Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
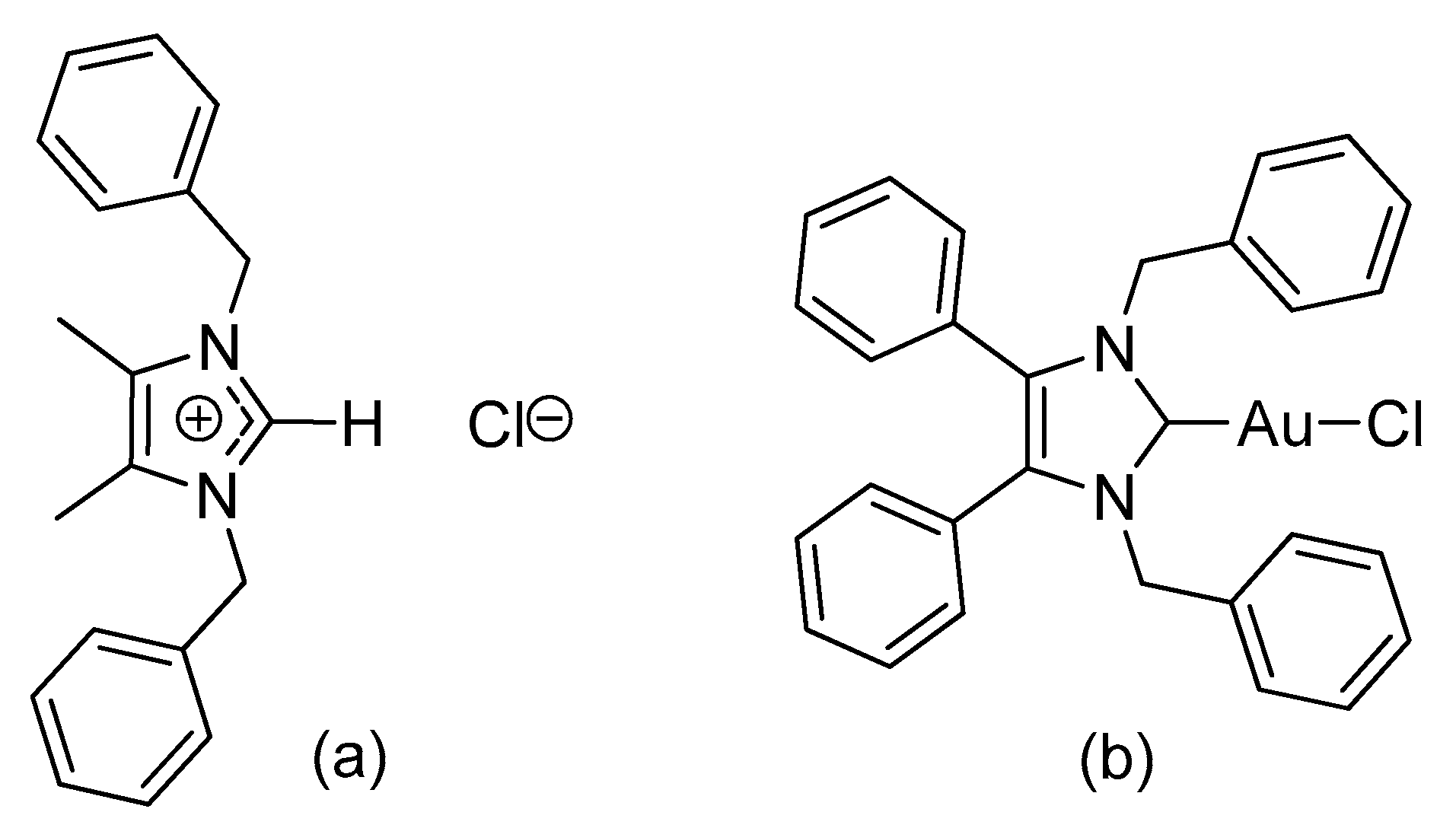
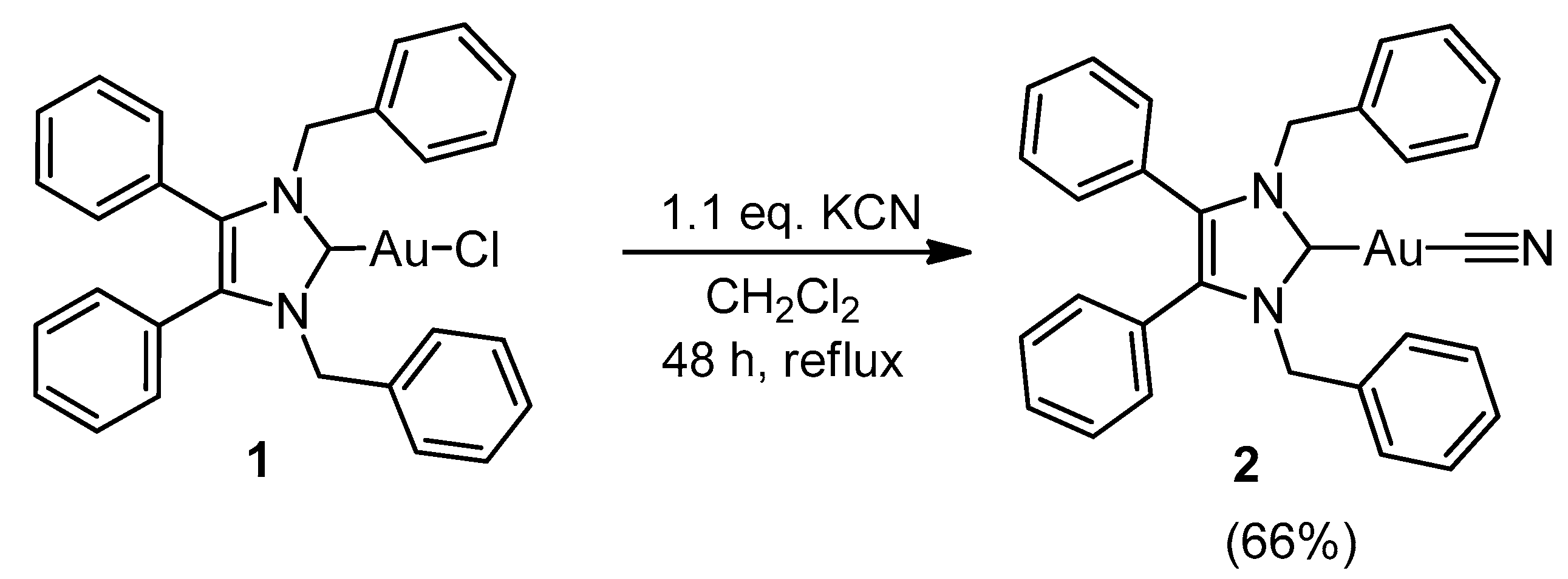
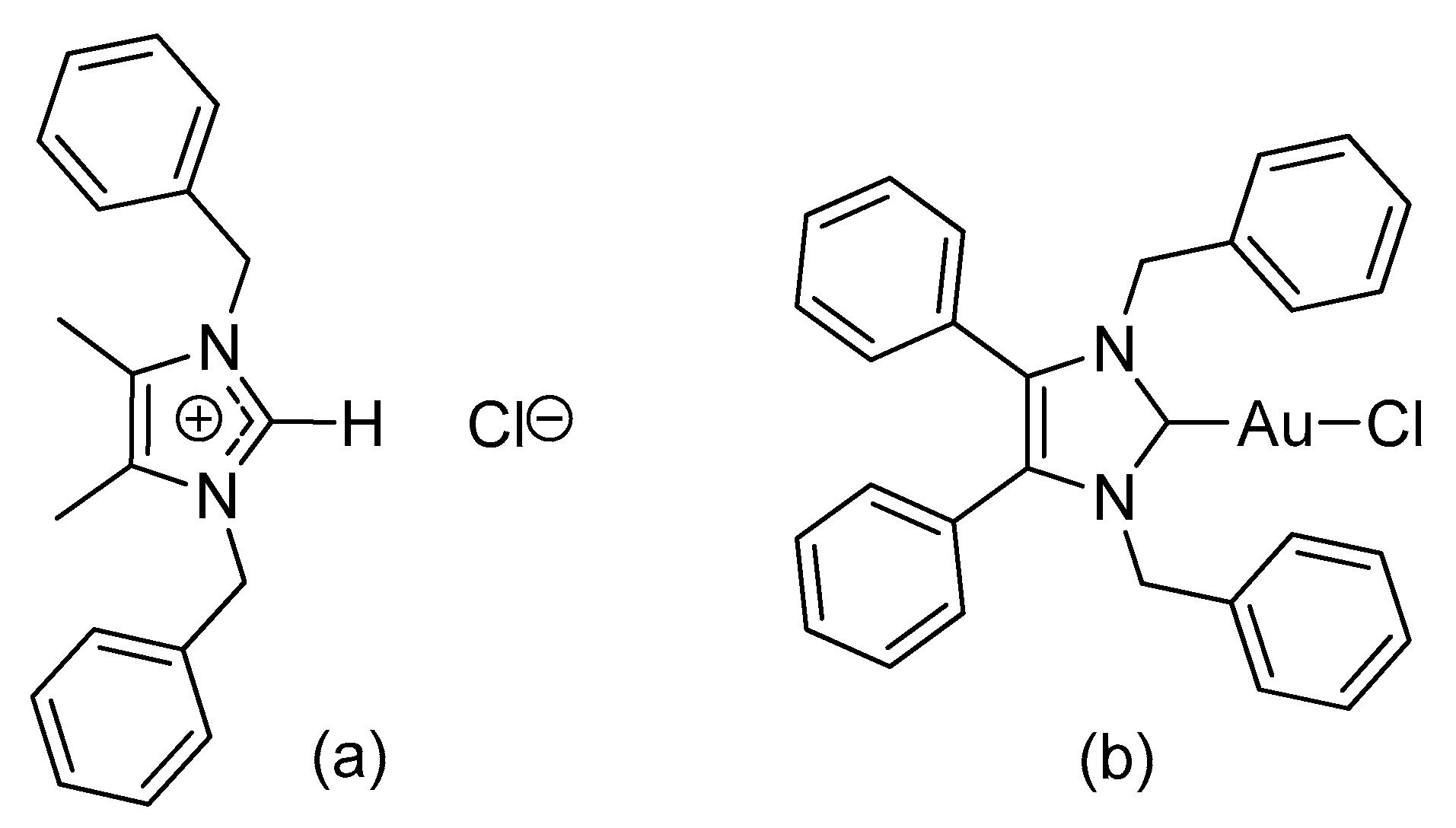
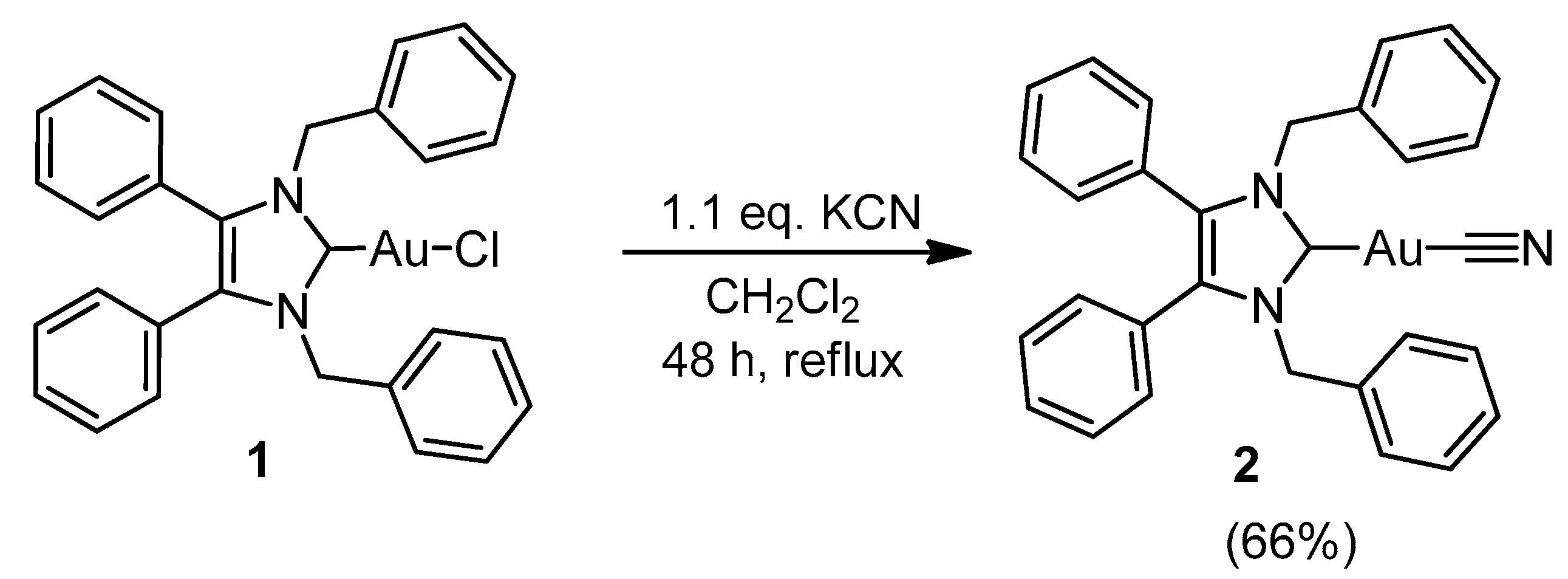
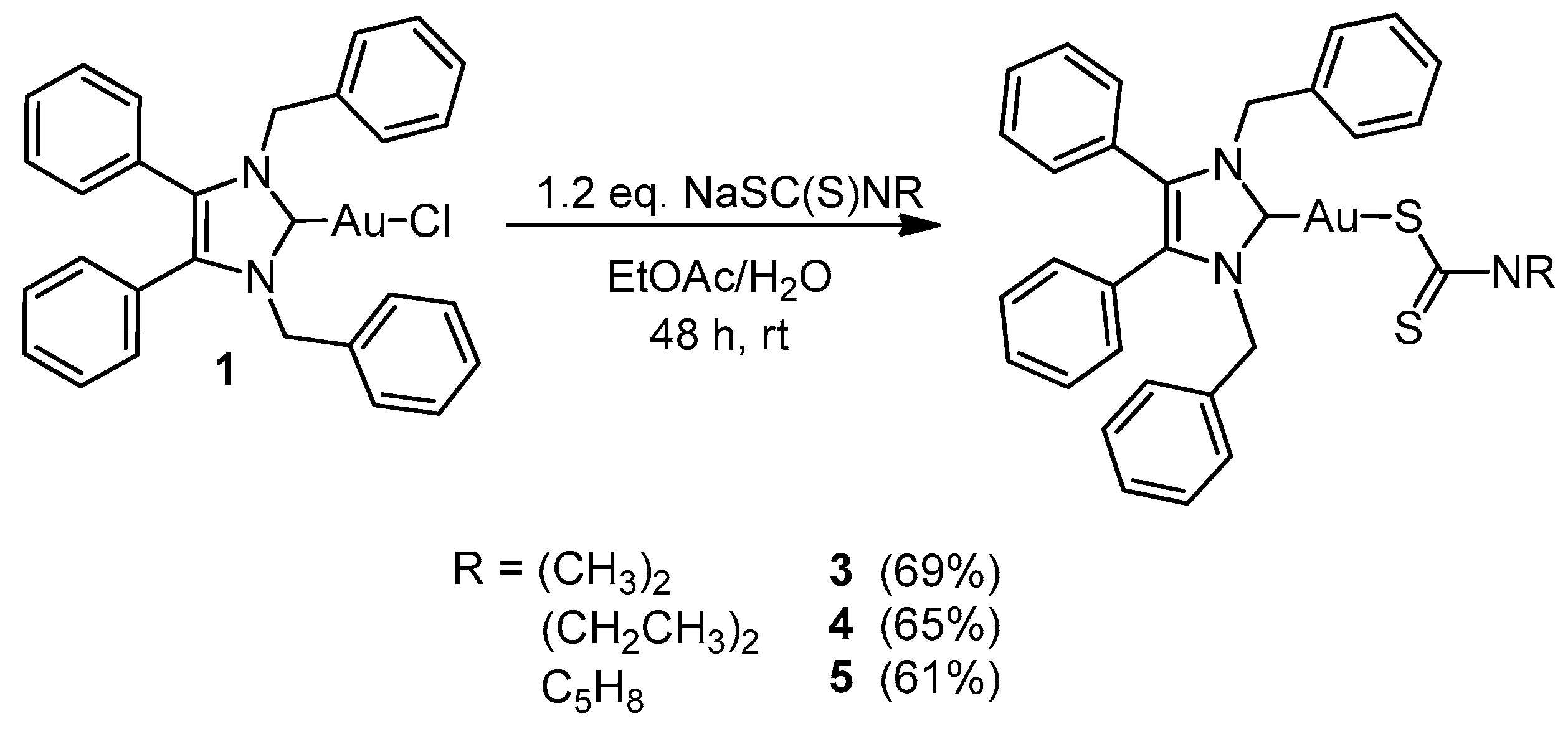
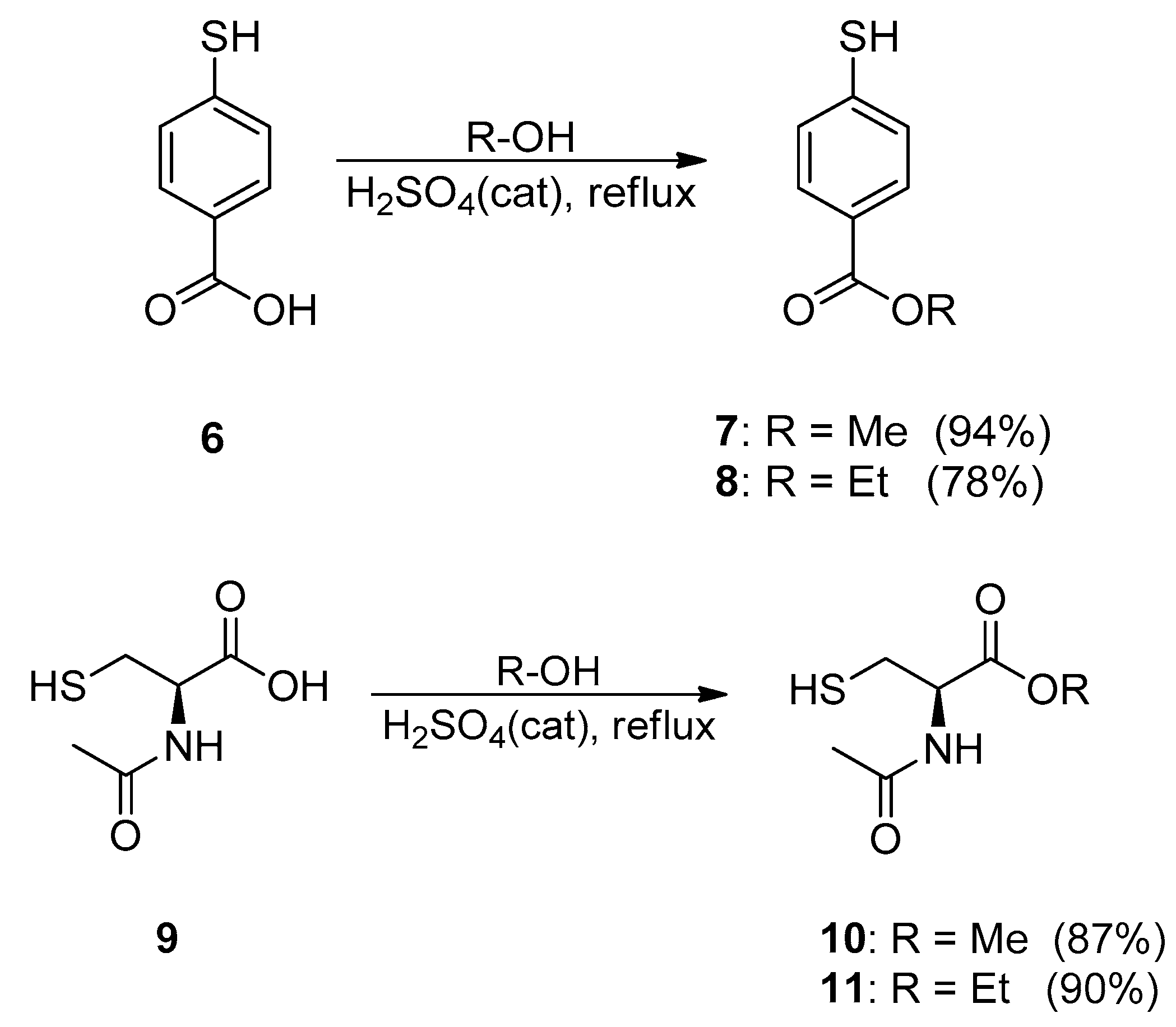
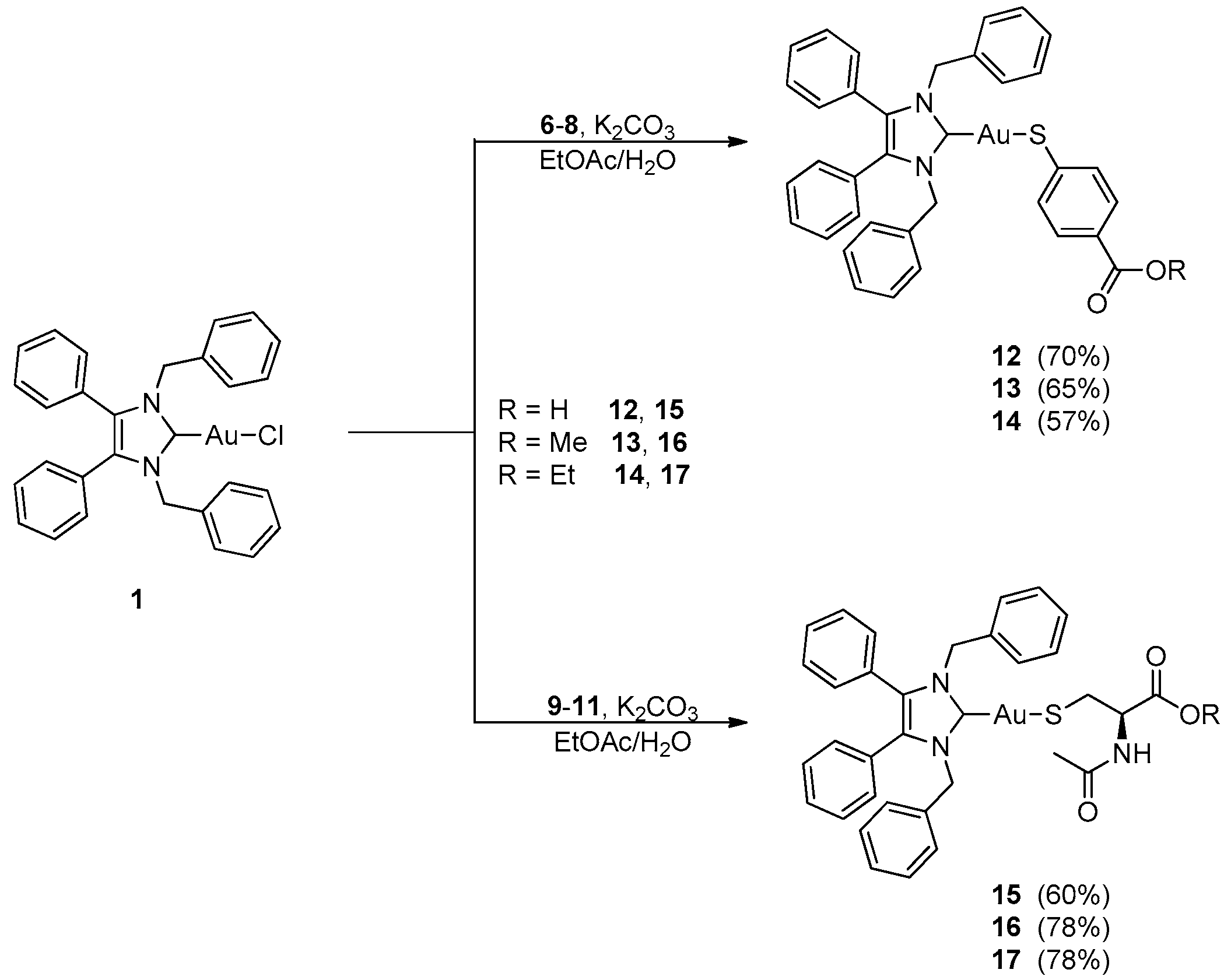
2.1. Synthesis and Characterisation
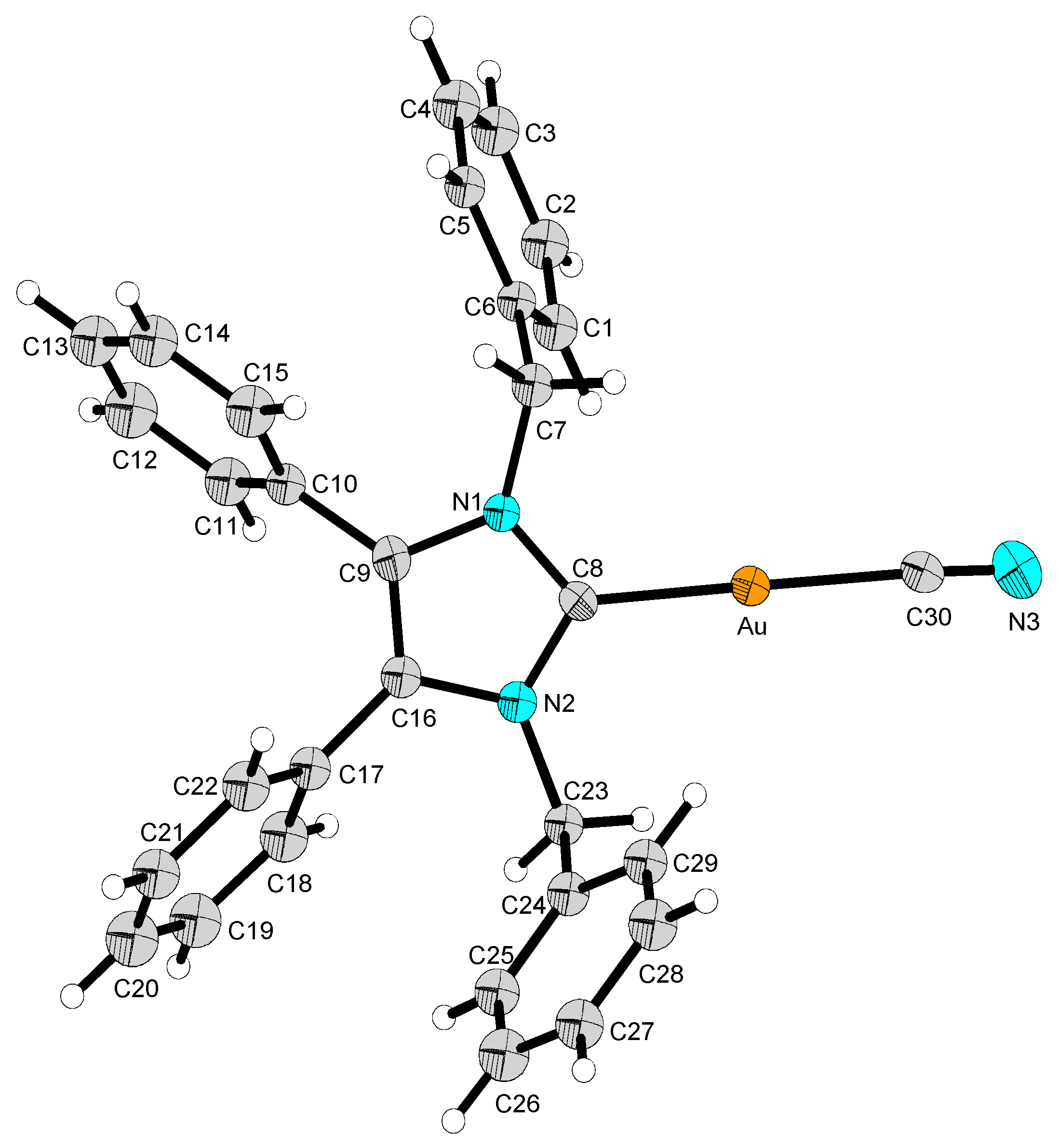
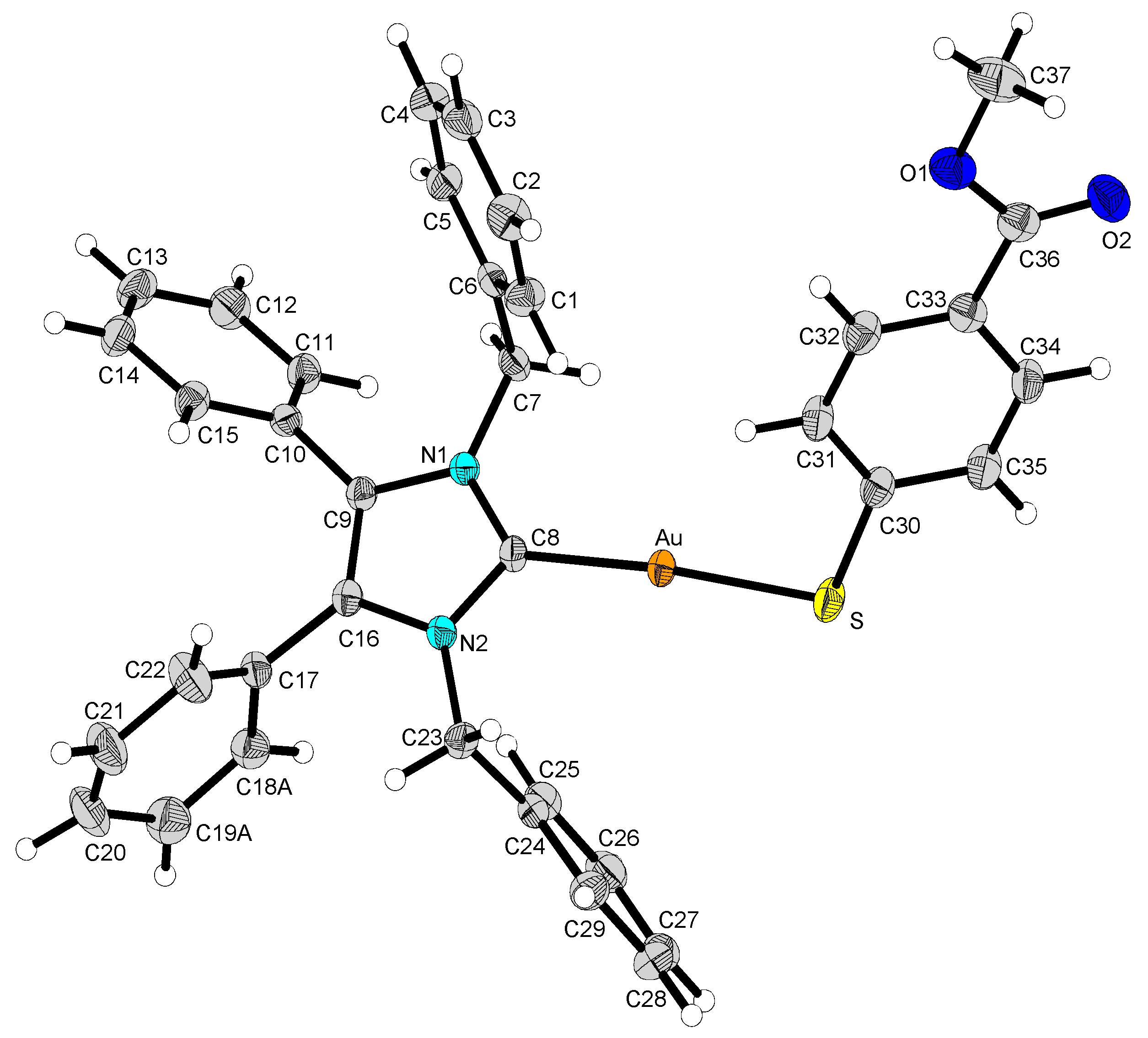
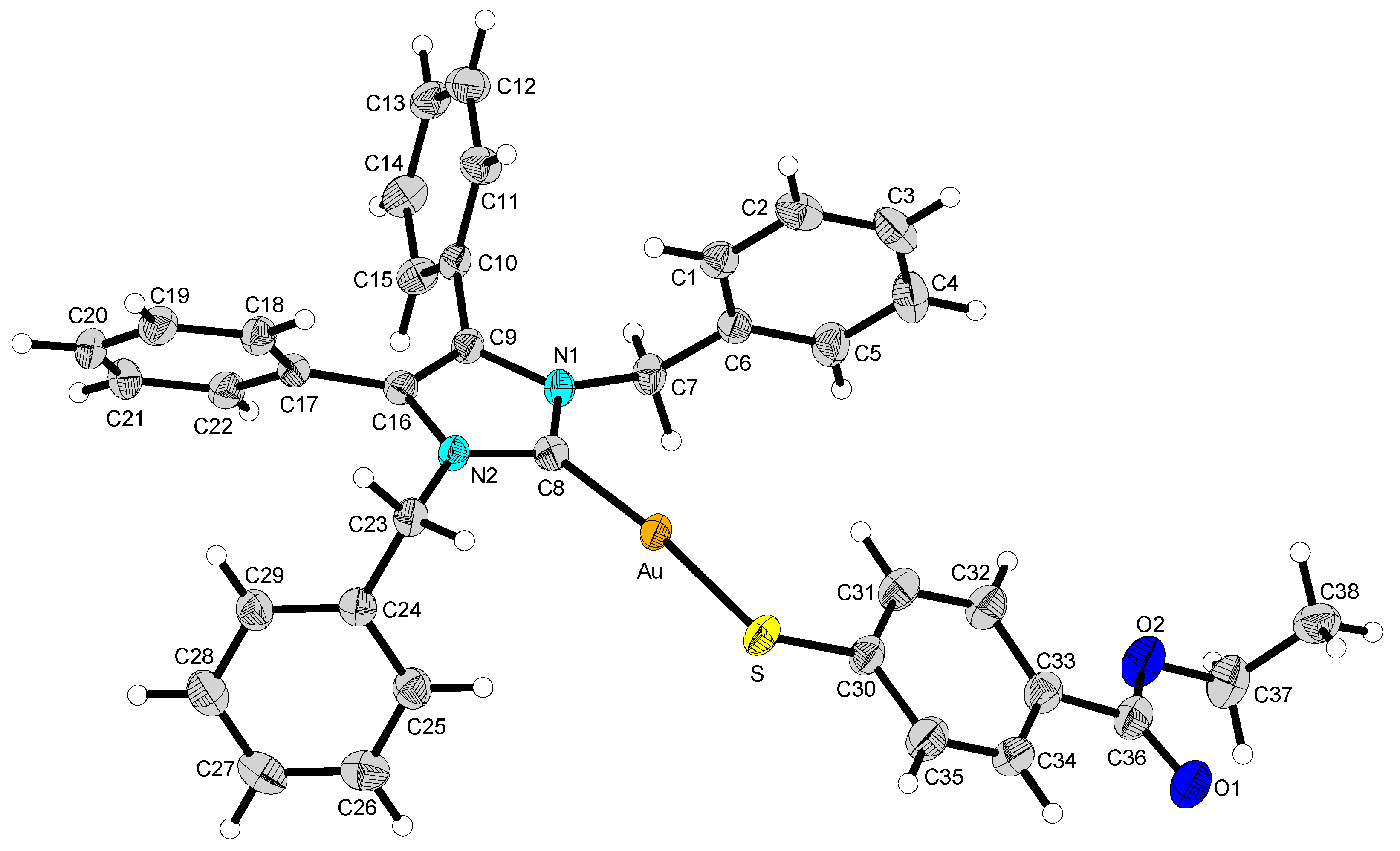
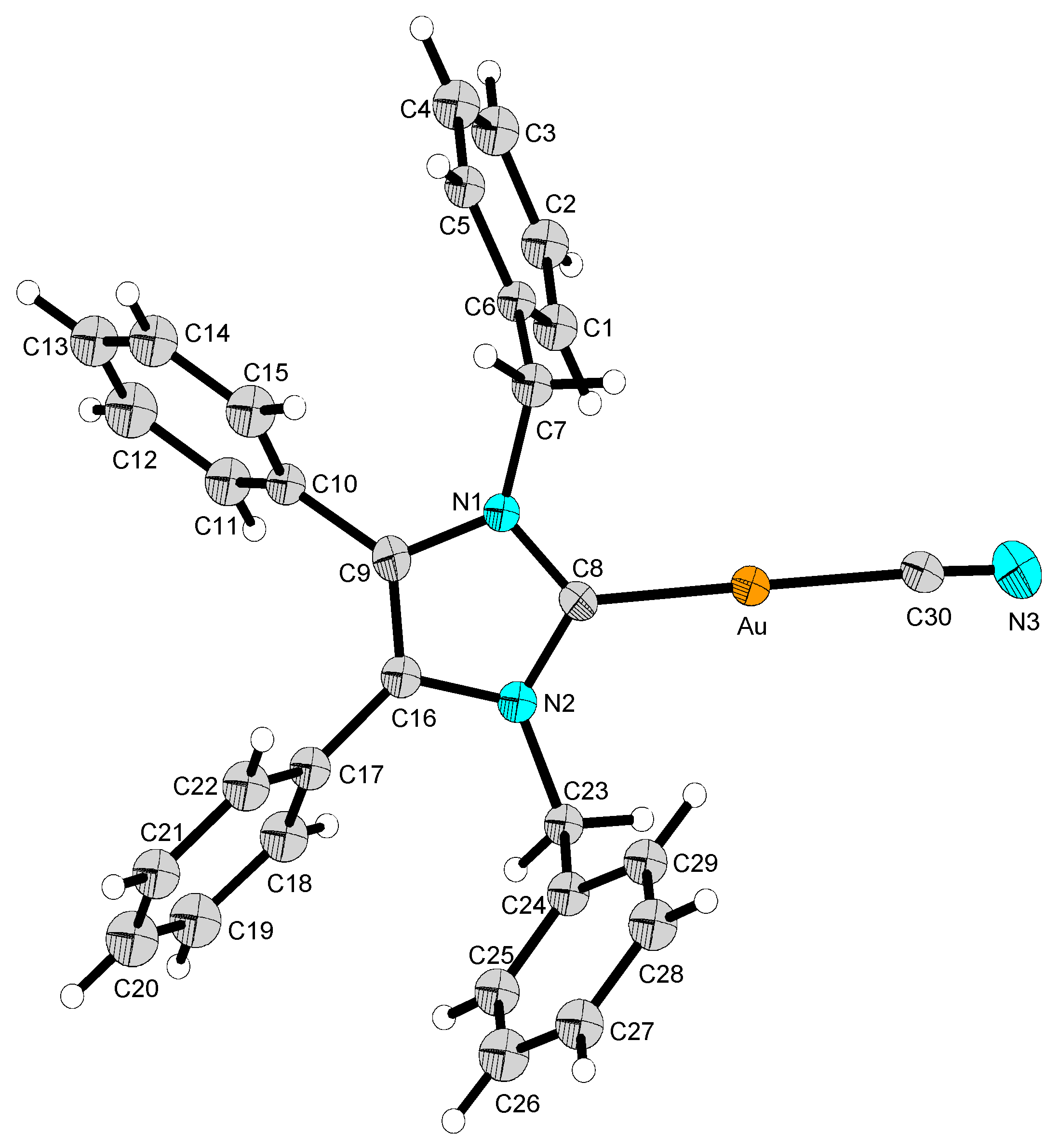
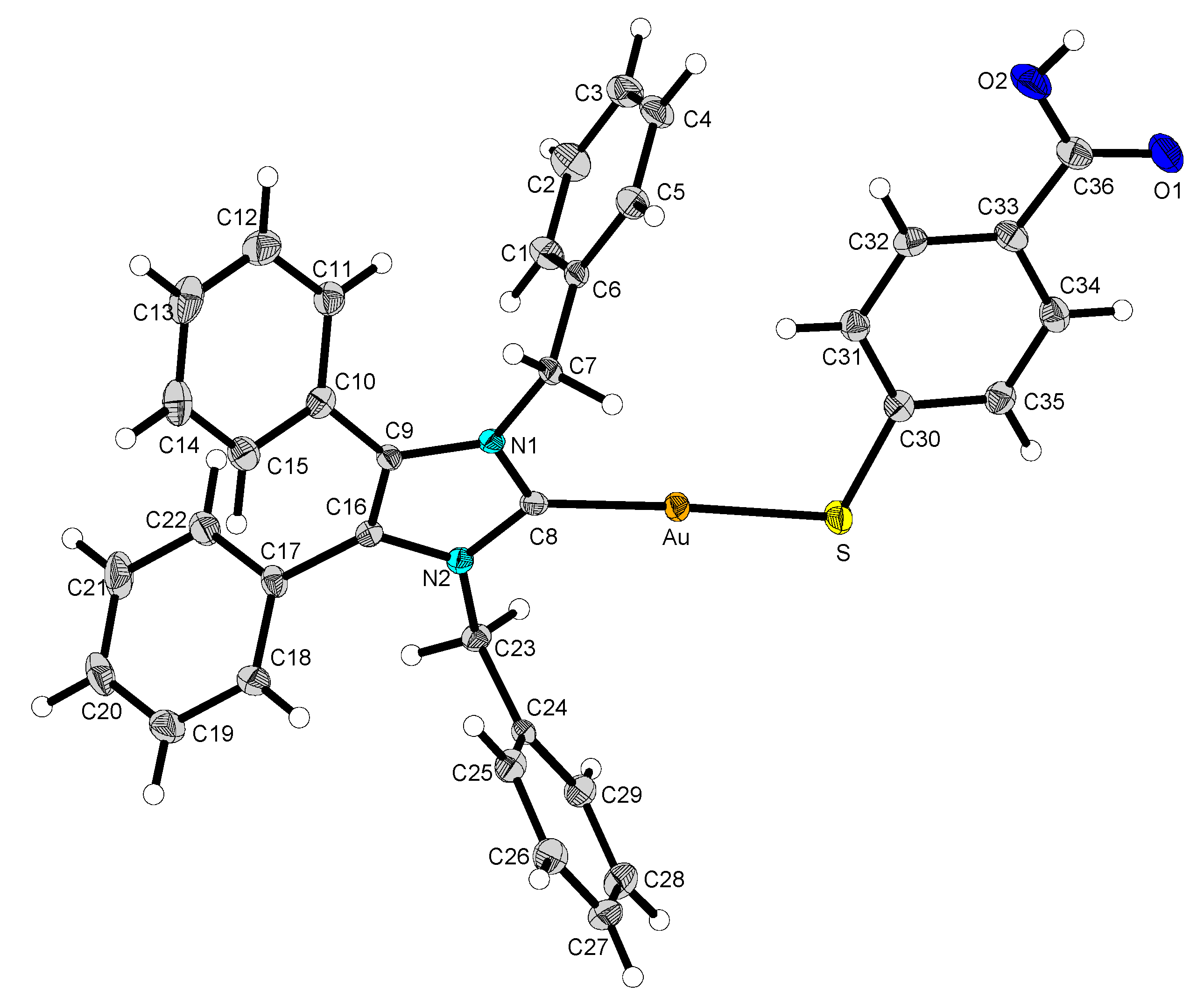
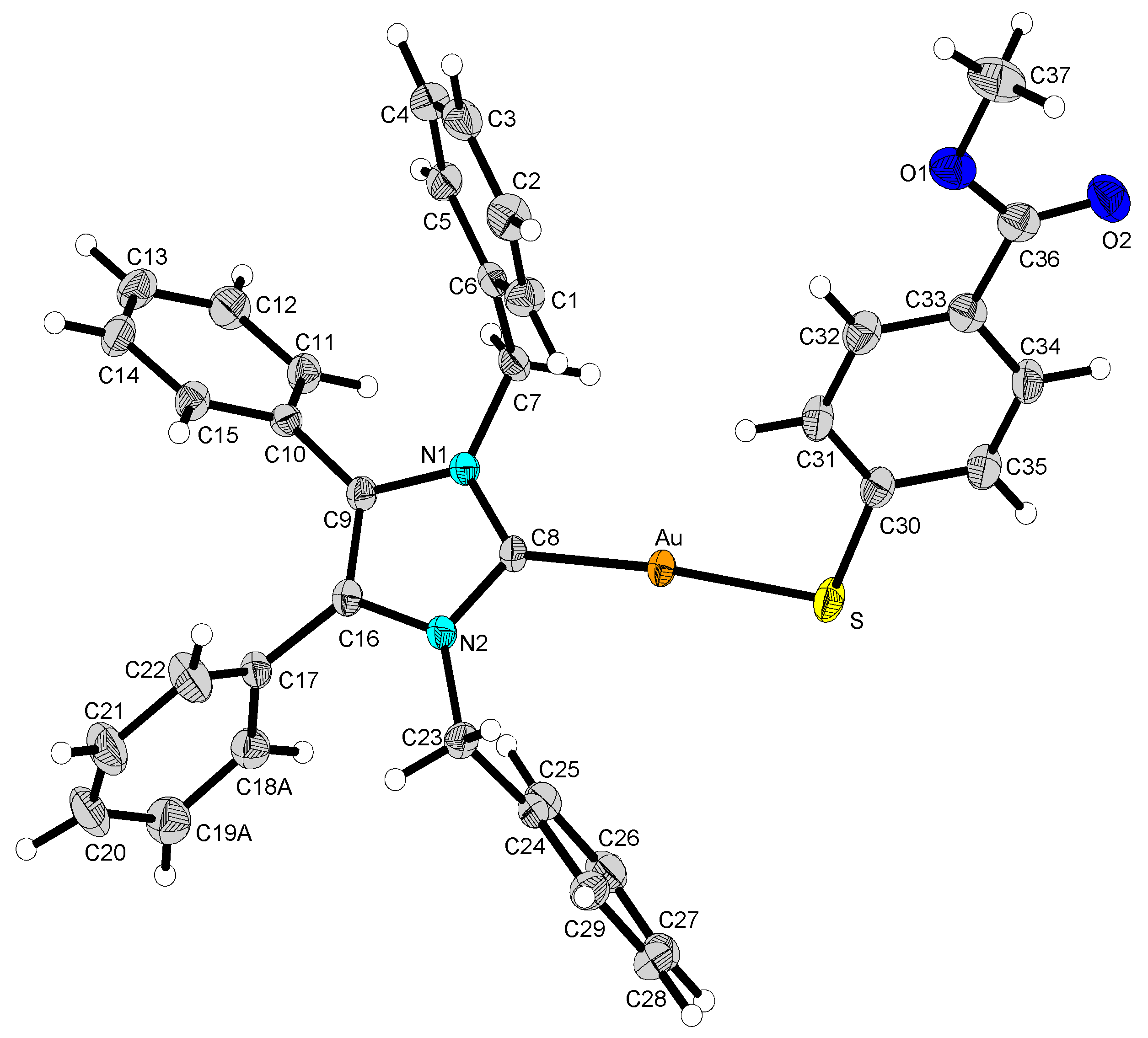
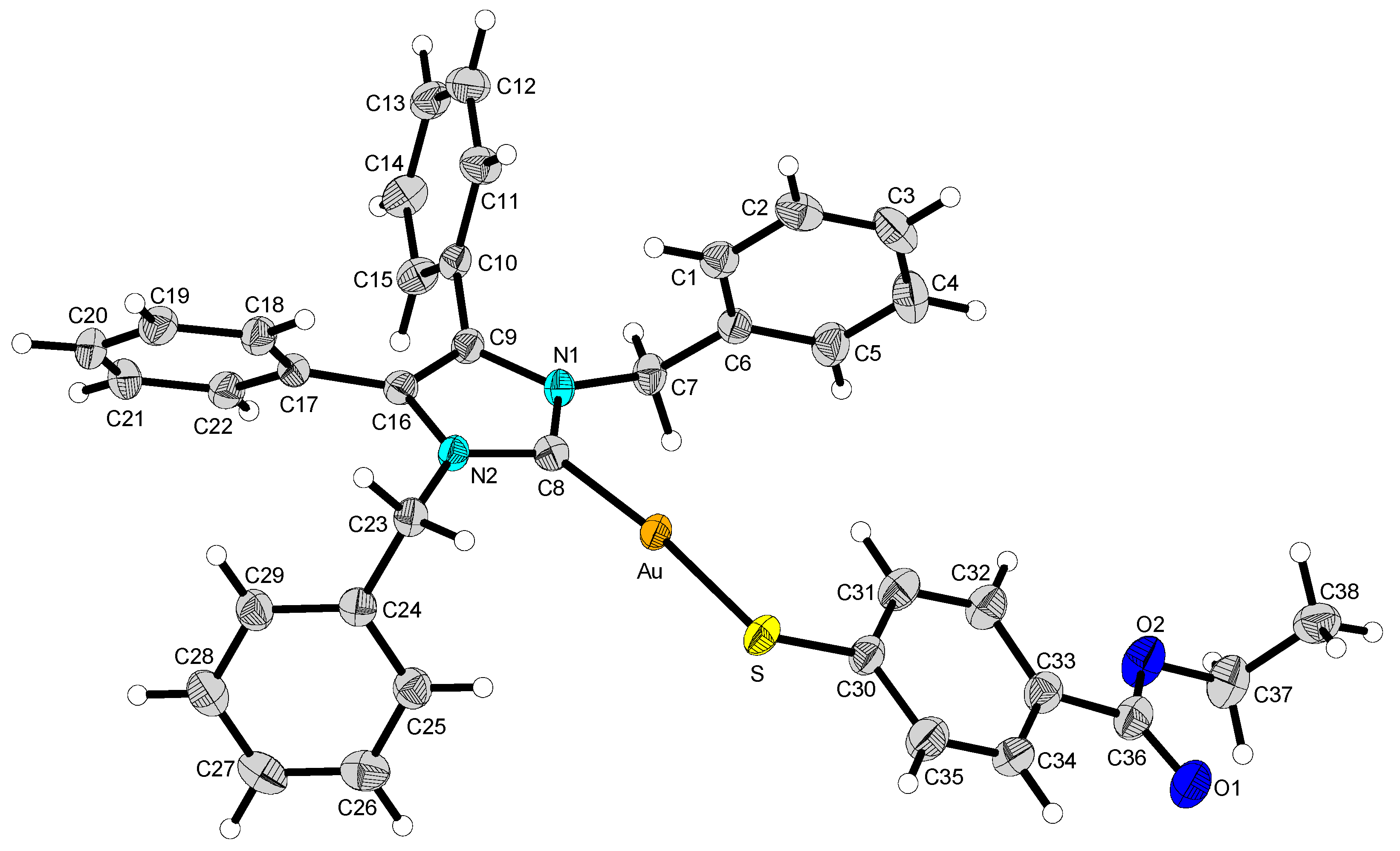
2.2. Structural Discussion
2.3. Biological Evaluation
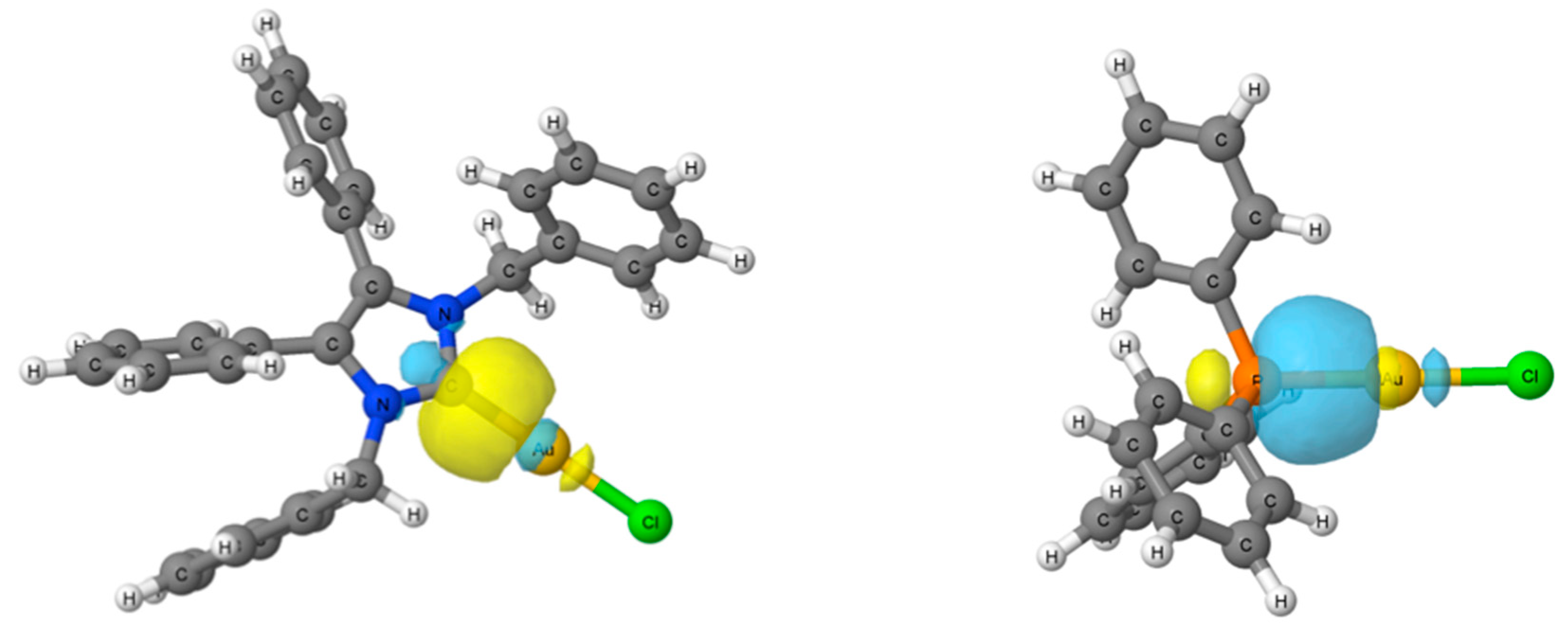
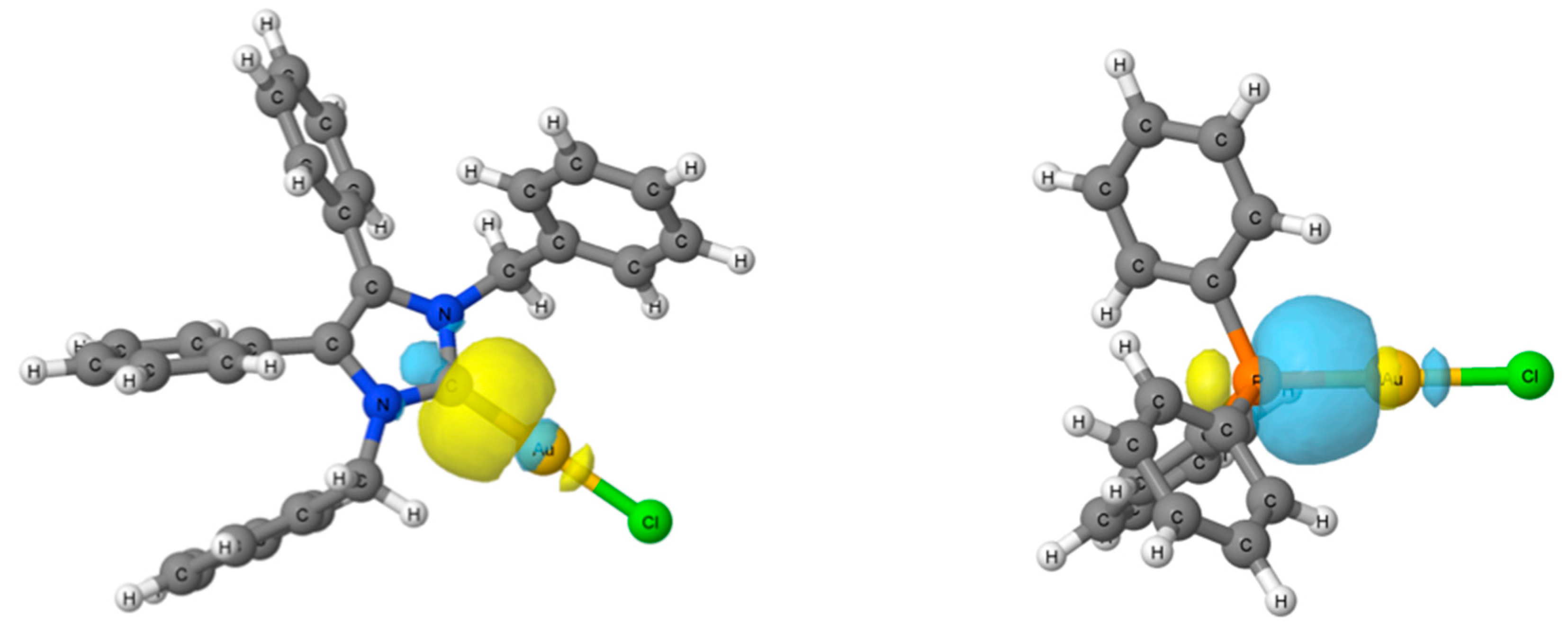
2.4. Computational Results
3. Materials and Methods
3.1. General Conditions
3.2. Synthesis
3.2.1. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Chloride (1)
3.2.2. (1,3-Dibenzyl-4,5-diphenyl-2-ylidene)gold(I) Cyanide (2)
3.2.3. General Procedure for NHC-Au(I) Complexes 3–5
(1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Dimethyldithiocarbamate (3)
(1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Diethyldithiocarbamate (4)
