
Discovery of Lipid Peroxidation Inhibitors from Bacopa Species Prioritized through Multivariate Data Analysis and Multi-Informative Molecular Networking

 , , and
, , and
Abstract
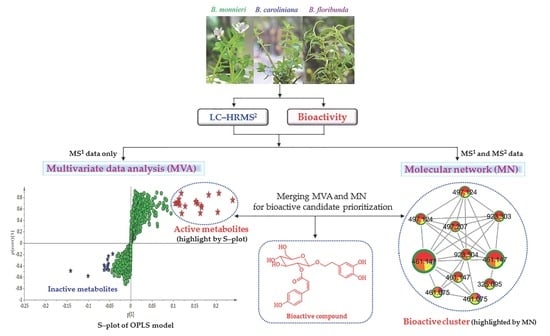
:
1. Introduction
2. Results and Discussion
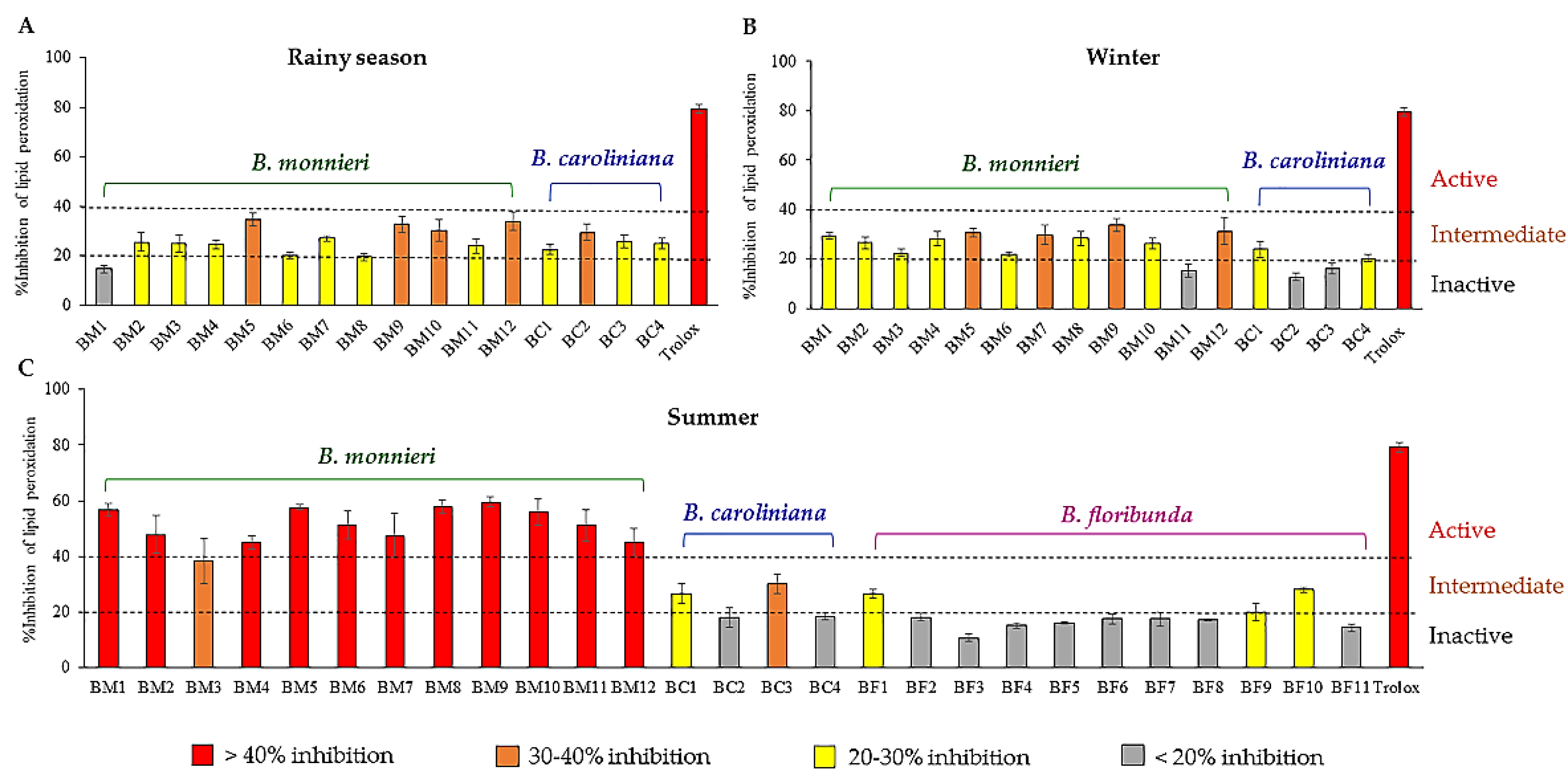
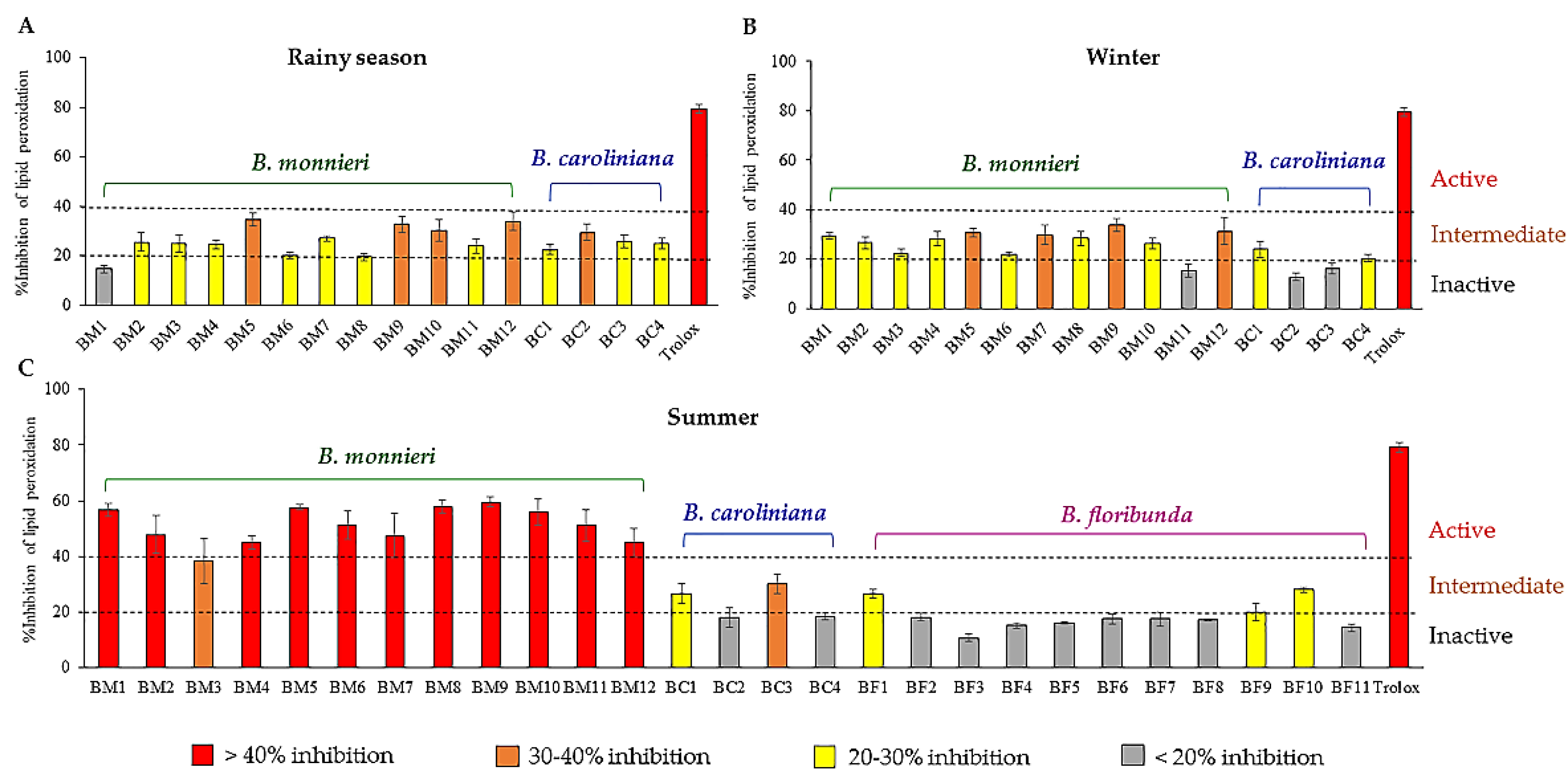
2.1. Lipid Peroxidation Inhibitory Activity Evaluation of the Extracts
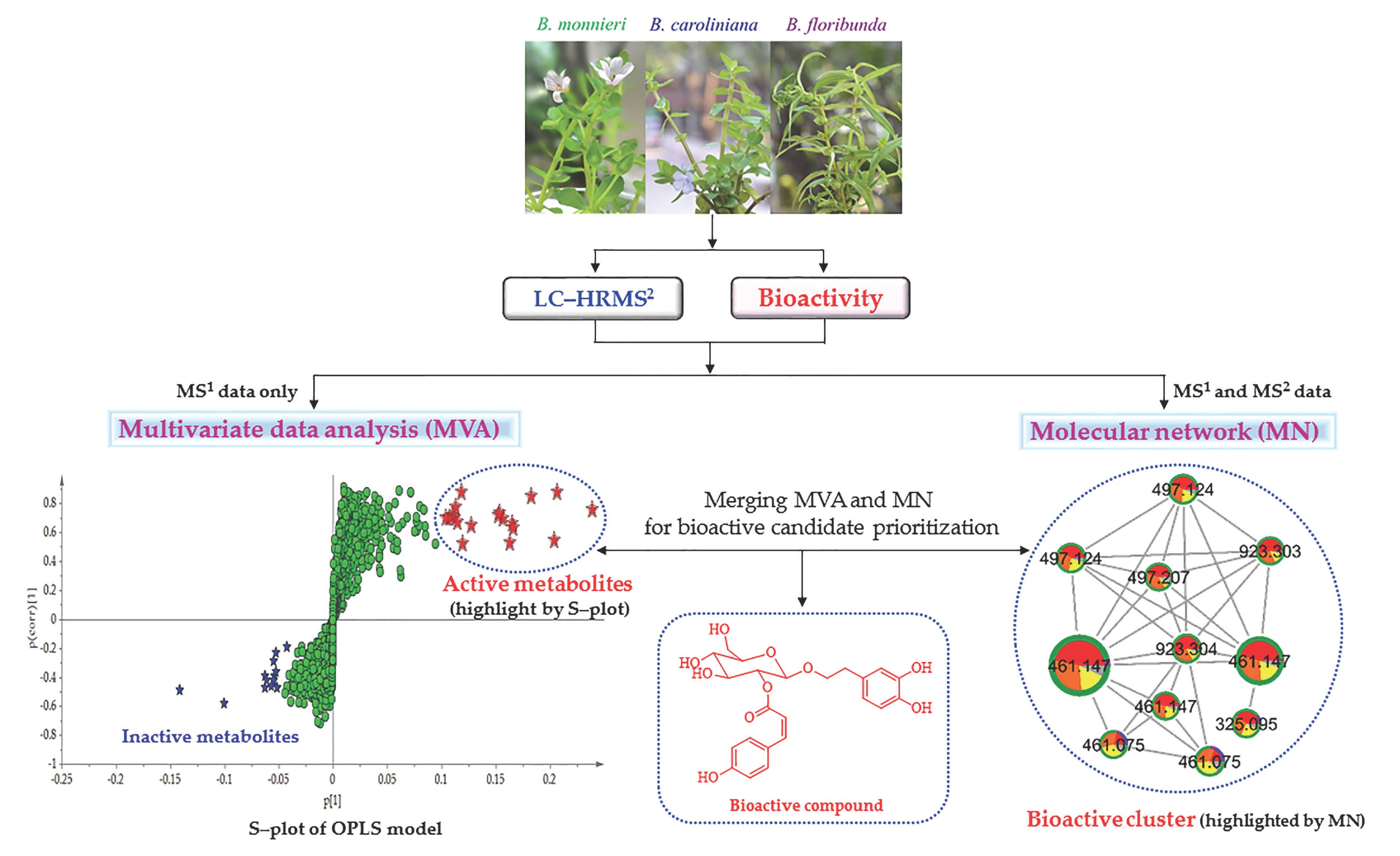
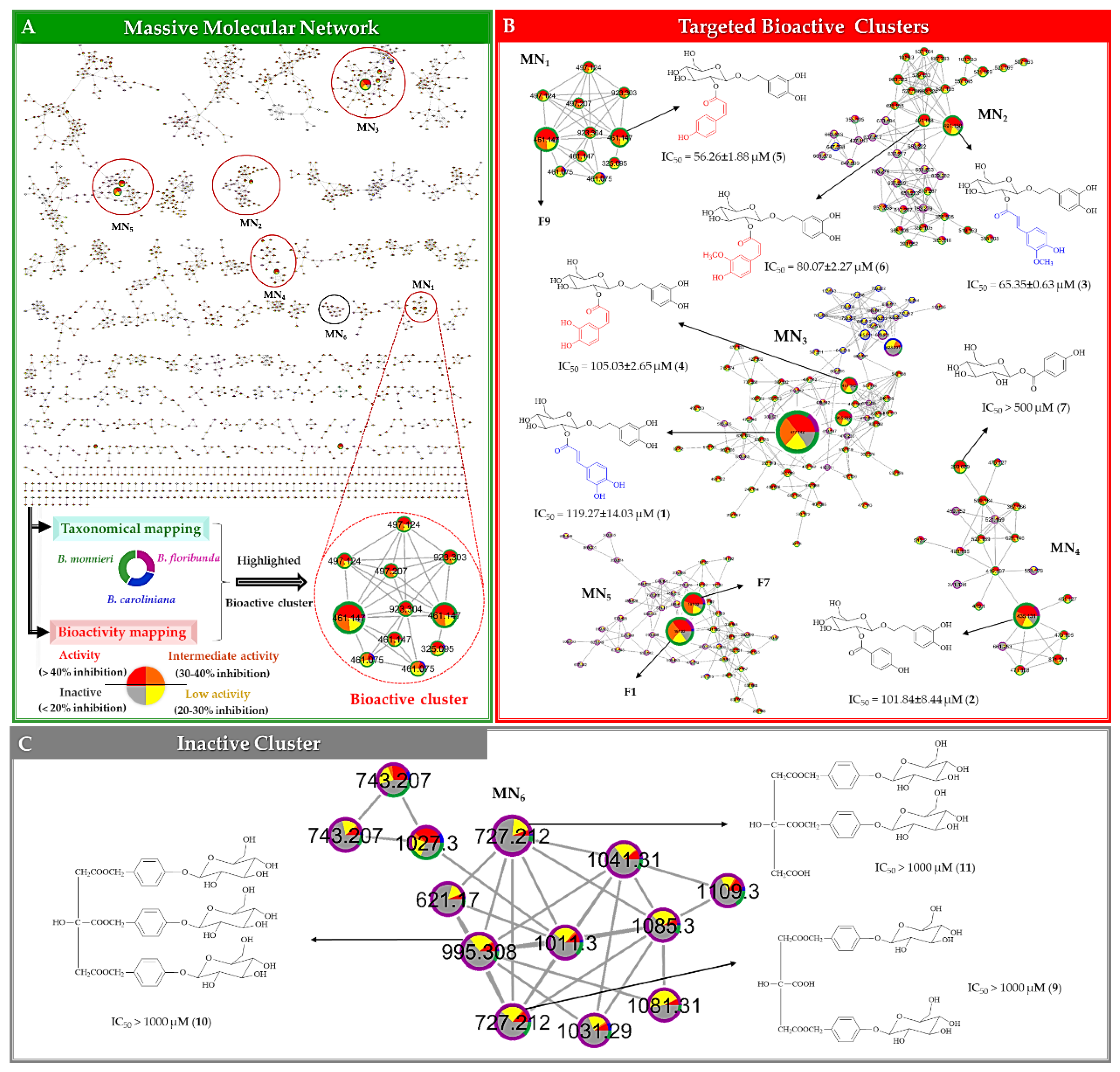
2.2. Potential Bioactive Metabolites Prioritized through Multivariate Statistical Analysis and Molecular Networking
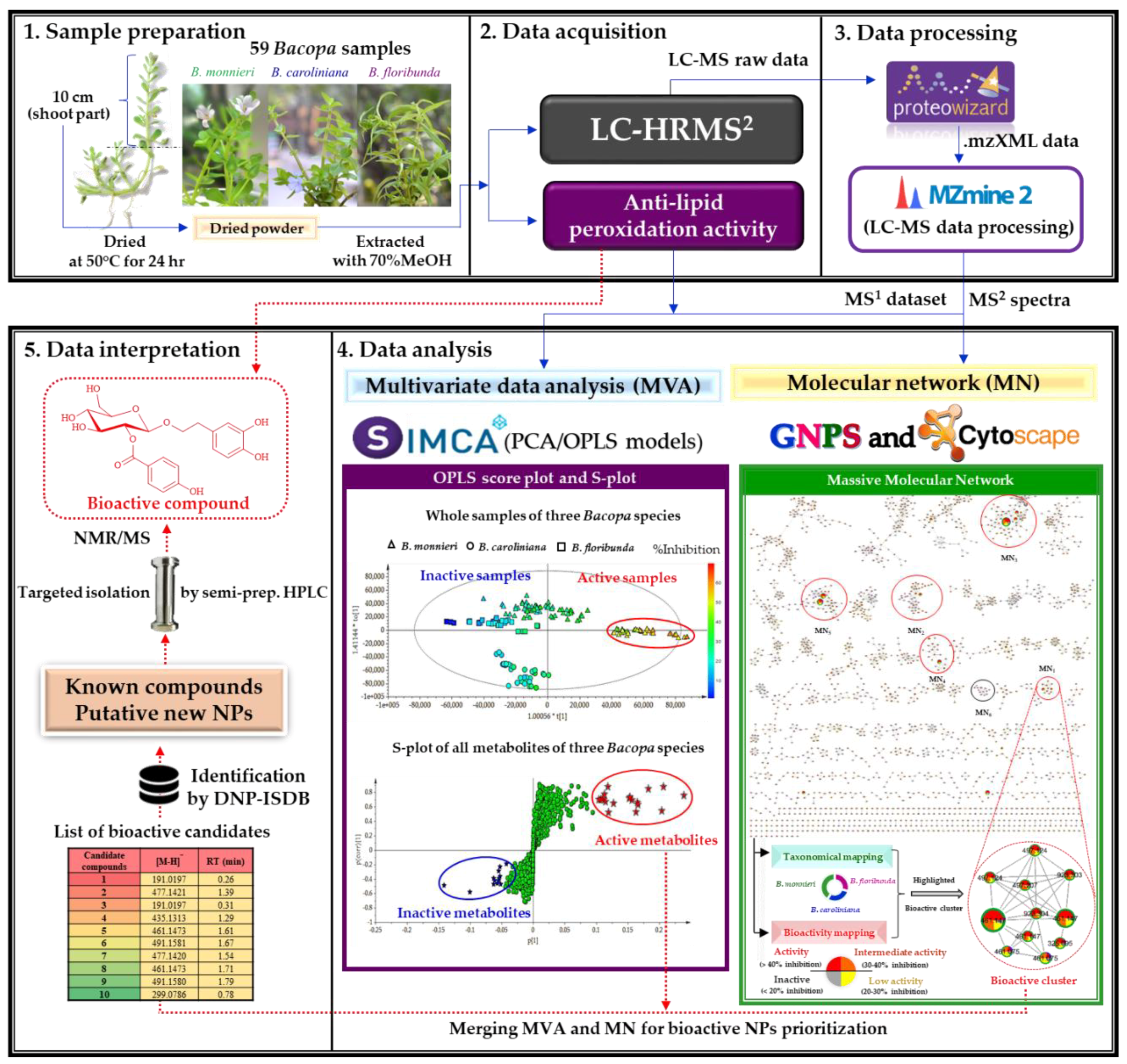
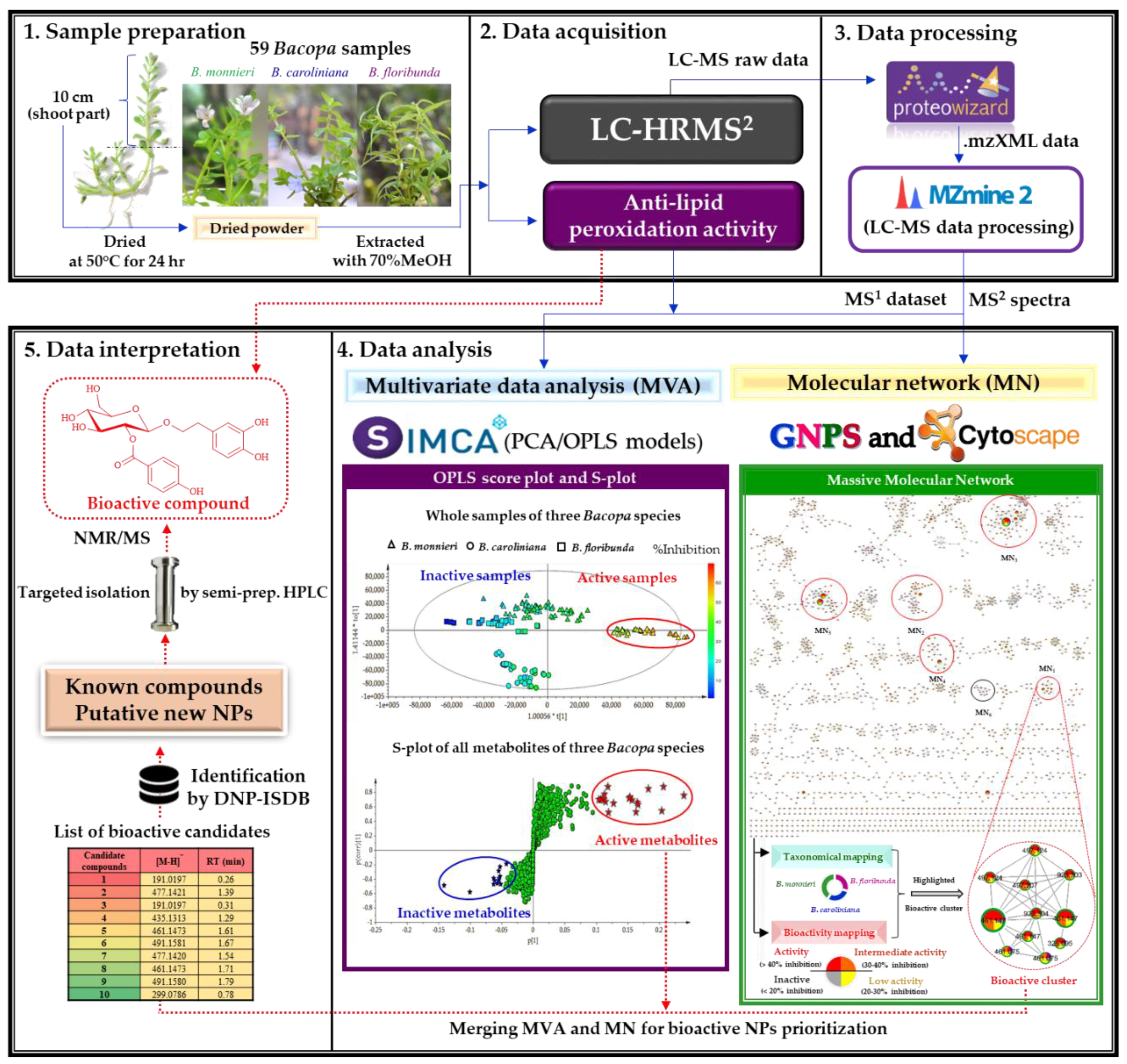
2.2.1. Organization and Pre–Treatment of the Metabolite Profiling Data
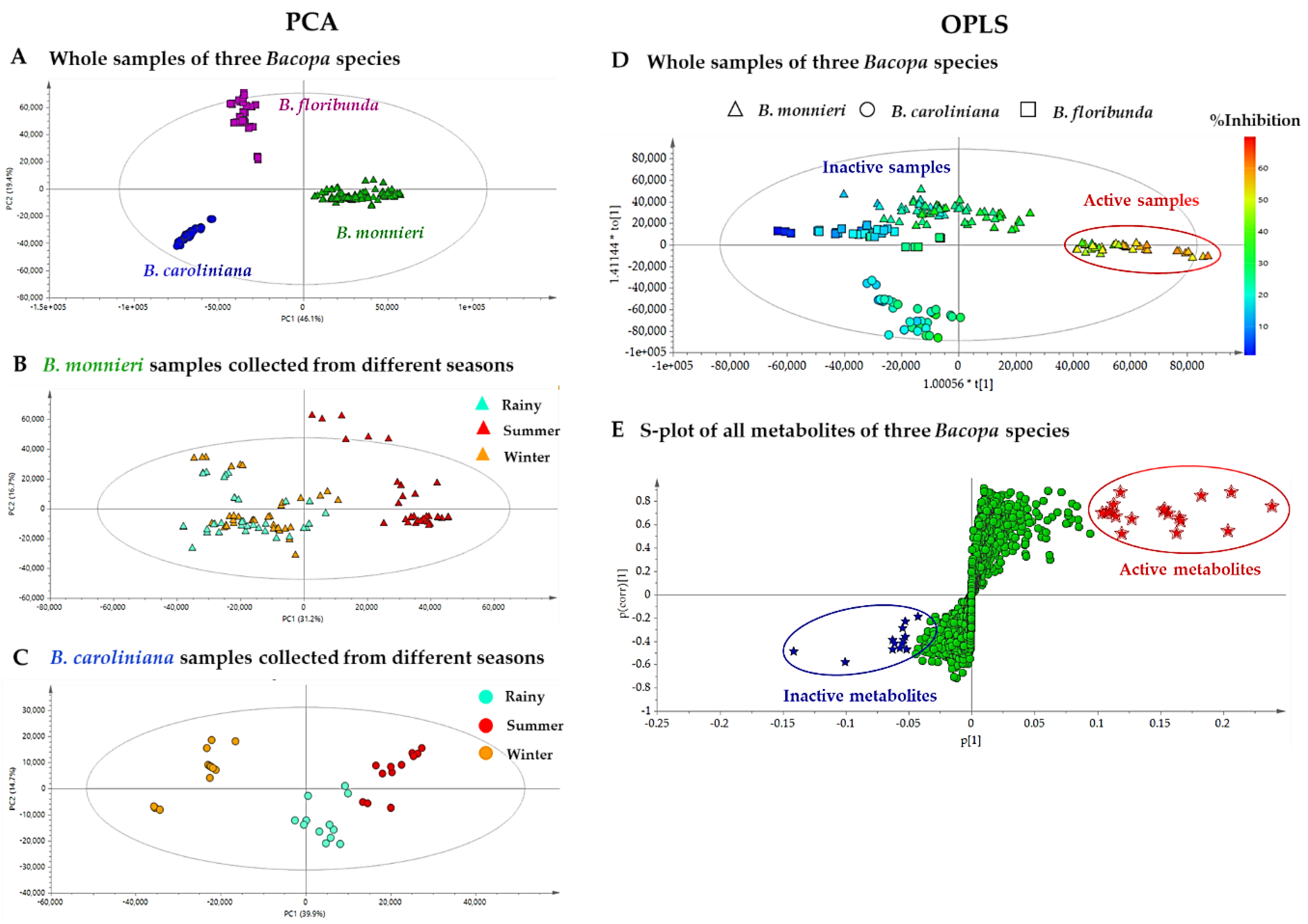
2.2.2. Multivariate Data Analysis
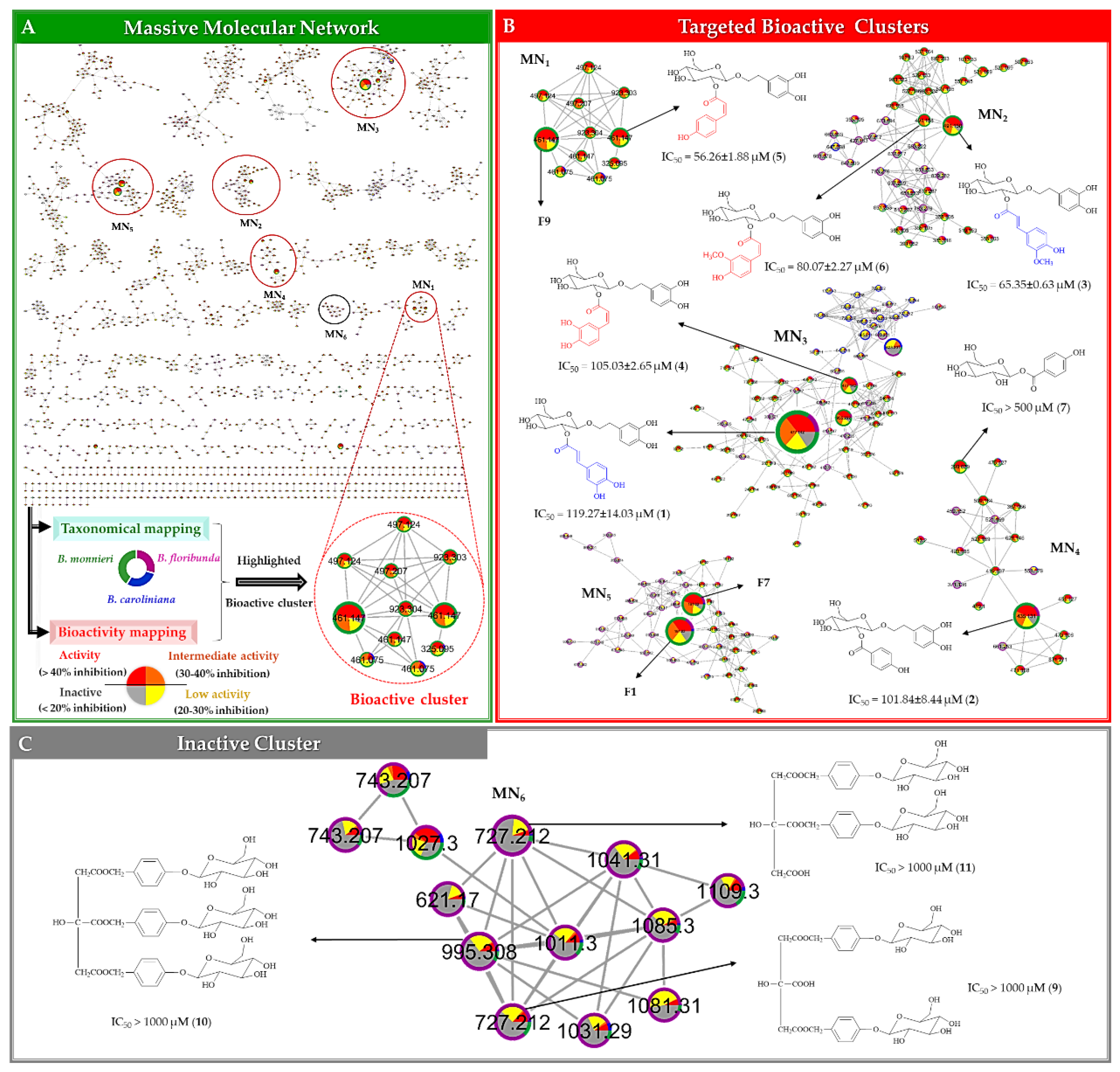
2.2.3. Multi-Informational Molecular Map
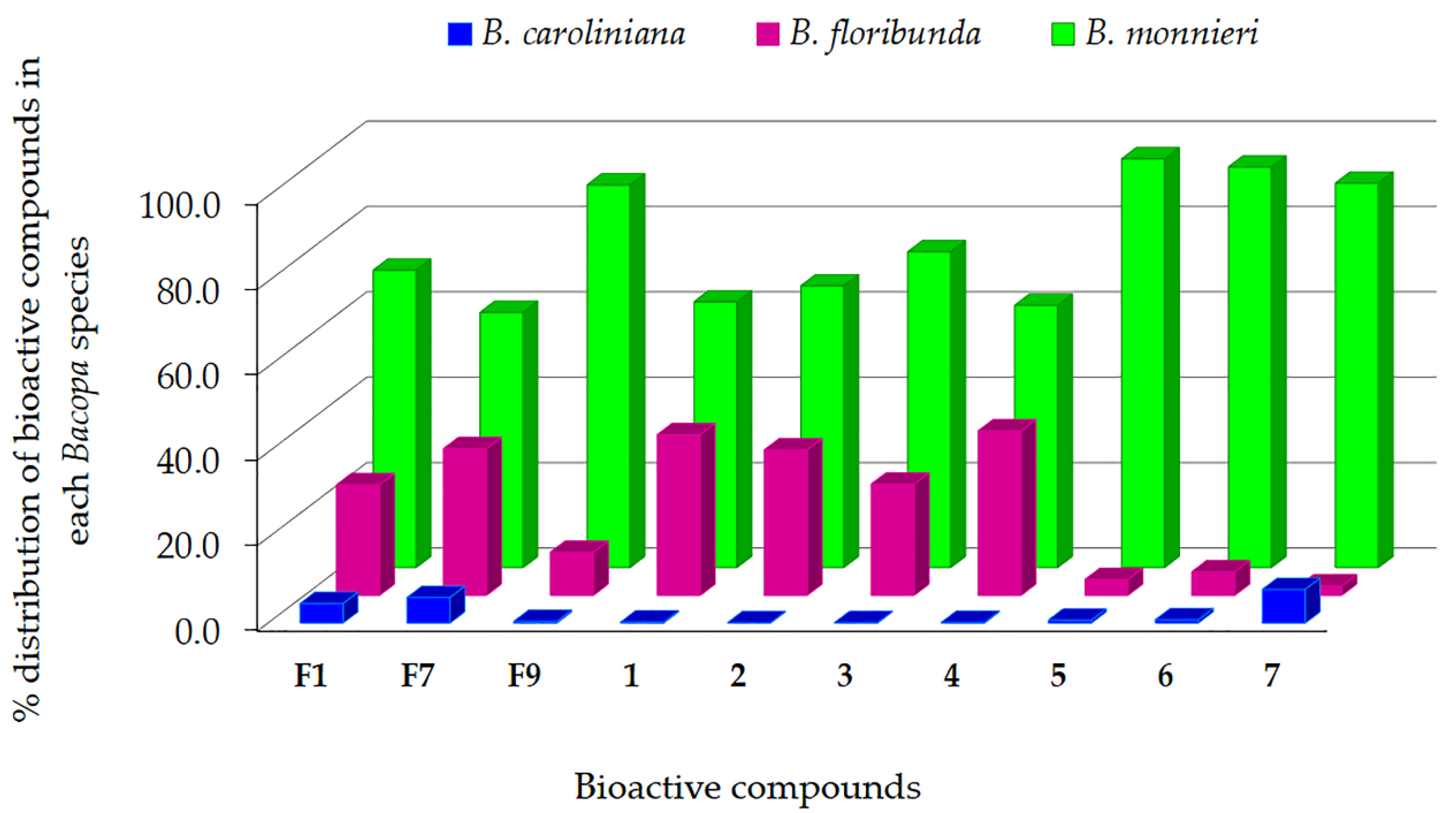
