Photochemical and Structural Studies on Cyclic Peptide Models

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
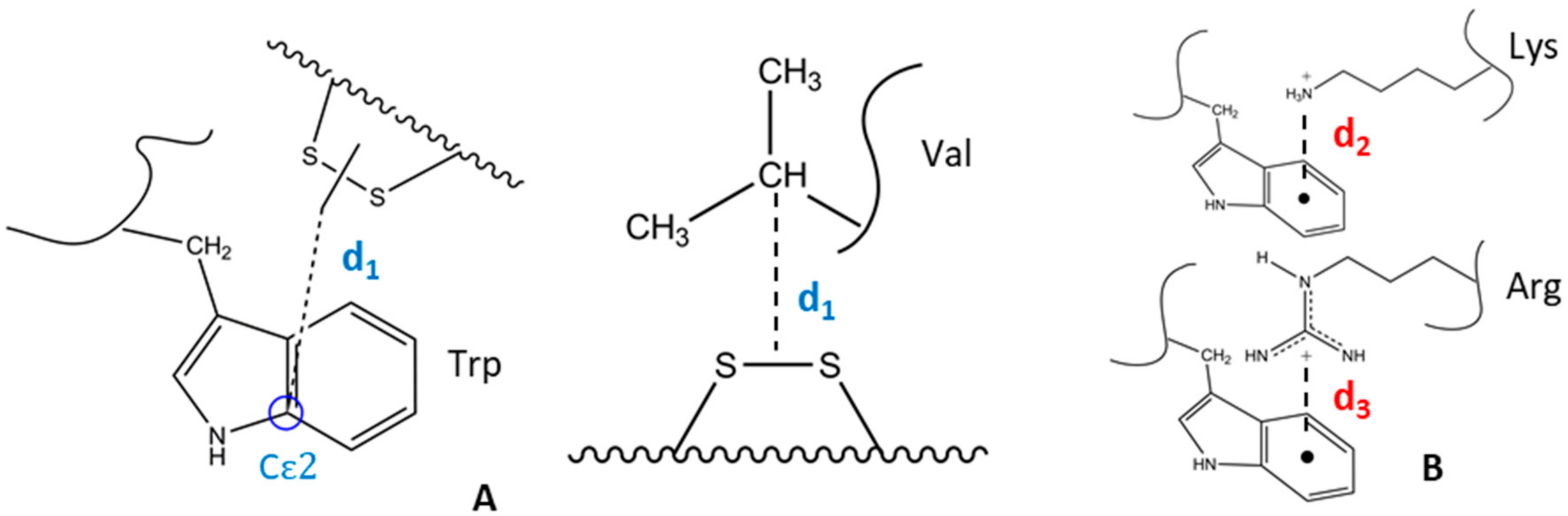
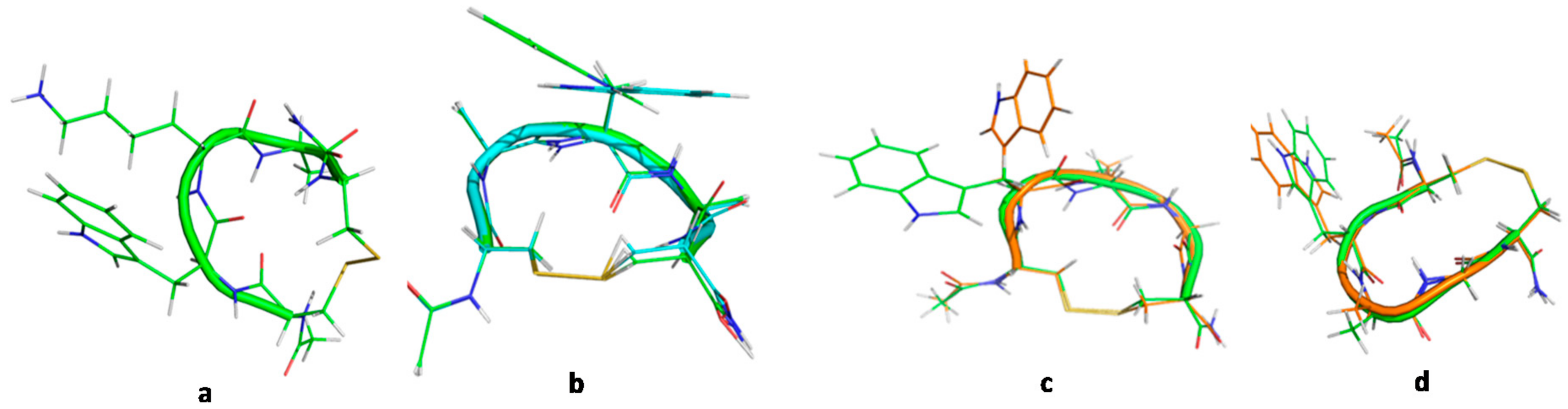
2.1. Design of Cyclic Peptide Models and Molecular Mechanics Calculations
2.2. Determination of Key Distances by NMR and MD Analyses
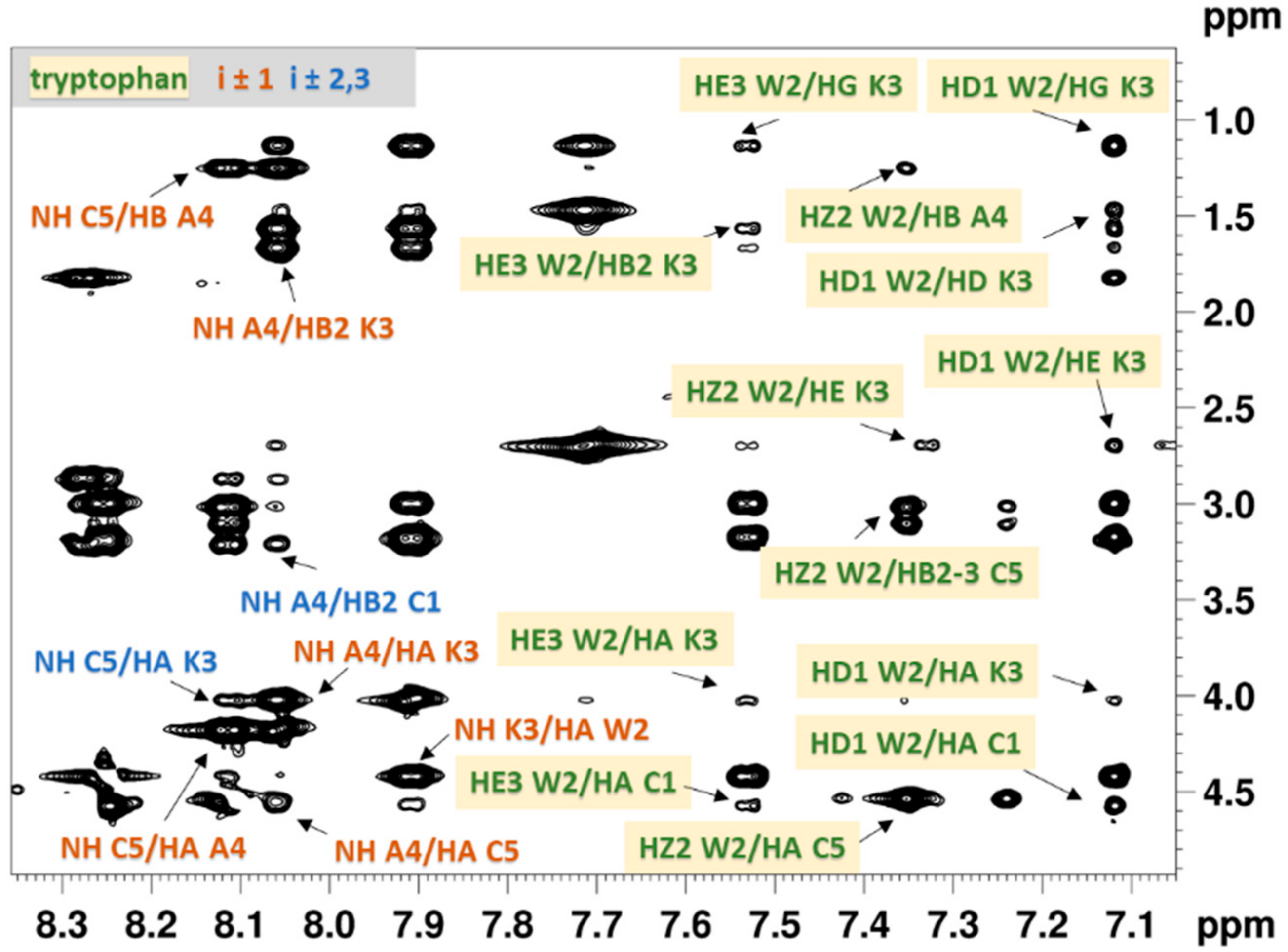
2.2.1. NMR Analysis
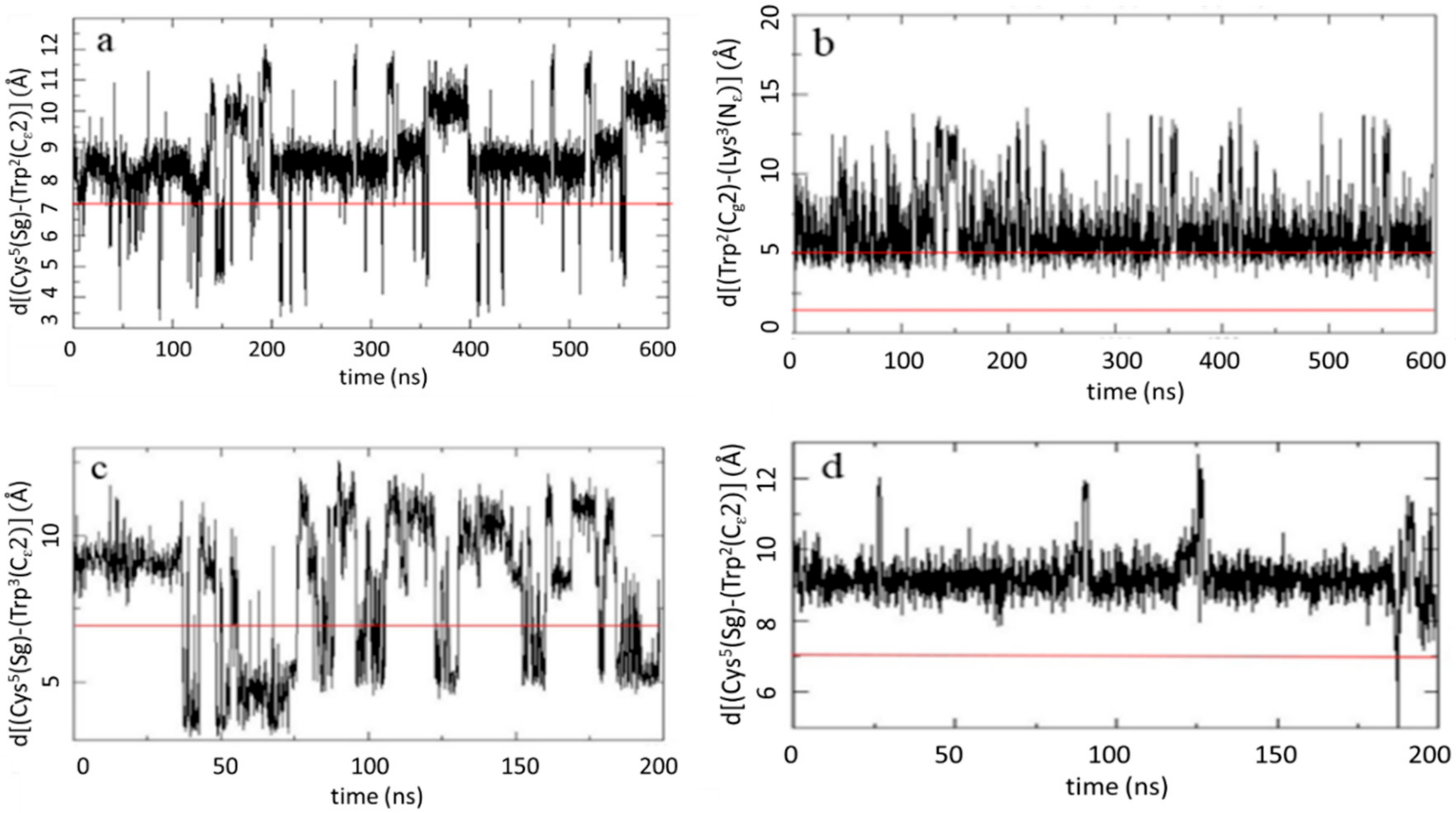
2.2.2. MD Analysis of Key Interatomic Distances
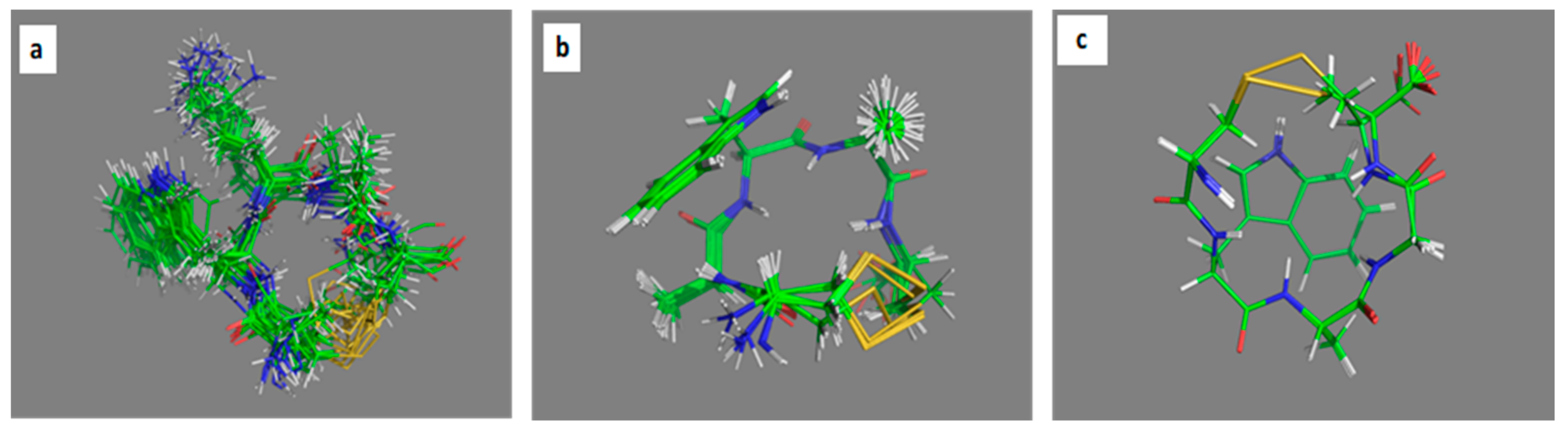
2.3. Comparative MD Analysis of Conformational Ensembles in Dimethyl Sulfoxide and Water
2.4. Analysis of Hydrogen Bonds
2.5. NMR and MD Analysis of Trp Side-Chain Conformation
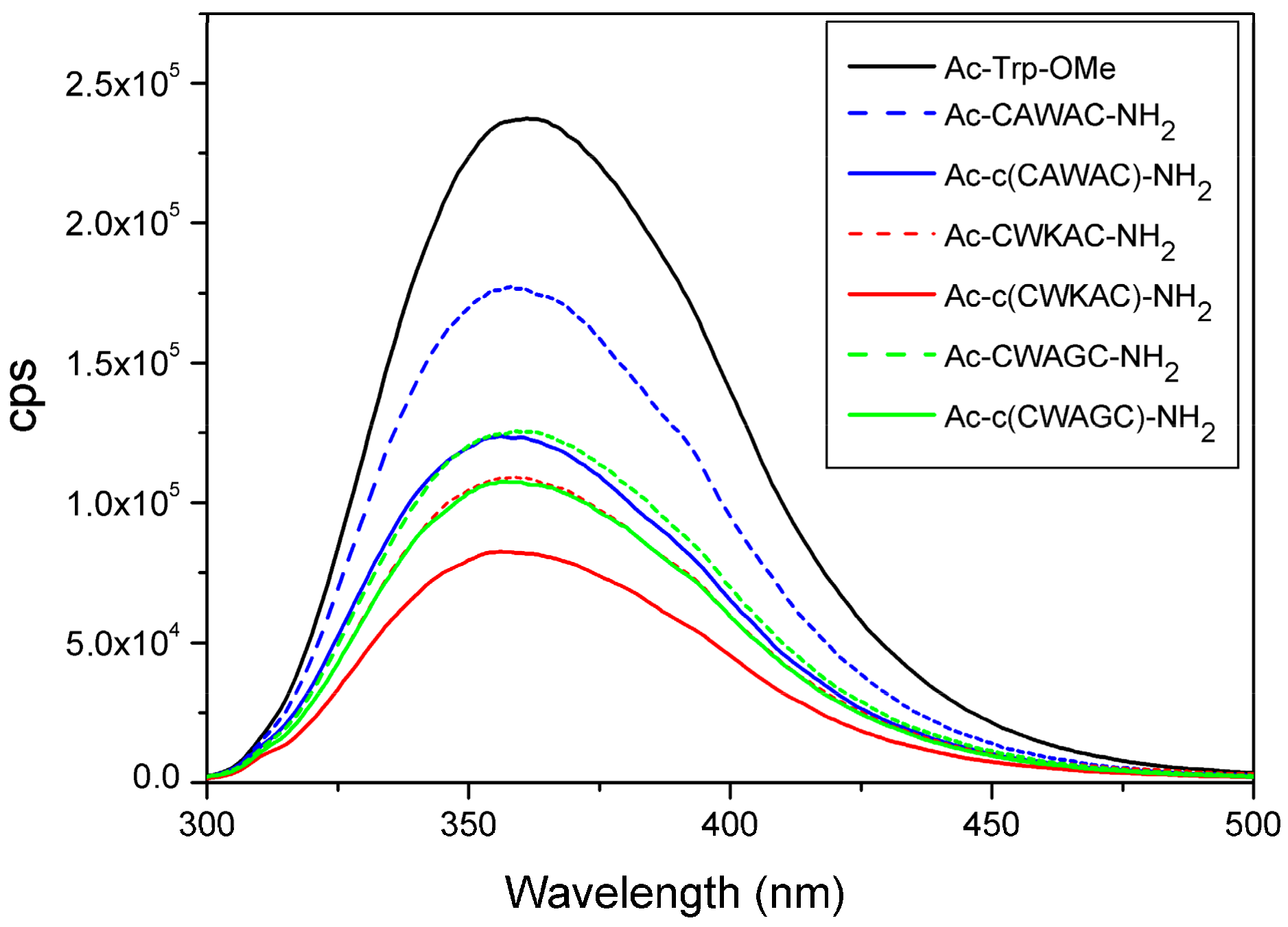
2.6. Quenching Effect of Disulfide Bridges in Peptide Models
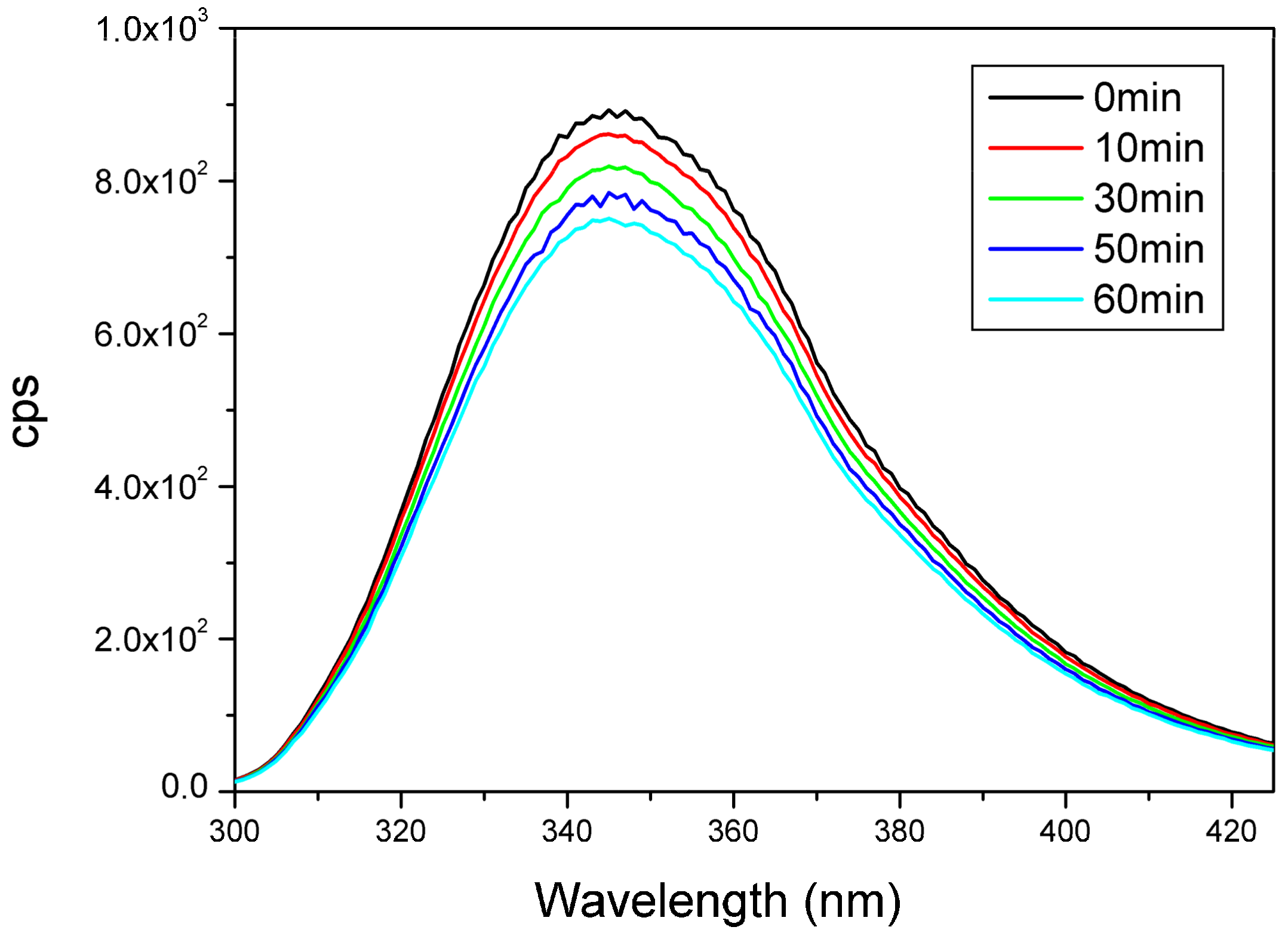
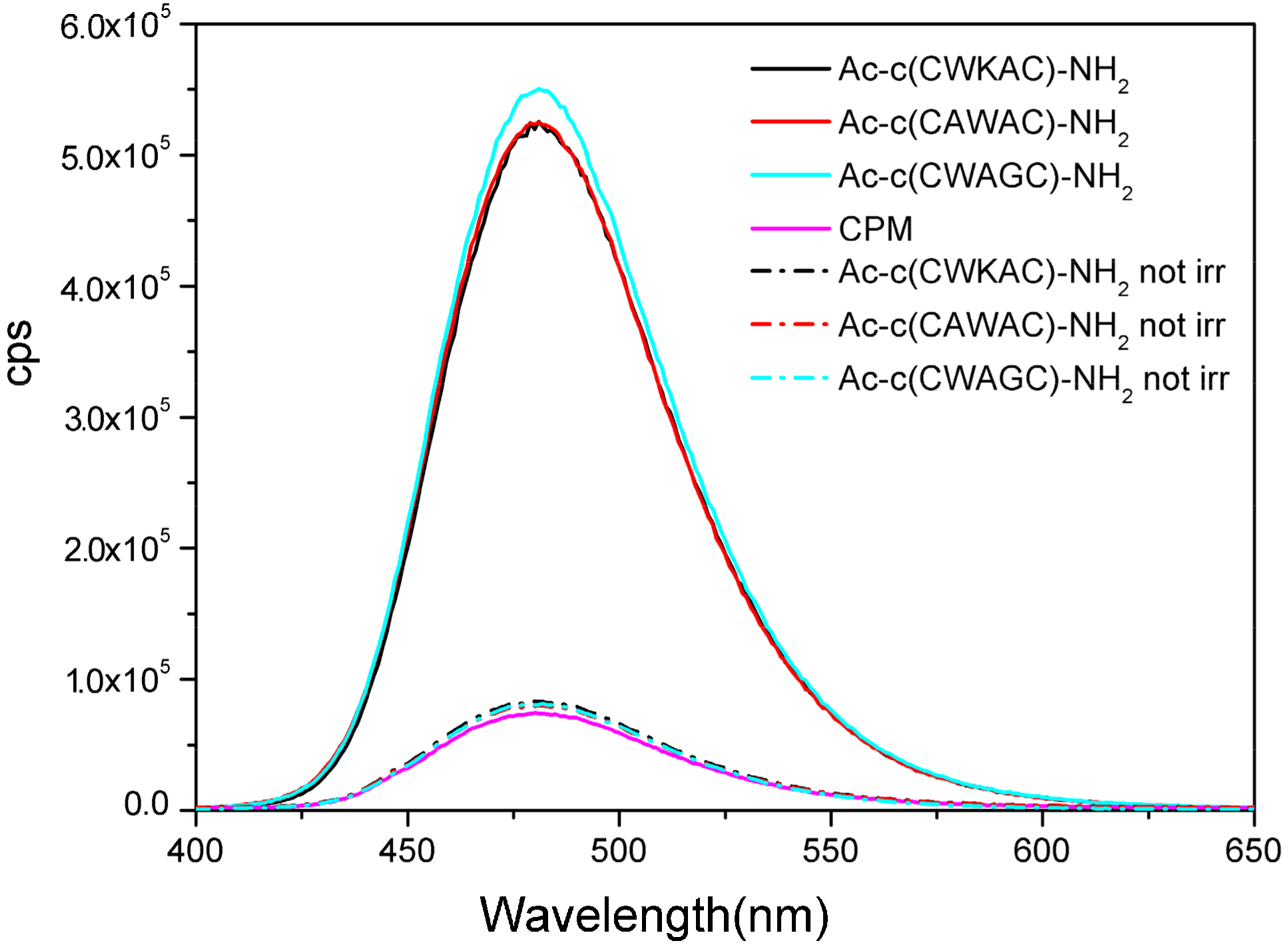
2.7. UV Irradiation of Peptides Monitored by Measuring the Amount of Evolving Thiol Groups with Fluorescence Spectroscopy
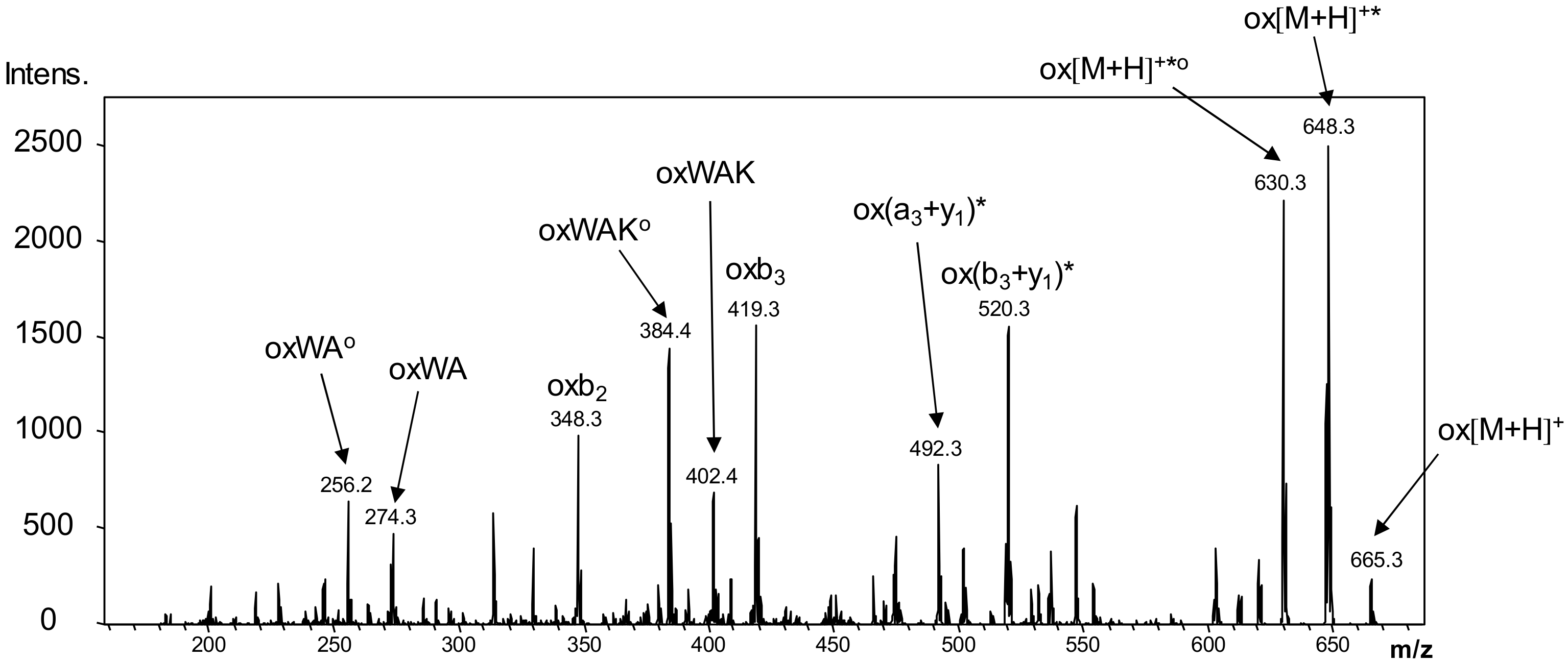
2.8. LC-MS Measurements
3. Experimental
3.1. Materials
3.2. High-Performance Liquid Cromatography (HPLC)
3.3. LC-MS
3.4. LC-MS/MS
3.5. MM Calculation
3.6. NMR Spectroscopy and Structure Calculation
3.6.1. Molecular Dynamics Simulations
3.6.2. MD Analysis
3.7. Synthesis of Model Peptides
Cyclization of Linear Peptide Precursors through Disulfide Bond in Solution
3.8. Fluorescence Spectroscopic Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snabe, T.; Roder, G.A.; Neves-Petersen, M.T.; Buus, S.; Petersen, S.B. Oriented coupling of major histocompatibility complex (MHC) to sensor surfaces using light assisted immobilisation technology. Biosens. Bioelectron. 