N1-Propargylguanosine Modified mRNA Cap Analogs: Synthesis, Reactivity, and Applications to the Study of Cap-Binding Proteins

 , , and
, , and
Abstract
:
1. Introduction
2. Results
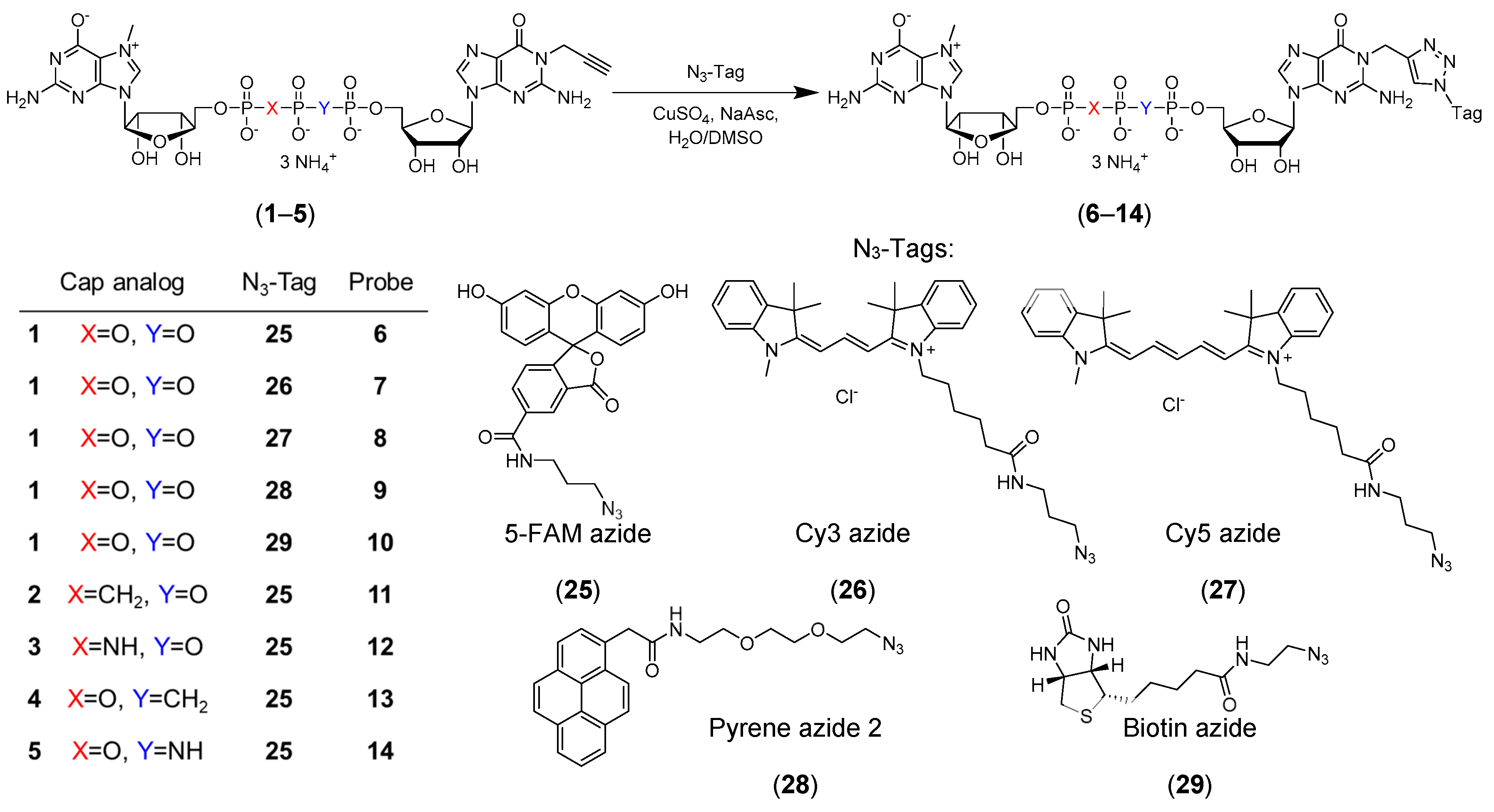
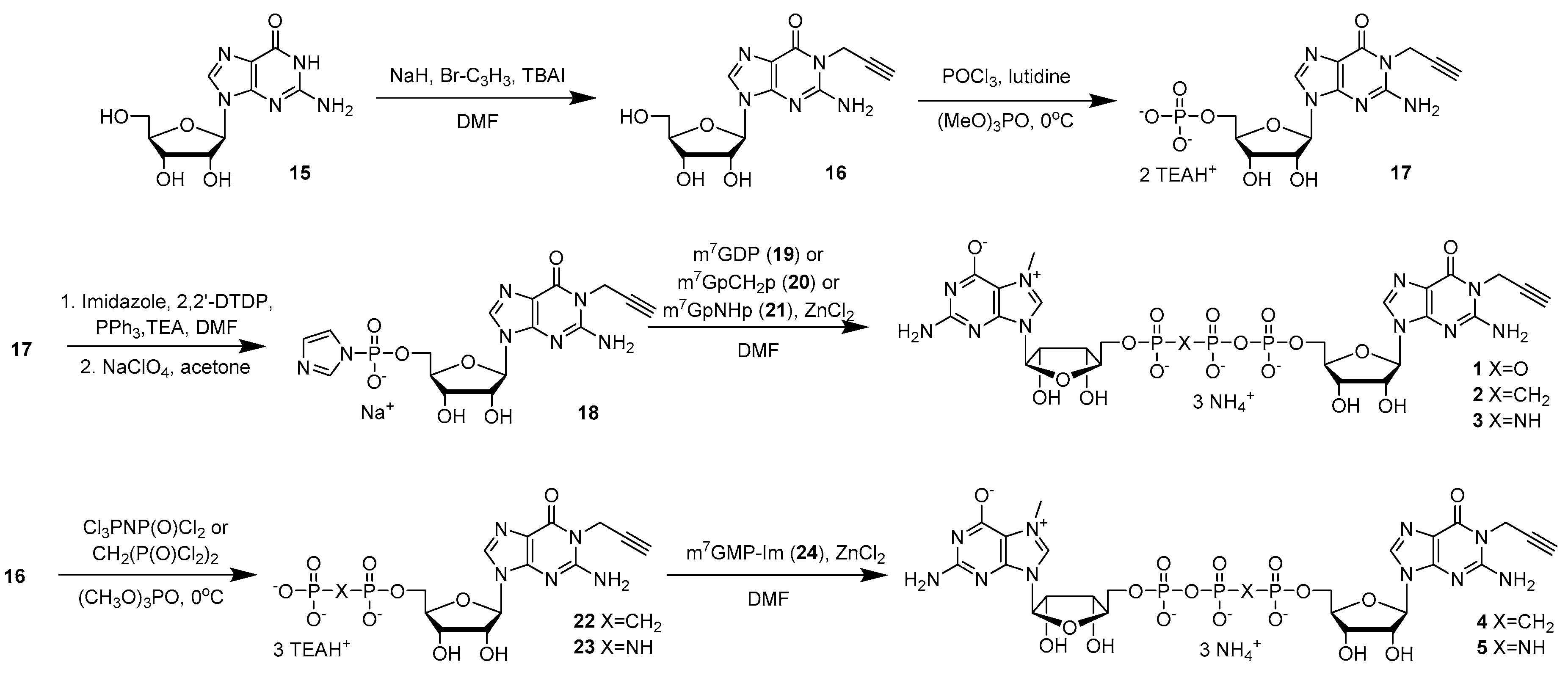
2.1. Cap Analogs Synthesis
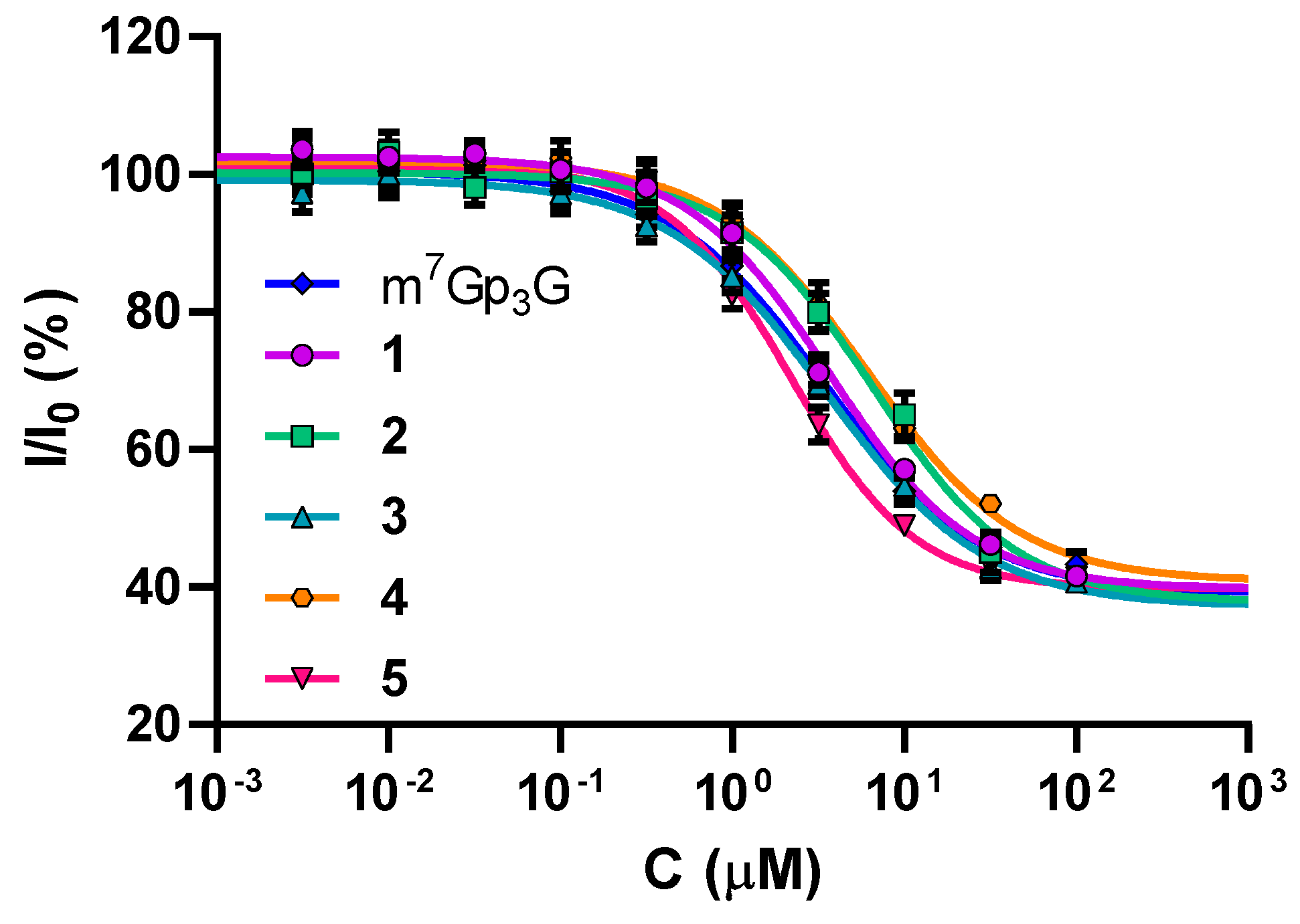
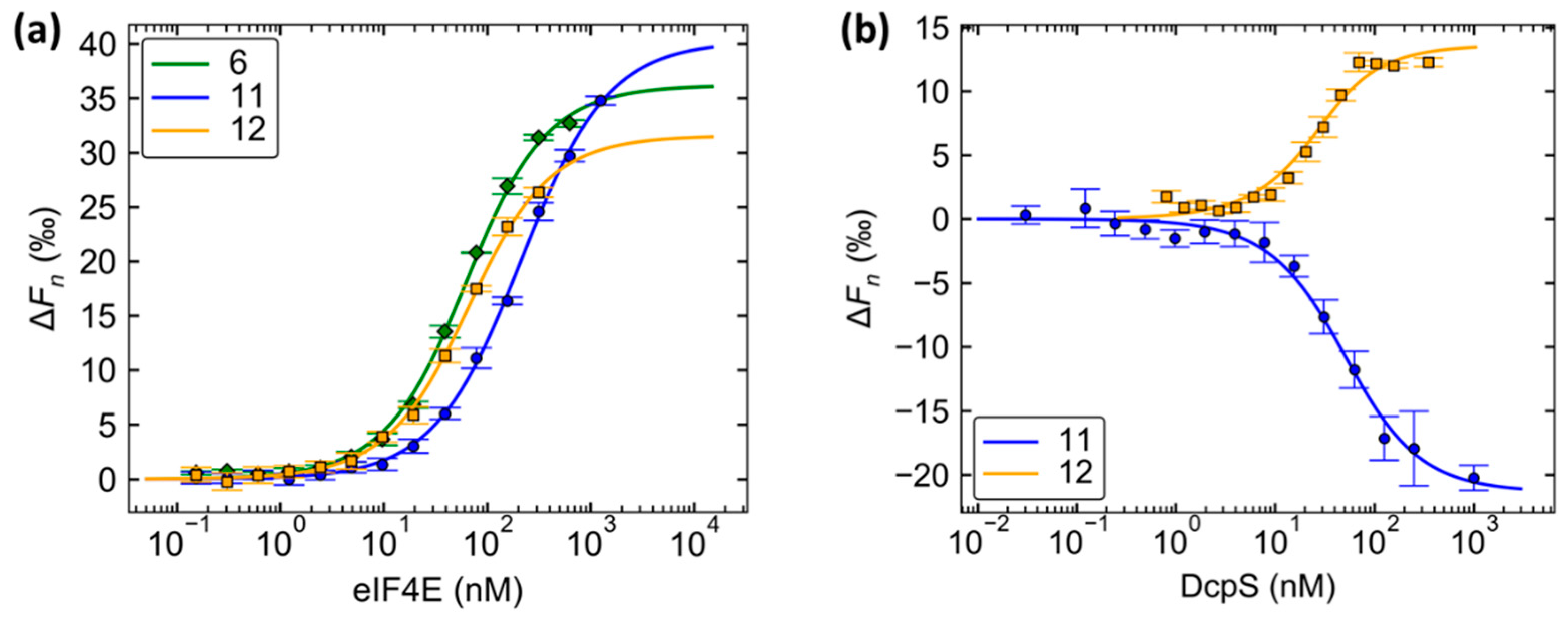
2.2. Binding Affinities to eIF4E
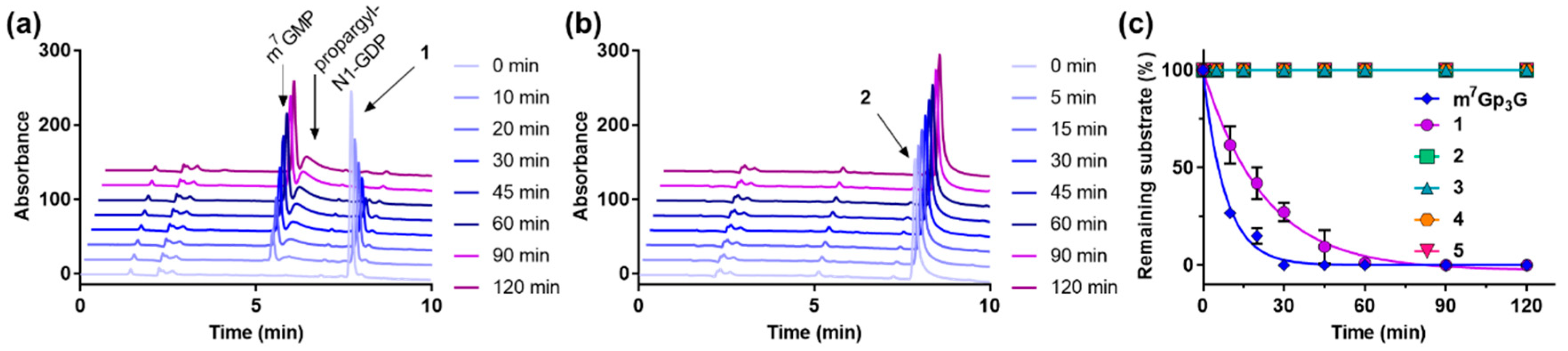
2.3. Enzymatic Degradation Studies
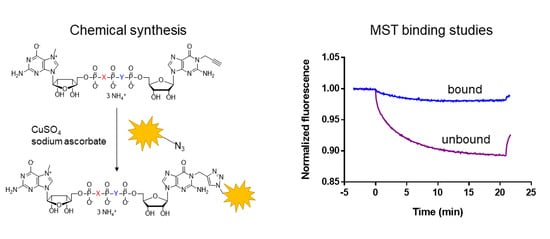
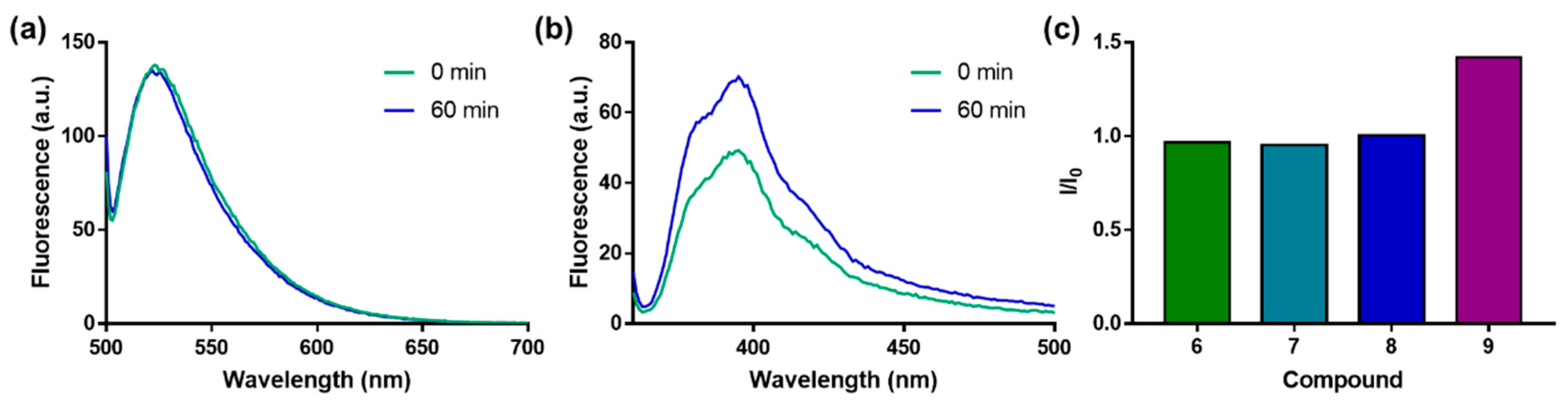
2.4. Evaluation of N1 Fluorescently Labeled Cap Analogs as Probes for Microscale Thermophoresis
3. Discussion
4. Materials and Methods
4.1. Starting Materials and Chemical Reagents
4.2. Chromatography and Optical Density Measurements
4.3. Analytical and Preparative Reversed-Phase (RP) HPLC
- Method A–eluant A was 0.05 M ammonium acetate buffer (pH 5.9); eluant B was a 1:1 (v/v) eluant A:MeOH mixture; eluant B gradient was 0–100% over 15 min; column was Supelcosil LC-18-T;
- Method B–eluant A was 0.05 M ammonium acetate buffer (pH 5.9); eluant B was 1:1 (v/v) eluant A:MeOH; eluant B gradient was 0–50% over 15 min; column was Supelcosil LC-18-T;
- Method C–eluant A was 0.05 M ammonium acetate buffer (pH 5.9); eluant B was acetonitrile; eluant B gradient was 0–100% over 15 min; column was Supelcosil LC-8;
- Method D–eluant A was 0.05 M ammonium acetate buffer (pH 5.9); eluant B is acetonitrile; eluant B gradient was 0–50% over 15 min; column was Supelcosil LC-8;
- Method E–eluant A was 0.1 M phosphate buffer (pH 6.0); eluant B was 1:1 (v/v) eluant A:MeOH; eluant B gradient was 0–50% over 15 min; column was Supelcosil LC-18-T.
4.4. NMR and MS Analyses