2.2.4. Merging MVA and MN for Bioactive Candidate Prioritization
2.3. DNP-ISDB Dereplication and Purification of Bioactive Candidates
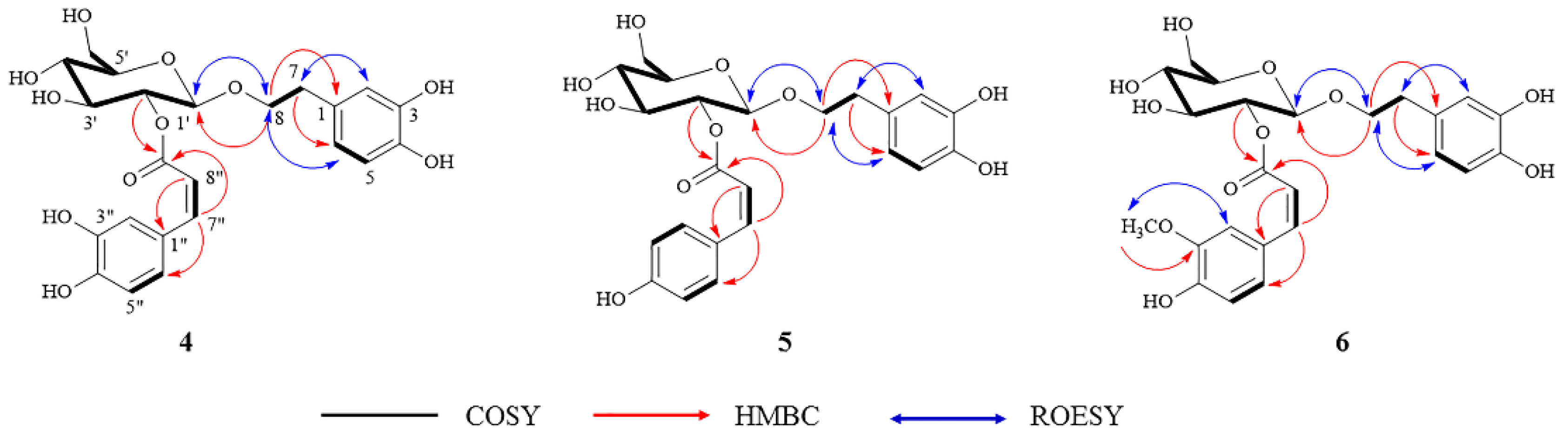
2.4. Identification and Structure Elucidation of Compounds 1–11
2.5. Evaluation of the Anti-Lipid Peroxidation Activity of the Isolated Compounds
3. Materials and Methods
3.1. Chemicals and Plant Materials
3.2. Sample Preparation
3.3. TBAR Assay
3.4. UHPLC-ESI-HRMS2 Analysis
3.5. MZmine data preprocessing
3.6. Multivariate Analysis
3.7. Molecular Networking Analyses
3.8. Purification of Candidate Bioactive Compounds
3.8.1. Extraction Procedure
3.8.2. Purification Methods
3.8.3. Identification Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allard, P.-M.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.-L. Integration of molecular networking and In-Silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication strategies in natural product research: How many tools and methodologies behind the same concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Yuliana, N.D.; Khatib, A.; Choi, Y.H.; Verpoorte, R. Metabolomics for bioactivity assessment of natural products. Phytother. Res. 2011, 25, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; AnandhiSenthilkumar, H.; Wu, S.-b.; Liu, B.; Guo, Z.-y.; Fata, J.E.; Kennelly, E.J.; Long, C.-l. Comparative UPLC-QTOF-MS-based metabolomics and bioactivities analyses of Garcinia oblongifolia. J. Chromatogr. B 2016, 1011, 179–195. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, G.; Pizza, C.; Piacente, S.; Montoro, P. Combination of LC–MS based metabolomics and antioxidant activity for evaluation of bioactive compounds in Fragaria vesca leaves from Italy. J. Pharm. Biomed. Anal. 2018, 150, 233–240. [Google Scholar] [CrossRef]
- Ayouni, K.; Berboucha-Rahmani, M.; Kim, H.K.; Atmani, D.; Verpoorte, R.; Choi, Y.H. Metabolomic tool to identify antioxidant compounds of Fraxinus angustifolia leaf and stem bark extracts. Ind. Crops. Prod. 2016, 88, 65–77. [Google Scholar] [CrossRef]
- Caesar, L.K.; Kellogg, J.J.; Kvalheim, O.M.; Cech, N.B. Opportunities and Limitations for Untargeted Mass Spectrometry Metabolomics to Identify Biologically Active Constituents in Complex Natural Product Mixtures. J. Nat. Prod. 2019, 82, 469–484. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based plant metabolomics: Where do we stand, where do we go? Trends Biotechnol. 2011, 29, 267–275. [Google Scholar] [CrossRef]
- Wolfender, J.-L.; Rudaz, S.; Hae Choi, Y.; Kyong Kim, H. Plant Metabolomics: From Holistic Data to Relevant Biomarkers. Curr. Med. Chem. 2013, 20, 1056–1090. [Google Scholar]
- Wolfender, J.-L.; Nuzillard, J.-M.; van der Hooft, J.J.J.; Renault, J.-H.; Bertrand, S. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography–High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem. 2019, 91, 704–742. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [Google Scholar] [CrossRef]
- Olivon, F.; Allard, P.-M.; Koval, A.; Righi, D.; Genta-Jouve, G.; Neyts, J.; Apel, C.; Pannecouque, C.; Nothias, L.-F.; Cachet, X.; et al. Bioactive natural products prioritization using massive multi-informational molecular networks. Acs Chem. Biol. 2017, 12, 2644–2651. [Google Scholar] [CrossRef]
- Naman, C.B.; Rattan, R.; Nikoulina, S.E.; Lee, J.; Miller, B.W.; Moss, N.A.; Armstrong, L.; Boudreau, P.D.; Debonsi, H.M.; Valeriote, F.A.; et al. Integrating molecular networking and biological assays to target the isolation of a cytotoxic cyclic octapeptide, samoamide A, from an American Samoan Marine Cyanobacterium. J. Nat. Prod. 2017, 80, 625–633. [Google Scholar] [CrossRef]
- Nothias, L.-F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-based molecular networking for the discovery of drug leads in natural product bioassay-guided fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef]
- Global Natural Product Social Molecular Networking. Available online: http://gnps.ucsd.edu (accessed on 10 Aprile 2018).