2006, 21, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Neves-Petersen, M.T.; Snabe, T.; Klitgaard, S.; Duroux, M.; Petersen, S.B. Photonic activation of disulfide bridges achieves oriented protein immobilization on biosensor surfaces. Protein Sci. 2006, 15, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, B.B.; Neves-Petersen, M.T.; Klitgaard, S.; Issinger, O.G.; Petersen, S.B. UV light blocks EGFR signalling in human cancer cell lines. Int. J. Oncol. 2007, 30, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Duroux, M.; Skovsen, E.; Neves-Petersen, M.T.; Duroux, L.; Gurevich, L.; Borrebaeck, C.A.K.; Wingren, C.; Petersen, S.B. Light-induced immobilisation of biomolecules as an attractive alternative to micro-droplet dispensing-based arraying technologies. Proteomics 2008, 8, 1113. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Basu, S.; Mandal, P.C. UV radiation effects on flavocytochrome b2 in dilute aqueous solution. J. Photochem. Photobiol. B 2000, 59, 54–63. [Google Scholar] [CrossRef]
- Vanhooren, A.; Devreese, B.; Vanhee, K.; Van Beeumen, J.; Hanssens, I. Photoexcitation of tryptophan groups induces reduction of two disulfide bonds in goat α-lactalbumin. Biochemistry 2002, 41, 11035–11043. [Google Scholar] [CrossRef] [PubMed]
- Creed, D. The photophysics and photochemistry of the near-UV absorbing amino acids–I. Tryptophan and its simple derivatives. Photochem. Photobiol. 1984, 39, 537–562. [Google Scholar] [CrossRef]
- Asmus, K.D.; Bahnemann, D.; Bonifačić, M.; Gillis, H.A. Free radical oxidation of organic sulphur compounds in aqueous solution. Faraday Discuss. 1977, 63, 213–225. [Google Scholar] [CrossRef]
- Hoffman, M.Z.; Hayon, E. One-electron reduction of the disulfide linkage in aqueous solution. Formation, protonation, and decay kinetics of the RSSR-radical. J. Am. Chem. Soc. 1972, 94, 7950–7957. [Google Scholar] [CrossRef]