4.5. Chemical Syntheses
4.5.1. P1-(N1-Propargylguanosin-5′-yl) P3-(N7-methylguanosin-5′-yl) triphosphate (m7Gp3G-N1-propargyl) (1)
4.5.2. P1-(N7-Methylguanosin-5′-yl) P3-(N1-propargylguanosin-5′-yl) 1,2-methylenetriphosphate (m7GpCH2ppG-N1-propargyl) (2)
4.5.3. P1-(N7-Methylguanosin-5′-yl) P3-(N1-propargylguanosin-5′-yl) 1,2-imidotriphosphate (m7GpNHppG-N1-propargyl) (3)
4.5.4. P1-(N7-Methylguanosin-5′-yl) P3-(N1-propargylguanosin-5′-yl) 2,3-methylenetriphosphate (m7GppCH2pG-N1-propargyl) (4)
4.5.5. P1-(N7-Methylguanosin-5′-yl) P3-(N1-propargylguanosin-5′-yl) 2,3-imidotriphosphate (m7GppNHpG-N1-propargyl) (5)
4.5.6. Probe 6 (m7Gp3G-N1-5FAM)
4.5.7. Probe 7 (m7Gp3G-N1-Cy3)
4.5.8. Probe 8 (m7Gp3G-N1-Cy5)
4.5.9. Probe 9 (m7Gp3G-N1-Py)
4.5.10. Probe 10 (m7Gp3G-N1-biotin)
4.5.11. Probe 11 (m7GpCH2ppG-N1-5FAM)
4.5.12. Probe 12 (m7GpNHppG-N1-5FAM)
4.5.13. Probe 13 (m7GppCH2pG-N1-5FAM)
4.5.14. Probe 14 (m7GppNHpG-N1-5FAM)
4.5.15. N1-propargylguanosine (16)
4.5.16. N1-Propargylguanosine-5′-O-monophosphate (propargyl-N1-GMP) (17)
4.5.17. N1-Propargylguanosine 5′-O-monophosphate P-imidazolide (propargyl-N1-GMP-Im) (18)
4.5.18. N1-Propargylguanosine 5′-O-(1,2-methylenediphosphate) (propargyl-N1-GpCH2p) (22)
4.5.19. N1-Propargylguanosine 5′-O-(1,2-imidodiphosphate) (propargyl-N1-GpNHp) (23)
4.6. Determination of the eIF4E–Cap Complex Dissociation Constants KD
4.7. Susceptibility to DcpS Hydrolysis
4.8. UV-Vis and Fluorescence Measurements
4.9. hDcpS and SVPDE Hydrolysis Monitoring
4.10. Microscale Thermophoresis Direct Binding Affinity for meIF4E and hDcps
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rottman, F.; Shatkin, A.J.; Perry, R.P.; Perry, R.N.A. Methylated Nucleotides at the 5’ Termini of Messenger RNAs : Possible Implications for Processing. Cell 1974, 3, 197–199. [Google Scholar] [CrossRef]