- Olivon, F.; Elie, N.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MetGem Software for the Generation of Molecular Networks Based on the t-SNE Algorithm. Anal. Chem. 2018, 90, 13900–13908. [Google Scholar] [CrossRef]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef]
- Tem, S. Thai Plant Names, 2014 ed.; The Forest Herbarium, Royal Forest Department: Bangkok, Thailand, 2014. [Google Scholar]
- Mukherjee, G.D.; Dey, C.D. Clinical trial on Brahmi. I. J. Exp. Med. Sci. 1966, 10, 5–11. [Google Scholar]
- Vollala, V.R.; Upadhya, S.; Nayak, S. Effect of Bacopa monniera Linn. (brahmi) extract on learning and memory in rats: A behavioral study. J. Vet. Behav. 2010, 5, 69–74. [Google Scholar] [CrossRef]
- Saraf, M.K.; Prabhakar, S.; Khanduja, K.L.; Anand, A. Bacopa monniera attenuates scopolamine-induced impairment of spatial memory in mice. Evid. Based Complement. Alternat. Med. 2011, 2011, 10. [Google Scholar] [CrossRef]
- Nathan, P.J.; Clarke, J.; Lloyd, J.; Hutchison, C.W.; Downey, L.; Stough, C. The acute effects of an extract of Bacopa monniera (Brahmi) on cognitive function in healthy normal subjects. Hum. Psychopharmacol. Clin. Exp. 2001, 16, 345–351. [Google Scholar] [CrossRef]
- Stough, C.; Lloyd, J.; Clarke, J.; Downey, L.A.; Hutchison, C.W.; Rodgers, T.; Nathan, P.J. The chronic effects of an extract of Bacopa monniera (Brahmi) on cognitive function in healthy human subjects. Psychopharmacology 2001, 156, 481–484. [Google Scholar] [CrossRef]
- Peth-Nui, T.; Wattanathorn, J.; Muchimapura, S.; Tong-Un, T.; Piyavhatkul, N.; Rangseekajee, P.; Ingkaninan, K.; Vittaya-areekul, S. Effects of 12-week Bacopa monnieri consumption on attention, cognitive processing, working memory, and functions of both cholinergic and monoaminergic systems in healthy elderly volunteers. Evid. Based Complement. Alternat. Med. 2012, 2012, 606424. [Google Scholar] [CrossRef]
- Kongkeaw, C.; Dilokthornsakul, P.; Thanarangsarit, P.; Limpeanchob, N.; Norman Scholfield, C. Meta-analysis of randomized controlled trials on cognitive effects of Bacopa monnieri extract. J. Ethnopharmacol. 2014, 151, 528–535. [Google Scholar] [CrossRef]
- Roodenrys, S.; Booth, D.; Bulzomi, S.; Phipps, A.; Micallef, C.; Smoker, J. Chronic effects of Brahmi (Bacopa monnieri) on human memory. Neuropsychopharmacology 2002, 27, 279–281. [Google Scholar] [CrossRef]
- Morgan, A.; Stevens, J. Does Bacopa monnieri Improve Memory Performance in Older Persons? Results of a Randomized, Placebo-Controlled, Double-Blind Trial. J. Altern. Complementary Med. 2010, 16, 753–759. [Google Scholar] [CrossRef]
- Gour, S.; Tembhre, M. Cholinergic inhibitory effects of bacopa monnieri and acephate in the kidney of rat. Int. J. Curr. Adv. Res. 2018, 7, 14136–14141. [Google Scholar]
- Teschke, R.; Bahre, R. Severe hepatotoxicity by Indian Ayurvedic herbal products: A structured causality assessment. Ann. Hepatol. 2009, 8, 258–266. [Google Scholar] [CrossRef]
- Sumathi, T.; Nongbri, A. Hepatoprotective effect of Bacoside-A, a major constituent of Bacopa monniera Linn. Phytomedicine 2008, 15, 901–905. [Google Scholar] [CrossRef]
- Menon, B.R.; Rathi, M.A.; Thirumoorthi, L.; Gopalakrishnan, V.K. Potential Effect of Bacopa monnieri on Nitrobenzene Induced Liver Damage in Rats. Indian J. Clin. Biochem. 2010, 25, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Dhawan, B.N. Neuropsychopharmacological effects of the ayurvedic nootropic Bacopa monnieri Linn. Indian J. Pharmacol. 1997, 29, 359–365. [Google Scholar]
- Russo, A.; Borrelli, F. Bacopa monniera, a reputed nootropic plant: An overview. Phytomedicine 2005, 12, 305–317. [Google Scholar] [CrossRef]
- Deepak, M.; Sangli, G.K.; Arun, P.C.; Amit, A. Quantitative determination of the major saponin mixture bacoside A in Bacopa monnieri by HPLC. Phytochem. Anal. 2005, 16, 24–29. [Google Scholar] [CrossRef]
- Ganzera, M.; Gampenrieder, J.; Pawar, R.S.; Khan, I.A.; Stuppner, H. Separation of the major triterpenoid saponins in Bacopa monnieri by high-performance liquid chromatography. Anal. Chim. Acta 2004, 516, 149–154. [Google Scholar] [CrossRef]
- Murthy, P.B.; Raju, V.R.; Ramakrisana, T.; Chakravarthy, M.S.; Kumar, K.V.; Kannababu, S.; Subbaraju, G.V. Estimation of twelve bacopa saponins in Bacopa monnieri extracts and formulations by high-performance liquid chromatography. Chem. Pharm. Bull. (Tokyo) 2006, 54, 907–911. [Google Scholar] [CrossRef]
- Phrompittayarat, W.; Wittaya-Areekul, S.; Jetiyanon, K.; Putalun, W.; Tanaka, H.; Ingkaninan, K. Determination of saponin glycosides in Bacopa monnieri by reversed phase high performance liquid chromatography. Thai Pharm. Health Sci. J. 2007, 2, 26–32. [Google Scholar]
- Bhandari, P.; Kumar, N.; Singh, B.; Singh, V.; Kaur, I. Silica-based monolithic column with evaporative light scattering detector for HPLC analysis of bacosides and apigenin in Bacopa monnieri. J. Sep. Sci. 2009, 32, 2812–2818. [Google Scholar] [CrossRef]
- British Pharmacopoeia Commission. The British Pharmacopoeia 2016; The Stationery Office: London, UK, 2016; Volume 1. [Google Scholar]
- Mathur, S.; Sharma, S.; Gupta, P.M.; Kumar, S. Evaluation of an Indian germplasm collection of the medicinal plant Bacopa monnieri (L.) Pennell by use of multivariate approaches. Euphytica 2003, 133, 255–265. [Google Scholar] [CrossRef]
- Bansal, M. Diversity among wild accessions of Bacopa monnieri (L.) Wettst. and their morphogenetic potential. Acta Physiol. Plant. 2014, 36, 1177–1186. [Google Scholar] [CrossRef]
- Bansal, M.; Reddy, M.S.; Kumar, A. Seasonal variations in harvest index and bacoside A contents amongst accessions of Bacopa monnieri (L.) Wettst. collected from wild populations. Physiol. Mol. Biol. Plants 2016, 22, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Phrompittayarat, W.; Jetiyanon, K.; Wittaya-Areekul, S.; Putalun, W.; Tanaka, H.; Khan, I.; Ingkaninan, K. Influence of seasons, different plant parts, and plant growth stages on saponin quantity and distribution in Bacopa monnieri. SJST 2011, 33, 193–199. [Google Scholar]