(1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) Pyrrolidinedithiocarbamate (5)
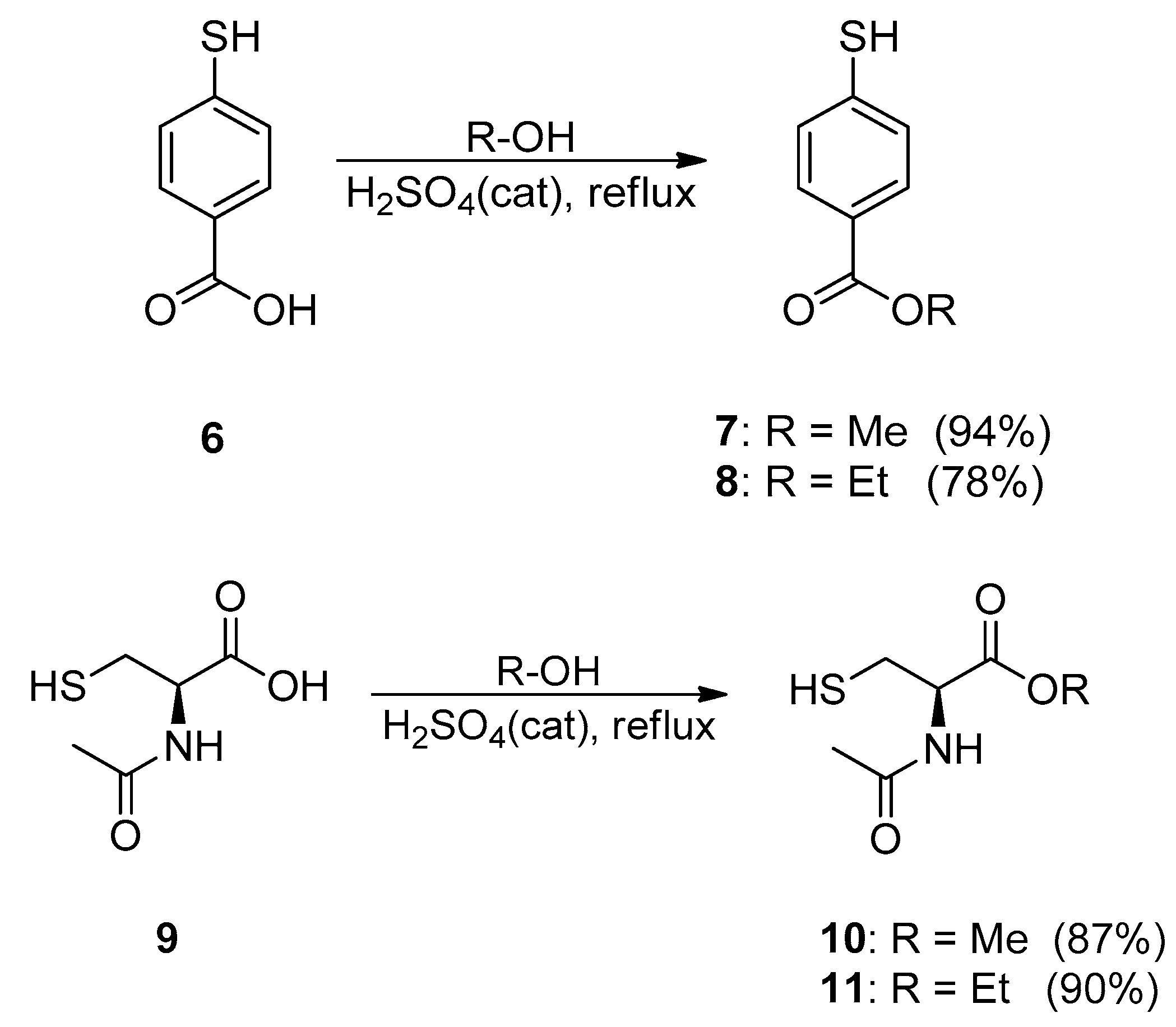
3.2.4. General Procedure for 7–8, 10–11
Methyl-p-mercaptobenzoate (7)
Ethyl-p-mercaptobenzoate (8)
N-Acetyl-l-cysteine Methyl Ester (10)
N-acetyl-l-cysteine Ethyl Ester (11)
3.2.5. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I) p-Mercaptobenzoic Acid (12)
3.2.6. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-methyl-p-mercaptobenzoate (13)
3.2.7. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-ethyl-p-mercaptobenzoate (14)
3.2.8. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-N-acetyl-l-cysteine (15)
3.2.9. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-N-acetyl-l-cysteine Methyl Ester (16)
3.2.10. (1,3-Dibenzyl-4,5-diphenylimidazol-2-ylidene)gold(I)-N-acetyl-l-cysteine Ethyl Ester (17)
3.3. Structure Determination
3.4. MTT-Based Proliferation Assay
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, D.H.; Smith, W.E. The chemistry of the gold drugs used in the treatment of rheumatoid arthritis. Chem. Soc. Rev. 1980, 9, 217–240. [Google Scholar] [CrossRef]