- Quiocho, F.A.; Hu, G.; Gershon, P.D. Structural basis of mRNA cap recognition by proteins. Curr. Opin. Struct. Biol. 2000, 10, 78–86. [Google Scholar] [CrossRef]
- Niedzwiecka, A.; Marcotrigiano, J.; Stepinski, J.; Jankowska-Anyszka, M.; Wyslouch-Cieszynska, A.; Dadlez, M.; Gingras, A.-C.; Mak, P.; Darzynkiewicz, E.; Sonenberg, N.; et al. Biophysical studies of eIF4E cap-binding protein: Recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J. Mol. Biol. 2002, 319, 615–635. [Google Scholar] [CrossRef]
- Zuberek, J.; Wyslouch-Cieszynska, A.; Niedzwiecka, A.; Dadlez, M.; Stepinski, J.; Augustyniak, W.; Gingras, A.-C.; Zhang, Z.; Burley, S.K.; Sonenberg, N.; et al. Phosphorylation of eIF4E attenuates its interaction with mRNA 5 cap analogs by electrostatic repulsion: Intein-mediated protein ligation strategy to obtain phosphorylated protein. RNA 2003, 9, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, Y.; Sugiyama, A.; Asai, A.; Okazaki, T.; Kuchino, Y.; Kerr, S.J. Elevated levels of eukaryotic translation initiation factor eIF-4E, mRNA in a broad spectrum of transformed cell lines. Cancer Lett. 1995, 91, 247–252. [Google Scholar] [CrossRef]
- De Benedetti, A.; Harris, A.L. eIF4E expression in tumors: Its possible role in progression of malignancies. Int. J. Biochem. Cell Biol. 1999, 31, 59–72. [Google Scholar] [CrossRef]
- Ghosh, B.; Benyumov, A.; Ghosh, P. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS Chem. Biol. 2009, 4, 367–377. [Google Scholar] [CrossRef]
- Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Ziemniak, M.; Darzynkiewicz, E.; Jemielity, J. Phosphorothioate analogs of m7GTP are enzymatically stable inhibitors of cap-dependent translation. Bioorg. Med. Chem. Lett. 2009, 19, 1921–1925. [Google Scholar] [CrossRef]
- Karaki, S.; Andrieu, C.; Ziouziou, H.; Rocchi, P. The Eukaryotic Translation Initiation Factor 4E (eIF4E) as a Therapeutic Target for Cancer. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Elsevier Inc.: San Diego, CA, USA, 2015; Volume 101, pp. 1–26. [Google Scholar]
- Singh, J.; Salcius, M.; Liu, S.-W.; Staker, B.L.; Mishra, R.; Thurmond, J.; Michaud, G.; Mattoon, D.R.; Printen, J.; Christensen, J.; et al. DcpS as a Therapeutic Target for Spinal Muscular Atrophy. ACS Chem. Biol. 2008, 3, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balintová, J.; Welter, M.; Marx, A. Antibody–nucleotide conjugate as a substrate for DNA polymerases. Chem. Sci. 2018, 9, 7122–7125. [Google Scholar] [CrossRef] [PubMed]
- Hardt, N.; Hacker, S.M.; Marx, A. Synthesis and fluorescence characteristics of ATP-based FRET probes. Org. Biomol. Chem. 2013, 11, 8298–8305. [Google Scholar] [CrossRef]
- Wanat, P.; Walczak, S.; Wojtczak, B.A.; Nowakowska, M.; Jemielity, J.; Kowalska, J. Ethynyl, 2-Propynyl, and 3-Butynyl C-Phosphonate Analogues of Nucleoside Di- and Triphosphates: Synthesis and Reactivity in CuAAC. Org. Lett. 2015, 17, 3062–3065. [Google Scholar] [CrossRef]
- Wanat, P.; Kasprzyk, R.; Kopcial, M.; Sikorski, P.J.; Strzelecka, D.; Jemielity, J.; Kowalska, J. ExciTides: NTP-derived probes for monitoring pyrophosphatase activity based on excimer-to-monomer transitions. Chem. Commun. 2018, 54, 9773–9776. [Google Scholar] [CrossRef] [PubMed]