- Purdie, J.W.; Gillis, H.A.; Klassen, N.V. The pulse radiolysis of penicillamine and penicillamine disulfide in aqueous solution. Can. J. Chem. 1973, 51, 3132–3142. [Google Scholar] [CrossRef]
- Prompers, J.J.; Hilbers, C.W.; Pepermans, H.A.M. Tryptophan mediated photoreduction of disulfide bond causes unusual fluorescence behaviour of Fusarium solani pisi cutinase. FEBS Lett. 1999, 456, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Neves-Petersen, M.T.; Gryczynski, Z.; Lakowicz, J.; Fojan, P.; Pedersen, S.; Petersen, E.; Bjørn Petersen, S. High probability of disrupting a disulphide bridge mediated by an endogenous excited tryptophan residue. Protein Sci. 2002, 11, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.L.; Hageman, M.J.; Thamann, T.J.; Barròn, L.B.; Schöneich, C. Solid-state photodegradation of bovine somatotropin (bovine growth hormone): Evidence for tryptophan-mediated photooxidation of disulfide bonds. J. Pharm. Sci. 2003, 92, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Vanhooren, A.; Illyes, E.; Majer, Z.; Hanssens, I. Fluorescence contributions of the individual Trp residues in goat α-lactalbumin. Biochim. Biophys. Acta 2006, 1764, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.Z.; Sheng, Y.B.; Xie, J.B.; Wang, W. Photoexcitation of tryptophan groups induced reduction of disulfide bonds in hen egg white lysozyme. J. Mol. Struct. 2008, 882, 101–106. [Google Scholar] [CrossRef]
- Illyés, E.; Staelens, S.; Vanhooren, A.; Deckmyn, H.; Hanssens, I.; Majer, Z. Role of the Trp-disulfide triads in the UV light induced degradation of a monoclonal antibody scFv. Biomed. Res. Int. 2014, 4, 367–385. [Google Scholar] [CrossRef]
