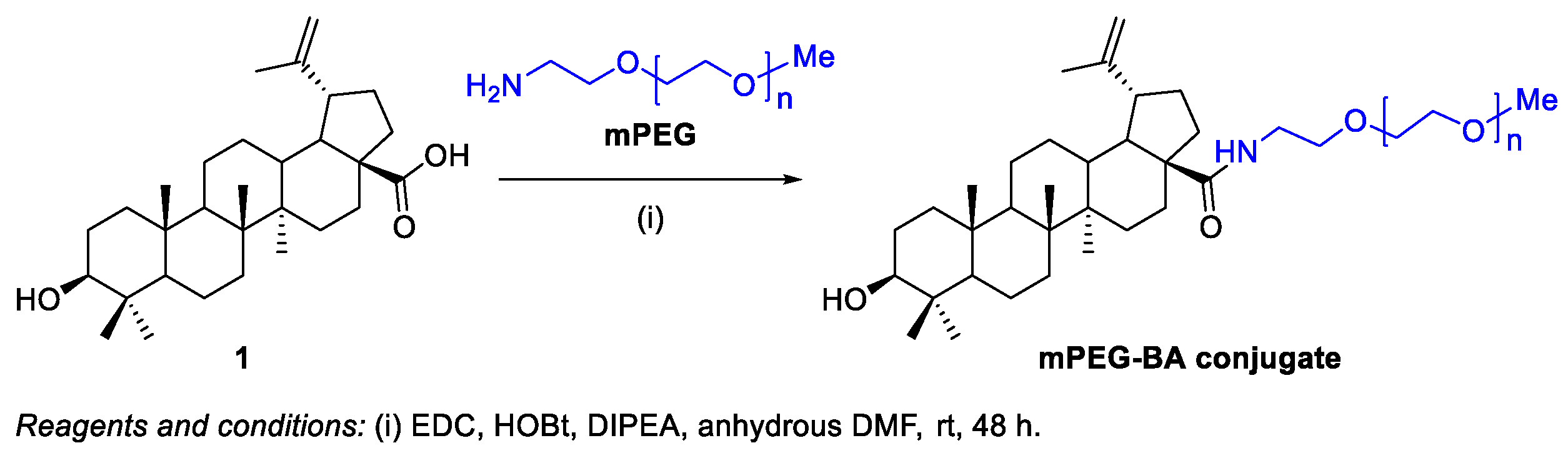
- Saneja, A.; Sharma, L.; Dubey, R.D.; Mintoo, M.J.; Singh, A.; Kumar, A.; Sangwan, P.L.; Tasaduq, S.A.; Singh, G.; Mondhe, D.M.; Gupta, P.N. Synthesis, characterization and augmented anticancer potential of PEG-betulinic acid conjugate. Mater. Sci. Eng. C 2017, 73, 616–626. [Google Scholar] [CrossRef] [PubMed]
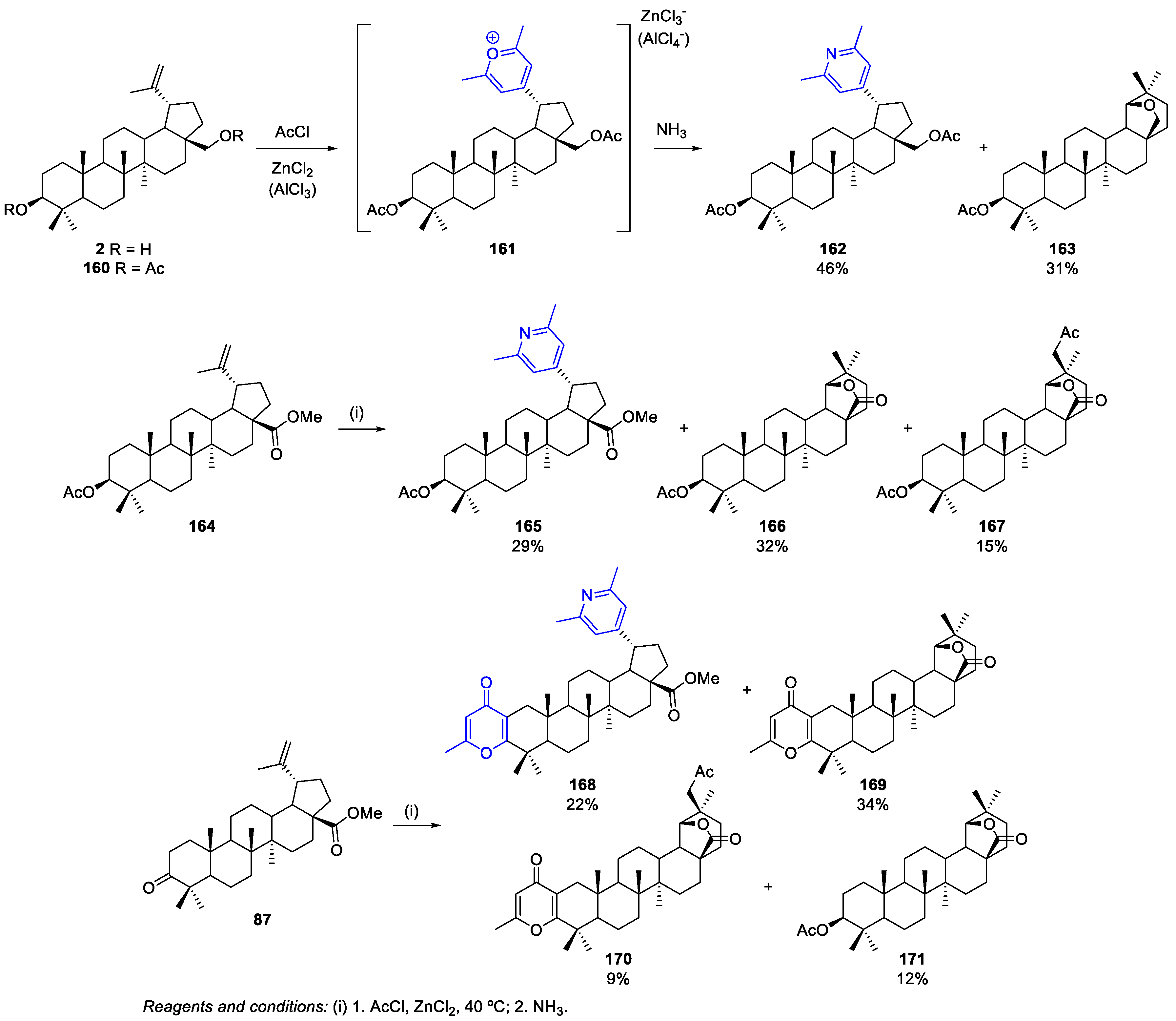
- Antimonova, A.N.; Petrenko, N.I.; Shakirov, M.M.; Shul’ts, E.E. Synthesis of 19-(2,6-Dimethylpyrid-4-yl)-20,29,30-trinorlupanes. Chem. Nat. Compd. 2014, 50, 305–310. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Keiser, J.; Vargas, M.; Gubaidullin, R.R.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Odinokov, V.N. Synthesis and activity of new triphenylphosphonium derivatives of betulin and betulinic acid against Schistosoma mansoni in vitro and in vivo. Bioorg. Med. Chem. 2014, 22, 6297–6304. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’skii, Y.P.; Bel’skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. MedChemComm 2017, 8, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Ghosh, A.; Ghosh, S.; Shil, S.; Bothra, A.K.; Ghosh, P. 3-Epihydroxy lup-20(29)-en-19(28)-olide: Partial synthesis, antitopoisomerase activity, and 3D molecular docking. Med. Chem. Res. 2016, 25, 1087–1095. [Google Scholar] [CrossRef]
- Pokorny, J.; Krajcovicova, S.; Hajduch, M.; Holoubek, M.; Gurska, S.; Dzubak, P.; Volna, T.; Popa, I.; Urban, M. Triterpenic azines, a new class of compounds with selective cytotoxicity to leukemia cells CCRF-CEM. Future Med. Chem. 2018, 10, 483–491. [Google Scholar] [CrossRef] [PubMed]
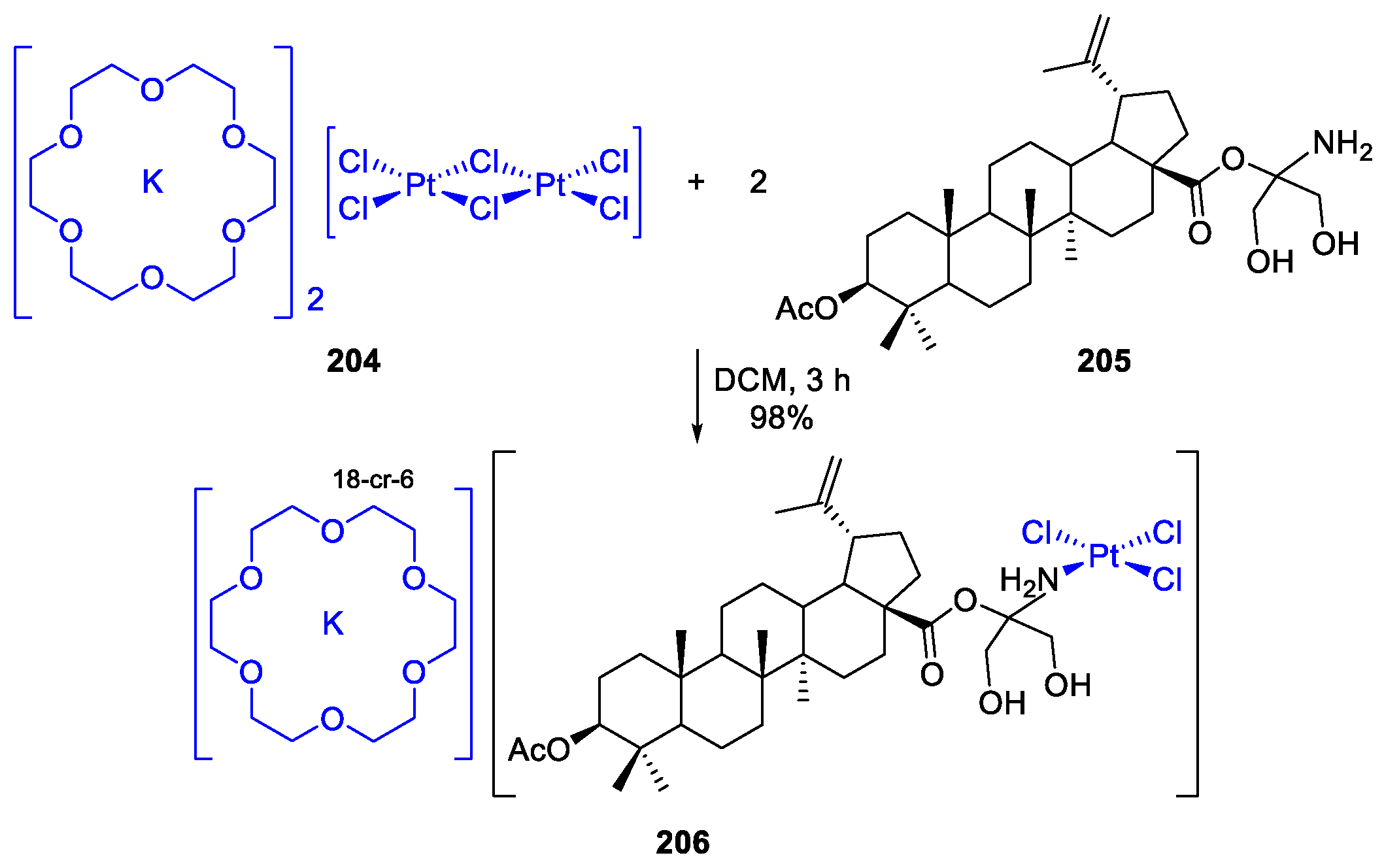
- Kaluđerović, G.; Bulatović, M.; Krajnović, T.; Paschke, R.; B. Zmejkovski, B.; Maksimović-Ivanić, D.; Mijatović, S. (18-Crown-6)potassium(I) Trichlorido[28-acetyl-3-(tris-(hydroxylmethyl)amino-ethane)betulinic ester-κN]platinum(II): Synthesis and In Vitro Antitumor Activity. Inorganics 2017, 5, 56. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, J.L.C.; Freire, C.S.R.; Silvestre, A.J.D.; Silva, A.M.S. Recent Developments in the Functionalization of Betulinic Acid and Its Natural Analogues: A Route to New Bioactive Compounds. Molecules 2019, 24, 355. https://doi.org/10.3390/molecules24020355
Sousa JLC, Freire CSR, Silvestre AJD, Silva AMS. Recent Developments in the Functionalization of Betulinic Acid and Its Natural Analogues: A Route to New Bioactive Compounds. Molecules. 2019; 24(2):355. https://doi.org/10.3390/molecules24020355
Chicago/Turabian StyleSousa, Joana L. C., Carmen S. R. Freire, Armando J. D. Silvestre, and Artur M. S. Silva. 2019. "Recent Developments in the Functionalization of Betulinic Acid and Its Natural Analogues: A Route to New Bioactive Compounds" Molecules 24, no. 2: 355. https://doi.org/10.3390/molecules24020355