- Kasprzyk, R.; Starek, B.J.; Ciechanowicz, S.; Kubacka, D.; Kowalska, J.; Jemielity, J. Fluorescent turn-on probes for the development of binding and hydrolytic activity assays for mRNA cap-recognising proteins. Chem. Eur. J. 2019, 25, 6728–6740. [Google Scholar] [CrossRef] [PubMed]
- Hacker, S.M.; Hintze, C.; Marx, A.; Drescher, M. Monitoring enzymatic ATP hydrolysis by EPR spectroscopy. Chem. Commun. 2014, 50, 7262–7264. [Google Scholar] [CrossRef] [Green Version]
- Junker, A.; Renn, C.; Dobelmann, C.; Namasivayam, V.; Jain, S.; Losenkova, K.; Irjala, H.; Duca, S.; Balasubramanian, R.; Chakraborty, S.; et al. Structure−Activity Relationship of Purine and Pyrimidine Nucleotides as Ecto-5′-Nucleotidase (CD73) Inhibitors. J. Med. Chem. 2019, 62, 3677–3695. [Google Scholar] [CrossRef]
- Walczak, S.; Nowicka, A.; Kubacka, D.; Fac, K.; Wanat, P.; Mroczek, S.; Kowalska, J.; Jemielity, J. A novel route for preparing 5′ cap mimics and capped RNAs: Phosphate-modified cap analogues obtained via click chemistry. Chem. Sci. 2017, 8, 260–267. [Google Scholar] [CrossRef]
- Mamot, A.; Sikorski, P.J.; Warminski, M.; Kowalska, J.; Jemielity, J. Azido-Functionalized 5′ Cap Analogues for the Preparation of Translationally Active mRNAs Suitable for Fluorescent Labeling in Living Cells. Angew. Chem. Int. Ed. 2017, 56, 15628–15632. [Google Scholar] [CrossRef]
- Kozarski, M.; Kubacka, D.; Wojtczak, B.A.; Kasprzyk, R.; Baranowski, M.R.; Kowalska, J. 7-Methylguanosine monophosphate analogues with 5′-(1,2,3-triazoyl) moiety: Synthesis and evaluation as the inhibitors of cNIIIB nucleotidase. Bioorg. Med. Chem. 2018, 26, 191–199. [Google Scholar] [CrossRef]
- Holstein, J.M.; Muttach, F.; Schiefelbein, S.H.H.; Rentmeister, A. Dual 5′ Cap Labeling Based on Regioselective RNA Methyltransferases and Bioorthogonal Reactions. Chem. Eur. J. 2017, 23, 6165–6173. [Google Scholar] [CrossRef]