- Eisler, R. Chrysotherapy: A synoptic review. Inflamm. Res. 2003, 52, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, F.; Muller-Bunz, H.; Smith, R.; Streciwilk, W.; Zhu, X.; Tacke, M. Novel ruthenium(II) and gold(I) NHC complexes: Synthesis, characterization, and evaluation of their anticancer properties. Organometallics 2013, 32, 5551–5560. [Google Scholar] [CrossRef]
- Baker, M.V.; Barnard, P.J.; Berners-Price, S.J.; Brayshaw, S.K.; Hickey, J.L.; Skelton, B.W.; White, A.H. Synthesis and structural characterisation of linear Au(I) N-Heterocyclic carbene complexes: New analogues of the Au(I) phosphine drug auranofin. J. Organomet. Chem. 2005, 690, 24–25. [Google Scholar] [CrossRef]
- Zhao, W.; Ferro, V.; Baker, M.V. Carbohydrate-N-heterocyclic carbene metal complexes: Synthesis, catalysis and biological studies. Coord. Chem. Rev. 2007, 339, 1–16. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Patil, S.A.; Patil, R.; Keri, R.S.; Balakrishna, G.R.; Tacke, M. N-heterocylic carbene metal complexes as bio-organometallic antimicrobial and anticancer drugs. Future Med. Chem. 2015, 7, 1305–1333. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, M.; Santini, C.; Pellei, M. Recent advances in medicinal applications of coinage-metal (Cu and Ag) N-heterocyclic carbene complexes. Curr. Top. Med. Chem. 2016, 16, 2995–3017. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L.A.; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W.S.; Wolber, G.; et al. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010, 53, 8608–8618. [Google Scholar] [CrossRef] [PubMed]