- Bhandari, P.; Kumar, N.; Singh, B.; Kaul, V.K. Bacosterol Glycoside, a New 13,14-Seco-steroid Glycoside from Bacopa monnieri. Chem. Pharm. Bull. (Tokyo) 2006, 54, 240–241. [Google Scholar] [CrossRef]
- Bhandari, P.; Kumar, N.; Gupta, A.P.; Singh, B.; Kaul, V.K. A rapid RP-HPTLC densitometry method for simultaneous determination of major flavonoids in important medicinal plants. J. Sep. Sci. 2007, 30, 2092–2096. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Sarkar, T.; Nakane, T.; Kawahara, N.; Masuda, K. New Phenylethanoid Glycosides from Bacopa monniera. Chem. Pharm. Bull. (Tokyo) 2002, 50, 1616–1618. [Google Scholar] [CrossRef]
- Ohta, T.; Nakamura, S.; Nakashima, S.; Oda, Y.; Matsumoto, T.; Fukaya, M.; Yano, M.; Yoshikawa, M.; Matsuda, H. Chemical structures of constituents from the whole plant of Bacopa monniera. J. Nat. Med. 2016, 70, 404–411. [Google Scholar] [CrossRef]
- Limpeanchob, N.; Jaipan, S.; Rattanakaruna, S.; Phrompittayarat, W.; Ingkaninan, K. Neuroprotective effect of Bacopa monnieri on beta-amyloid-induced cell death in primary cortical culture. J. Ethnopharmacol. 2008, 120, 112–117. [Google Scholar] [CrossRef]
- Katajamaa, M.; Miettinen, J.; Orešič, M. MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 2006, 22, 634–636. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Challal, S.; Queiroz, E.F.; Debrus, B.; Kloeti, W.; Guillarme, D.; Gupta, M.P.; Wolfender, J.L. Rational and Efficient Preparative Isolation of Natural Products by MPLC-UV-ELSD based on HPLC to MPLC Gradient Transfer. Planta Med. 2015, 81, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Guillarme, D.; Nguyen, D.T.T.; Rudaz, S.; Veuthey, J.-L. Method transfer for fast liquid chromatography in pharmaceutical analysis: Application to short columns packed with small particle. Part I: Isocratic separation. Eur. J. Pharm. Biopharm. 2007, 66, 475–482. [Google Scholar] [CrossRef]
- Queiroz, E.F.; Alfattani, A.; Afzan, A.; Marcourt, L.; Guillarme, D.; Wolfender, J.-L. Utility of dry load injection for an efficient natural products isolation at the semi-preparative chromatographic scale. J. Chromatogr. A 2019, 1598, 85–91. [Google Scholar] [CrossRef]
- Tabata, M.; Umetani, Y.; Ooya, M.; Tanaka, S. Glucosylation of phenolic compounds by plant cell cultures. Phytochemistry 1988, 27, 809–813. [Google Scholar] [CrossRef]
- Bianco, A.; Mazzei, R.A.; Melchioni, C.; Romeo, G.; Scarpati, M.L.; Soriero, A.; Uccella, N. Microcomponents of olive oil—III. Glucosides of 2(3,4-dihydroxy-phenyl)ethanol. Food Chem. 1998, 63, 461–464. [Google Scholar] [CrossRef]
- Jer-Huei, L.; Yi-Chu, L.; Jiing-Ping, H.; Kuo-Ching, W. Parishins B and C from rhizomes of Gastrodia elata. Phytochemistry 1996, 42, 549–551. [Google Scholar] [CrossRef]
- Saluk-Juszczak, J.; Olas, B.; Nowak, P.; Staroń, A.; Wachowicz, B. Protective effects of D-glucaro-1,4-lactone against oxidative modifications in blood platelets. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 422–428. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- ChemViz: Cheminformatics Plugin for Cytoscape. Available online: http://www.cgl.ucsf.edu/cytoscape/chemViz/ (accessed on 10 July 2018).
- Global Natural Product Social Molecular Networking. Available online: https://gnps.ucsd.edu/ProteoSAFe/status.jsp? (accessed on 19 June 2018).
- MASSIVE datasets. Available online: ftp://massive.ucsd.edu/MSV000083989 (accessed on 18 June 2019).
Sample Availability: Samples of the compounds 1–11 are available upon request from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | m/z | RT (min) | p[1] Value* | Selected Bioactive Cluster | Chemical Formula | Δ ppm | Dereplicated Compounds (MSMS Based Identification)** | Isolated Compounds (NMR Identification) | Anti-Lipid Peroxidation Activity (IC50 (µM)) |
|---|---|---|---|---|---|---|---|---|---|
| F1 | 191.0197[M − H]¯ | 0.25 | 0.239 | MN5 | C6H8O7 | 0.14 | Idaric acid-1,4-lactone | ||
| F2 | 217.0487[M + Cl]¯ | 0.23 | 0.207 | NS | C6H14O6 | −1.20 | Adduct of F18 | ||
| F3 | 477.1420[M − H]¯ | 1.38 | 0.204 | MN3 | C23H26O11 | −3.91 | Plantainoside Bb | Plantainoside B (1) | 119.27 ± 14.03 |
| F4 | 377.0873[M − H]¯ | 0.24 | 0.183 | NS | C18H18O9 | 1.08 | 4,8-Dihydroxy-1,2,3,6,7-pentamethoxy-9H-xanthen-9-one | ||
| F5 | 315.1099[M − H]¯ | 0.60 | 0.166 | NS | C14H20O8 | −4.31 | 2-(3,5-Dihydroxyphenyl)ethanol-3'-O-β-d-glucopyranoside | 3,4-dihydroxyphenethyl glucoside (8) | >500 |
| F6 | 435.1313[M − H]¯ | 1.28 | 0.165 | MN4 | C21H24O10 | −3.74 | Monnieraside IIIa | Monnieraside III (2) | 101.84 ± 8.44 |
| F7 | 191.0197[M − H]¯ | 0.31 | 0.163 | MN5 | C6H8O7 | 0.14 | Idaric acid-1,4-lactone isomer | ||
| F8 | 955.2917[2M − H]− | 1.38 | 0.156 | MN3 | C23H26O11 | −4.91 | Dimer of F3 | ||
| F9 | 461.1473[M − H]¯ | 1.60 | 0.154 | MN1 | C23H26O10 | −4.29 | 8-O-(6’-O-trans-Coumaroyl-β-d-glucopyranosyl)-3,4-dihydroxyphenylethanol | ||
| F10 | 491.1580[M − H]¯ | 1.67 | 0.152 | MN2 | C24H28O11 | −4.51 | Monnieraside IIa | Monnieraside II (3) | 65.35 ± 0.63 |
| F11 | 1063.4467[M − H]¯ | 3.07 | 0.128 | NS | C49H76O23S | −3.92 | Unidentifiedc | ||
| F12 | 477.1420[M − H]¯ | 1.53 | 0.119 | MN3 | C23H26O11 | −3.91 | Plantainoside Bb | Monnieraside IVe (4) | 105.03 ± 2.65 |
| F13 | 219.0458[M − H]¯ | 0.23 | 0.118 | NS | C15H8O2 | −3.41 | Unidentifiedc | ||
| F14 | 461.1473[M − H]¯ | 1.71 | 0.115 | MN1 | C23H26O10 | −4.29 | 8-O-(6’-O-trans-Coumaroyl-β-d-glucopyranosyl)-3,4-dihydroxyphenylethanol | Monnieraside Ve (5) | 56.26 ± 1.88 |
| F15 | 491.1579[M − H]¯ | 1.78 | 0.113 | MN2 | C24H28O11 | −4.31 | Monnieraside IIa | Monnieraside VIe (6) | 80.07 ± 2.27 |
| F16 | 977.4458[M − H]¯ | 3.00 | 0.112 | NS | C46H74O20S | −3.75 | Bacopaside Ia | >1000f | |
| F17 | 299.0786[M − H]¯ | 0.78 | 0.110 | MN4 | C13H16O8 | −4.54 | 4-Hydroxybenzoyl glucoseb | 4-hydroxybenzoyl glucose (7) | >500 |
| F18 | 181.0717[M − H]¯ | 0.23 | 0.107 | NS | C6H14O6 | −0.21 | Mannitold | ||
| F19 | 631.2274[M − H]¯ | 0.60 | 0.104 | NS | C46H32O3 | 0.74 | Unidentifiedc | ||
| F20 | 497.1243[M + Cl]¯ | 1.58 | 0.033 | MN1 | C23H26O10 | −4.63 | Adduct of F9 | ||