- Schulz, D.; Holstein, J.M.; Rentmeister, A. A chemo-enzymatic approach for site-specific modification of the RNA cap. Angew. Chem. Int. Ed. 2013, 52, 7874–7878. [Google Scholar] [CrossRef] [PubMed]
- Holstein, J.M.; Schulz, D.; Rentmeister, A. Bioorthogonal site-specific labeling of the 5′-cap structure in eukaryotic mRNAs. Chem. Commun. 2014, 50, 4478–4481. [Google Scholar] [CrossRef] [PubMed]
- Piecyk, K.; Jankowska-Anyszka, M. Chemical conjugation of an mRNA cap analogue with a cell-penetrating peptide as a potential membrane permeable translation inhibitor. Tetrahedron Lett. 2014, 55, 606–609. [Google Scholar] [CrossRef]
- Piecyk, K.; Lukaszewicz, M.; Darzynkiewicz, E.; Jankowska-Anyszka, M. Triazole-containing monophosphate mRNA cap analogs as effective translation inhibitors. RNA 2014, 20, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Hienzsch, A.; Deiml, C.; Reiter, V.; Carell, T. Total synthesis of the hypermodified RNA bases wybutosine and hydroxywybutosine and their quantification together with other modified RNA bases in plant materials. Chem. Eur. J. 2013, 19, 4244–4248. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. A novel method for phosphorylation of nucleosides to 5′-nucleotides. Tetrahedron Lett. 1967, 8, 5065–5068. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Hashimoto, M. Synthesis of oligothymidylates and nucleoside cyclic phosphates by oxidation - reduction condensation. J. Am. Chem. Soc. 1972, 94, 8528–8532. [Google Scholar] [CrossRef]
- Kalek, M.; Jemielity, J.; Grudzien, E.; Zuberek, J.; Bojarska, E.; Cohen, L.S.; Stepinski, J.; Stolarski, R.; Davis, R.E.; Rhoads, R.E.; et al. Synthesis and biochemical properties of novel mRNA 5′ cap analogs resistant to anzymatic hydrolysis. Nucleos. Nucleot. Nucl. 2005, 24, 615–621. [Google Scholar] [CrossRef]