- Muenzner, J.K.; Biersack, B.; Albrecht, A.; Rehm, T.; Lacher, U.; Milius, W.; Casini, A.; Zhang, J.; Ott, I. Ferrocenyl-coupled N-heterocyclic carbene complexes of gold (I): A successful approach to multinuclear anticancer drugs. Chem. Eur. J. 2016, 22, 18953–18962. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Zheng, B.L.; He, K.; Zheng, Q.Y. Imidazole alkaloids from lepidium meyenii. J. Nat. Prod. 2003, 66, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Riduan, S.N.; Zhang, Y. Imidazolium salts and their polymeric materials for biological applications. Chem. Soc. Rev. 2013, 42, 9055–9070. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Mönchgesang, S.; Sheldrick, W.S.; Wölfl, S.; Ott, I. Comparative in vitro evaluation of N-heterocyclic carbene gold(I) complexes of the benzimidazolylidene type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gust, R. Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs. Coord. Chem. Rev. 2016, 329, 191–213. [Google Scholar] [CrossRef]
- Ott, I. On the Medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.N.; To, W.P.; Low, K.H.; Che, C.M. A binuclear gold(I) complex with mixed bridging diphosphine and bis(N-heterocyclic carbene) ligands shows favorable thiol reactivity and inhibits tumor growth and angiogenesis in vivo. Angew. Chem. Int. Ed. 2014, 53, 5810–5814. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Lum, C.T.; Lok, C.; Zhang, J.; Che, C. Chemical biology of anticancer gold(III) and gold(I) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Marzo, I.; Cativiela, C.; Laguna, A.; Gimeno, M.C. Highly cytotoxic bioconjugated gold(I) complexes with cysteine-containing dipeptides. Chem. Eur. J. 2015, 21, 11088–11095. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayana, D.N. Vibrational Spectroscopy: Theory and Applications, 1st ed.; New Age International (P) Ltd.: New Delhi, India, 2004. [Google Scholar]
- Jamaluddin, N.A.; Baba, I. Synthesis and structural characterization of new dithiocarbamate complexes from Sb(III) and Bi(III). AIP Conf. Proc. 2013, 1571, 789–794. [Google Scholar]
- Durgaprasad, G.; Sathyanarayana, D.N.; Patel, C.C. Normal coordinate analysis of dialkylditlhioearbamate and its selenium analogue. Can. J. Chem. 1969, 47, 631–635. [Google Scholar] [CrossRef]
- Pakiari, A.H.; Jamshidi, Z. Nature and strength of M−S Bonds (M = Au, Ag, and Cu) in binary alloy gold clusters. J. Phys. Chem. A 2010, 114, 9212–9221. [Google Scholar] [CrossRef] [PubMed]