- Mozziconacci, O.; Williams, T.D.; Kerwin, B.A.; Schöneich, C. Reversible intramolecular hydrogen transfer between protein cysteine thiyl radicals and αC–H bonds in insulin: Control of selectivity by secondary structure. J. Phys. Chem. B 2008, 112, 15921–15932. [Google Scholar] [CrossRef] [PubMed]
- Nauser, T.; Casi, G.; Koppenol, W.H.; Schöneich, C. Intramolecular addition of cysteine thiyl radicals to phenylalanine in peptides: Formation of cyclohexadienyl type radicals. Chem. Commun. (Camb) 2005, 27, 3400–3402. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Volkin, D.B.; Mach, H.; Middaugh, C.R. Degradative covalent reactions important to protein stability. Mol. Biotechnol. 1997, 8, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Kerwin, B.A.; Schöneich, C. Photolysis of an intrachain peptide disulfide bond: Primary and secondary processes, formation of H2S, and hydrogen transfer reactions. J. Phys. Chem. B 2010, 114, 3668–3688. [Google Scholar] [CrossRef] [PubMed]
- Ioerger, T.R.; Du, C.; Linthicum, D.S. Conservation of cys–cys trp structural triads and their geometry in the protein domains of immunoglobulin superfamily members. Mol. Immunol. 1999, 36, 373–386. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Pal, D.; Chakrabarti, P. Disulfide bonds, their stereospecific environment and conservation in protein structures. Protein Eng. Des. Sel. 2004, 17, 795–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, U.; Pal, D.; Chakrabarti, P. Environment of tryptophan side chains in proteins. Proteins 2000, 38, 288–300. [Google Scholar] [CrossRef]