| F21 | 461.0749[M − H]¯ | 1.56 | 0.003 | MN1 | C21H18O12 | −5.09 | 3',4',5,7-Tetrahydroxyflavone5-O-β-d-glucurono-pyranoside | ||
| F22 | 497.1243[M + Cl]¯ | 1.71 | 0.029 | MN1 | C23H26O10 | −4.63 | Adduct of F14 | ||
| F23 | 521.1685[M − H]¯ | 1.66 | 0.053 | MN2 | C25H30O12 | −3.93 | Aucubigenin-10-O-(4-hydroxy-3-methoxy-cinnamoyl), 1-O-β-D-glucopyranosideb | ||
| F24 | 983.3235[2M − H]¯ | 1.67 | 0.084 | MN2 | C24H28O11 | −4.53 | Dimer of F10 | ||
| F25 | 527.1347[M + Cl]¯ | 1.67 | 0.030 | MN2 | C24H28O11 | −4.05 | Adduct of F10 | ||
| F26 | 341.0893[M − H]¯ | 0.66 | 0.064 | MN3 | C15H18O9 | −4.37 | Chaenorrhinosideb | ||
| F27 | 477.1421[M − H]¯ | 1.62 | 0.043 | MN3 | C23H26O11 | −3.90 | Plantainoside Bb | ||
| F28 | 871.2706[2M − H]¯ | 1.29 | 0.077 | MN4 | C21H24O10 | −4.57 | Dimer of F6 | ||
| F29 | 471.1084[M + Cl]¯ | 1.29 | 0.033 | MN4 | C21H24O10 | −4.36 | Adduct of F6 | ||
| F30 | 599.1642[2M − H]¯ | 0.79 | 0.068 | MN4 | C13H16O8 | −4.07 | Dimer of F17 | ||
| F31 | 421.0046[M − H]¯ | 0.31 | 0.008 | MN5 | C17H10O13 | 0.63 | Unidentifiedc | ||
| F32 | 405.0308[M − H]¯ | 0.31 | 0.057 | MN5 | C14H14O14 | 0.69 | Unidentifiedc | ||
| F33 | 191.0198[M − H]¯ | 0.38 | 0.027 | MN5 | C6H8O7 | −0.38 | Idaric acid-1,4-lactone | ||
| Three isolated inactive compounds selected from inactive cluster (MN6) of the MN and in S-plot of MVA | |||||||||
| F34 | 727.2125[M − H]¯ | 1.20 | −0.026 | MN6 | C32H40O19 | −4.67 | Unidentifiedc | Parishin C (9) | >1000 |
| F35 | 995.3078[M − H]¯ | 1.40 | −0.037 | MN6 | C45H56O25 | −4.03 | Parishin Ad | Parishin A (10) | >1000 |
| F36 | 727.2123[M − H]¯ | 1.12 | −0.060 | MN6 | C32H40O19 | −4.40 | Unidentifiedc | Parishin B (11) | >1000 |
| Position | Monnieraside IV (4) | Monnieraside V (5) | Monnieraside VI (6) | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 131.5 | 131.3 | 131.2 | |||
| 2 | 117.0 | 6.61, d (2.1) | 116.8 | 6.62, d (2.1) | 116.6 | 6.61, d (2.1) |
| 3 | 146.0 | 145.8 | 145.8 | |||
| 4 | 144.6 | 144.4 | 144.3 | |||
| 5 | 116.3 | 6.61, d (8.1) | 116.1 | 6.61, d (8.1) | 116.2 | 6.60, d (8.1) |
| 6 | 121.4 | 6.49, dd (8.1, 2.1) | 121.1 | 6.50, dd (8.1, 2.1) | 121.1 | 6.48, dd (8.1, 2.1) |
| 7 | 36.5 | 2.68, t (7.2) | 36.3 | 2.68, m | 36.3 | 2.67, t (7.1) |
| 8 | 71.9 | 3.64, dt (9.8, 7.2) 4.00, dt (9.8, 7.2) | 71.5 | 3.63, dt (9.4, 7.3) 4.01, dt (9.4, 6.9) | 71.6 | 3.63, dt (9.8, 7.1) 4.00, dt (9.8, 7.1) |
| 1' | 102.3 | 4.45, d (8.1) | 102.0 | 4.44, d (8.1) | 102.1 | 4.46, d (8.0) |
| 2' | 74.9 | 4.79, dd (9.3, 8.1) | 74.6 | 4.79, dd (9.3, 8.1) | 74.7 | 4.79, dd (9.3, 8.0) |
| 3' | 76.2 | 3.51, t (9.3) | 75.9 | 3.50, t (9.3) | 76.0 | 3.52, t (9.3) |
| 4' | 71.8 | 3.38, t (9.3) | 71.5 | 3.38, t (9.3) | 71.5 | 3.38, t (9.3) |
| 5' | 78.1 | 3.29 (overlapped) | 77.9 | 3.28 (overlapped) | 77.9 | 3.30 (overlapped) |
| 6' | 62.6 | 3.69, dd (12.0, 5.7) 3.88, dd (12.0, 2.3) | 62.4 | 3.69, dd (11.9, 5.8) 3.88, dd (11.9, 1.8) | 62.4 | 3.69, dd (12.0, 5.8) 3.88, dd (12.0, 2.3) |
| 1" | 128.2 | 127.6 | 127.9 | |||
| 2" | 118.7 | 7.38, d (2.1) | 133.4 | 7.60, d (8.7) | 114.8 | 7.72, d (2.0) |
| 3" | 145.7 | 115.5 | 6.74, d (8.7) | 148.0 | ||
| 4" | 148.3 | 159.8 | 149.1 | |||
| 5" | 115.7 | 6.72, d (8.2) | 115.5 | 6.74, d (8.7) | 115.4 | 6.76, d (8.2) |
| 6" | 125.1 | 7.08, dd (8.2, 2.1) | 133.4 | 7.60, d (8.7) | 126.4 | 7.12, dd (8.2, 2.0) |
| 7" | 145.1 | 6.81, d (12.8) | 144.6 | 6.88 d (12.7) | 145.1 | 6.87, d (12.8) |
| 8" | 116.7 | 5.73, d (12.8) | 116.6 | 5.76, d (12.7) | 116.5 | 5.77, d (12.8) |
| 9" | 167.4 | 167.3 | 167.1 | |||
| OCH3 | 56.2 | 3.85, s | ||||
| No. | Code | Bacopa spp. | Sources |
|---|---|---|---|
| 1 | BM1 | B. monnieri | Perth, Australia |
| 2 | BM2 | B. monnieri | Wat Phra Sri Mahathat, Bangkok, Thailand |
| 3 | BM3 | B. monnieri | Samphan garden, Nakhon Pathom, Thailnd |
| 4 | BM4 | B. monnieri | Naresuan University, Phitsanulok, Thailand |
| 5 | BM5 | B. monnieri | Kasetsart University, Bangkok, Thailand |
| 6 | BM6 | B. monnieri | Nakhon Nayok, Thailand |
| 7 | BM7 | B. monnieri | Chatuchak Market, Bangkok, Thailand |
| 8 | BM8 | B. monnieri | Ayutthaya, Thailand |
| 9 | BM9 | B. monnieri | Fukuoka, Japan (originated in India) |
| 10 | BM10 | B. monnieri | Siriraj hospital, Bangkok, Thailand |
| 11 | BM11 | B. monnieri | Chatuchak Market, Bangkok, Thailand |
| 12 | BM12 | B. monnieri | Phetchabun, Thailand (originated in India) |
| 13 | BC1 | B. caroliniana | Naresuan University, Phitsanulok, Thailand |
| 14 | BC2 | B. caroliniana | Nakhon Nayok, Thailand |
| 15 | BC3 | B. caroliniana | Chiang Mai, Thailand |
| 16 | BC4 | B. caroliniana | Bangkok, Thailand |
| 17 | BF1 | B. floribunda | Sakolnakorn, Thailand |
| 18–27 | BF2 to BF11 | B. floribunda | Plant tissue cultures obtained from Department of Biology, Faculty of Science, Naresuan University |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saesong, T.; Allard, P.-M.; Ferreira Queiroz, E.; Marcourt, L.; Nuengchamnong, N.; Temkitthawon, P.; Khorana, N.; Wolfender, J.-L.; Ingkaninan, K. Discovery of Lipid Peroxidation Inhibitors from Bacopa Species Prioritized through Multivariate Data Analysis and Multi-Informative Molecular Networking. Molecules 2019, 24, 2989. https://doi.org/10.3390/molecules24162989
Saesong T, Allard P-M, Ferreira Queiroz E, Marcourt L, Nuengchamnong N, Temkitthawon P, Khorana N, Wolfender J-L, Ingkaninan K. Discovery of Lipid Peroxidation Inhibitors from Bacopa Species Prioritized through Multivariate Data Analysis and Multi-Informative Molecular Networking. Molecules. 2019; 24(16):2989. https://doi.org/10.3390/molecules24162989
Chicago/Turabian StyleSaesong, Tongchai, Pierre-Marie Allard, Emerson Ferreira Queiroz, Laurence Marcourt, Nitra Nuengchamnong, Prapapan Temkitthawon, Nantaka Khorana, Jean-Luc Wolfender, and Kornkanok Ingkaninan. 2019. "Discovery of Lipid Peroxidation Inhibitors from Bacopa Species Prioritized through Multivariate Data Analysis and Multi-Informative Molecular Networking" Molecules 24, no. 16: 2989. https://doi.org/10.3390/molecules24162989