- Kalek, M.; Jemielity, J.; Darzynkiewicz, Z.M.; Bojarska, E.; Stepinski, J.; Stolarski, R.; Davis, R.E.; Darzynkiewicz, E. Enzymatically sTable 5’ mRNA cap analogs: Synthesis and binding studies with human DcpS decapping enzyme. Bioorg. Med. Chem. 2006, 14, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Rydzik, A.M.; Kulis, M.; Lukaszewicz, M.; Kowalska, J.; Zuberek, J.; Darzynkiewicz, Z.M.; Darzynkiewicz, E.; Jemielity, J. Synthesis and properties of mRNA cap analogs containing imidodiphosphate moiety--fairly mimicking natural cap structure, yet resistant to enzymatic hydrolysis. Bioorg. Med. Chem. 2012, 20, 1699–1710. [Google Scholar] [CrossRef]
- Zuberek, J.; Jemielity, J.; Jablonowska, A.; Stepinski, J.; Dadlez, M.; Stolarski, R.; Darzynkiewicz, E. Influence of electric charge variation at residues 209 and 159 on the interaction of eIF4E with the mRNA 5′ terminus. Biochemistry 2004, 43, 5370–5379. [Google Scholar] [CrossRef]
- Tomasz, J.; Vaghefi, M.M.; Ratsep, P.C.; Willis, R.C.; Robins, R.K.; Vaghefi, M.M.; Ratsep, P.C.; Willis, R.C.; Robins, R.K.; Vaghefi, M.M.; et al. Nucleoside imidodiphospbates synthesis and biological activities. Nucleic Acids Res. 1988, 16, 8645–8664. [Google Scholar] [CrossRef] [Green Version]
- Nikolovska-Coleska, Z.; Wang, R.; Fang, X.; Pan, H.; Tomita, Y.; Li, P.; Roller, P.P.; Krajewski, K.; Saito, N.G.; Stuckey, J.A.; et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004, 332, 261–273. [Google Scholar] [CrossRef]