- Abaskharon, R.M.; Gai, F. Direct measurement of the tryptophan-mediated photocleavage kinetics of a protein disulfide bond. Phys. Chem. Chem. Phys. 2016, 18, 9602–9607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, M.; Snabe, T.; Thiagarajan, V.; Petersen, S.B.; Campos, S.R.; Baptista, A.M.; Neves-Petersen, M.T. Photonic activation of plasminogen induced by low dose UVB. PLoS ONE 2015, 10, e0116737. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. Cation-π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA. 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Petsko, G.A. Amino-aromatic interactions in proteins. FEBS Lett. 1986, 203, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Slutsky, M.M.; Marsh, E.N.G. Cation–π interactions studied in a model coiled-coil peptide. Protein Sci. 2004, 13, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Sippel, T.O. Microfluorometric analysis of protein thiol groups with coumarinylphenylmaleimide. J. Histochem. Cytochem. 1981, 29, 1377–1381. [Google Scholar] [CrossRef] [PubMed]
- Atherton, H.F.E.; Diana Harkiss, C.; Logan, J.; Sheppard, R.C.; Williams, B.J. A mild procedure for solid phase peptide synthesis: Use of fluorenylmethoxycarbonylamino-acids. J. Chem. Soc. Chem. Commun. 1978, 537–539. [Google Scholar] [CrossRef]
- Andreu, F.A.D.; Sole, N.A.; Munrou, M.C.; Ferrer, M.; Barany, G. Peptide Synthesis Protocols; Pennington, M.W., Dunn, B.M., Eds.; Human Press: Totowa, NJ, USA, 1994; Volume 35. [Google Scholar]
- Knapp, K.; Illyés, E.; Csík, G.; Majer, Z. Role of cation-pi interaction in the photolysis of disulphide bridge containing cyclic model peptides. In European peptide Symposium; Kokotos, V.C.-K.G., Matsoukas, J., Eds.; Peptides: Athens, Greece, 2012; pp. 678–699. [Google Scholar]