- Kokkin, D.L.; Zhang, R.; Steimle, T.C.; Wyse, I.A.; Pearlman, B.W.; Varberg, T.D. Au–S bonding revealed from the characterization of diatomic gold sulfide, AuS. J. Phys. Chem. A 2015, 119, 11659–11667. [Google Scholar] [CrossRef] [PubMed]
- Hormann, A.L.; Bennett, D.W.; Shaw, C.F.; Reiff, W.M. Solid-state structure and solution equilibria of cyano(Triethylphosphine)gold(I). Inorg. Chem. 1986, 25, 3953–3957. [Google Scholar] [CrossRef]
- Tacke, M.; Dada, O.; O’Beirne, C.; Zhu, X.; Müller-Bunz, H. The non-isomorphous crystal structures of NHC–Au–Cl and NHC–Au–Br (NHC is 1,3-dibenzyl-4,5-diphenylimidazol-2-ylidene). Acta Crystallogr. Sect. C Struct. Chem. 2016, 72, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Dada, O.; Curran, D.; O’Beirne, C.; Müller-Bunz, H.; Zhu, X.; Tacke, M. Synthesis and cytotoxicity studies of novel NHC–gold(I) pseudohalides and thiolates. J. Organomet. Chem. 2017, 840, 30–37. [Google Scholar] [CrossRef]
- Novoa, A.; Eierhoff, T.; Topin, J.; Varrot, A.; Barluenga, S.; Imberty, A.; Roemer, W.; Winssinger, N. A LecA ligand identified from a galactoside-conjugate array inhibits host cell invasion by Pseudomonas aeruginosa. Angew. Chem. 2014, 53, 8885–8889. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Cohen, S.M. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem. Commun. 2012, 48, 5259–5261. [Google Scholar] [CrossRef] [PubMed]
- Sala, O.; Santschi, N.; Jungen, S.; Lüthi, H.P.; Iannuzzi, M.; Hauser, N.; Togni, A. S-trifluoromethylation of thiols by hypervalent iodine reagents: A joint experimental and computational study. Chem. Eur. J. 2016, 22, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, G.J.L.; Grayson, E.J.; Thompson, S.; Chalker, J.M.; Errey, J.C.; El Oualid, F.; Claridge, T.D.W.; Davis, B.G. From disulfide-to thioether-linked glycoproteins. Angew. Chem. 2008, 47, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, R.K.; Picard, M.A.; Beyreuther, K.; Wiessler, M. Synthesis of antioxidative and anti-inflammatory drugs glucoconjugates. Carbohydr. Res. 2000, 325, 72–80. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A 1995, 51, 887–897. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. supplementary functions for gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio effective core potentials for molecular calculations: Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gassian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 15 April 2018).