- Tso, S.-C.; Chen, Q.; Vishnivetskiy, S.A.; Gurevich, V.V.; Iverson, T.M.; Brautigam, C.A. Using two-site binding models to analyze microscale thermophoresis data. Anal. Biochem. 2018, 540–541, 64–75. [Google Scholar] [CrossRef]
- Brautigam, C.A. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 2015, 562, 109–133. [Google Scholar]
Sample Availability: Samples of the compounds 1–14 are available from the authors on request. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | KD (nM) |
|---|---|---|
| 1 | 3.57 ± 0.25 | 305 ± 45 |
| 2 | 6.37 ± 0.73 | 548 ± 95 |
| 3 | 3.48 ± 0.22 | 297 ± 43 |
| 4 | 6.04 ± 0.46 | 520 ± 78 |
| 5 | 2.16 ± 0.20 | 182 ± 29 |
| m7Gp3G | 3.46 ± 0.30 | 294 ± 46 |
| Compound | meIF4E | hDcpS | ||
|---|---|---|---|---|
| KD [nM] | Confidence Interval | KD [nM] | Confidence Interval | |
| 6 | 50 | (43, 58) | n.d. | n.d. |
| 11 | 201 | (184, 220) | 38 | (28, 51) |
| 12 | 53 | (46, 62) | 14 | (7, 24) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopcial, M.; Wojtczak, B.A.; Kasprzyk, R.; Kowalska, J.; Jemielity, J. N1-Propargylguanosine Modified mRNA Cap Analogs: Synthesis, Reactivity, and Applications to the Study of Cap-Binding Proteins. Molecules 2019, 24, 1899. https://doi.org/10.3390/molecules24101899
Kopcial M, Wojtczak BA, Kasprzyk R, Kowalska J, Jemielity J. N1-Propargylguanosine Modified mRNA Cap Analogs: Synthesis, Reactivity, and Applications to the Study of Cap-Binding Proteins. Molecules. 2019; 24(10):1899. https://doi.org/10.3390/molecules24101899
Chicago/Turabian StyleKopcial, Michal, Blazej A. Wojtczak, Renata Kasprzyk, Joanna Kowalska, and Jacek Jemielity. 2019. "N1-Propargylguanosine Modified mRNA Cap Analogs: Synthesis, Reactivity, and Applications to the Study of Cap-Binding Proteins" Molecules 24, no. 10: 1899. https://doi.org/10.3390/molecules24101899