- Kishore, R.; Mathew, M.K.; Balaram, P. A fluorescent peptide model for the thioredoxin active site. FEBS Lett. 1983, 159, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Cline, D.J.; Thorpe, C.; Schneider, J.P. Structure-based design of a fluorimetric redox active peptide probe. Anal. Biochem. 2004, 325, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.; Milkowski, J.; Varga, S.; Denkewalter, R.; Hirschmann, R. Acetamidomethyl. A novel thiol protecting group for cysteine. J. Am. Chem. Soc. 1972, 94, 5456–5461. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Sharov, V.; Williams, T.D.; Kerwin, B.A.; Schöneich, C. Peptide cysteine thiyl radicals abstract hydrogen atoms from surrounding amino acids: The photolysis of a cystine containing model peptide. J. Phys. Chem. B 2008, 112, 9250–9257. [Google Scholar] [CrossRef] [PubMed]
- Bane, J.M. Novel Cys Crosslinks and Trp Side Chain Cleavages In Proteins And Peptides Exposed to Light. Ph.D. Thesis, University of Kansas, Lawrence, KS, USA, 2015. [Google Scholar]
- Bax, A.D.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Uhrı́n, D.; Batta, G.; Hruby, V.J.; Barlow, P.N.; Kövér, K.E. Sensitivity-and gradient-enhanced hetero (ω1) half-filtered TOCSY experiment for measuring long-range heteronuclear coupling constants. J. Magn. Reson. 1998, 130, 155–161. [Google Scholar] [CrossRef] [PubMed]
- R Bothner-By, A.A.; Stephens, R.L.; Lee, J.; Warren, C.D.; Jeanloz, R.W. Structure determination of a tetrasaccharide: Transient nuclear Overhauser effects in the rotating frame. J. Am. Chem. Soc. 1984, 106, 811–813. [Google Scholar] [CrossRef]
- Bax, A.D.; Davis, D.G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. [Google Scholar] [CrossRef]
- Kay, L.; Keifer, P.; Saarinen, T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992, 114, 10663–10665. [Google Scholar] [CrossRef]