Sample Availability: Samples of the compounds 1–5 and 12–17 are available from the authors. |





{kind=link}
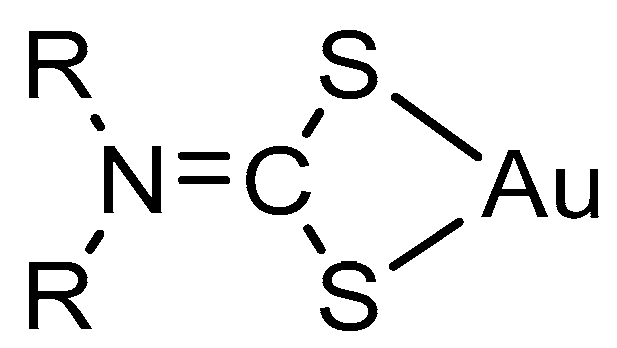{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 12 | 13 | 14 | |
|---|---|---|---|---|
| Empirical Formula | C30H24AuN3 | C36H29N2O2SAu | C37H31N2O2SAu | C38H33N2O2SAu |
| Formula Weight (g·mol−1) | 623.49 | 750.54 | 764.66 | 778.69 |
| Temperature (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P21/m (#11) | P (#2) | P (#2) | C2/c (#15) |
| Unit cell dimensions | ||||
| a (Å) | 12.8150(7) | 9.3234(3) | 8.89830(6) | 26.1234(3) |
| b (Å) | 6.4797(3) | 10.4210(3) | 12.12378(8) | 10.2154(1) |
| c (Å) | 15.6802(8) | 16.0388(5) | 15.5000(1) | 23.9038(3) |
| α (°) | 90 | 75.663(3) | 103.1637(6) | 90 |
| β (°) | 112.268(6) | 85.553(2) | 105.3420(6) | 100.387(1) |
| γ (°) | 90 | 83.264(3) | 98.8340(6) | 90 |
| Volume (Å3) | 1204.94(12) | 1497.46(8) | 1528.942(19) | 6274.45(12) |
| Z | 2 | 2 | 2 | 8 |
| Density (calcd) (mg/m3) | 1.718 | 1.665 | 1.661 | 1.649 |
| Absorption coefficient (mm−1) | 11.641 | 5.018 | 9.964 | 9.723 |
| F (000) | 608 | 740 | 756 | 3088 |
| Crystal size (mm3) | 0.255 × 0.034 × 0.026 | 0.194 × 0.121 × 0.082 | 0.248 × 0.193 × 0.120 | 0.113 × 0.035 × 0.010 |
| θ (°) | 3.727 to 77.196 | 2.90 to 29.59 | 3.845 to 76.876 | 3.44 to 76.91 |
| Index ranges | −16 ≤ h ≤ 15 | −12 ≤ h ≤ 12 | −11 ≤ h ≤ 11 | −32 ≤ h ≤ 32 |
| −8 ≤ k ≤ 8 | −13 ≤ k ≤ 13 | −15 ≤ k ≤ 15 | −12 ≤ k ≤ 12 | |
| −19 ≤ l ≤ 19 | −21 ≤ l ≤ 21 | −19 ≤ l ≤ 19 | −30 ≤ l ≤ 28 | |
| Reflections collected | 24,420 | 20,276 | 34,268 | 39,269 |
| Independent reflections Rint | 2760(0.1335) | 7306(0.0341) | 6409(0.0268) | 6584(0.0339) |
| Completeness to θmax (%) | 99.8 | 99.2 | 100.0 | 99.4 |
| Absorption correction | Gaussian | Gaussian | Gaussian | Gaussian |
| Max and min transmission | 0.788 and 0.291 | 0.714 and 0.472 | 0.455 and 0.227 | 0.913 and 0.516 |
| Refinement method | Full-matrix Least-squares on F2 | Full-matrix Least-squares on F2 | Full-matrix Least-squares on F2 | Full-matrix Least-squares on F2 |
| Data/restraints/parameters | 2760/0/147 | 7306/0/380 | 6409/0/389 | 6584/0/398 |
| Goodness-of-fit on F2 | 1.139 | 1.046 | 1.085 | 1.041 |
| Final R indices [I > 2σ(I)] | R1 = 0.0442, wR2 = 0.0983 | R1 = 0.0255, wR2 = 0.0437 | R1 = 0.0178, wR2 = 0.0439 | R1 = 0.0266, wR2 = 0.0674 |
| R indices (all data) | R1 = 0.0464, wR2 = 0.0998 | R1 = 0.0324, wR2 = 0.0465 | R1 = 0.0187, wR2 = 0.0442 | R1 = 0.0305, wR2 = 0.0701 |
| Largest diff. peak and hole | 1.982 and −1.411 | 1.009 and −0.762 | 0.639 and −0.859 | 1.508 and −1.500 |
| 2 | 12 | 13 | 14 | |
|---|---|---|---|---|
| Au–C(8) | 2.031(8) | 2.012(3) | 2.008(2) | 2.008(3) |
| Au–C(30) | 2.026(9) | |||
| C(30)–N(3) | 1.113(12) | |||
| Au–S(1) | 2.2856(7) | 2.2851(6) | 2.3012(8) | |
| S(1)–C(30) | 1.751(3) | 1.755(2) | 1.735(3) | |
| C(36)–O(1) | 1.249(3) | 1.211(3) | 1.215(5) | |
| C(36)–O(2) | 1.304(3) | 1.349(3) | 1.337(5) | |
| O(2)–C(37) | 1.442(3) | 1.450(5) |
| 2 | 12 | 13 | 14 | |
|---|---|---|---|---|
| C(8)–Au–S | 177.48(8) | 175.20(6) | 173.45(9) | |
| C(8)–Au–C(30) | 179.6(4) | |||
| Au–S–C(30) | 108.40(10) | 109.44(8) | 108.83(12) | |
| Au–C(30)–N(3) | 177.0(8) | |||
| S(1)–C(30)–S(2) | ||||
| O(1)–C(36)–O(2) | 122.8(3) | 122.9(2) | 123.4(3) | |
| O(1)–C(36)–C(33) | 120.8(3) | 125.2(2) | 124.2(4) | |
| O(2)–C(36)–C(33) | 116.4(3) | 111.9(2) | 112.4(3) | |
| C(36)–O(2)–C(37) | 115.4(2) | 117.0(3) |
| HCT-116wt | HCT-116 p53−/− | MCF-7topo | |
|---|---|---|---|
| 2 | 14.8 ± 1.9 | - | 10.8 ± 0.9 |
| 3 | 1.5 ± 0.1 | - | 0.28 ± 0.03 |
| 4 | 8.0 ± 0.1 | 3.8 ± 0.4 | 0.36 ± 0.03 |
| 5 | 6.2 ± 0.3 | 2.0 ± 0.6 | 1.5 ± 0.3 |
| 12 | 5.5 ± 0.1 | 2.7 ± 0.2 | 5.4 ± 0.5 |
| 13 | 18.1 ± 6.5 | 9.5 ± 0.6 | 21.3 ± 3.4 |
| 14 | 6.8 ± 0.2 | 7.9 ± 0.2 | 13.2 ± 3.7 |
| 15 | 4.5 ± 1.2 | 6.6 ± 0.3 | 7.1 ± 0.3 |
| 16 | 2.8 ± 0.1 | 4.5 ± 0.6 | 6.3 ± 0.5 |
| 17 | 2.9 ± 0.1 | 3.7 ± 0.2 | 5.4 ± 0.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curran, D.; Dada, O.; Müller-Bunz, H.; Rothemund, M.; Sánchez-Sanz, G.; Schobert, R.; Zhu, X.; Tacke, M. Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules 2018, 23, 2031. https://doi.org/10.3390/molecules23082031
Curran D, Dada O, Müller-Bunz H, Rothemund M, Sánchez-Sanz G, Schobert R, Zhu X, Tacke M. Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A. Molecules. 2018; 23(8):2031. https://doi.org/10.3390/molecules23082031
Chicago/Turabian StyleCurran, Danielle, Oyinlola Dada, Helge Müller-Bunz, Matthias Rothemund, Goar Sánchez-Sanz, Rainer Schobert, Xiangming Zhu, and Matthias Tacke. 2018. "Synthesis and Cytotoxicity Studies of Novel NHC*-Gold(I) Complexes Derived from Lepidiline A" Molecules 23, no. 8: 2031. https://doi.org/10.3390/molecules23082031