- Kontaxis, G.; Stonehouse, J.; Laue, E.D.; Keeler, J. The sensitivity of experiments which use gradient pulses for coherence-pathway selection. J. Magn. Reson. Ser. A 1994, 111, 70–76. [Google Scholar] [CrossRef]
- Andersen, N.H.; Neidigh, J.W.; Harris, S.M.; Lee, G.M.; Liu, Z.; Tong, H. Extracting information from the temperature gradients of polypeptide NH chemical shifts. 1. The importance of conformational averaging. J. Am. Chem. Soc. 1997, 119, 8547–8561. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley & Sons: Hoboken, NY, USA, 1986. [Google Scholar]
- Keller, R.L.J. Optimizing the Process of Nuclear Magnetic Resonance Spectrum Analysis and Computer Aided Resonance Assignment. Ph.D. Thesis, Swiss Federal Institute of Techonology Zürich, Zürich, Switzerland, 2004. [Google Scholar]
- Guerry, P.; Duong, V.D.; Herrmann, T. CASD-NMR 2: Robust and accurate unsupervised analysis of raw NOESY spectra and protein structure determination with UNIO. J. Biomol. NMR 2015, 62, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 2002, 24, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, T.; Güntert, P.; Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002, 319, 209–227. [Google Scholar] [CrossRef]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Gr. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Pall, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In International Conference on Exascale Applications and Software; Springer: Cham, Switherland, 2014; pp. 3–27. [Google Scholar]
- Fox, T.; Kollman, P.A. Application of the RESP methodology in the parametrization of organic solvents. J. Phys. Chem. B 1998, 102, 8070–8079. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- DeLano, W.L. DeLano Scientific LLC, San Carlos, CA, USA, 2002. Available online: https://pymol.org/2/ (accessed on 26 March 2018).
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Edit. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Calvert, J.G.; Pitts, N.J., Jr. Photochemistry; John Wiley and Sons: New York, NY, USA, 1967. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Constitution | MWcalc. Monoisotopic (Measured M + H+) | ||
|---|---|---|---|---|
| Ac-c(CAXAC)-NH2 | ||||
| X=V | 8.26 | - | C19H32N6O6S2 | 504.2 (505.3) |
| X=W | 5.49 | - | C25H33N7O6S2 | 591.2 (592.3) |
| Ac-c(CXAGC)-NH2 | ||||
| X=V | 6.60 | - | C18H30N6O6S2 | 491.2 (492.3) |
| X=W | 9.61 | - | C24H31N7O6S2 | 577.2 (578.3) |
| Ac-c(CXKAC)-NH2 | ||||
| X=V | 7.76 | - | C22H39N7O6S2 | 561.2 (562.3) |
| X=W | 7.95 | 7.82 | C28H40N8O6S2 | 648.2 (649.2) |
| Ac-c(CWKAC)-NH2 | ||||||
|---|---|---|---|---|---|---|
| Unordered | Type I β-turn | Type II β-turn | Type IV β-Turn | Inv. γ-Turn | 310-Helix | |
| H2O | 49% | 5% | 21% | 18% | 1% | 6% |
| DMSO | 58% | 2% | 21% | 12% | - | 7% |
| Ac-c(CAWAC)-NH2 | ||||||
| Unordered | Type I β-Turn | Type IV β-Turn | Type VIII β-Turn | Inv. γ-turn | 310-Helix | |
| H2O | 60% | 1% | 30% | 1% | 8% | - |
| DMSO | 74% | 5% | 15% | - | 2% | 4% |
| Ac-c(CWAGC)-NH2 | ||||||
| Unordered | Type i β-Turn | Type ii β-Turn | Type iv β-Turn | Inv. Γ-Turn | - | |
| H2O | 45% | 44% | - | 11% | - | - |
| DMSO | 33% | - | 30% | 30% | 7% | - |
| NH-Chemical Shift Temperature Gradient (ppb/K) in Model Peptides | |||||
|---|---|---|---|---|---|
| Ac-c(CWKAC)-NH2 | Cys1(NH) | Trp2(NH) | Lys3(NH) | Ala4(NH) | Cys5(NH) |
| −5.50 | −5.80 | −1.70 | −5.40 | −5.70 | |
| Ac-c(CAWAC)-NH2 | Cys1(NH) | Ala2(NH) | Trp3(NH) | Ala4(NH) | Cys5(NH) |
| −5.90 | −5.40 | −1.60 | −5.50 | −5.50 | |
| Ac-c(CWAGC)-NH2 | Cys1(NH) | Trp2(NH) | Ala3(NH) | Gly4(NH) | Cys5(NH) |
| −5.80 | −4.90 | −5.60 | −4.80 | −5.60 | |
| Gauche (+) | Gauche (−) | Trans | ||||
|---|---|---|---|---|---|---|
| Ac-c(CWKAC)-NH2 | water | DMSO | water | DMSO | water | DMSO |
| MD | 39% | 25% | 8% | 16% | 53% | 59% |
| NMR | - | 32% | - | 25% | - | 43% |
| Ac-c(CAWAC)-NH2 | Gauche (+) | Gauche (−) | Trans | |||
| MD | 62% | 78% | 16% | 11% | 22% | 12% |
| NMR | - | - | - | - | - | - |
| Ac-c(CWAGC)-NH2 | Gauche (+) | Gauche (−) | Trans | |||
| MD | 93% | 14% | 4% | 65% | 3% | 21% |
| NMR | - | 29% | - | 58% | - | 12% |
| Peptide 280 nm | I (480 nm)/cps a | c/μM b | SH% c | d(SS-W)/Å | d(Lys-W)/Å |
|---|---|---|---|---|---|
| Ac-c(CWKAC)-NH2 | 519565 | 2.04 | 5.73 | 7.95 | 7.83 |
| Ac-c(CAWAC)-NH2 | 523460 | 2.06 | 5.88 | 5.49 | - |
| Ac-c(CWAGC)-NH2 | 548670 | 2.16 | 6.17 | 9.61 | - |
| Ac-c(CVKAC)-NH2 | 179549 | 0.71 | 2.02 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, T.M.; Knapp, K.; Illyés, E.; Timári, I.; Schlosser, G.; Csík, G.; Borics, A.; Majer, Z.; Kövér, K.E. Photochemical and Structural Studies on Cyclic Peptide Models. Molecules 2018, 23, 2196. https://doi.org/10.3390/molecules23092196
Nagy TM, Knapp K, Illyés E, Timári I, Schlosser G, Csík G, Borics A, Majer Z, Kövér KE. Photochemical and Structural Studies on Cyclic Peptide Models. Molecules. 2018; 23(9):2196. https://doi.org/10.3390/molecules23092196
Chicago/Turabian StyleNagy, Tamás Milán, Krisztina Knapp, Eszter Illyés, István Timári, Gitta Schlosser, Gabriella Csík, Attila Borics, Zsuzsa Majer, and Katalin E. Kövér. 2018. "Photochemical and Structural Studies on Cyclic Peptide Models" Molecules 23, no. 9: 2196. https://doi.org/10.3390/molecules23092196